
CO-enabled rhenium hydride catalyst for directed C(sp2)–H bond alkylation with olefins†‡
Hongming
Jin
a,
Zhengbo
Zhu
a,
Ning
Jin
a,
Jin
Xie
a,
Yixiang
Cheng
a and
Chengjian
Zhu
*ab
aState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, P. R. China. E-mail: cjzhu@nju.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Shanghai 200032, P. R. China
First published on 3rd February 2015
Abstract
We report the first example of CO-enabled rhenium hydride complex-catalyzed inter- and intra-molecular hydroarylation of activated alkenes, with a broad reaction scope. α,β-Unsaturated ketones, esters and amides are all suitable for this transformation. It affords a novel and convenient protocol to construct 3-methylindolones. In addition, mechanistic investigations suggest that a rhenium carbonyl hydride species may act as a key intermediate.
Introduction
The transition metal-catalyzed insertion reaction of unsaturated compounds into C–H bonds has increasingly been regarded as an efficient and atom-economic strategy to construct C–C bonds.1,2 In the past decade, C–H transformations catalyzed by rhenium carbonyl compounds have attracted considerable attention.3 Despite outstanding achievements, the development of simple, accessible and novel rhenium catalysts to accomplish these transformations is still highly desirable for chemical researchers.4a Based on previous work in rhenium chemistry, it was found that rhenium hydride complexes, which could be easily prepared from cheap potassium perrhenate,4b,c have significant advantages in handling, storage and availability. In 2009, Moehring's group obtained an ortho-metalated compound through rhenium hydride complex-promoted sp2 C–H activation.5 Recently, rhenium hydride complexes were also reported to be excellent and elegant catalysts for α-C–H activation of carbonyl compounds6a and dehydrogenation of alcohols.6b,c However, to the best of our knowledge, instances of rhenium hydride complex-promoted inert C(sp2)–H functionalization are very rare. Herein, we report a CO-enabled rhenium hydride catalyst for directed C(sp2)–H bond activation (Scheme 1). Moreover, the intramolecular transformation affords a novel and efficient strategy to construct 3-methylindolone frameworks,7a,b which are essential building blocks in organic synthesis.7c,d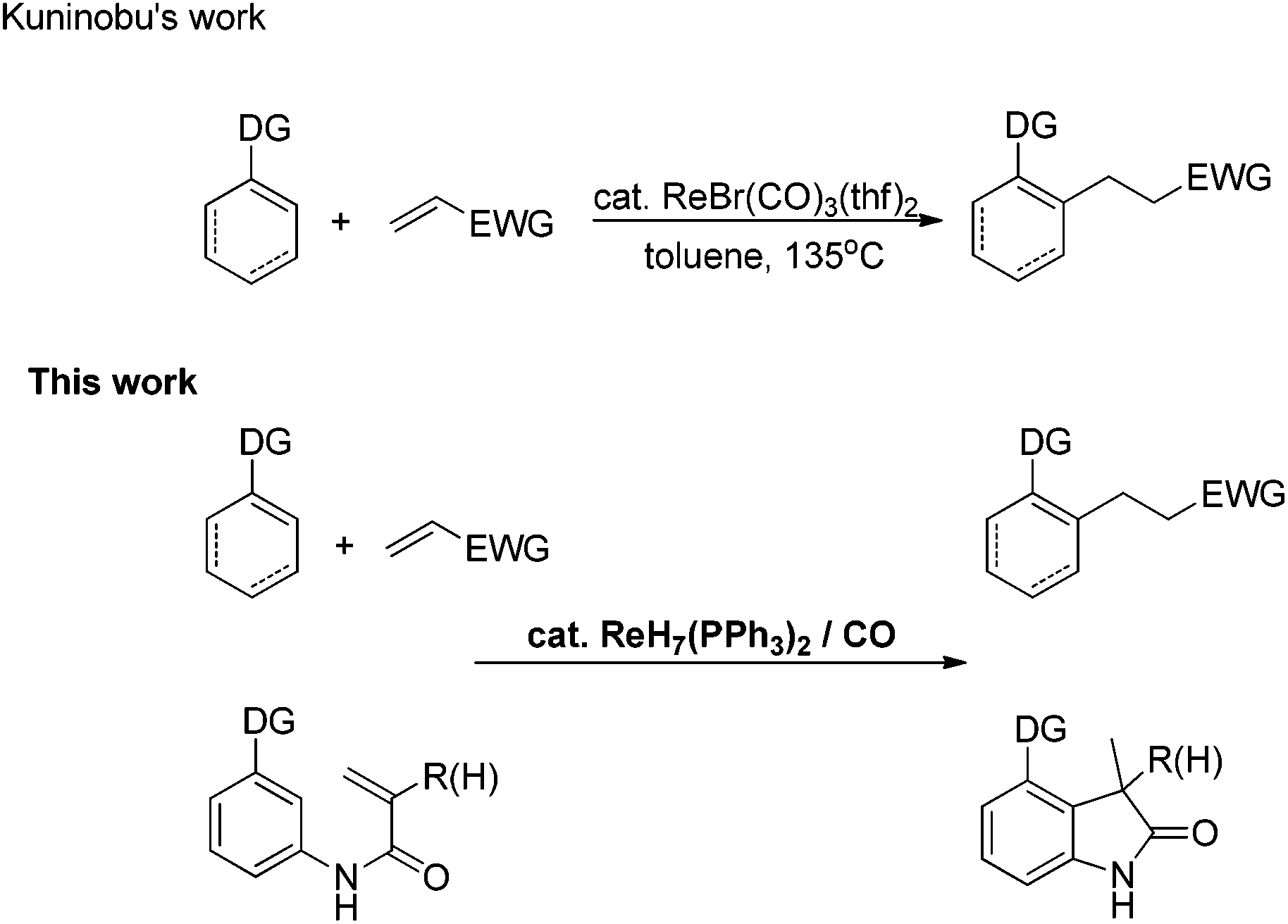 | ||
| Scheme 1 Re-catalyzed sp2 C–H bond alkylation with olefins. | ||
Results and discussion
Initially, the reaction of 2-phenylpyridine 1a and pent-1-en-3-one 2a was chosen as a model reaction to optimise the reaction conditions (Table 1). The influence of different atmospheres was evaluated in the presence of 5 mol% ReH7(PPh3)2 at 150 °C in THF. To our delight, although the transformation did not proceed under N2, Ar, or air atmospheres (entries 1–3), 3aa could be obtained in 68% yield when CO was used (entry 4). During the testing of several commercial ligands (entries 5–8), we found that PPh3 showed higher efficiency than P(tolyl)3, but the other phosphine ligands led to unsatisfactory results. Furthermore, the yield of 3aa could be improved to 83% by using toluene as the solvent (entry 9). Notably, increasing the catalyst loading to 10 mol% provided the desired product in 92% yield (entry 10). It should be noted that the di-substituted product was not observed, even though pent-1-en-3-one 2a was present in excess. However, the product was only obtained in 58% yield when 1 equiv. 2a was used (entry 11). Thus, the optimal reaction conditions were 10 mol% ReH7(PPh3)2 catalyst, a 1.5 atm CO atmosphere and toluene as the solvent.|
|
||||
|---|---|---|---|---|
| Entry | L | Solvent | Atmosphere | Yield (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.6 mmol), ReH7L2 (5 mol%), CO (1.5 atm) and solvent (0.5 mL) at 150 °C for 24 h. Isolated yields are reported. The reactions were carried out in a 10 mL sealed tube. b Catalyst formula: ReH7L. c 10 mol% ReH7(PPh3)2 was used. d 0.2 mmol 2a was used. | ||||
| 1 | PPh3 | THF | N2 | 0 |
| 2 | PPh3 | THF | Ar | 0 |
| 3 | PPh3 | THF | Air | 0 |
| 4 | PPh3 | THF | CO | 68 |
| 5b | dppe | THF | CO | 0 |
| 6b | dppp | THF | CO | 0 |
| 7 | PCy3 | THF | CO | 0 |
| 8 | P(tolyl)3 | THF | CO | 62 |
| 9 | PPh3 | Toluene | CO | 83 |
| 10 | PPh 3 | Toluene | CO | 92 |
| 11d | PPh3 | Toluene | CO | 58 |
| 12 | PPh3 | CH3CN | CO | 0 |
| 13 | PPh3 | Anisole | CO | 74 |
| 14 | PCy3 | 1,4-Dioxane | CO | 31 |
With the optimized conditions in hand, the scope of the substrates was investigated. Firstly, substrates with various kinds of substituent were applied in the transformation (Table 2). To our delight, the reaction proceeded smoothly with high yield, regardless of the presence of electron-donating or electron-withdrawing groups. The sp2 C–H bonds of simple cycloalkenes were also successfully employed to explore the scope of substrates, giving excellent yields (3ia–3ka). Moreover, heterocycles such as thiophene and furan (3la–3na) were also easily alkylated to produce a series of functional molecules. Functional groups such as methoxy (3ba), trifluoromethyl (3fa), ester (3ha) and formyl (3wa) were all tolerated, which was beneficial for further decoration of the products. Meanwhile, when m-substituted arenes were employed (3va, 3wa), high regioselectivity was revealed, favoring functionalization of the less hindered C–H bond. Secondly, different directing groups were examined. The reaction tolerated a number of pyridine derivatives (3oa–3qa), and pyridines with electron-withdrawing substituents could lead to higher yields. Pyrimidine and purine, which widely exist in biological molecules and natural products, were also demonstrated to be excellent directing groups (3ra–3ua).
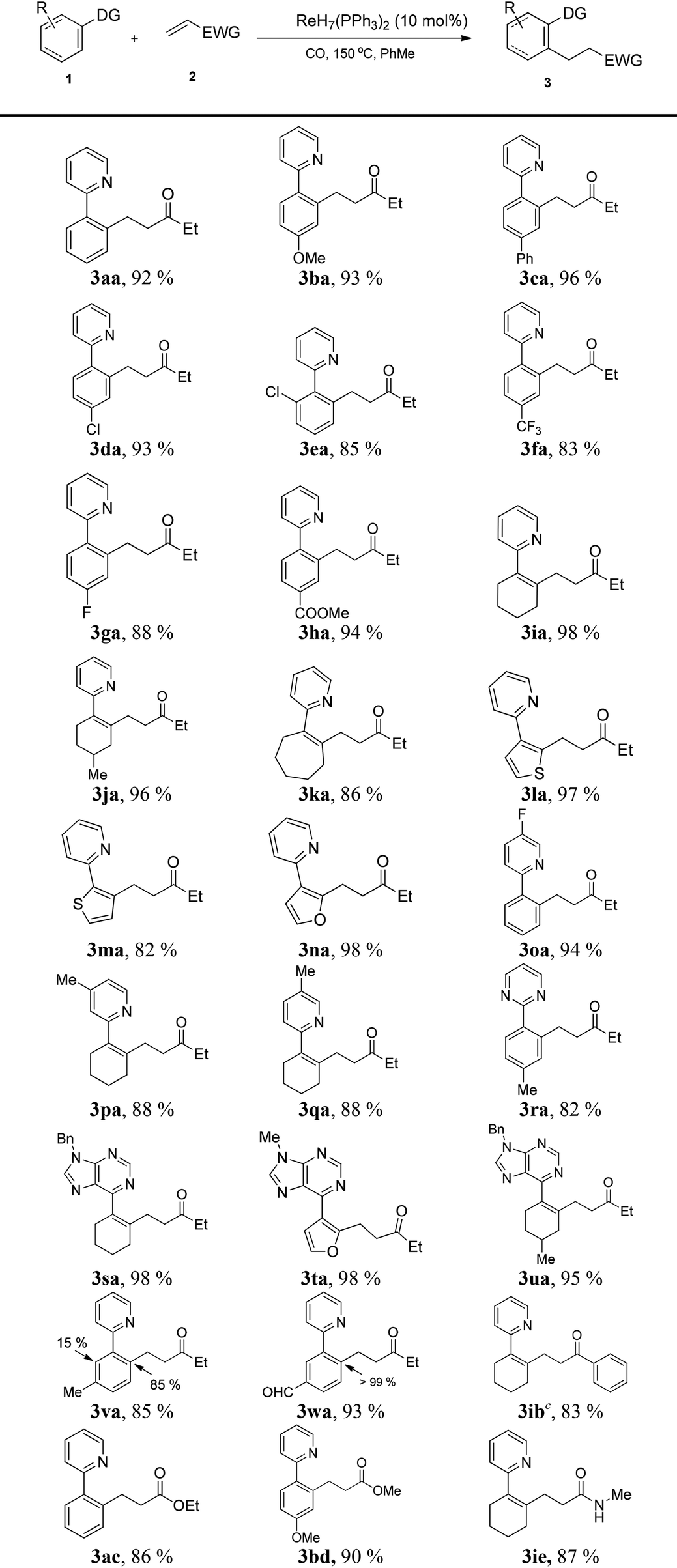
We further explored the range of α,β-unsaturated compounds. Phenylvinylketone was also suitable for this transformation (3ib). Particularly, the reactions with less electrophilic α,β-unsaturated esters (3ac, 3bd) and an α,β-unsaturated amide (3ie) proceeded smoothly with moderate yields.
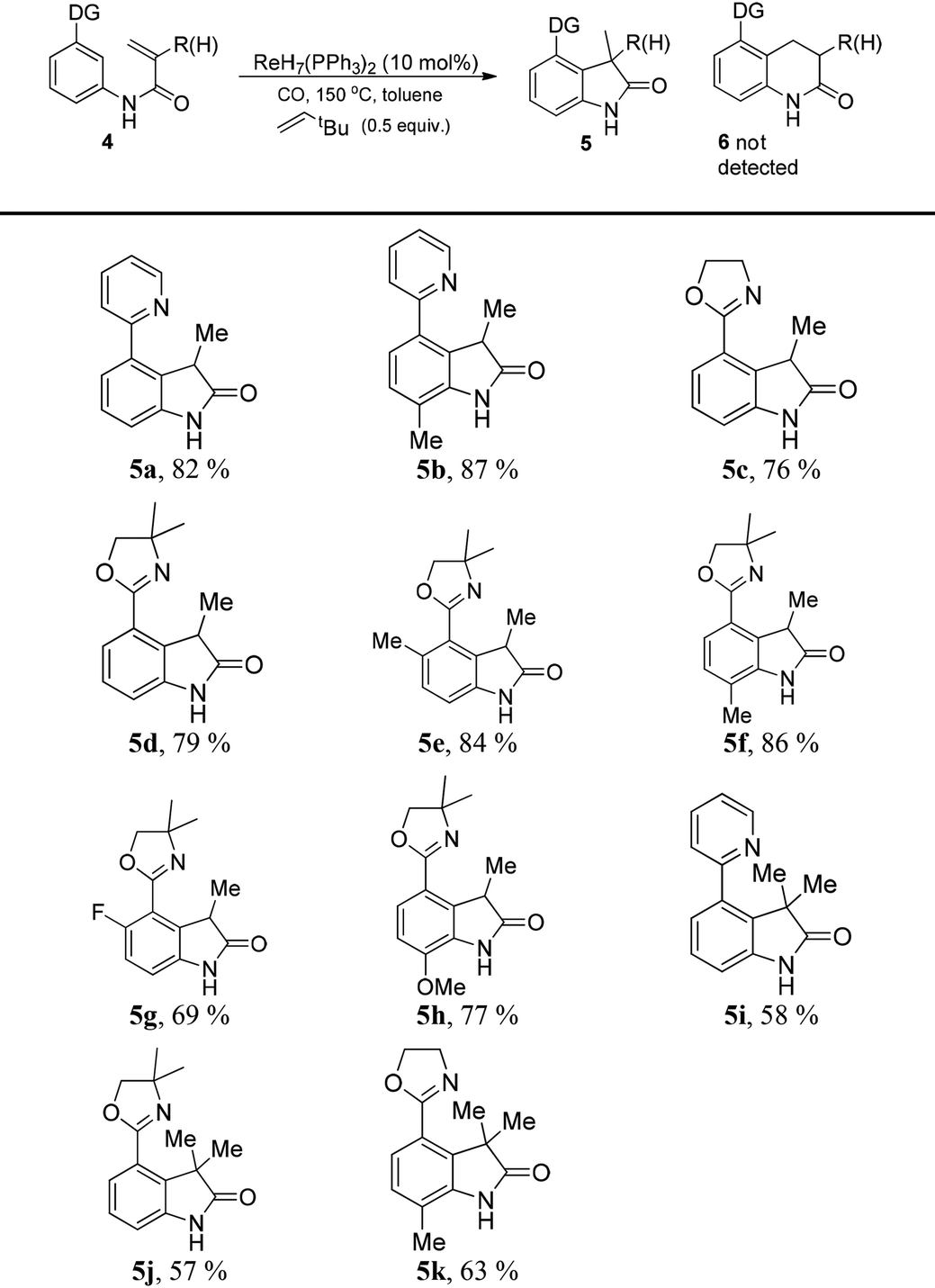
In order to apply the rhenium-catalyzed C–H activation to the construction of heterocyclic frameworks, the intramolecular transformation was surveyed. Interestingly, the sp2 C–H bond selectively inserted into the α-site of the double bond to yield indolones (5), and the product 6 was not detected. Additionally, 3,3-dimethylbut-1-ene was utilized as a hydrogen acceptor, protecting the double bonds of substrates from reduction by in situ-generated reductive H species.8 The catalytic system was applied to the synthesis of a series of indolones (Table 3). Different substituted products were obtained in moderate to excellent yields. To convert these frameworks to more general structures, removable directing groups have been considered. Oxazolines, as benign directing groups, can be reduced to aldehydes or turned into carboxylic acids by hydrolysis, which favours further functionalization.9
To gain insight into the Re-catalyzed C–H alkylation reaction,10a several experiments were carried out to elucidate the mechanism. Firstly, rhenium-catalyzed ortho-directed C–H deuteration with D2O indicated that a C–H bond metalation step is involved (Scheme 2a).10b Furthermore, H/D scrambling was observed during the reaction of [D]2-1a with 2a, which suggested the C–H bond metalation to be reversible (Scheme 2b).2a,d,i In order to monitor the influence of the hydrogen in the catalyst, ReD7(PPh3)2 was used (Scheme 2c and 2d). H/D scrambling was observed at the ortho-positions of the phenylpyridine moiety. The similar results indicated that the hydrogen in the catalyst had little influence on the mechanistic experiments ([D]n-3aa), and also demonstrated that the C–H bond activation was reversible,11a in stark contrast to rhenium carbonyl compound-catalyzed C–H activation, where it was found to be the rate determining step11b (Scheme 2e). In addition, apparent H/D exchange was observed when a mixture of [D]2-1a and 3aa was stirred under standard conditions (Scheme 2f). This suggested that a key Re–H species might enable the H/D transfer between different substrates (Scheme 3a).
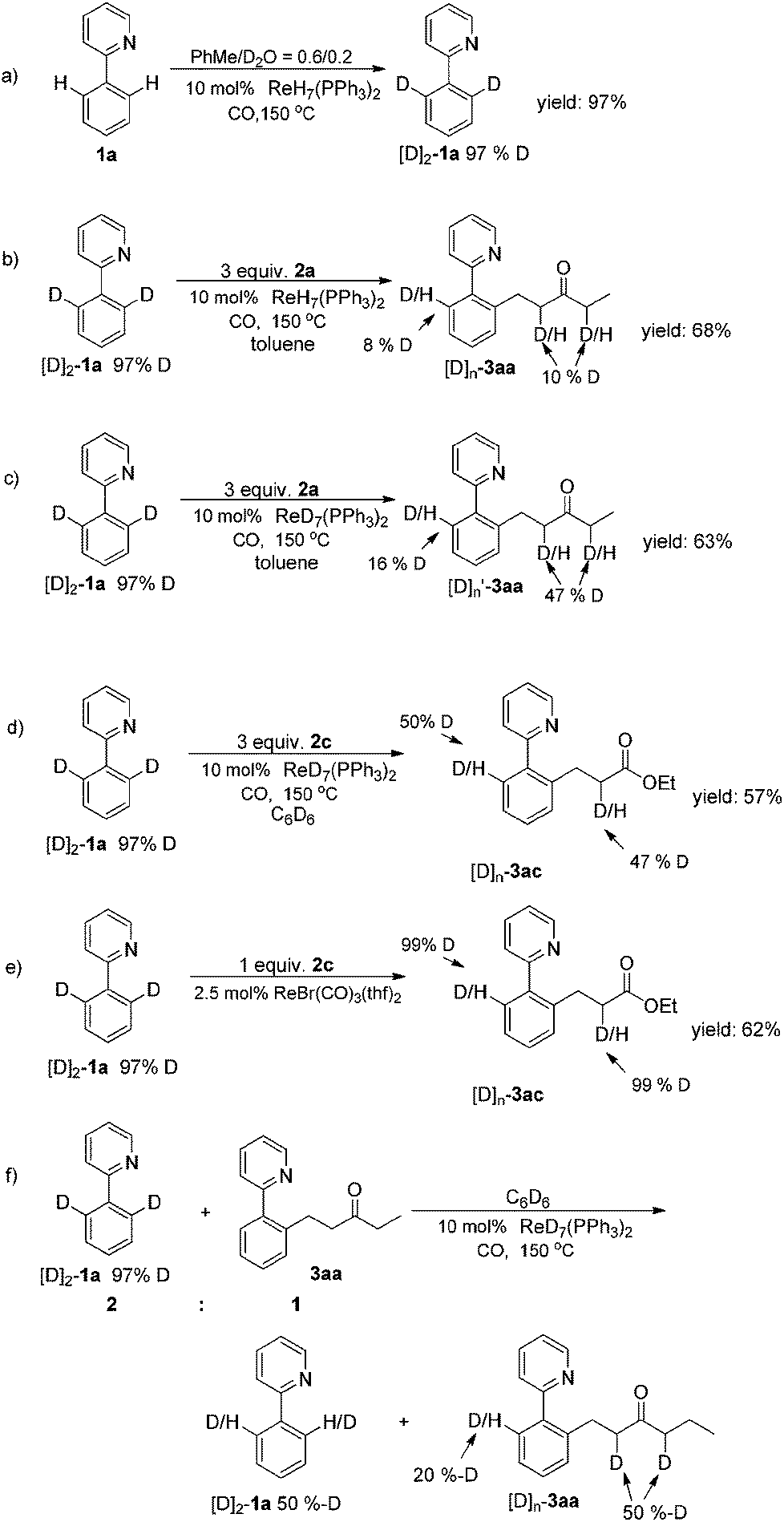 | ||
| Scheme 2 Mechanistic experiments. | ||
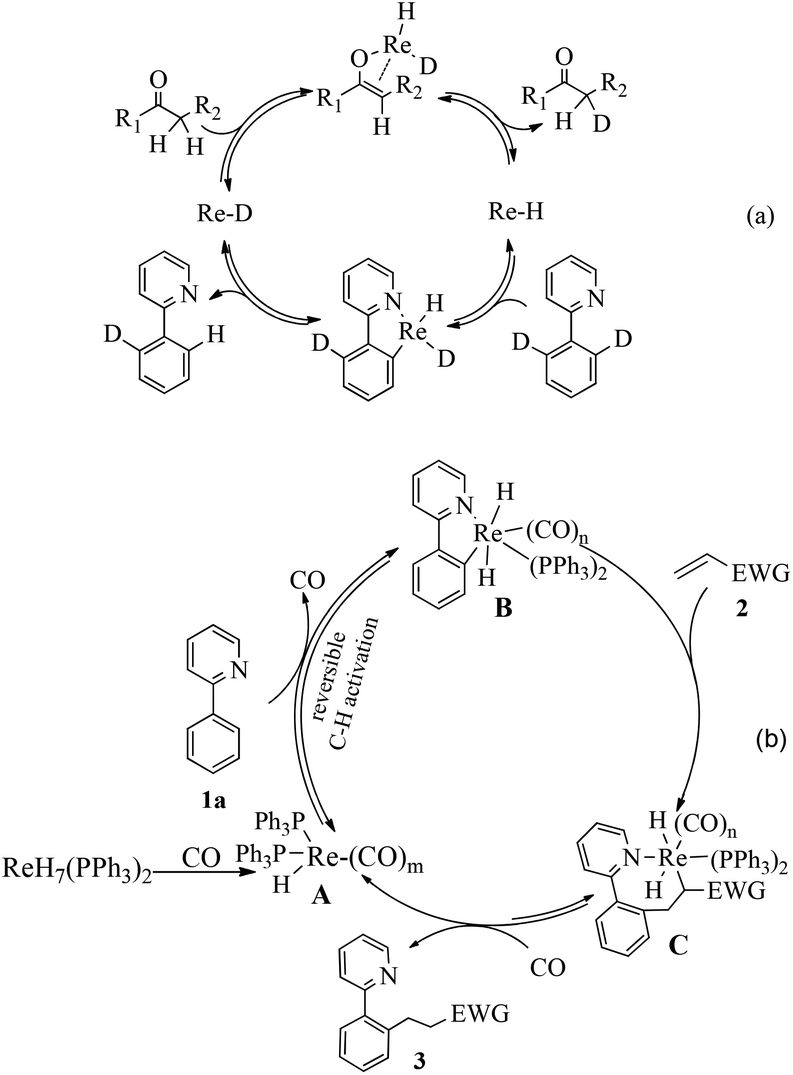 | ||
| Scheme 3 Proposed mechanism. | ||
Finally, a tentative mechanism has been proposed based on these experiments and previous work (Scheme 3b). Initially, the rhenium hydride catalyst reacts with CO to generate Re–H species A.12 Subsequently, reversible C–H metalation generates the intermediate B, which could be stabilized by CO coordination at high temperature. Then, the alkene 2 inserts into the Re–C bond, giving intermediate C. The intermediate C undergoes reductive elimination to recover Re–H species A.
Summary
In summary, we have reported the first example of CO-enabled rhenium hydride compound-catalyzed directed sp2 C–H alkylation. A variety of directing groups and substrates are tolerated by this catalytic system. The intramolecular transformation also provides a new and efficient protocol to construct indolone frameworks. The investigation of the mechanism suggested that a Re–H species might play a key role in the catalytic cycle, which provides further insight into rhenium-catalyzed C–H functionalization. Further studies on transformations catalyzed by rhenium hydride complexes are under way in our laboratory.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (21172106, 21074054 and 21372114), the National Basic Research Program of China (2010CB923303), the Research Fund for the Doctoral Program of Higher Education of China (20120091110010) for their financial support, and National Science Fund for Talent Training in Basic Science (no. J1103310).Notes and references
- Recent reviews: (a) X. Chen, K. M. Engle, D. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (c) Y. Kuninobu and K. Takai, Chem. Rev., 2011, 111, 1938 CrossRef CAS PubMed; (d) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (e) S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886 RSC; (f) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed.
- For selected recent reports on transition metal-catalyzed sp2 C–H alkylation, see: (a) L. Yang, B. Qian and H. Huang, Chem. – Eur. J., 2012, 18, 9511 CrossRef CAS PubMed; (b) Z. Shi, C. Grohmann and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 5393 CrossRef CAS PubMed; (c) H. Wang, N. Schröder and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 5386 CrossRef CAS PubMed; (d) M. Schinkel, I. Marek and L. Ackermann, Angew. Chem., Int. Ed., 2013, 52, 3977 CrossRef CAS PubMed; (e) N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877 CrossRef CAS PubMed; (f) K. Gao and N. Yoshikai, Angew. Chem., Int. Ed., 2011, 50, 6888 CrossRef CAS PubMed; (g) T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 2207 CrossRef CAS PubMed; (h) B. Li, Z. Wu, Y. Gu, C. Sun, B. Wang and Z.-J. Shi, Angew. Chem., Int. Ed., 2011, 50, 1109 CrossRef CAS PubMed; (i) L. Jiao and T. Bach, Angew. Chem., Int. Ed., 2013, 52, 6080 CrossRef CAS PubMed; (j) G. Deng, L. Zhao and C. Li, Angew. Chem., Int. Ed., 2008, 47, 6278 CrossRef CAS PubMed; (k) S. Yu and X. Li, Org. Lett., 2014, 16, 1200 CrossRef CAS PubMed; (l) Z.-M. Sun, J. Zhang, R. S. Manan and P. Zhao, J. Am. Chem. Soc., 2010, 132, 6935 CrossRef CAS PubMed; (m) L. Yang, C. A. Correia and C. Li, Org. Biomol. Chem., 2011, 9, 7176 RSC; (n) B. Zhou, P. Ma, H. Chen and C. Wang, Chem. Commun., 2014, 50, 14558 RSC.
- For selected reports on C–H activation catalyzed by rhenium carbonyl complexes, see: (a) Y. Kuninobu, Y. Nishina, M. Shouho, K. Takai and A. K. Takai, Angew. Chem., Int. Ed., 2006, 45, 2766 CrossRef CAS PubMed; (b) Y. Kuninobu, T. Matsuk and K. Takai, J. Am. Chem. Soc., 2009, 131, 9914 CrossRef CAS PubMed; (c) Y. Kuninobu, Y. Tokunaga, A. Kawata and K. Takai, J. Am. Chem. Soc., 2006, 128, 202 CrossRef CAS PubMed; (d) D. Xia, Y. Wang, Z. Du, Q. Zheng and C. Wang, Org. Lett., 2012, 14, 588 CrossRef CAS PubMed; (e) Q. Tang, D. Xia, X. Jin, Q. Zhang, X. Sun and C. Wang, J. Am. Chem. Soc., 2013, 135, 4628 CrossRef CAS PubMed; (f) Y. Fukumoto, M. Daijo and N. Chatani, J. Am. Chem. Soc., 2012, 134, 8762 CrossRef CAS PubMed; (g) Y. Kuninobu, P. Yu and K. Takai, Org. Lett., 2010, 12, 4274 CrossRef CAS PubMed; (h) Y. Kuninobu, Y. Fujii, T. Matsuki, Y. Nishina and K. Takai, Org. Lett., 2009, 11, 2711 CrossRef CAS PubMed; (i) S. Sueki, Y. Guo, M. Kanai and Y. Kuninobu, Angew. Chem., Int. Ed., 2013, 52, 11879 CrossRef CAS PubMed.
- (a) The syntheses of rhenium carbonyl compounds from perrhenate usually require dangerous elevated pressures (100–300 atm), high temperatures and long reaction times (up to 2 days); (b) J. Chatt and R. S. Coffey, J. Chem. Soc. A, 1969, 1963 RSC; (c) The price of potassium perrhenate is $3.3 per g.
- (a) G. A. Moehring, C. C. Williams, J. Buford, M. Kaviani, J. Sulko and P. E. Fanwick, Inorg. Chem., 1998, 37, 3848 CrossRef CAS PubMed; (b) Y. Jimenez, A. M. Strepka, M. D. Borgohain, P. A. Hinojosa and G. A. Moehring, Inorg. Chim. Acta, 2009, 362, 3259 CrossRef CAS PubMed.
- (a) H. Takaya, M. Ito and H. Murahashi, J. Am. Chem. Soc., 2009, 131, 10824 CrossRef CAS PubMed; (b) P. P. M. Schleker, R. Honeker, J. Klankermayer and W. Leitner, ChemCatChem, 2013, 5, 1762 CrossRef CAS; (c) H. Jin, J. Xie, C. Pan, Z. Zhu, Y. Cheng and C. Zhu, ACS Catal., 2013, 3, 2195 CrossRef CAS.
- (a) F. Zhou, Y. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381 CrossRef CAS; (b) C. Liu, D. Liu, W. Zhang, L. Zhou and A. Lei, Org. Lett., 2013, 15, 6166 CrossRef CAS PubMed; (c) K. Shen, X. Liu, G. Wang, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2011, 50, 4684 CrossRef CAS PubMed; (d) D. Sarraf, N. Richy and J. Vidal, J. Org. Chem., 2014, 79, 10945 CrossRef CAS PubMed.
- Less reductive hydrogen species derived from the decomposition of the rhenium hydride complex under standard conditions.
- (a) J. I. Levin and S. M. Weinreb, Tetrahedron Lett., 1982, 23, 2347 CrossRef CAS; (b) M. S. Babu and K. M. L. Rai, Tetrahedron Lett., 2004, 45, 7969 CrossRef PubMed.
- (a) Y. Kuninobu, Y. Nishina, K. Okaguchi, M. Shouho and K. Takai, Bull. Chem. Soc. Jpn., 2008, 81, 1393 CrossRef CAS; (b) S. Ma, G. Villa, P. S. Thuy-Boun, A. Homs and J.-Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 734 CrossRef CAS PubMed.
- (a) A large negative kinetic isotope effect is shown in the ESI‡; (b) Note that Kuninobu and Takai also investigated the KIE in the ortho-alkylation of 2-phenylpyridine with ethyl acrylate using rhenium carbonyl catalysis (see ref. 10a). In contrast to our system, they suggested that C–H oxidative addition was the rate determining step (KIE = 2.6).
- M. Freni, D. Giusto and V. Valenti, J. Inorg. Nucl. Chem., 1965, 27, 755 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Ei-ichi Negishi for his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Preparation of substrates, characterization data, 1H and 13C NMR, and HRMS. See DOI: 10.1039/c4qo00329b |
| This journal is © the Partner Organisations 2015 |