
4-Alkenyl-5H-1,2,3-oxathiazole 2,2-dioxides in catalytic and enantioselective [4 + 2] cycloaddition through iminium activation. Straightforward access to the trans-decaline framework and to densely functionalized cyclohexanes†
Iker
Riaño
,
Uxue
Uria
,
Luisa
Carrillo
*,
Efraim
Reyes
and
Jose L.
Vicario
*
Department of Organic Chemistry II, UPV/EHU. P.O. Box 644, 48080 Bilbao, Spain. E-mail: marisa.carrillo@ehu.es; joseluis.vicario@ehu.es; Fax: +34 94 601 2748; Tel: +34 94 601 5454
First published on 26th January 2015
Abstract
We have employed 4-alkenyl-5H-1,2,3-oxathiazole 2,2-dioxides as useful reagents able to generate in situ an electron-rich diene intermediate that has the potential to undergo [4 + 2] cycloaddition with α,β-unsaturated aldehydes through the iminium/enamine activation. Under the optimized reaction conditions that comprise the use of O-TMS diphenylprolinol as an organocatalyst and a Brønsted base as a cocatalyst, a series of [4 + 2] cycloadducts has been prepared as single diastereoisomers with very high enantiomeric excess. Moreover, the cyclic sulfamidate entity can play the role of a masked β-aminoalcohol moiety in which these adducts can be converted by simple diastereoselective reduction/hydrolysis.
Introduction
Cyclic sulfamidates represent a very versatile family of reagents with multiple applications in organic synthesis.1 Among them, cyclic sulfamidate imines have also received some deal of attention as useful masked imine reagents with enhanced electrophilicity. In fact, most of the reports found in the literature regarding the synthetic application of five-membered cyclic sulfamidate imines (5H-1,2,3-oxathiazole 2,2-dioxides) have focused exclusively on the reactivity of the azomethine function towards nucleophiles or under hydrogenation conditions (see Fig. 1).2 In contrast, only a few examples exist in the literature showing the potential reactivity of the highly acidic protons at the 5-position of this particular class of heterocycles.3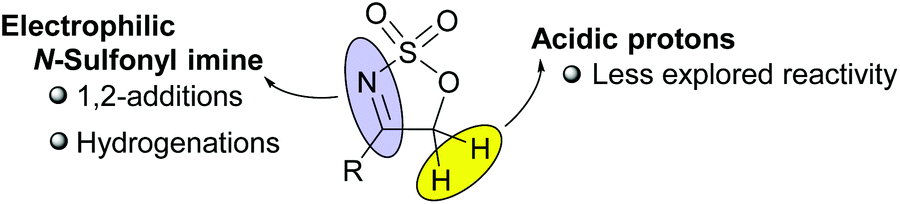 | ||
| Fig. 1 Reactivity of 5H-1,2,3-oxathiazole 2,2-dioxides. | ||
With this situation in mind, and as a part of our ongoing program devoted to the development of new methodological approaches to carry out cycloaddition reactions in a catalytic and enantioselective manner using organocatalysis as the vehicle for stereocontrol,4 we turned our attention to the potential of 4-alkenyl-5H-1,2,3-oxathiazole 2,2-dioxides as very convenient reagents to be used as precursors of functionalized dienes in Diels–Alder-type reactions (Scheme 1). In particular, we envisaged that the presence of these already mentioned two highly acidic protons at the 5-position of the oxathiazole moiety together with a 4-alkenyl substituent would favour the presence of an enamine tautomer that can be regarded as a particular electron-rich diene reagent. As a consequence, this was expected to show expedient potential to undergo formal [4 + 2] cycloaddition5 in the presence of an α,β-unsaturated aldehyde as the electron-poor dienophile counterpart, leading to the formation of a highly substituted cyclohexene cycloadduct that would also presumably isomerize to the corresponding imine tautomer. This would result in the formation of a final product with five contiguous stereogenic centres in one single step. Considering this reaction design, application of the LUMO-lowering effect associated with iminium ion formation6 also prompted us to explore the use of a chiral secondary amine as a suitable catalyst for this transformation.
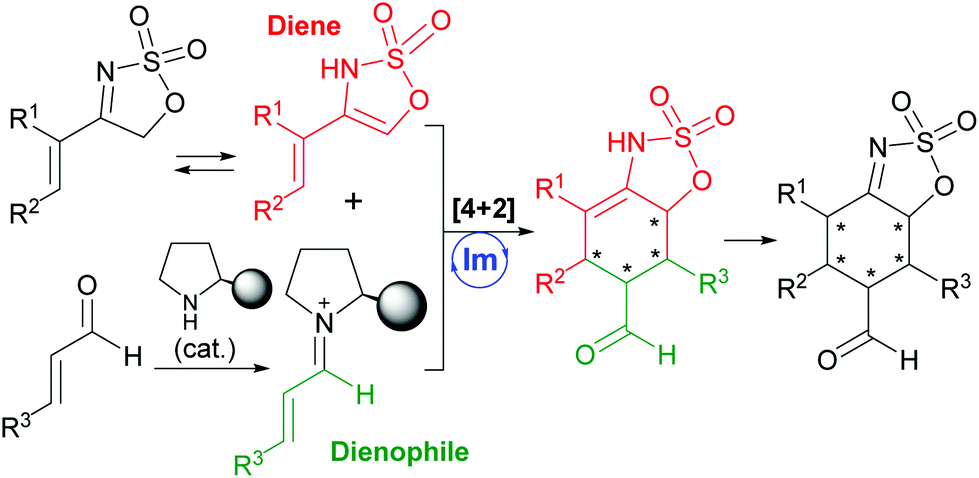 | ||
| Scheme 1 Reaction design. | ||
We started with the evaluation of the best catalyst for the projected transformation using the reaction between cyclic sulfamidate imine 1a and crotonaldehyde (2a) as a model system (Table 1). We first surveyed the viability of the proposed reaction using the archetypal O-TMS diphenylprolinol 3a as the catalyst which is recognized as a privileged architecture to activate enals through iminium ion intermediates.7 The reaction between 1a and 2a in the presence of 3a did not produce any adduct after 24 h either in the absence of a cocatalyst (entry 1) or in the presence of PhCO2H, which is known to facilitate the iminium ion formation step (entry 2). On the other hand, when a Brønsted base such as DABCO was incorporated as the cocatalyst, cycloadduct 4a was isolated in 55% yield as a single diastereoisomer of very high enantiopurity (entry 3). This indicated that a Brønsted base was absolutely necessary for the generation of the putative electron-rich diene from cyclic sulfamidate imine 1a. We next studied the use of other catalysts, observing that modifying the O-protecting group of the diphenylprolinol scaffold to the smaller methyl (entry 4) or the bulkier triphenylsilyl (entry 6) provided similar results in terms of yield and stereocontrol, although becoming somewhat slower in the case of catalyst 3d. The simple diphenylprolinol 3c was unable to provide any product (entry 5) and catalyst 3e bearing more sterically demanding aryl substituent also failed, even after prolonged reaction times (entry 7). Removal of the silyloxy/alkoxy group from the catalyst structure maintained the activity but decreased the enantiocontrol of the process (entry 8). In all cases one single diastereoisomer was detected by NMR analysis of the crude reaction mixture.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Additive | Solvent | T (°C) | Yielda (%) | eeb (%) |
| a Yield of the pure product after flash column chromatography purification. In all cases 1H-NMR analysis of the crude reaction mixture showed the presence of one single diastereoisomer. b Determined by HPLC in a chiral stationary phase (see ESI). c Reaction carried out for 48 h. | ||||||
| 1 | 3a | None | CHCl3 | r.t. | <5 | — |
| 2 | 3a | PhCO2H | CHCl3 | r.t. | <5 | — |
| 3 | 3a | DABCO | CHCl3 | r.t. | 55 | 92 |
| 4 | 3b | DABCO | CHCl3 | r.t. | 50 | 93 |
| 5 | 3c | DABCO | CHCl3 | r.t. | <5 | — |
| 6c | 3d | DABCO | CHCl3 | r.t. | 47 | 95 |
| 7c | 3e | DABCO | CHCl3 | r.t. | <5 | — |
| 8 | 3f | DABCO | CHCl3 | r.t. | 51 | 66 |
| 9 | 3a | DBU | CHCl3 | r.t. | 24 | 90 |
| 10 | 3a | Et3N | CHCl3 | r.t. | 34 | 91 |
| 11 | 3a | DMAP | CHCl3 | r.t. | 45 | 91 |
| 12 | 3a | DABCO | CH2Cl2 | r.t. | 49 | 94 |
| 13 | 3a | DABCO | THF | r.t. | 33 | 93 |
| 14 | 3a | DABCO | Et2O | r.t. | 20 | 93 |
| 15 | 3a | DABCO | iPrOH | r.t. | 16 | 96 |
| 16 | 3a | DABCO | Toluene | r.t. | 30 | 92 |
| 17 | 3a | DABCO | CHCl3 | 50 | 45 | 90 |
| 18c | 3a | DABCO | CHCl3 | −30 | 66 | 96 |
| 19c | 3a | DABCO | CHCl3 | −50 | <5 | — |
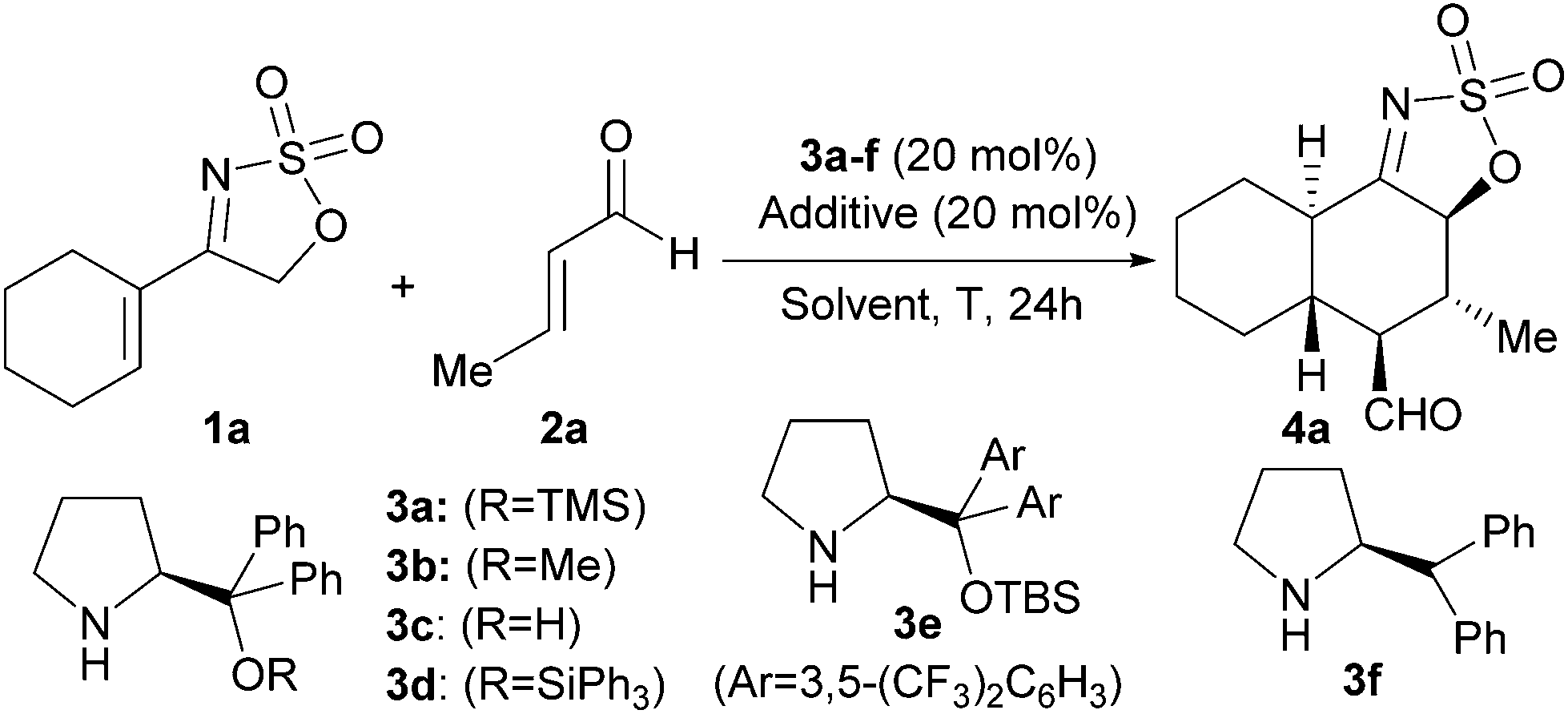
We next evaluated the use of other Brønsted bases as possible cocatalysts but none furnished better results than those observed with DABCO (entries 9–11 vs. entry 3). The effect of the solvent was also studied, observing that dichloromethane performed in similar terms as chloroform (entry 12 vs. entry 3), while the use of more polar solvents (entries 13–15) or a less polar one like toluene (entry 16) affected significantly the yield of the reaction, although it had little effect on the enantioselectivity. We finally evaluated the effect of temperature on the reaction, observing that heating had a deleterious effect on the yield due to product decomposition (entry 17) but lowering the temperature to −30 °C increased the yield to 66%, although requiring a longer reaction time (entry 18). Finally, when the reaction was carried out at an even lower temperature, the rate was importantly affected observing very little product formation after 48 h (entry 19).
Once the best protocol for the reaction had been found, we proceeded next to evaluate the scope of the reaction with regard to the use of other α,β-unsaturated aldehydes in the reaction with cyclic sulfamidate imine 1a as an electron rich diene precursor, which gives direct access to products with the trans-decaline framework and containing multiple stereocentres and different functionalities. As can be seen in Table 2, the reaction proceeded in a satisfactory way in most of the cases studied, furnishing cycloadducts 4a–i in moderate to good yields, as single diastereoisomers and enantioselectivities over 90% ee.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Product | R | T (°C) | Yielda (%) | eeb (%) |
| a Yield of the pure product after flash column chromatography purification. In all cases 1H-NMR analysis of the crude reaction mixture showed the presence of one single diastereoisomer. b Determined by HPLC in a chiral stationary phase (see ESI). c Compound 4h was isolated as the corresponding hemiaminal, formed after the spontaneous intramolecular addition of carbamate to the formyl moiety. d Compound isolated after in situ reduction (NaBH4) of the formyl and azomethine groups. e Reaction carried out on a 0.5 g scale of 1a. | |||||
| 1 | 4a | Me | −30 | 66 | 96 |
| 2 | 4b | nPr | −30 | 67 | 97 |
| 3 | 4b | nPr | r.t. | 71 | 96 |
| 4 | 4c | Et | r.t. | 66 | 95 |
| 5 | 4d | nBu | r.t. | 67 | 95 |
| 6 | 4e | iBu | r.t. | 65 | 97 |
| 7 | 4f | nC5H11 | r.t. | 61 | 95 |
| 8 | 4g | nC6H13 | r.t. | 57 | 95 |
| 9c | 4h | CH2CH2NHCbz | r.t. | 61 | 94 |
| 10d | 4i | Ph | r.t. | 30 | 96 |
| 11e | 4b | nPr | r.t. | 67 | 95 |
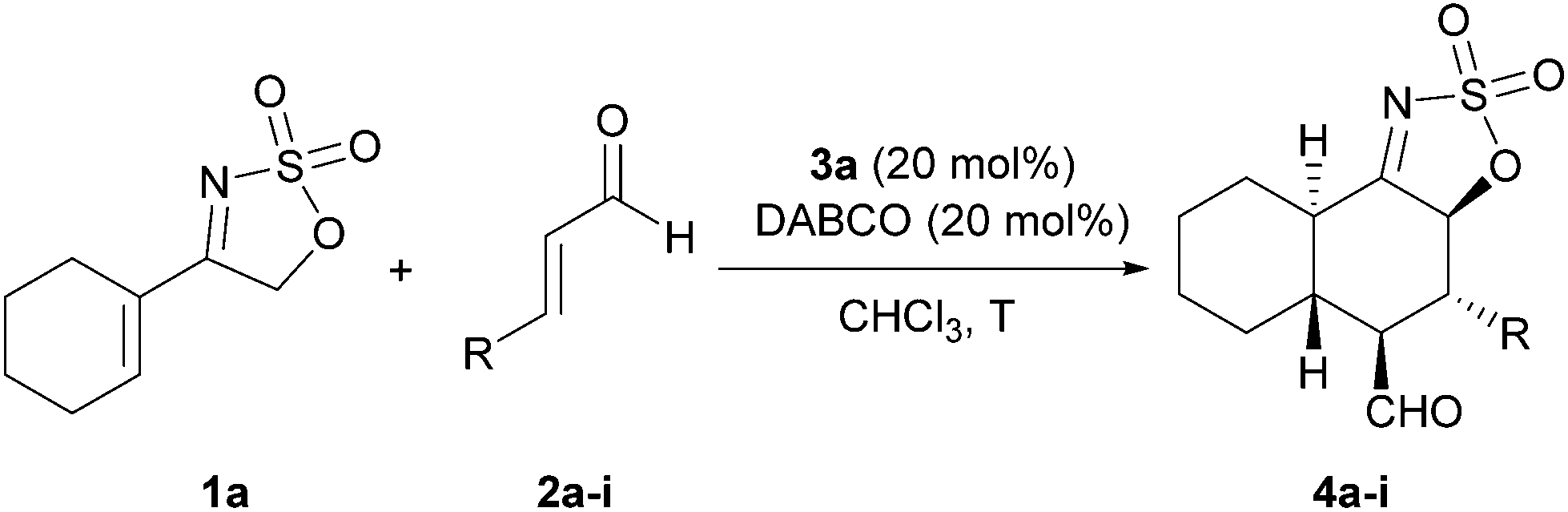
It should be highlighted that both the yield and the diastereoselectivity of the process did not become significantly affected as the size of the chain increased, also observing that moving from the smallest methyl (entry 1) to propyl required a higher temperature (see entry 2 vs. 3). In the same line, the incorporation of longer linear or β-ramified alkyl chains did not affect significantly the yield of the reaction (entries 4–8). Remarkably, we could also use a functionalized enal with success (entry 9) to demonstrate the functional group compatibility of the methodology. On the other hand, moving to an aromatic enal like cinnamaldehyde turned to a very slow reaction that affected significantly the yield of the process most likely due to product decomposition after stirring for such a long reaction time (entry 10). Despite this, adduct 4i was isolated in high diastereo- and enantioselectivity after extensive reduction of both the formyl and the azomethine moieties that resulted in the formation of a more stable compound that could be purified by flash column chromatography. In this case, the yield of the cycloaddition process could not be improved using higher temperatures. Finally, we also checked the performance of the process at a higher scale by carrying out the reaction between 1a and 2b using 0.5 g of the former. As can be seen in entry 11 of Table 2, the reaction also proceeded satisfactorily providing 4b in a comparable yield and enantioselection as those obtained in entry 3.
We next surveyed the performance of other more conformationally flexible cyclic sulfamidate imines with the results shown in Table 3. As can be seen from this table, the reaction of the two substrates 1b and 1c occurred satisfactorily with aromatic enals, regardless of the electronic nature of the aryl substituent.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Product | R1 | R2 | dr | Yielda (%) | eeb (%) |
| a Yield of the pure product after flash column chromatography purification. In all cases NMR of the crude reaction mixture showed the presence of one single diastereoisomer. b Determined by HPLC in a chiral stationary phase (see ESI). | ||||||
| 1 | 5a | Me | Ph | >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
66 | 98 |
| 2 | 5b | Me | 4-MeC6H4 | >20:1 |
59 | >99 |
| 3 | 5c | Me | 4-MeOC6H4 | >20:1 |
65 | >99 |
| 4 | 5d | Me | 4-AcO-3-MeOC6H3 | >20:1 |
72 | >99 |
| 5 | 5e | Me | 3,5-(MeO)2C6H3 | >20:1 |
71 | 99 |
| 6 | 5f | Me | 4-BrC6H4 | >20:1 |
67 | 96 |
| 7 | 5g | Me | 4-NO2C6H4 | >20:1 |
73 | 97 |
| 8 | 5h | Me | 4-CNC6H4 | >20:1 |
73 | 98 |
| 9 | 5i | Me | 4-CF3C6H4 | >20:1 |
67 | 98 |
| 10 | 5j | Me | Me | >20:1 |
36 | 91 |
| 11 | 5k | Et | Ph | >20:1 |
57 | 96 |
| 12 | 5l | Et | 4-MeOC6H4 | 10:1 |
66 | 99 |
| 13 | 5m | Et | 3,5-(MeO)2C6H3 | 8:1 |
61 | 99 |
| 14 | 5n | Et | 4-BrC6H4 | 8:1 |
62 | 97 |
| 15 | 5o | Et | 4-NO2C6H4 | 13:1 |
50 | 94 |
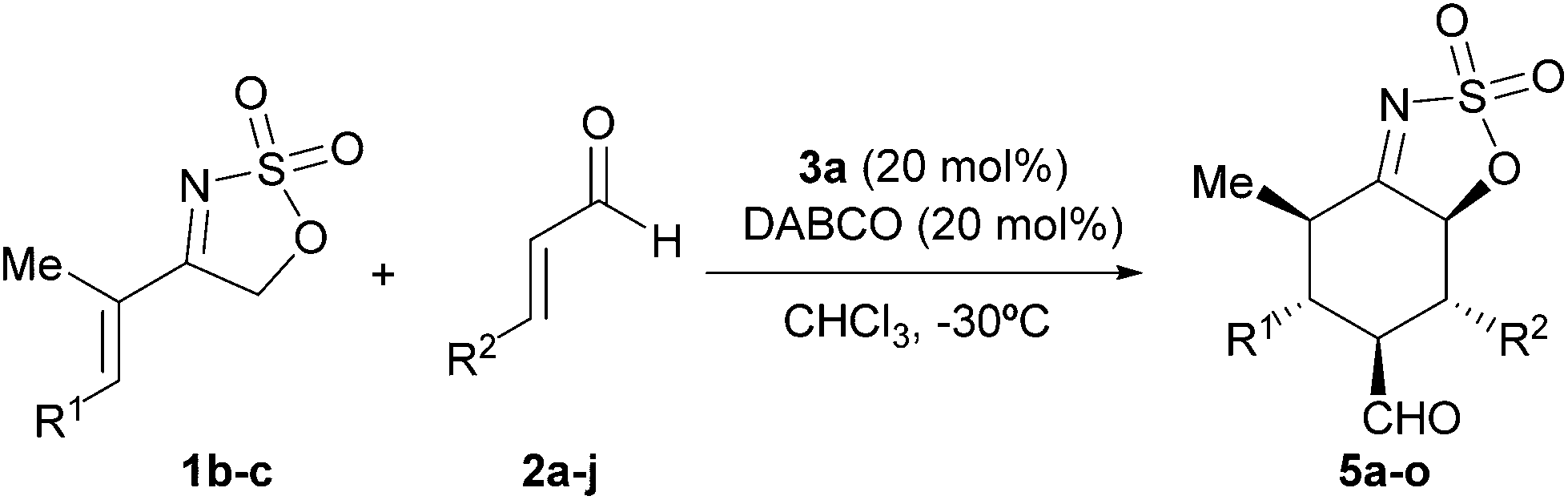
In particular, cinnamaldehyde (entry 1) and other β-aryl substituted α,β-unsaturated aldehydes containing both electron-rich (entries 2–5) and electron-poor (entries 6–9) substituents provided the corresponding [4 + 2] cycloadducts in good to high yields and excellent enantioselectivities when reacted with substrate 1b. In all these cases, adducts 5a–i were isolated as single diastereoisomers. However, the same reaction with crotonaldehyde afforded 5j in poor yield (entry 10), albeit with high enantioselectivity. A similar behavior was observed when the related imine 1c was employed (entries 11–15), although in these cases the presence of minor amounts of the corresponding 5,6-cis diastereoisomer was observed.
We also decided to explore the synthetic possibilities for the 5H-1,2,3-oxathiazole 2,2-dioxide scaffold to behave as a masked aminoalcohol moiety, which is an interesting motif with multiple applications in synthetic organic chemistry being found as part of the structure of many useful chiral building blocks or as chiral auxiliaries or ligands.8 Using adduct 4a as the model compound, we could easily obtain densely functionalized and stereodefined β-aminoalcohol 7 following the reaction sequence shown in Scheme 2. According to this design, we initially carried out the diastereoselective reduction of the azomethine moiety in 4a, which also took place with the concomitant reduction of the formyl group, leading to the quantitative formation of 6 as a single diastereoisomer as indicated by 1H-NMR analysis of the crude reaction mixture. Next, LAH-mediated reduction of the sulfonimidate group, followed by hydrolytic treatment, delivered aminoalcohol 7 without any epimerization of the stereocentres initially generated during the [4 + 2] cycloaddition.
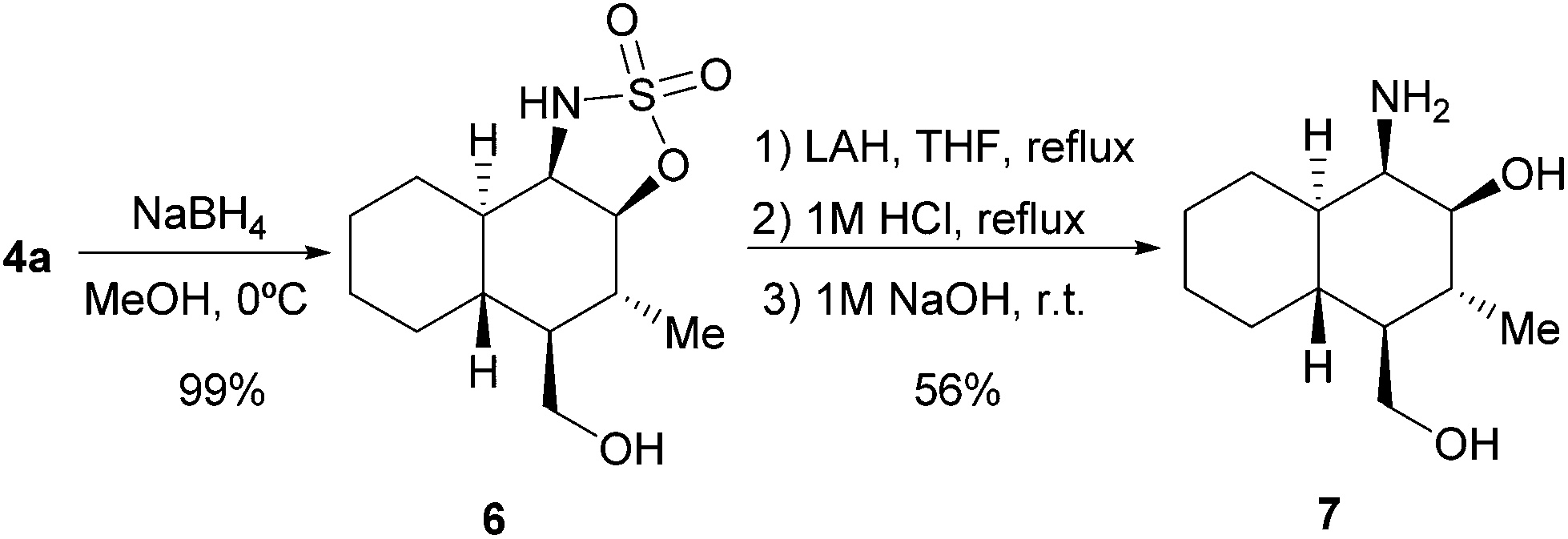 | ||
| Scheme 2 Synthesis of β-aminoalcohol 7 from adduct 4a. | ||
The absolute configuration of the products obtained in the 3a-catalyzed [4 + 2] cycloaddition was established by single-crystal X-ray analysis of compound 4a (Fig. 2).9 According to the absolute stereostructure obtained for this compound, the configuration of all other compounds 4b–i and 5a–o was established based on mechanistic analogy.
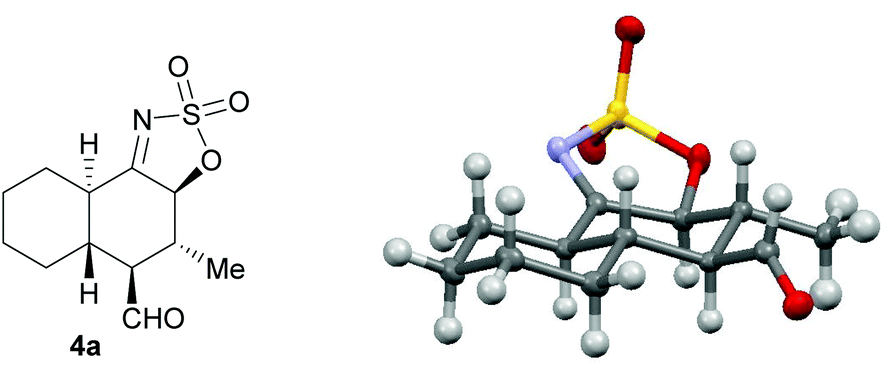 | ||
| Fig. 2 Crystal structure of compound 4a. | ||
A plausible mechanistic proposal for this transformation is given in Scheme 3. In view of the absolute configuration of the final adducts 4 and 5 and, in particular, after observing the relative arrangement between the two stereocentres created at the two terminal positions of the original diene reagent, the possibility of a concerted Diels–Alder cycloaddition pathway seems very unlikely to occur.10 As a consequence, a more plausible proposal would involve a stepwise Michael/Michael cascade reaction involving the iminium/enamine manifold, as shown in Scheme 3. In this sense, activation of the sulfamidate imine 1 by DABCO would deliver an active nucleophilic reagent with the ability to undergo Michael reaction with the α,β-unsaturated iminium ion intermediate generated after condensation between the enal and the catalyst. This initial Michael reaction would generate two initial stereocentres, in which the absolute configuration is in agreement with the approach of the nucleophile to the Michael acceptor through its less hindered Re face11 and presenting the electron-rich enamine moiety and the electron-poor alkene electrophile in an antiperiplanar arrangement between each other. Once the initial Michael reaction had taken place, an enamine intermediate is generated with potential to undergo a second intramolecular Michael reaction with the remaining α,β-unsaturated N-sulfonylimine moiety. This step involves the formation of two other new stereocentres in which the absolute stereochemistry is also consistent with an approach of the electrophile to the (E)-enamine nucleophile through its less hindered face12 and leading to an anti-relative arrangement between the substituents. Finally, the catalyst releasing by hydrolysis and imine/enamine tautomerization would account for the formation of the final cyclohexane adducts. This last tautomerization also leads to the formation of a new stereocentre, whose configuration is presumably driven by thermodynamic issues, as the most stable diastereoisomer in which all substituents of the cyclohexane scaffold are placed in equatorial positions is formed at the end.
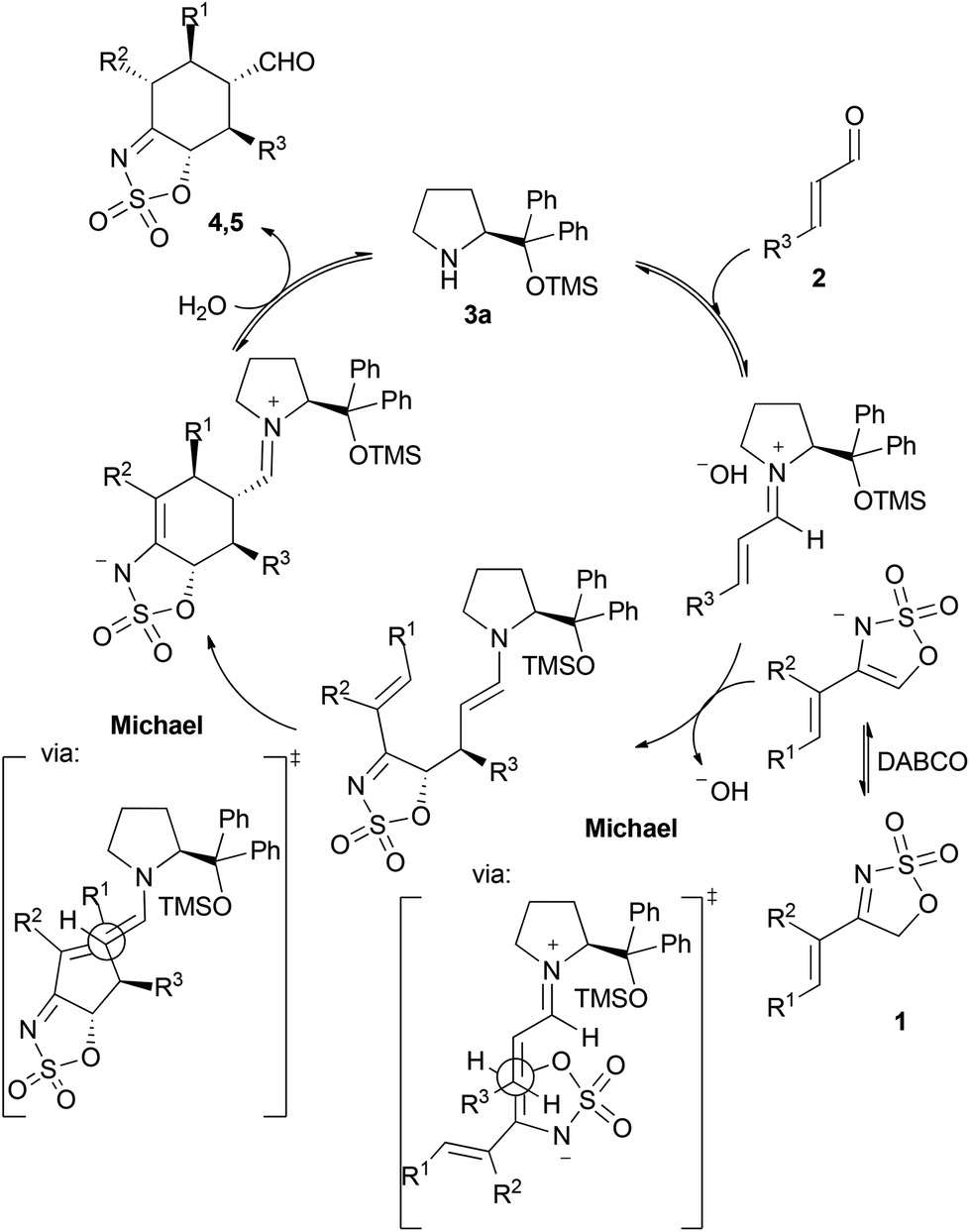 | ||
| Scheme 3 A plausible reaction pathway for the reaction. | ||
Conclusions
In conclusion, we have demonstrated that 4-alkenyl-5H-1,2,3-oxathiazole 2,2-dioxides behave as suitable polyfunctionalized diene precursors that can be generated in situ in the presence of DABCO and which subsequently react with enals in the presence of a chiral secondary amine catalyst to furnish the corresponding [4 + 2] cycloaddition products. This allows the preparation of densely functionalized trans-decalines and cyclohexanes using a variety of different α,β-unsaturated aldehydes and cyclic sulfamidate imines, which are obtained as highly stereopure materials. Moreover, the 5H-1,2,3-oxathiazole 2,2-dioxide entity could be used as a masked β-aminoalcohol moiety, which can be expressed from the obtained cycloadducts by a simple diastereoselective reduction/hydrolysis sequence.Acknowledgements
The authors thank the Spanish MINECO (CTQ2011-22790 and Juan de la Cierva contract to U. U.), the Basque Government (Grupos IT328-10 and OSALAN14/02) and UPV/EHU (UFI QOSYC 11/22 and a fellowship to I. R.) for financial support.Notes and references
- Reviews: (a) A. Megia-Fernández, J. Morales-Sanfrutos, F. Hernández-Mateo and F. Santoyo-González, Curr. Org. Chem., 2011, 15, 401 CrossRef; (b) J. F. Bower, J. Rujirawanich and T. Gallagher, Org. Biomol. Chem., 2010, 8, 1505 RSC; (c) R. E. Meléndez and W. D. Lubell, Tetrahedron, 2003, 59, 2581 CrossRef.
- (a) Y.-J. Chen, Y.-H. Chen, C.-G. Feng and G.-Q. Lin, Org. Lett., 2014, 16, 3400 CrossRef CAS PubMed; (b) T. Nishimura, Y. Ebe, H. Fujimoto and T. Hayashi, Chem. Commun., 2013, 49, 5504 RSC; (c) Y. Luo, H. B. Hepburn, N. Chotsaeng and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 8309 CrossRef CAS PubMed; (d) H.-K. Lee, S. Kang and E. B. Choi, J. Org. Chem., 2012, 77, 5454 CrossRef CAS PubMed; (e) J. Han, S. Kang and H.-K. Lee, Chem. Commun., 2011, 47, 4004 RSC; (f) S. A. Lee, S. H. Kwak and K.-I. Lee, Chem. Commun., 2011, 47, 2372 RSC; (g) S. Kang, J. Han, E. S. Lee, E. B. Choi and H.-K. Lee, Org. Lett., 2010, 12, 4184 CrossRef CAS PubMed; (h) S. Chang and E. E. Lee, Synthesis, 2010, 2361 CAS; (i) Y.-Q. Wang, C.-B. Yu, D.-W. Wang, X.-B. Wang and Y.-G. Zhou, Org. Lett., 2008, 10, 2071 CrossRef CAS PubMed.
- (a) D. Majee, S. Biswas, S. M. Mobin and S. Samanta, Tetrahedron Lett., 2014, 55, 4553 CrossRef CAS PubMed; (b) D. Majee, A. Srivastava, S. M. Mobin and S. Samanta, RSC Adv., 2013, 3, 11502 RSC.
- (a) J. I. Martínez, L. Villar, U. Uria, L. Carrillo, E. Reyes and J. L. Vicario, Adv. Synth. Catal., 2014, 356, 3627 CrossRef; (b) L. Prieto, G. Talavera, U. Uria, E. Reyes, J. L. Vicario and L. Carrillo, Chem. – Eur. J., 2014, 20, 2145 CrossRef CAS PubMed; (c) G. Talavera, E. Reyes, J. L. Vicario, L. Carrillo and U. Uria, Adv. Synth. Catal., 2013, 355, 653 CrossRef CAS; (d) A. Orue, E. Reyes, J. L. Vicario, L. Carrillo and U. Uria, Org. Lett., 2012, 14, 3740 CrossRef CAS PubMed; (e) S. Reboredo, E. Reyes, J. L. Vicario, D. Badia, L. Carrillo, A. de Cózar and F. P. Cossío, Chem. – Eur. J., 2012, 18, 7179 CrossRef CAS PubMed; (f) M. Fernández, E. Reyes, J. L. Vicario, D. Badía and L. Carrillo, Adv. Synth. Catal., 2012, 354, 317 CrossRef; (g) M. Fernández, J. L. Vicario, E. Reyes, L. Carrillo and D. Badía, Chem. Commun., 2012, 48, 2092 RSC; (h) G. Talavera, E. Reyes, J. L. Vicario and L. Carrillo, Angew. Chem., Int. Ed., 2012, 51, 4104 CrossRef CAS PubMed; (i) S. Reboredo, J. L. Vicario, D. Badía, L. Carrillo and E. Reyes, Adv. Synth. Catal., 2011, 353, 3307 CrossRef CAS; (j) E. Reyes, G. Talavera, J. L. Vicario, D. Badía and L. Carrillo, Angew. Chem., Int. Ed., 2009, 48, 5701 CrossRef CAS PubMed.
- Some general reviews: (a) P. T. Parvatkar, H. K. Kaam and S. G. Tilve, Tetrahedron, 2014, 70, 2857 CrossRef CAS PubMed; (b) S. Carosso and M. J. Miller, Org. Biomol. Chem., 2014, 12, 7445 RSC; (c) G. Masson, C. Lalli, M. Benohoud and G. Dagousset, Chem. Soc. Rev., 2013, 42, 902 RSC; (d) J. P. Miller, Adv. Chem. Res., 2013, 18, 179 CAS; (e) J. A. Funel and S. Abele, Angew. Chem., Int. Ed., 2013, 52, 3822 CrossRef CAS PubMed; (f) M. Juhl and D. Tanner, Chem. Soc. Rev., 2009, 38, 2983 RSC; (g) E. J. Corey, Angew. Chem., Int. Ed., 2002, 41, 1650 CrossRef CAS; (h) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668 CrossRef CAS. For specific reviews covering organocatalytic [4 + 2] cycloadditions see: (i) P. Merino, E. Marques-Lopez, T. Tejero and R. P. Herrera, Synthesis, 2010, 1 CrossRef CAS PubMed; (j) J. Shen and C. H. Tan, Org. Biomol. Chem., 2008, 6, 3229 RSC.
- (a) K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243 CrossRef CAS. For some reviews exclusively focused on the iminium activation approach see: (b) A. Erkkila, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416 CrossRef PubMed; (c) G. Lelais and D. W. C. MacMillan, Aldrichimica Acta, 2006, 39, 79 CAS.
- (a) S. Meninno and A. Lattanzi, Chem. Commun., 2013, 49, 3821 RSC; (b) K. L. Jensen, G. Dickmeiss, H. Jiang, L. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248 CrossRef CAS PubMed; (c) C. Palomo and A. Mielgo, Angew. Chem., Int. Ed., 2006, 45, 7876 CrossRef CAS PubMed; (d) A. Mielgo and C. Palomo, Chem. – Asian J., 2008, 3, 922 CrossRef CAS PubMed; (e) L.-W. Xu, L. Li and Z.-H. Shi, Adv. Synth. Catal., 2010, 352, 243 CrossRef CAS.
- (a) C. Weng, H. Zhang, X. Xiong, X. Lu and Y. Zhou, Asian J. Org. Chem., 2014, 26, 3761 CAS; (b) C. Kouklovsky, in Comprehensive Chirality, ed. E. M. Carreira and H. Yamamoto, Elsevier, 2012, vol. 3, pp. 486–527 Search PubMed; (c) G. Della Sala, A. Russo and A. Lattanzi, Curr. Org. Chem., 2011, 15, 2147 CrossRef CAS; (d) C. A. de Parrodi and E. Juaristi, Synlett, 2006, 2699 CrossRef PubMed; (e) M. Lait, D. A. Rankic and B. A. Keay, Chem. Rev., 2007, 107, 767 CrossRef PubMed; (f) S. C. Bergmeier, Tetrahedron, 2000, 56, 2561 CrossRef CAS; (g) D. J. Ager, I. Prakash and S. R. Schaad, Chem. Rev., 1996, 96, 835 CrossRef CAS PubMed.
- CCDC 1034779 contains the supplementary crystallographic data for this paper.
- The only possibility for a concerted Diels–Alder type mechanism would require epimerization of the stereocentre containing the sulfonate substituent. Due to the known acidity of this proton, this is also a plausible possibility that we have also currently under study.
- See for example: (a) Y. Hayashi, D. Okamura, T. Yamazaki, Y. Ameda, H. Gotoh, S. Tsuzuki, T. Uchimaru and D. Seebach, Chem.– Eur. J., 2014, 20, 17077 CrossRef CAS PubMed; (b) U. Groselj, D. Seebach, D. M. Badine, W. B. Schweizer, A. K. Beck, I. Krossing, P. Klose, Y. Hayashi and T. Uchimaru, Helv. Chim. Acta, 2009, 92, 1225 CrossRef CAS; (c) D. Seebach, R. Gilmour, U. Groselj, G. Deniau, C. Sparr, M.-O. Ebert, A. K. Beck, L. B. McCusker, D. Sisak and T. Uchimaru, Helv. Chim. Acta, 2010, 93, 603 CrossRef CAS; (d) M. Nielsen, D. Worgull, T. Zweifel, B. Gschwend, S. Bertelsen and K. A. Jorgensen, Chem. Commun., 2011, 47, 632 RSC.
- See for example: (a) M. B. Schmid, K. Zeitler and R. M. Gschwind, Angew. Chem., Int. Ed., 2010, 49, 4997 CrossRef CAS PubMed; (b) M. B. Schmid, K. Zeitler, R. M. Gschwind, M. B. Schmid, K. Zeitler and R. M. Gschwind, Chem. Sci., 2011, 2, 1793 RSC; (c) M. B. Schmid, K. Zeitler and R. M. Gschwind, J. Am. Chem. Soc., 2011, 133, 7065 CrossRef CAS PubMed; (d) M. B. Schmid, K. Zeitler and R. M. Gschwind, Chem. Sci., 2011, 2, 1793 RSC; (e) J. Bures, A. Armstrong and D. G. Blackmond, J. Am. Chem. Soc., 2012, 134, 6741 CrossRef CAS PubMed; (f) J. Bures, A. Armstrong and D. G. Blackmond, Chem. Sci., 2012, 3, 1273 RSC; (g) P. Diner, A. Kjærsgaard, M. A. Lie and K. A. Jørgensen, Chem. – Eur. J., 2008, 14, 122 CrossRef CAS PubMed . See also ref. 7b and 11d.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation of all new compounds, copies of 1H and 13C-NMR spectra and chiral HPLC traces. CCDC 1034779 (4a). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00316k |
| This journal is © the Partner Organisations 2015 |