
Cationic iridium-catalyzed C–H alkylation of 2-substituted pyridine N-oxides with acrylates†‡
Takanori
Shibata
*ab and
Hideaki
Takano
a
aDepartment of Chemistry and Biochemistry, School of Advanced Science and Engineering, Waseda University, Shinjuku, Tokyo 169-8555, Japan. E-mail: tshibata@waseda.jp; Fax: (+81) 3-5286-8098
bJST, ACT-C, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 11th February 2015
Abstract
The reaction of 2-arylmethyl-, 2-aryl-, and 2-alkyl substituted pyridine N-oxides with acrylates proceeded in the presence of a cationic Ir-rac-BINAP catalyst under heating conditions. Various 2,6-disubstituted pyridine N-oxides were obtained by C–H alkylation at the C6-position.
Introduction
The transition metal-catalyzed synthetic transformations initiated by C–H bond cleavage have become a core strategy in organic synthesis.1 Among various types of C–H bond functionalization, ortho-selective arene C–H bond activation using directing groups is an established protocol. In particular, C–H alkylation using alkenes is more atom-economical than using alkyl halides and alkyl metallic species, because there is no atom loss through transformation. From a historical point of view, Lewis's work of hydroxyl-directed Ru-catalyzed reaction using ethylene was the first example,2 while, from the synthetic point of view, Murai's work of carbonyl-directed Ru-catalyzed reaction using vinylsilanes is a monumental achievement.3 Later, Rh- and Ir-catalyzed carbonyl-directed ortho-C–H alkylations of arenes followed.4,5 The imino group is a potent directing group, and Rh-, Ru- and Co-catalyzed reactions with alkenes were reported.6–8 The pyridyl group is more versatile: Co-, Ir- and Ru-catalyzed C–H alkylations were disclosed9–11 and an enantioselective variant for the creation of planar chirality of ferrocenes was also achieved.12 Efficient ortho-alkylation of phenol and anisole was also reported using a rhenium catalyst and a yttrium catalyst, respectively.13,14 The 8-quinolylcarbamoyl group was used as an efficient N,N-bidentate directing group in C–H alkylation.15In contrast, pyridine N-oxides and their derivatives are fascinating substrates in the direct C–H functionalization, because of their high reactivity at the C2 position and facile removal of the oxygen atom by reduction.16 Fagnou pioneeringly reported Pd-catalyzed C–H arylation of pyridine N-oxides by aryl halides.17 He further developed site-selective sp3 C–H arylation of 2-methylpyridine N-oxides, which gave 2-arylmethyl pyridine N-oxides (Scheme 1-a).18 Recently, cross hydrogenative coupling was achieved with the use of relatively active C–H bonds in heteroaromatics.19 However, as for the C–H alkylation of pyridine N-oxides using alkenes, there is only one example: Chang disclosed Rh-catalyzed reaction with electron-deficient alkenes in the presence of a stoichiometric amount of a base (Scheme 1-b).20,21 In this article, we show a cationic iridium-catalyzed C–H alkylation of 2-substituted pyridine N-oxides with acrylates (Scheme 1-c).
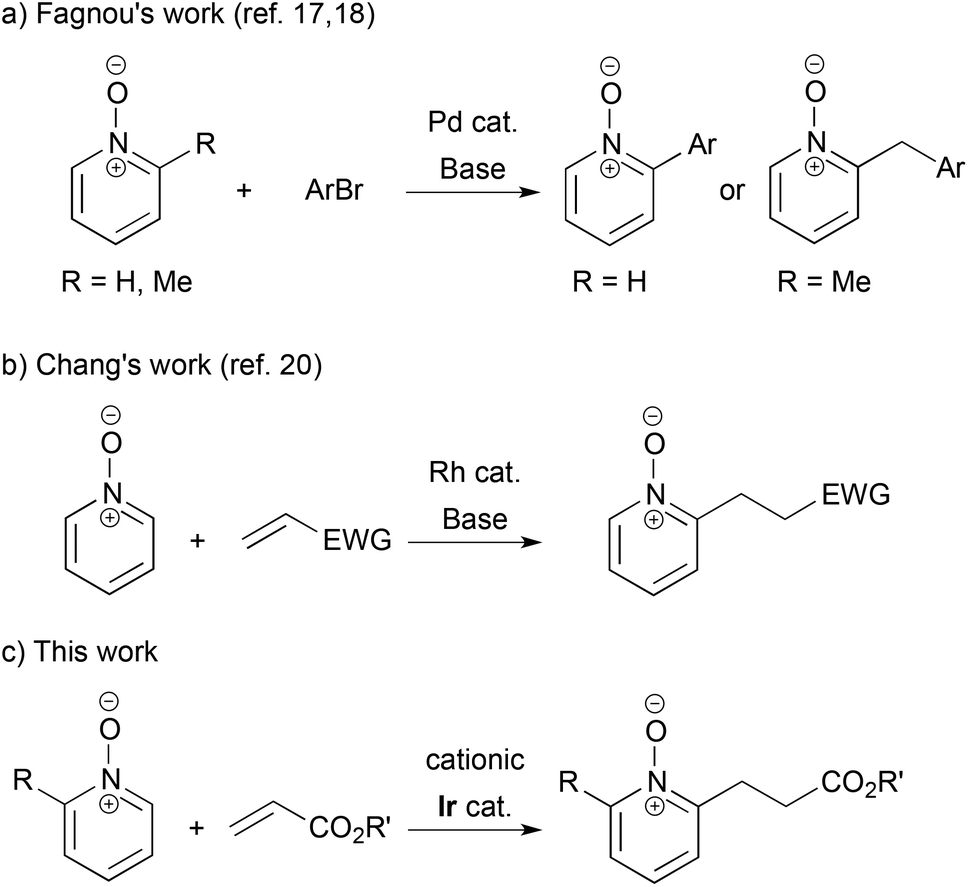 | ||
| Scheme 1 C–H bond functionalization of pyridine N-oxides with aryl bromides and alkenes. | ||
Results and discussion
We recently reported the cationic Rh-catalyzed reaction of quinoline N-oxides with diphenylacetylene, where C-8 position-selective alkenylation proceeded due to the directing effect of the N-oxide moiety, and no C-2 alkenylated product was observed.22 We got interested in the site-selectivity of N-oxides, and examined the reaction of 2-benzylpyridine N-oxide (1a)18,23 with diphenylacetylene in the presence of a cationic Rh- or Ir-rac-BINAP catalyst, but no alkenylated products were detected (entries 1 and 2 in Table 1). When the reaction with ethyl acrylate was conducted, C-6 alkylated product 3a was obtained in the presence of the Ir catalyst in low yield, but the alkylated product at the benzylic position could not be detected at all (entries 3 and 4).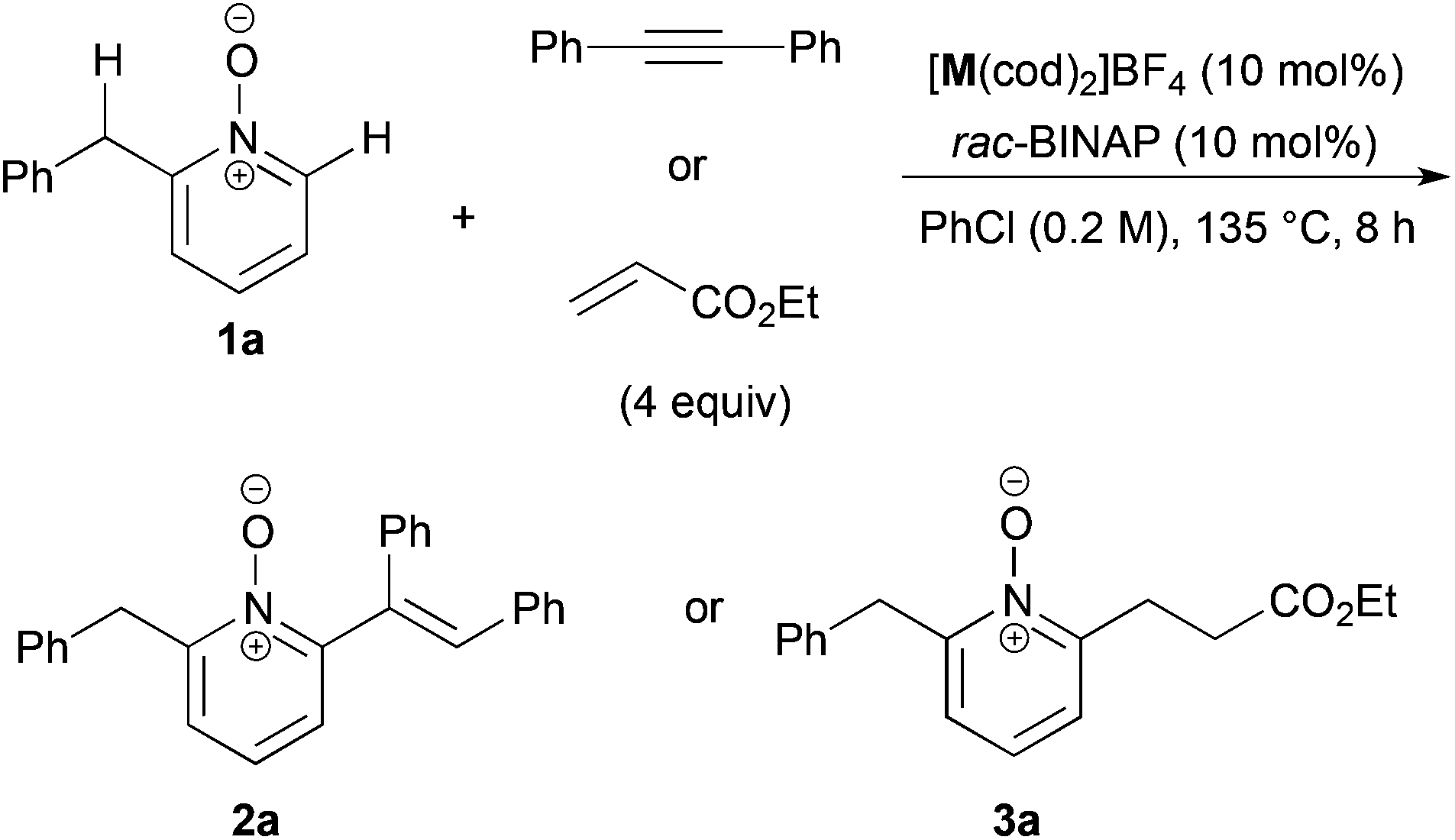
We next optimized the reaction conditions (Table 2). The effect of counter anion of iridium was significant: when tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (BARF) was used, the yield drastically improved to 65% (entry 2). After further ligand screening, a biaryl skeleton was found to be important, and rac-BINAP was used for further investigations (entries 3–5). A slight increase of yield was observed when the reaction was examined in a screw-capped Schlenk flask in place of glass-stoppered one, due to the low boiling point of ethyl acrylate (entry 6). The reaction was sensitive to reaction temperature: the best yield of 88% was achieved at 120 °C after prolonged reaction time (entry 7). The reaction also proceeded at 100 °C, but the yield was moderate after 48 h (entry 9). The Rh counterpart was ineffective in this reaction (entry 10).
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Temp. (°C) | Time (h) | Yield (%) |
| a Triflate was used as a counter anion of the Ir catalyst. b Not detected. c Screw-capped Schlenk flask was used in place of glass-stoppered one. d [Rh(cod)2]BARF was used in place of [Ir(cod)2]BARF. | ||||
| 1a | rac-BINAP | 135 | 8 | 34 |
| 2 | rac-BINAP | 135 | 8 | 65 |
| 3 | BIPHEP | 135 | 8 | 32 |
| 4 | DPPP | 135 | 8 | NDb |
| 5 | PPh3 | 135 | 8 | NDb |
| 6c | rac-BINAP | 135 | 8 | 70 |
| 7c | rac-BINAP | 120 | 24 | 88 |
| 8c | rac-BINAP | 100 | 24 | 42 |
| 9c | rac-BINAP | 100 | 48 | 44 |
| 10c,d | rac-BINAP | 120 | 24 | ND |
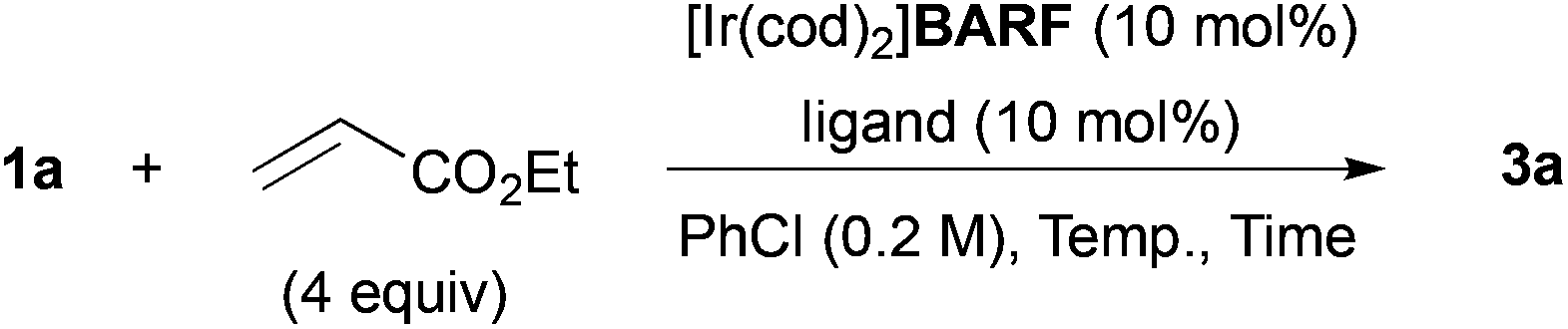
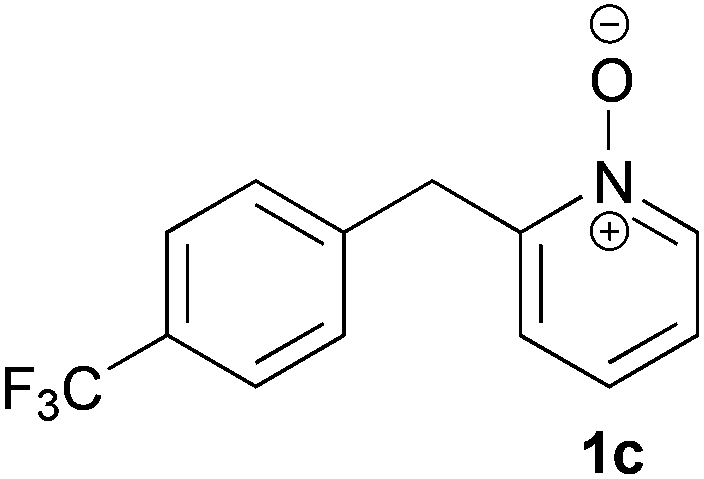
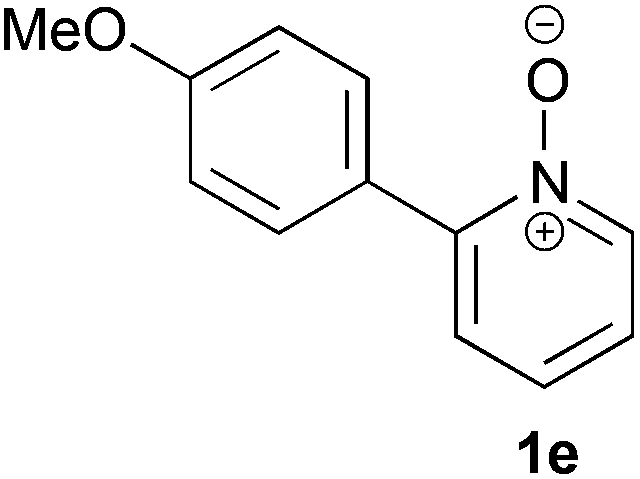
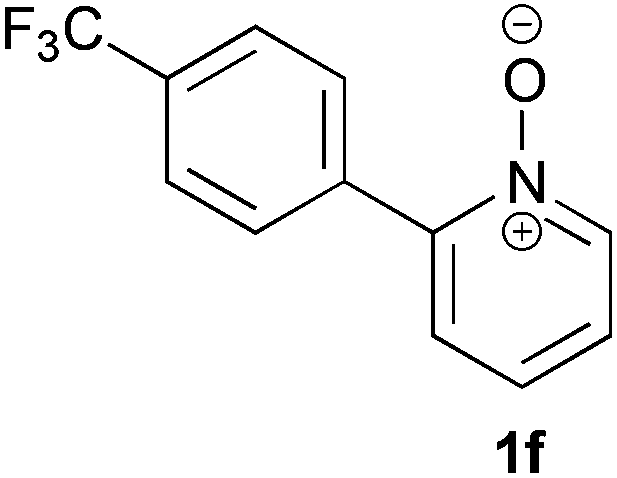
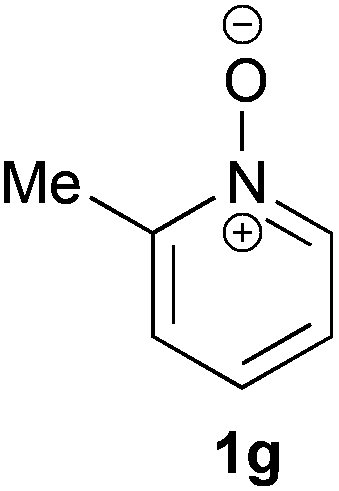
Under the reaction conditions of entry 7 in Table 2, several 2-substituted pyridine N-oxides 1 were subjected to the reaction with ethyl acrylate (Table 3). Both electron-donating and -withdrawing group-substituted 2-benzylpyridine N-oxides 1b and 1c could be transformed into the corresponding 2,6-disubstituted products 3b and 3c, respectively (entries 1 and 2). 2-Aryl-substituted pyridine N-oxides were also good substrates, and electron-donating and -withdrawing groups were tolerable at the para position (entries 3–5). 2-Methylpyridine N-oxide (1g) and 5,6,7,8-tetrahydroquinoline N-oxide (1h) also reacted smoothly to give the desired products in high yields (entries 6 and 7).24,25 Interestingly, the reaction using non-substituted pyridine N-oxide (1i) gave the desired monoalkylated product 3i, yet in low yield without the formation of a dialkylated product (entry 8). The reaction was messy and 1i was not completely consumed.26
|
|
|||
|---|---|---|---|
| Entry | N-Oxide | Time (h) | Yield (%) |
| 1 |
|
48 | 67 (3b) |
| 2 |
|
24 | 75 (3c) |
| 3 |

|
24 | 79 (3d) |
| 4 |
|
24 | 82 (3e) |
| 5 |
|
48 | 73 (3f) |
| 6 |
|
48 | 75 (3g) |
| 7 |
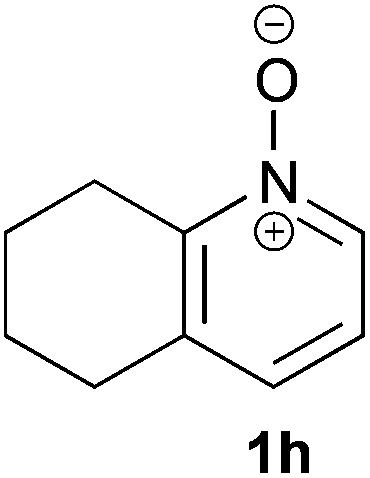
|
24 | 78 (3h) |
| 8 |
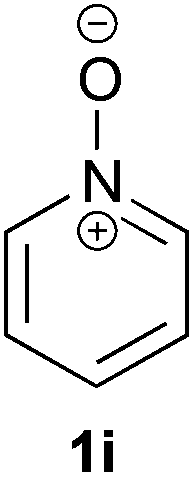
|
48 | 18 (3i) |
The reaction of N-oxide 1a with tert-butyl acrylate also proceeded (Scheme 2).27 But the reaction with methyl vinyl ketone and phenyl vinyl sulfone was messy and the yield of alkylated products 5a and 6a was low. When styrene was used, only a trace amount of product 7a was detected.
 | ||
| Scheme 2 Reaction with other alkenes. | ||
Reduction of alkylated product 3a using trichlorophosphine proceeded smoothly at room temperature to afford 2,6-unsymmetrically disubstituted pyridine 8 in high yield (Scheme 3).
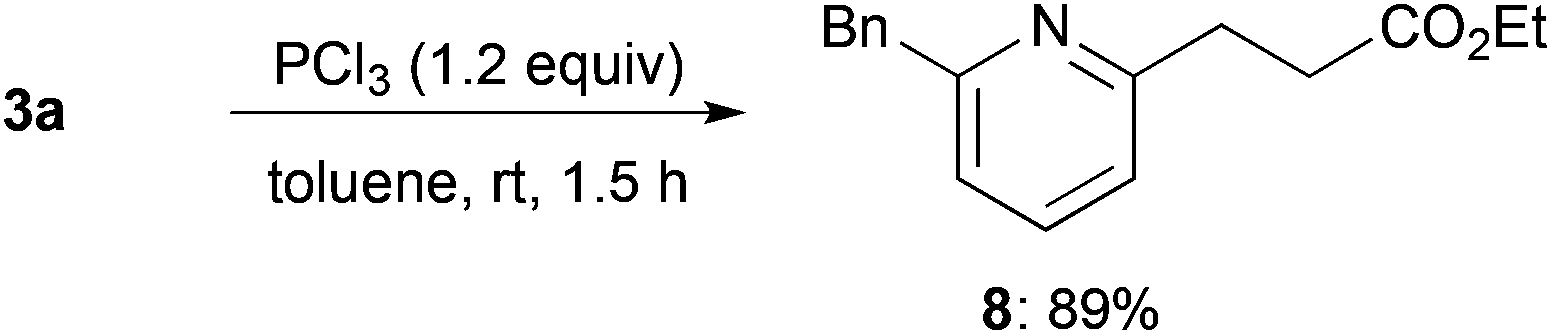 | ||
| Scheme 3 Reduction of alkylated product 3a. | ||
As a preliminary mechanism study, N-oxides were subjected to otherwise similar conditions, in the presence of excess amounts of D2O and in the absence of ethyl acrylate, then D-content of the recovered N-oxides was measured (Scheme 4). In the case of 1a, a high degree of deuteration at the C2 position and a low degree of deuteration at the benzylic position were observed. Also in the case of 1i, deuteration was observed. These results suggest that C–H bond cleavage adjacent to the N-oxide surely occurred whether there is a substituent at the C2 position or not.
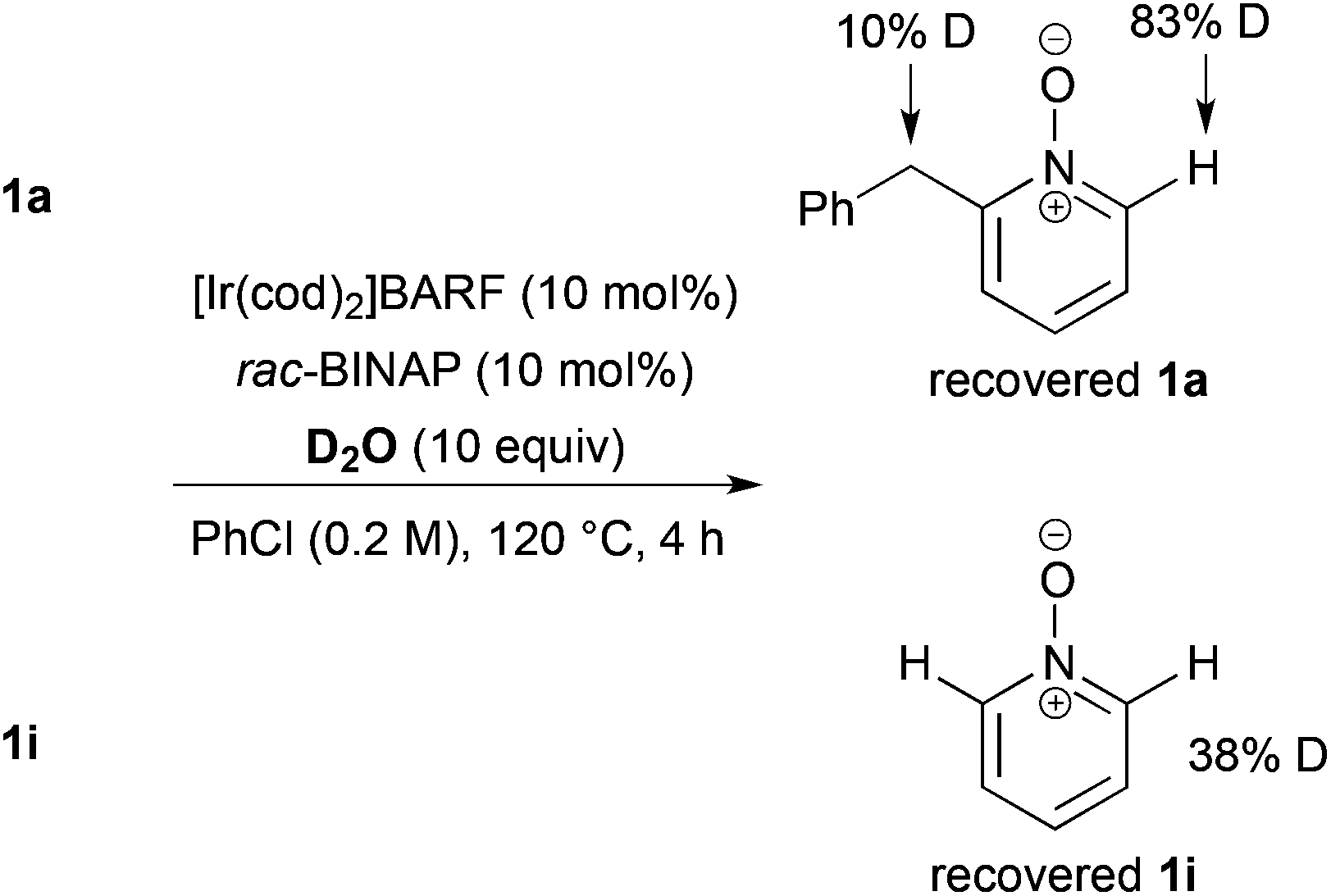 | ||
| Scheme 4 Reaction in the presence of D2O. | ||
We next examined the reaction of deuterated 1a-D with p-bromostyrene (Scheme 5). Although no C6-alkylated product was detected, but deuterium incorporation into both α- and β-positions of the recovered styrene was ascertained. These results indicate that C–H bond cleavage and alkene insertion proceeded, but subsequent reductive elimination did not proceed in the reaction with styrene.
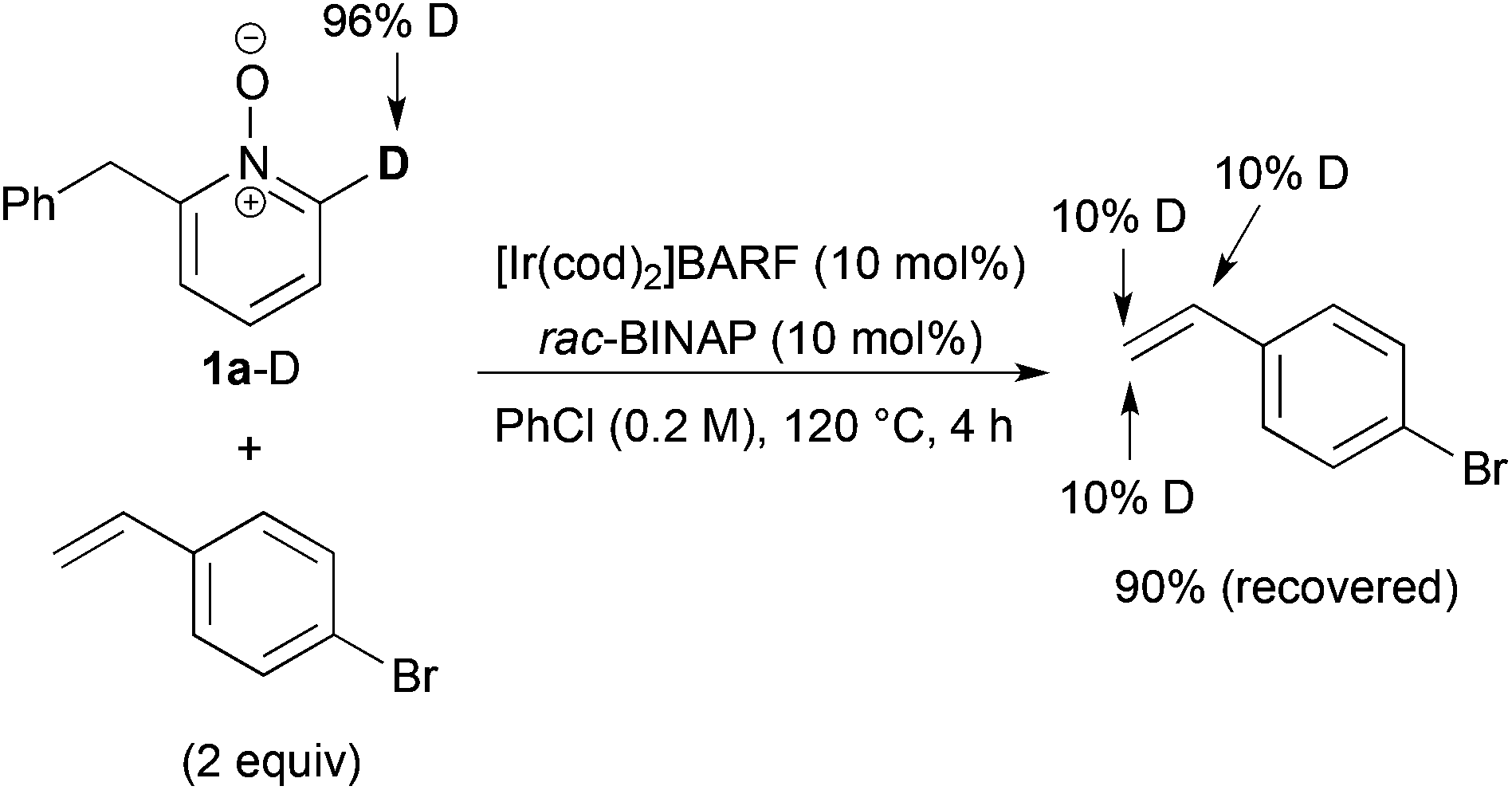 | ||
| Scheme 5 Reaction of deuterated N-oxide 1a-D with p-bromostyrene. | ||
Finally, we measured the kinetic isotope effect (KIE) of the present cationic Ir-catalyzed C–H alkylation by two parallel experiments using 1a and 1a-D, respectively (Scheme 6). The reactions were quenched after 4 h. The KIE value was ca. 1.0 based on the yield ratio of 3a-D/3a. Moreover, the deuterium incorporation into both α- and β-positions of the ester moiety of the product 3a-D was observed. These results suggest that both C–H bond cleavage and alkene insertion are reversible steps, and that the reductive elimination would be a rate-determining step, which is contrasting to the C–H alkylation using the combination of a neutral Rh catalyst and a base (KIE value = 3.2).20
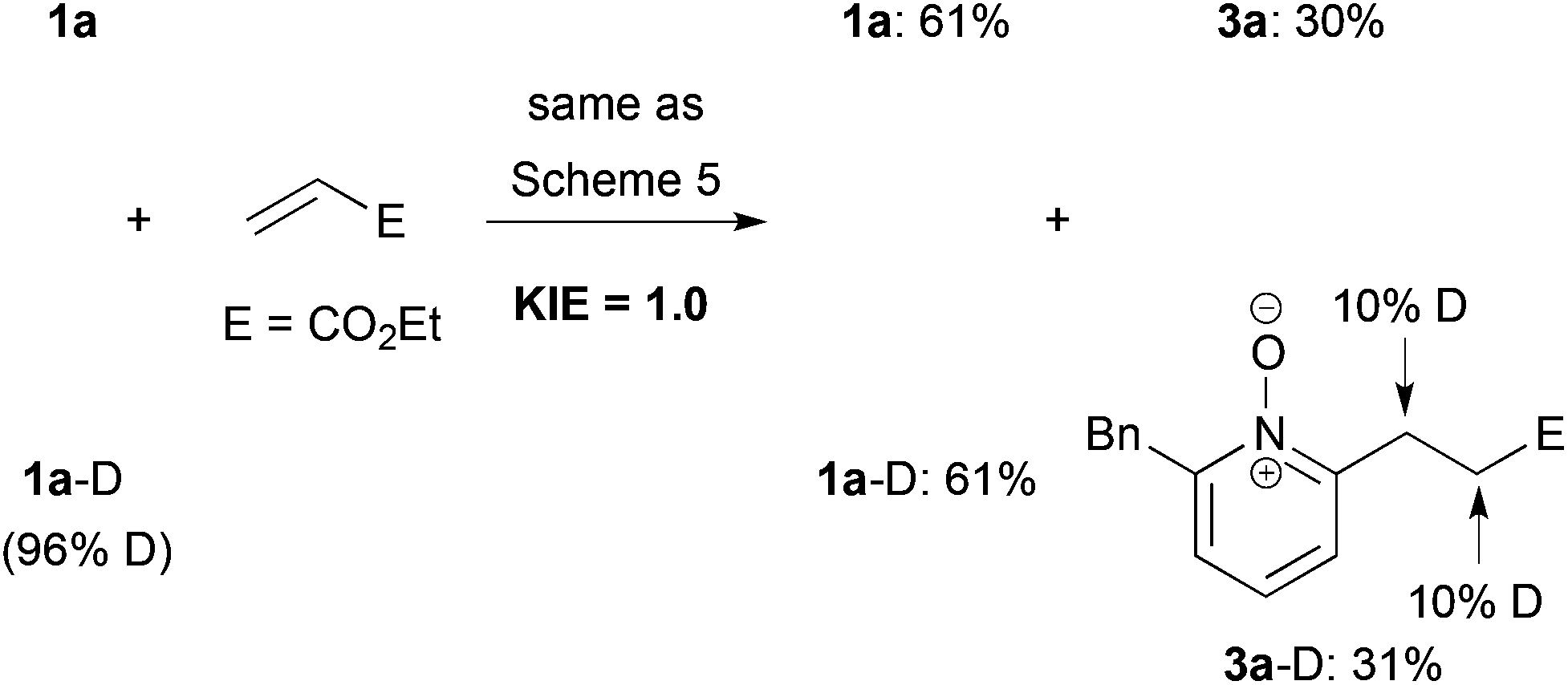 | ||
| Scheme 6 KIE measurement by two parallel experiments. | ||
The proposed mechanism is depicted in Scheme 7. We assumed that the C–H bond cleavage is assisted by the coordination of N-oxide to the Ir center and intermediate A was formed.28 Subsequent hydroiridation to acrylate provides intermediate B, but the pathway of carboiridation cannot be excluded. Finally, reductive elimination gives a linear alkylated product and only the final step may be irreversible.
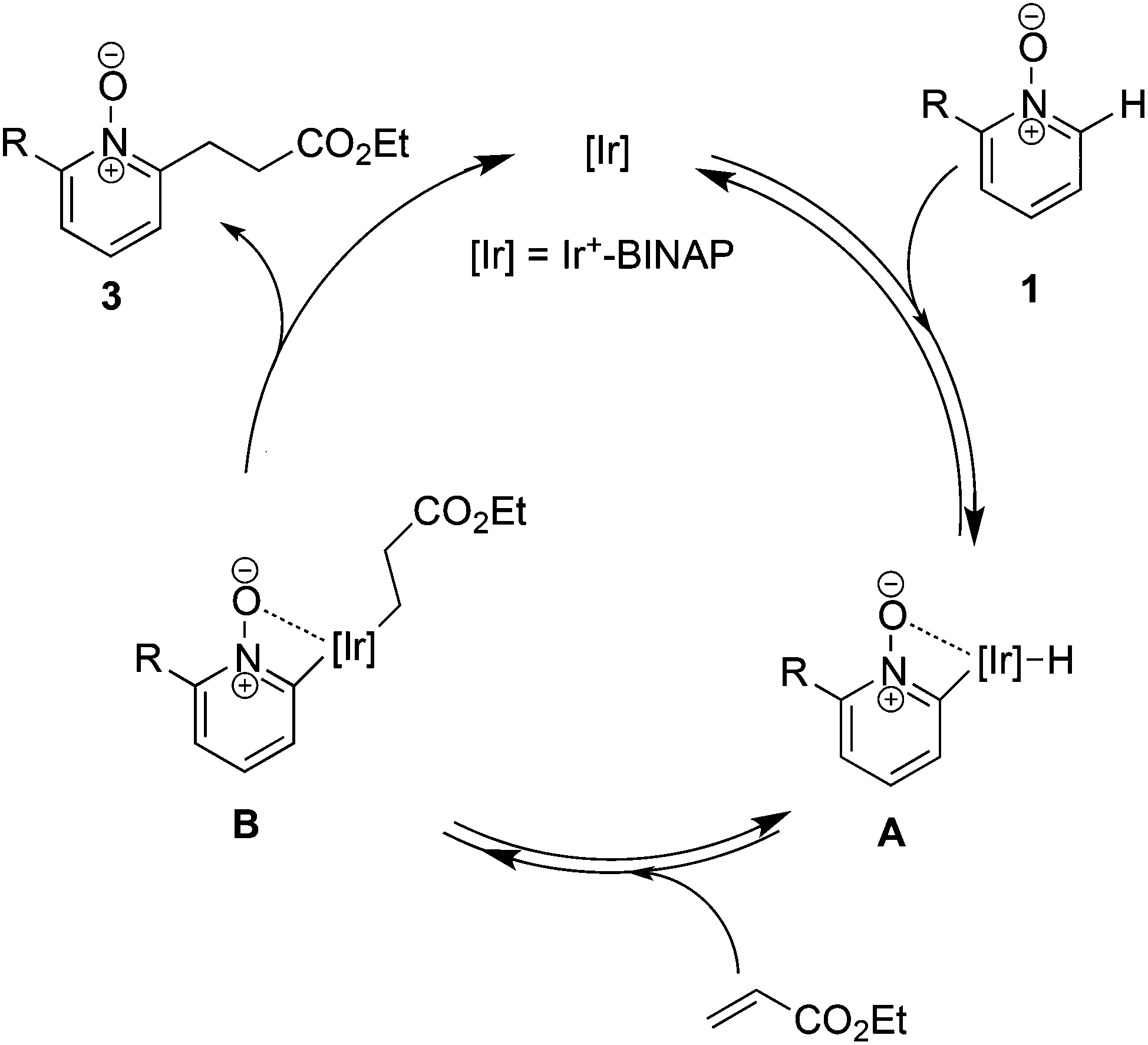 | ||
| Scheme 7 Proposed mechanism of C–H alkylation. | ||
Conclusions
The cationic Ir catalyst combining with a rac-BINAP ligand realized C–H alkylation of 2-substituted pyridine N-oxides with acrylates. Pd-catalyzed C–H arylation and subsequent Ir-catalyzed C–H alkylation provides a facile synthetic protocol for the preparation of 2,6-unsymmetrically disubstituted pyridine derivatives. The preliminary mechanism study revealed that the steps of C–H bond cleavage and alkene insertion are reversible.Acknowledgements
This work was supported by Grant-in-Aid for Scientific Research on Innovative Areas, “Molecular Activation Directed toward Straightforward Synthesis,” MEXT and JST, ACT-C, Japan. We also thank Mr Takayuki Wakaki (Waseda Univ.) for his findings at the preliminary stage. We are grateful to Umicore for generous support in [Ir(cod)2]BARF supply.Notes and references
- For recent reviews, see: (a) C–H Activation, ed. J.-Q. Yu and Z. Shi, Springer-Verlag, Berlin, Germany, 2010 Search PubMed; (b) O. Daugulis, Top. Curr. Chem., 2010, 292, 57 CrossRef CAS PubMed; (c) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (d) J. Wencel-Delord, T. Droege, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (e) K. M. Engle and J.-Q. Yu, Transition metal-catalyzed C-H functionalization. Synthetically enabling reactions for building molecular complexity, in Organic Chemistry, ed. K. Ding and L.-X. Dai, 2012, ch. 8, p. 279 Search PubMed; (f) H. Ibrar and T. Singh, Adv. Synth. Catal., 2014, 356, 1661 CrossRef; (g) M. E. Farmer, B. N. Laforteza and J.-Q. Yu, Bioorg. Med. Chem., 2014, 22, 4445 CrossRef CAS PubMed.
- N. L. Lewis and J. F. Smith, J. Am. Chem. Soc., 1986, 108, 2728 CrossRef.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef CAS.
- C. P. Lenges and M. Brookhart, J. Am. Chem. Soc., 1999, 121, 6616 CrossRef CAS.
- K. Tsuchikama, M. Kasagawa, Y. Hashimoto, K. Endo and T. Shibata, J. Organomet. Chem., 2008, 693, 3939 CrossRef CAS.
- (a) C.-H. Jun, J.-B. Hong, Y.-H. Kim and K. Y. Chung, Angew. Chem., Int. Ed., 2000, 39, 3440 CrossRef CAS; (b) S.-G. Lim, J.-A. Ahn and C.-H. Jun, Org. Lett., 2004, 6, 4687 CrossRef CAS PubMed.
- F. Kakiuchi, M. Yamauchi, N. Chatani and S. Murai, Chem. Lett., 1996, 25, 111 CrossRef.
- K. Gao and N. Yoshikai, Angew. Chem., Int. Ed., 2011, 50, 6888 CrossRef CAS PubMed.
- K. Gao and N. Yoshikai, J. Am. Chem. Soc., 2011, 133, 400 CrossRef CAS PubMed.
- S. Takebayashi and T. Shibata, Organometallics, 2012, 31, 4114 CrossRef CAS.
- M. Chinkel, I. Marek and L. Ackermann, Angew. Chem., Int. Ed., 2013, 52, 3977 CrossRef PubMed.
- T. Shibata and T. Shizuno, Angew. Chem., Int. Ed., 2014, 53, 5410 CrossRef CAS PubMed.
- Y. Kuninobu, T. Matsuki and K. Takai, J. Am. Chem. Soc., 2009, 131, 9914 CrossRef CAS PubMed.
- J. Oyamada and Z. Hou, Angew. Chem., Int. Ed., 2012, 51, 12828 CrossRef CAS PubMed.
- (a) R. Martinez, J.-P. Genet and S. Darses, Chem. Commun., 2008, 3855 RSC; (b) G. Rouquet and N. Chatani, Chem. Sci., 2013, 4, 2201 RSC.
- For a review, see: G. Yan, A. J. Borah and M. Yang, Adv. Synth. Catal., 2014, 356, 2375 CrossRef CAS.
- L.-C. Campeau, S. Rousseaux and K. Fagnou, J. Am. Chem. Soc., 2005, 127, 18020 CrossRef CAS PubMed.
- (a) L.-C. Campeau, D. J. Schipper and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 3266 CrossRef CAS PubMed; (b) D. J. Schipper, M. El-Salfiti, C. J. Whipp and K. Fagnou, Tetrahedron, 2009, 65, 4977 CrossRef CAS.
- (a) D. Mandal, A. D. Yamaguchi, J. Yamaguchi and K. Itami, J. Am. Chem. Soc., 2011, 133, 19660 CrossRef CAS PubMed; (b) K. Li, D. Zhao, J. Lan and J. You, Angew. Chem., Int. Ed., 2011, 50, 5365 CrossRef PubMed; (c) X. Gong, G. Song, H. Xhang and X. Li, Org. Lett., 2011, 13, 1766 CrossRef CAS PubMed; (d) W. Liu, Y. Li, Y. Wang and C. Kuang, Org. Lett., 2013, 15, 4682 CrossRef CAS PubMed.
- J. Ryu, S. H. Cho and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 3677 CrossRef CAS PubMed.
- Direct C–H alkylation of pyridine at the C-4 or C-2 position: (a) Y. Nakao, Y. Yamada, N. Kashihara and T. Hiyama, J. Am. Chem. Soc., 2010, 132, 13666 CrossRef CAS PubMed; (b) B.-T. Guan and Z. Hou, J. Am. Chem. Soc., 2011, 133, 18086 CrossRef CAS PubMed.
- T. Shibata and Y. Matsuo, Adv. Synth. Catal., 2014, 356, 1516 CrossRef CAS.
- W. Mai, J. Yuan, Z. Li, L. Yang, Y. Xiao, P. Mao and L. Qu, Synlett, 2012, 938 CrossRef CAS.
- The reaction of quinoline N-oxide was messy and the alkylated product could not be detected, but quinoline was detected as a reduced product.
- In each entry of Table 3, a small amount of reduced 2-substituted pyridine was obtained as a by-product.
- The reaction of 4-phenylpyridine N-oxide gave the corresponding alkylated product in low yield (4%).
- Substituted acrylates, such as ethyl methacrylate, ethyl cinnamate, and (E)-ethyl penta-2,4-dienoate, were inactive in this reaction.
- Y. A. Hernández, J. López-Serrano, M. Paneque, M. L. Poveda, F. Vattier, V. Salazar, E. Álvarez and E. Carmona, Chem. – Eur. J., 2011, 17, 9302 CrossRef PubMed.
Footnotes |
| † Dedicated to Professor Ei-ichi Negishi on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00355a |
| This journal is © the Partner Organisations 2015 |