
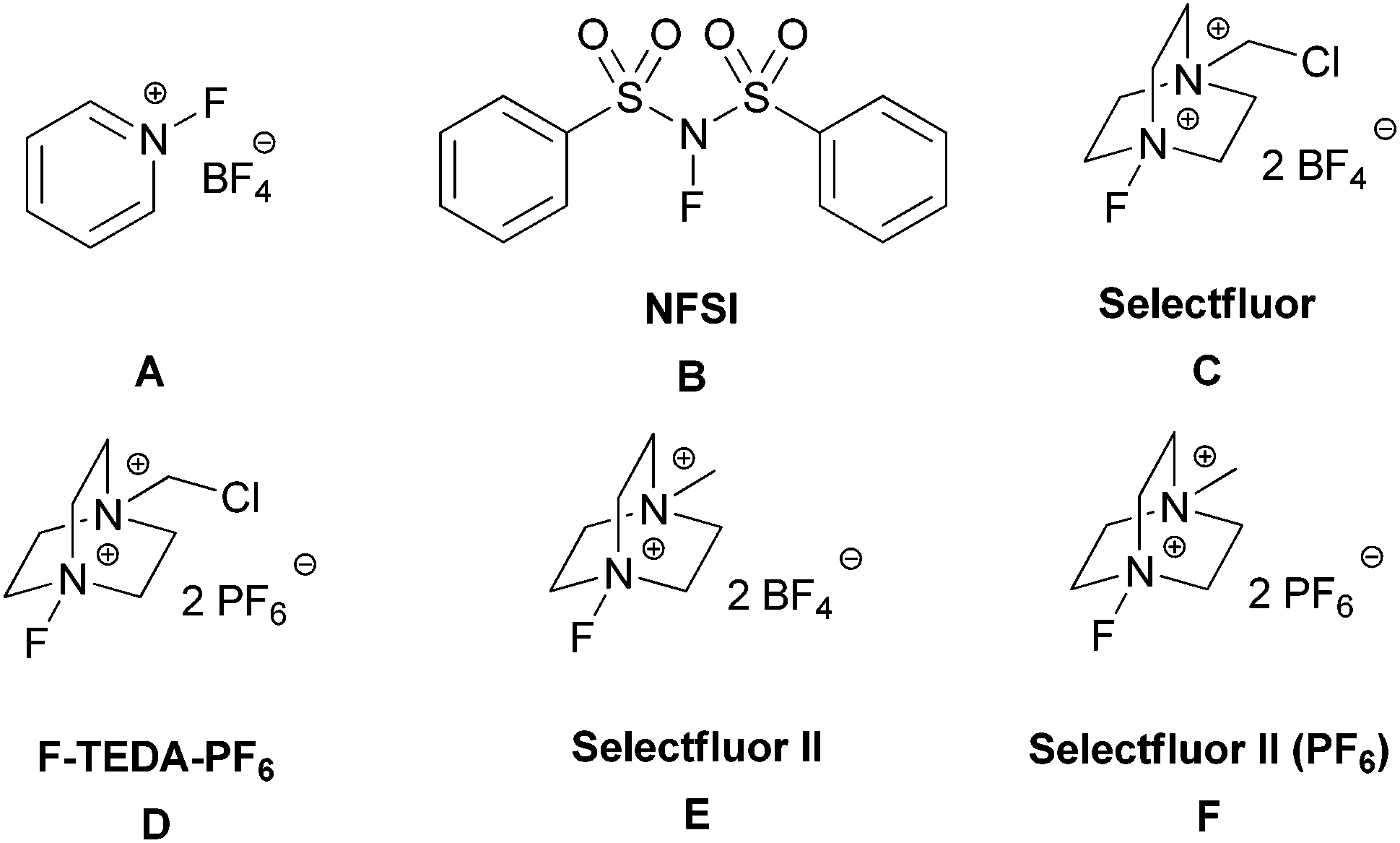
Transition-metal free oxidative aliphatic C–H fluorination†
Xiaofei
Zhang
,
Shuo
Guo
and
Pingping
Tang
*
State Key Laboratory and Institute of Elemento-Organic Chemistry, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin 300071, China. E-mail: ptang@nankai.edu.cn
First published on 16th April 2015
Abstract
A practical and highly selective fluorination of unactivated aliphatic C–H bonds without the use of transition metals has been developed. This C–H fluorination protocol is robust, operationally simple, scalable and compatible with moisture and air. Furthermore, the method exhibits a broad substrate scope and is applicable to late-stage fluorination of natural products and derivatives, which make it a valuable method for the synthesis of fluorinated complex molecules.
Fluorine substitution can beneficially influence the properties of different classes of functional molecules.1 For example, roughly 20% of commercially available pharmaceuticals and 30% of agrochemicals contain at least one fluorine atom. Incorporation of fluorine atoms into pharmaceuticals can result in favorable properties that are often beneficial for the pharmacodynamic profile of drugs such as increased metabolic stability, lipophilicity and overall biological activity. Despite the importance of fluorine's utility, chemical synthesis of C–F bonds is challenging due to the highly electronegative nature of fluorine and the high hydration energy of fluoride.2 Consequently, unactivated Csp3–H fluorination methods are rare and lack generality, practicality, and predictability.3
Traditional methods for the synthesis of alkyl fluorides through C–H bond functionalization suffer from poor selectivity and require the use of dangerous fluorinating reagents such as fluorine gas,4 cesium fluoroxysulfate5 or cobalt trifluoride.6 Enantioselective fluorination has been achieved through C–H bond functionalization adjacent to a carbonyl group.7 In addition to the recent success of Csp3–H fluorination in allylic and benzylic positions,8 methods for the transformation of unactivated Csp3–H bonds to Csp3–F bonds were also developed with transition metals.9 Alternative routes to aliphatic Csp3–H fluorination include employing a radical initiator10 or a photocatalyst.11 Despite the advances of these methods, the development of other general and practical methods for fluorination of alkanes are desirable. Herein, we report a highly selective, scalable and transition-metal-free aliphatic Csp3–H fluorination reaction. The reaction, which is operationally simple and proceeds under mild reaction conditions, can be performed for the late-stage fluorination of complex small molecules (Scheme 1).
 | ||
| Scheme 1 Transition-metal-free aliphatic Csp3–H fluorination reaction. | ||
Recently, we developed a silver-catalyzed oxidative activation of benzylic C–H bonds for the synthesis of difluoromethylated arenes.8f The transformation can be conducted on a gram scale to provide structurally diverse difluoromethylated arenes. Studying this reaction in which Na2S2O8 was believed to oxidize the silver(I) to a silver(II) species, we found that 20% of the difluoromethylated product was observed with Na2S2O8 and Selectfluor in the absence of a silver salt. Inspired by this result, we envisioned that unactivated Csp3–H fluorination could be obtained without the transition metal.
After extensive screening, we found that transition metal additives were not required for a productive reaction. The reaction of 4-methyl-2-pentyl benzoate (1f) with K2S2O8 (K2S2O8 99.99% from Alfa was used) and Selectfluor II (PF6) (F)12 in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (v/v) acetonitrile/H2O at 50 °C for 10 h under air led to the formation of the desired fluorinated compound (2f) in 70% NMR yield. As briefly illustrated in Table 1, various fluorinating reagents were evaluated (entries 1–6). Selectfluor II (PF6) (F), which is easily prepared from commercially available Selectfluor II (E), gave the best result. In addition, peroxydisulfate salts were evaluated and K2S2O8 gave the highest yield (entries 6–8). No fluorinated product was observed in anhydrous acetonitrile (entry 9) or in water (entry 10). The role of water is not clear. The control experiment was performed in the absence of K2S2O8 and no fluorinated product was observed (entry 11). The reaction gave the highest yield when conducted at 50 °C whereas a lower yield was observed for the reaction at high temperatures owing to the formation of more side products, and no difluorinated product was detected at high temperatures. The reaction is sensitive to the amount of K2S2O8 and Selectfluor II (PF6). After thoroughly optimizing the reaction conditions, reactions with 1.0 equiv. of K2S2O8 and 2.5 equiv. of Selectfluor II (PF6) were found to produce high yields of the desired product.
:2 (v/v) acetonitrile/H2O at 50 °C for 10 h under air led to the formation of the desired fluorinated compound (2f) in 70% NMR yield. As briefly illustrated in Table 1, various fluorinating reagents were evaluated (entries 1–6). Selectfluor II (PF6) (F), which is easily prepared from commercially available Selectfluor II (E), gave the best result. In addition, peroxydisulfate salts were evaluated and K2S2O8 gave the highest yield (entries 6–8). No fluorinated product was observed in anhydrous acetonitrile (entry 9) or in water (entry 10). The role of water is not clear. The control experiment was performed in the absence of K2S2O8 and no fluorinated product was observed (entry 11). The reaction gave the highest yield when conducted at 50 °C whereas a lower yield was observed for the reaction at high temperatures owing to the formation of more side products, and no difluorinated product was detected at high temperatures. The reaction is sensitive to the amount of K2S2O8 and Selectfluor II (PF6). After thoroughly optimizing the reaction conditions, reactions with 1.0 equiv. of K2S2O8 and 2.5 equiv. of Selectfluor II (PF6) were found to produce high yields of the desired product.
|
|
|||
|---|---|---|---|
| Entry | Fluorinating reagenta | Conditionsb | Yieldc (%) |
| a 2.5 equiv. of the fluorinating reagent was used. b 1.0 equiv. of the oxidant was used. c Yields were determined by 19F NMR with 1-fluoro-3-nitrobenzene as a standard. | |||
| 1 | A | K2S2O8, MeCN/H2O, 50 °C | 0 |
| 2 | B | K2S2O8, MeCN/H2O, 50 °C | 0 |
| 3 | C | K2S2O8, MeCN/H2O, 50 °C | 17 |
| 4 | D | K2S2O8, MeCN/H2O, 50 °C | 37 |
| 5 | E | K2S2O8, MeCN/H2O, 50 °C | 67 |
| 6 | F | K 2 S 2 O 8 , MeCN/H 2 O, 50 °C | 70 |
| 7 | F | Na2S2O8, MeCN/H2O, 50 °C | 57 |
| 8 | F | (NH4)2S2O8, MeCN/H2O, 50 °C | 55 |
| 9 | F | K2S2O8, MeCN, 50 °C | 0 |
| 10 | F | K2S2O8, H2O, 50 °C | 0 |
| 11 | F | MeCN/H2O, 50 °C | 0 |
|
|||

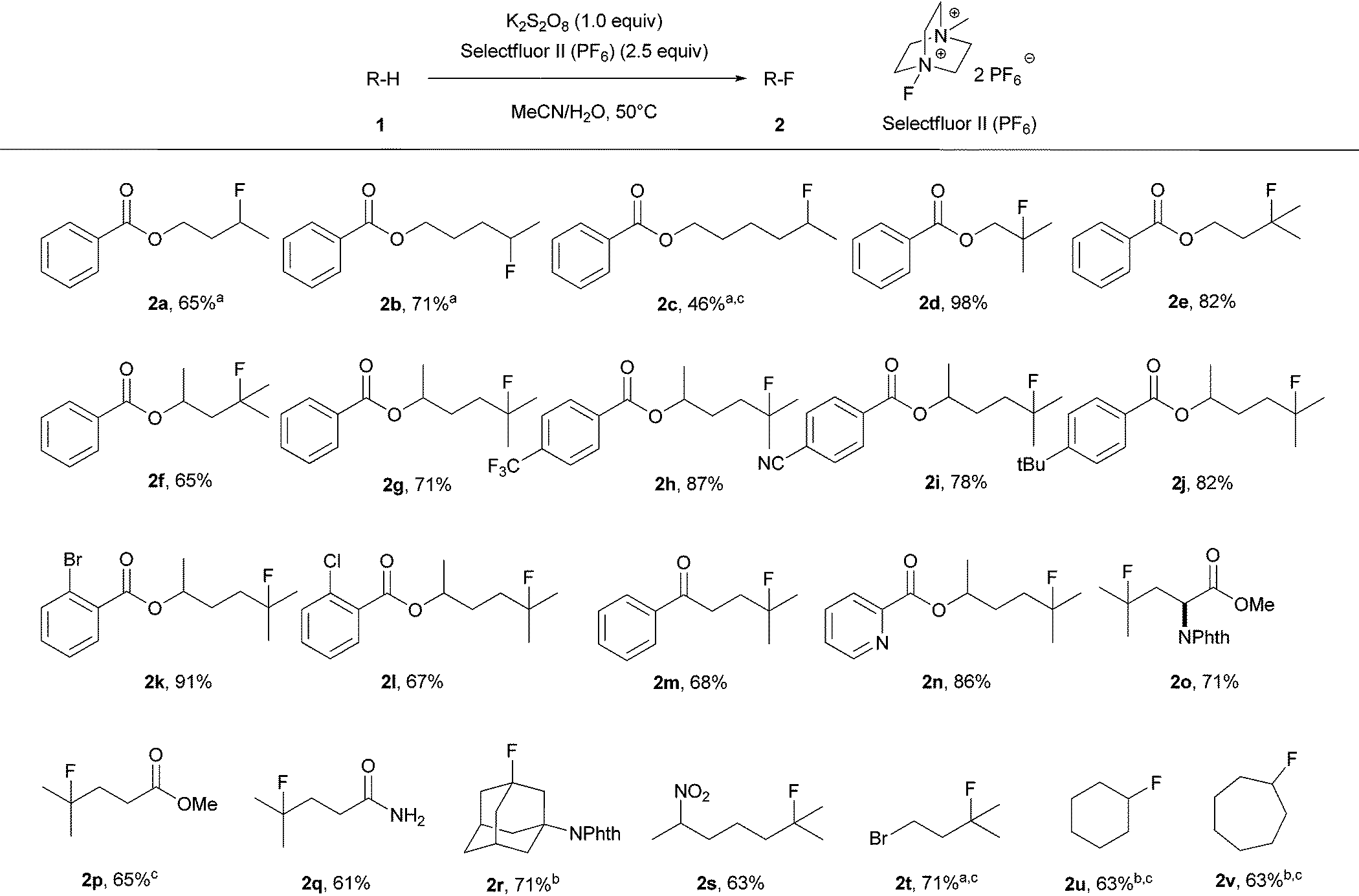
With the optimized conditions in hand, we then investigated the substrate scope as displayed in Table 2. A variety of simple molecules which have multiple unactivated Csp3–H bonds were smoothly transformed into the corresponding monofluorinated products with isolated yields ranging from 46% to 98%. Ketone, ester, amide, aromatic nitrile, chloride and bromide functionalities were all tolerated under the standard reaction conditions. Fluorination occurs at the carbon in accord with the relative radical stability. The selectivity of fluorination was observed at methines or methylenes which are remote from electron-withdrawing groups. For example, the transformation of butyl benzoate (1a) to the fluorinated product (2a) occurred predominantly at the methylene position remote from the carboxylic ester. However, further increasing the chain length resulted in decreased selectivity. For instance, transformation of hexyl benzoate (1c) to fluorinated products gave 80% yield of a mixture of regioisomers that were inseparable; the major isomer, 2c, was observed in 46% NMR yield. The reaction worked with a heteroaromatic substituted alkyl chain derivative (1n). Notably, an amino acid derivative was also successfully employed to provide the corresponding fluorinated compound with high selectivity and good yield (2o). With substrate (1r) and cycloalkanes (1u, 1v), an excess of the substrate was used due to polyfluorination products observed with 1 equiv. of the substrate; fluorination yields for the substrates were measured based on Selectfluor II (PF6). Product 2k was prepared on a gram scale under the standard reaction conditions in 83% isolated yield, proving the operational simplicity and practicality of our transition-metal-free fluorination method.
In order to demonstrate the utility of our method for late-stage fluorination of complex small molecules, we attempted to apply the system to more complex substrates. Gratifyingly, the fluorination of sclareolide (1w), a natural product with antifungal and cytotoxic activities, proceeded to form the fluorinated compound (2w) under standard reaction conditions in 69% isolated yield as the major isomer (Scheme 2A). The C3 fluorination product was observed in 10% yield determined by 19F NMR. Fluorination does not occur at the two methine positions due to more steric hindrance and deactivation of these positions, instead the selective fluorination was observed at the C2 methylene position (α:β > 30:1) which has less steric hindrance. It is worth mentioning that this high efficiency and selectivity for fluorinating sclareolide has not been previously achieved by any other fluorination method to date. In addition, other molecules derived from complex natural products were successfully fluorinated using our methodology. For example, a diterpene compound (1x) derived from gibberellic acid, a hormone found in plants, which displays five 3° C–H bonds along its pentacyclic skeleton, reacted smoothly to give 2x in 42% isolated yield (around 10% recovered starting material) (Scheme 2B) with 4.0 equiv. of K2S2O8. A number of minor, unidentified fluorination products were observed; however, fluorination at the C16 methine position occurred as the major product due to more electron-rich and less steric hindrance compared to the other 3° C–H bonds. In addition, a derivative (1y) of the anticancer drug taxol, which contains a double bond and a free hydroxyl group, proceeded with 4.0 equiv. of K2S2O8 and 2.5 equiv. of Selectfluor II (PF6) to provide the fluorinated product 2y in 33% isolated yield (around 50% recovered starting material) (Scheme 2C). The selective fluorination occurred at the methine position on the side chain due to the steric hindrance and deactivation of the available 3° C–H bonds on the rings of 1y.
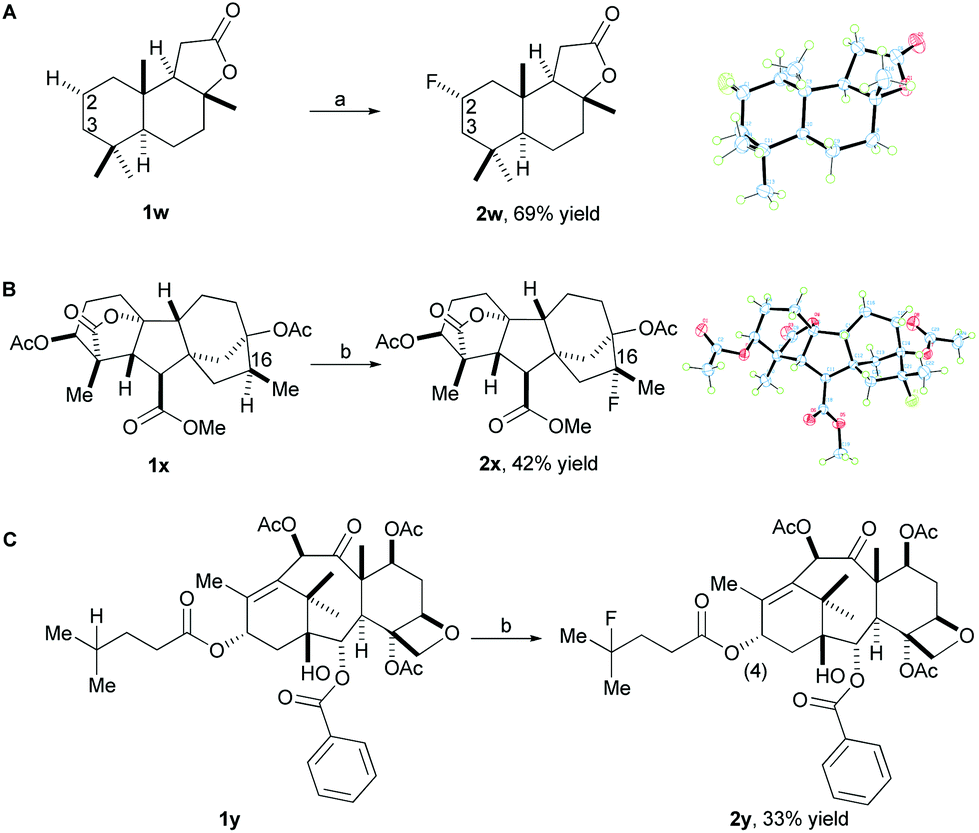 | ||
| Scheme 2 Late stage fluorination of complex molecules. Reaction conditions: (a) K2S2O8 (1.0 equiv.), Selectfluor II (PF6) (2.5 equiv.), MeCN/H2O, 50 °C. (b) K2S2O8 (4.0 equiv.), Selectfluor II (PF6) (2.5 equiv.), MeCN/H2O, 50 °C. | ||
Furthermore, we also compared our method with direct fluorination using diluted fluorine gas in nitrogen (10%) in acetonitrile13 at 0 °C with a range of substrates (1a, 1f, 1m, 1n, 1o, 1w) (see the ESI† for more details). In general, the reaction with fluorine gas gave poor selectivity compared to our method. For example, the aromatic fluorinated products were observed with poor selectivity when substrates 1a, 1f, 1m, and 1n were used.
Some preliminary mechanistic studies were conducted. The deuterium kinetic isotope effect (KIE) was evaluated by an intermolecular competition experiment of a 1:1 mixture of cyclohexane and cyclohexane-d12 in excess to give a modest KIE of 1.8, which suggests that the C–H bond cleavage might be the rate-limiting step (Scheme 3A).14 No fluorinated product was formed when the radical inhibitor butylated hydroxytoluene (BHT) or 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) was added. In addition, although a similar yield was found when the reaction was performed under air or under an inert atmosphere in a 2 mL sealed vial, no fluorinated product was observed under the O2 atmosphere. To gain further insights into the reaction mechanism, EPR (electron paramagnetic resonance) experiments were performed with the addition of a free radical spin trapping agent DMPO (5,5-dimethyl-1-pyrroline N-oxide). Some signals of unknown organic radicals were observed and the signals of the DMPO-OH˙ adducts were not detected,15 which suggested that hydroxyl free radicals may not be generated during the reaction process. Altogether, these observations indicated that a radial chain mechanism or single-electron transfer (SET) may be involved in this transformation.
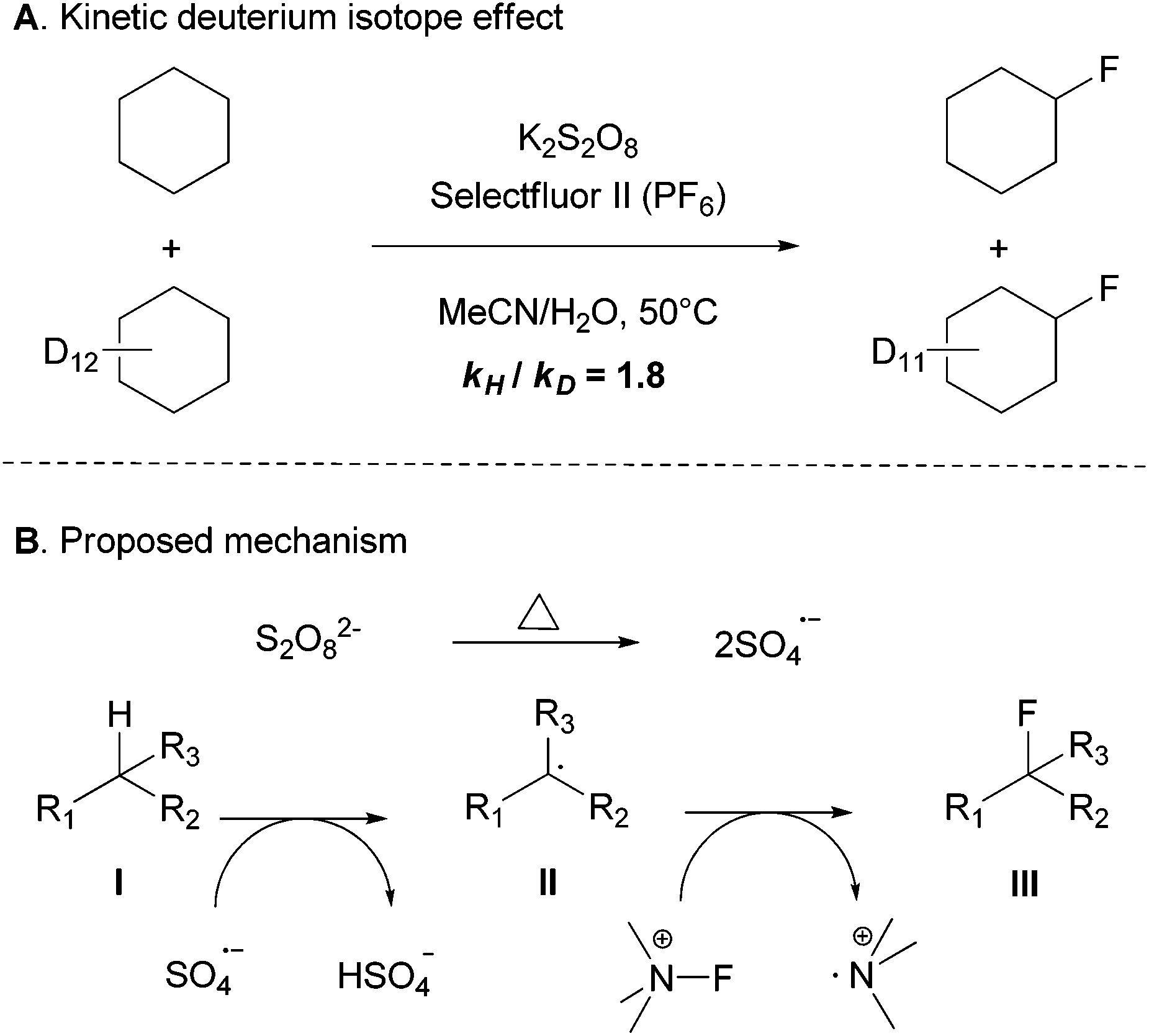 | ||
| Scheme 3 (A) Mechanism study and (B) proposed mechanism. | ||
The detailed mechanism is not clear. Based on our observations, we proposed the mechanism depicted in Scheme 3B, it is known that the peroxydisulfate anion decomposes into the sulfate radical anion,16 which could potentially oxidize the aliphatic C–H bond into a carbon radical (II). Selectfluor is also known to fluorinate alkyl radicals (II) to form C–F bonds (III).17
Conclusions
In conclusion, we are strongly encouraged by the excellent selectivity and efficient fluorination of complex small molecules without transition metals or the rigorous exclusion of water or air. Compared to other unactivated Csp3–H fluorination methods previously described in the literature, our approach has the limitation of excess use of reagents. However, given the practical and broad scope demonstrated in this study, we anticipate that this general transition-metal-free oxidative unactivated Csp3–H fluorination method will improve the ways in which complex small molecules and pharmaceuticals are currently fluorinated.Acknowledgements
We gratefully acknowledge the State Key Laboratory of Elemento-Organic Chemistry for generous start-up financial support. This work was supported by the “1000 Youth Talents Plan”, NSFC (21402098, 21421062) and the Natural Science Foundation of Tianjin (13JCYBJC36500). Liyan Wang and Yan Li are acknowledged to reproduce the experiment with substrate 1f. Prof. Tobias Ritter and Prof. Jennifer M. Murphy are greatly acknowledged for helpful suggestions.Notes and references
- (a) P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS PubMed; (b) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (c) C. Isanbor and D. O'Hagan, J. Fluorine Chem., 2006, 127, 303 CrossRef CAS; (d) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (e) R. Berger, G. Resnati, P. Metrangolo and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496 RSC.
- For selected reviews, see: (a) J. Emsley, Chem. Soc. Rev., 1980, 9, 91 RSC; (b) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (c) V. V. Grushin, Acc. Chem. Res., 2010, 43, 160 CrossRef CAS PubMed. For some examples to form C–F bonds: (d) T. Furuya, H. M. Kaiser and T. Ritter, Angew. Chem., 2008, 120, 6082 ( Angew. Chem. Int. Ed. , 2008 , 47 , 5993 ) CrossRef; (e) V. V. Grushin and W. J. Marshall, Organometallics, 2008, 27, 4825 CrossRef CAS; (f) X. Wang, T. S. Mei and J. Q. Yu, J. Am. Chem. Soc., 2009, 131, 7520 CrossRef CAS PubMed; (g) T. Wu, G. Yin and G. Liu, J. Am. Chem. Soc., 2009, 131, 16354 CrossRef CAS PubMed; (h) D. A. Watson, M. Su, G. Teverovskiy, Y. Zhang, J. García-Fortanet, T. Kinzel and S. L. Buchwald, Science, 2009, 325, 1661 CrossRef CAS PubMed; (i) W. Zhang, W. Huang and J. Hu, Angew. Chem., 2009, 121, 10042 ( Angew. Chem. Int. Ed. , 2009 , 48 , 9858 ) CrossRef; (j) Z. Zhang, F. Wang, X. Mu, P. Chen and G. Liu, Angew. Chem., 2013, 125, 7697 ( Angew. Chem. Int. Ed. , 2013 , 52 , 7549 ) CrossRef; (k) T. Truong, K. Klimovica and O. Daugulis, J. Am. Chem. Soc., 2013, 135, 9342 CrossRef CAS PubMed; (l) P. S. Fier and J. F. Hartwig, Science, 2013, 342, 956 CrossRef CAS PubMed; (m) H. Zhang, Y. Song, J. Zhao, J. Zhang and Q. Zhang, Angew. Chem., Int. Ed., 2014, 53, 11079 CrossRef CAS PubMed.
- J. Ma and S. Li, Org. Chem. Front., 2014, 1, 712 RSC.
- S. Rozen, Acc. Chem. Res., 1988, 21, 307 CrossRef CAS.
- S. Stavber and M. Zupan, Tetrahedron, 1989, 45, 2737 CrossRef CAS.
- J. Burdon, S. T. Ezmirly and T. N. Huckerby, J. Fluorine Chem., 1988, 40, 283 CrossRef CAS.
- For the selected reviews, see: (a) J. Ma and D. Cahard, Chem. Rev., 2004, 104, 6119 CrossRef CAS PubMed; (b) V. Bizet, T. Besset, J. Ma and D. Cahard, Curr. Top. Med. Chem., 2014, 14, 901 CrossRef CAS PubMed. For the latest selective examples: (c) P. Kwiatkowski, T. D. Beeson, J. C. Conrad and D. W. C. MacMillan, J. Am. Chem. Soc., 2011, 133, 1738 CrossRef CAS PubMed; (d) X. Yang, R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5225 CrossRef CAS PubMed; (e) Y. Lam and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 9556 CrossRef CAS PubMed.
- (a) M. Braun and A. G. Doyle, J. Am. Chem. Soc., 2013, 135, 12990 CrossRef CAS PubMed; (b) K. L. Hull, W. Q. Anani and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 7134 CrossRef CAS PubMed; (c) W. Liu and J. T. Groves, Angew. Chem., 2013, 125, 6140 ( Angew. Chem. Int. Ed. , 2013 , 52 , 6024 ) CrossRef; (d) S. Bloom, C. R. Pitts, R. Woltornist, M. G. Holl and T. Lectka, Org. Lett., 2013, 15, 1722 CrossRef CAS PubMed; (e) J. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494 CrossRef CAS PubMed; (f) P. Xu, S. Guo, L. Wang and P. Tang, Angew. Chem., 2014, 126, 6065 ( Angew. Chem. Int. Ed. , 2014 , 53 , 5955 ) CrossRef; (g) S. Bloom, J. L. Knippel, M. G. Holl, R. Barber and T. Lectka, Tetrahedron Lett., 2014, 55, 4576 CrossRef CAS; (h) J. Ma, W. Yi, G. Lu and C. Cai, Org. Biomol. Chem., 2015, 13, 2890 RSC.
- (a) T. Liang, C. Neumann and T. Ritter, Angew. Chem., 2013, 125, 8372 ( Angew. Chem. Int. Ed. , 2013 , 52 , 8214 ) CrossRef; (b) W. Liu, X. Huang, M. Cheng, R. J. Nielsen, W. A. Goddard III and J. T. Groves, Science, 2012, 337, 1322 CrossRef CAS PubMed; (c) S. Bloom, C. R. Pitts, D. C. Miller, N. Haselton, M. G. Holl, E. Urheim and T. Lectka, Angew. Chem., 2012, 124, 10732 ( Angew. Chem. Int. Ed. , 2012 , 51 , 10580 ) CrossRef; (d) S. D. Halperin, H. Fan, S. Chang, R. E. Martin and R. Britton, Angew. Chem., 2014, 126, 4778 ( Angew. Chem. Int. Ed. , 2014 , 53 , 4690 ) CrossRef; (e) J. Xia, Y. Ma and C. Chen, Org. Chem. Front., 2014, 1, 468 RSC; (f) Z. Li, L. Song and C. Li, J. Am. Chem. Soc., 2013, 135, 4640 CrossRef CAS PubMed; (g) C. R. Pitts, S. Bloom, R. Woltornist, D. J. Auvenshine, L. R. Ryzhkov, M. A. Siegler and T. Lectka, J. Am. Chem. Soc., 2014, 136, 9780 CrossRef CAS PubMed.
- (a) Y. Amaoka, M. Nagatomo and M. Inoue, Org. Lett., 2013, 15, 2160 CrossRef CAS PubMed; (b) C. R. Pitts, B. Ling, R. Woltornist, R. Liu and T. Lectka, J. Org. Chem., 2014, 79, 8895 CrossRef CAS PubMed.
- (a) S. Bloom, J. L. Knippel and T. Lectka, Chem. Sci., 2014, 5, 1175 RSC; (b) C. W. Kee, K. F. Chin, M. W. Wong and C. Tan, Chem. Commun., 2014, 50, 8211 RSC; (c) J. Xia, C. Zhu and C. Chen, Chem. Commun., 2014, 50, 11701 RSC.
- R. E. Banks, M. K. Besheesh, S. N. Mohialdin-Khaffaf and I. Sharif, J. Chem. Soc., Perkin Trans. 1, 1996, 2069 RSC.
- (a) S. Rozen, Acc. Chem. Res., 1988, 21, 307 CrossRef CAS; (b) R. D. Chambers, M. Parsons, G. Sandford and R. Bowden, Chem. Commun., 2000, 959 RSC; (c) R. D. Chambers, A. M. Kenwright, M. Parsons, G. Sandford and J. S. Moilliet, J. Chem. Soc., Perkin Trans. 1, 2002, 2190 RSC.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., 2012, 124, 3120 ( Angew. Chem. Int. Ed. , 2012 , 51 , 3066 ) CrossRef.
- K. Makino, A. Hagi, H. Ide, A. Murakami and M. Nishi, Can. J. Chem., 1992, 70, 2818 CrossRef CAS.
- D. A. House, Chem. Rev., 1962, 62, 185 CrossRef CAS.
- (a) P. T. Nyffeler, S. G. Durón, M. D. Burkhart, S. P. Vincent and C. H. Wong, Angew. Chem., 2004, 117, 196 ( Angew. Chem. Int. Ed. , 2004 , 44 , 192 ) CrossRef; (b) M. Rueda-Becerril, C. C. Sazepin, J. C. T. Leung, T. Okbinoglu, P. Kennepohl, J. Paquin and G. M. Sammis, J. Am. Chem. Soc., 2012, 134, 4026 CrossRef CAS PubMed; (c) F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401 CrossRef CAS PubMed; (d) T. J. Barker and D. L. Boger, J. Am. Chem. Soc., 2012, 134, 13588 CrossRef CAS PubMed; (e) C. Zhang, Z. Li, L. Zhu, L. Yu, Z. Wang and C. Li, J. Am. Chem. Soc., 2013, 135, 14082 CrossRef CAS PubMed; (f) M. Rueda-Becerril, O. Mahé, M. Drouin, M. B. Majewski, J. G. West, M. O. Wolf, G. M. Sammis and J. Paquin, J. Am. Chem. Soc., 2014, 136, 2637 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Data for new compounds and experimental procedures. CCDC 1007200 and 1007330. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00095e |
| This journal is © the Partner Organisations 2015 |