
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polycyclic imidazo[1,2-a]pyridine analogs – synthesis via oxidative intramolecular C–H amination and optical properties†
Łukasz
Kielesiński
a,
Mariusz
Tasior
a and
Daniel T.
Gryko
*ab
aInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: dtgryko@icho.edu.pl
bWarsaw University of Technology, Faculty of Chemistry, Noakowskiego 3, 00-664 Warsaw, Poland
First published on 15th October 2014
Abstract
A novel and straightforward approach to synthesize 5H-pyrido[2′,1′:2,3]imidazo[4,5-b]indoles has been developed. The key step is C–H amination of easily available 2-(2′-aminophenyl)imidazo[1,2-a]pyridines by the use of copper(II) triflate, trifluoroacetic acid and (diacetoxyiodo)benzene. The entire strategy consists of just four steps, starting from 2-aminopyridines and acetophenones, giving the target compounds in an overall yield of 20–35%. The optical properties of a library of π-expanded imidazo[1,2-a]pyridines were for the first time fully characterized, showing that these ladder-type compounds strongly absorb UV radiation and exhibit fluorescence in the 415–461 nm region.
Introduction
Imidazo[1,2-a]pyridines are a group of very important heterocycles possessing strong and diverse biological activity. Their structural motif can be found in several marketed drugs, such as anxiolytic alpidem,1 necopidem and saripidem and in drugs used for the treatment of insomnia and brain disorders (zolpidem).2 Their antiviral,3–5 antiparasitic,6 antibacterial,7 anti-inflammatory,8 analgesic and antipyretic9,10 properties are also well documented, as well as their ability to inhibit β-amyloid formation.11 Not surprisingly, methodology of their synthesis has attracted significant attention in the last decade.12–21 Besides the pharmacological importance, imidazo[1,2-a]pyridines exhibit interesting optical properties. In particular, they typically possess high fluorescence quantum yields.22–28With the advent of interest in ladder-type aromatic heterocycles,29,30 we reasoned that π-expanded imidazo[1,2-a]pyridines being analogues of recently explored systems such as indolo[3,2-b]indoles31,32 can offer new opportunities once an efficient synthetic methodology is developed. Indolo[3,2-b]indoles (1), benzofuroindoles (2) and benzothioindoles (3) were recently reported as highly active sex steroid hormone receptor modulators33 and anticancer agents (Fig. 1).34 Indolo[3,2-b]indoles and their analogues were also investigated in optoelectronics.35,36 Only three inefficient synthetic methodologies leading to our targeted 5H-pyrido[2′,1′:2,3]imidazo[4,5-b]indoles were reported: Cadogan cyclization,37–39 multicomponent Bienaymé reaction followed by N-arylation40,41 and ionic liquid promoted cyclization of N-methylisatin and 2-aminopyridine.42 The synthesis of a library of pyridoimidazoindoles and the analysis of the relationship between their structure and spectroscopic properties might open the door to their future optoelectronic applications. In this paper we propose a novel strategy towards this class of nitrogen containing heterocycles, with the key step being oxidative C–H bond amination.
 | ||
| Fig. 1 Comparison of the chemical structures of 5H-pyrido[2′,1′:2,3]imidazo[4,5-b]indoles, indoloindoles (1), benzofuroindoles (2) and benzothioindoles (3). | ||
Results and discussion
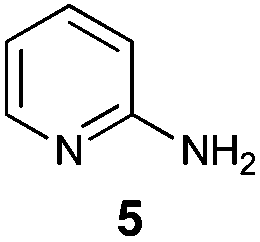
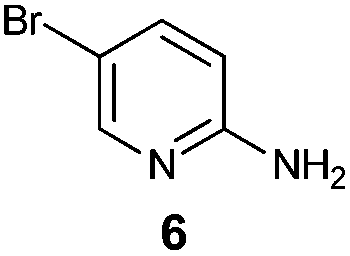
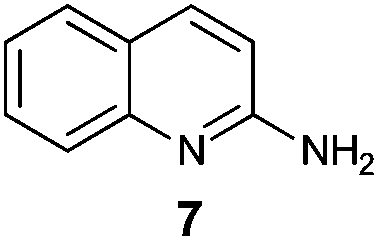
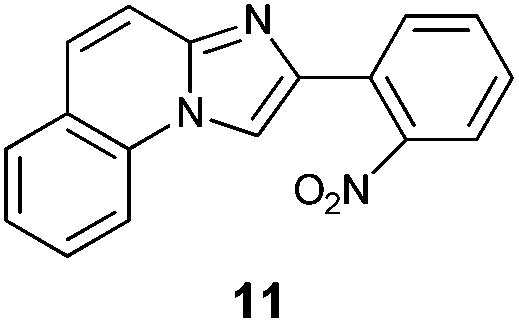
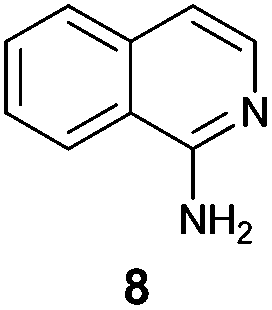
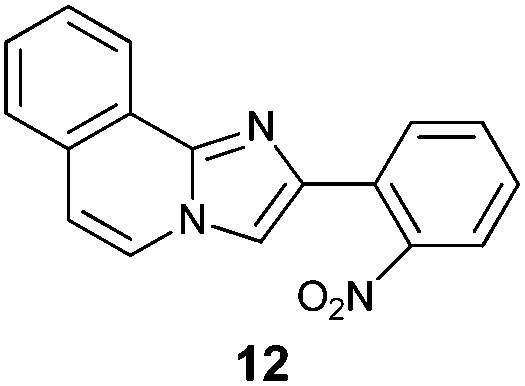
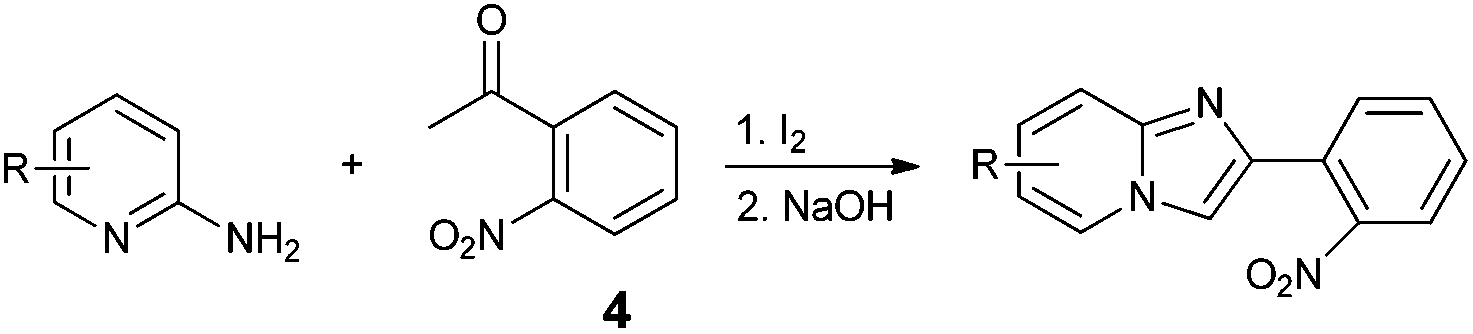
An analysis of possible synthetic routes towards 5H-pyrido[2′,1′:2,3]imidazo[4,5-b]indoles led us to the conclusion that oxidative C–H amination of easily available 2-(2′-aminophenyl) substituted imidazo[1,2-a]pyridines should be a straightforward strategy towards these compounds. This atom-economical process has attracted significant attention in recent years, because it avoids arene preactivation in the synthesis of industrially important heterocycles, e.g. carbazoles. The proposed mechanism for oxidative C–H amination requires the attacked carbon atom to possess a certain electron density. For this reason our strategy should benefit from the fact that the most electron-rich position in imidazo[1,2-a]pyridines is C-3, which remains unsubstituted if the classical Chichibabin method is employed. Thus, we synthesized a series of 2-(2′-nitrophenyl)imidazo[1,2-a]pyridines using the one-pot, tandem Ortoleva–King–Chichibabin process25 (Table 1). Both 2-aminopyridines and their expanded analogs gave the expected imidazopyridines 9–12 in good yield (24–69%).|
|
|||
|---|---|---|---|
| Entry | Amine | Product | Yield (%) |
| a Reaction conditions: (1) amine (20 mmol), 2-nitroacetophenone (10 mmol), I2 (10 mmol), 110 °C, 12 h; (2) NaOH aq. | |||
| 1 |
|
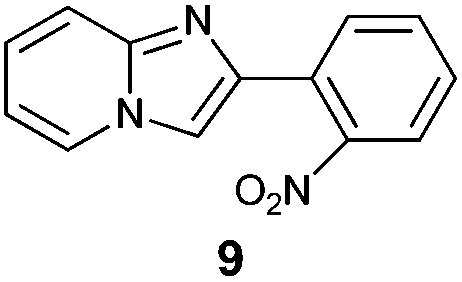
|
69 |
| 2 |
|
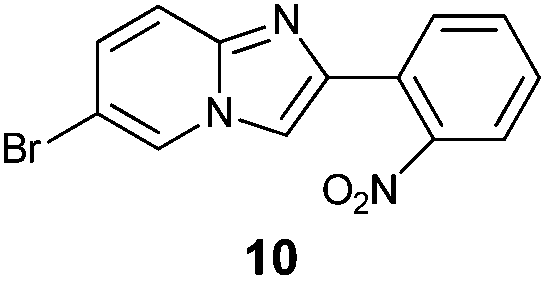
|
64 |
| 3 |
|
|
24 |
| 4 |
|
|
49 |
The previously reported reduction of nitroarenes using H2 and Pd on charcoal did not work in our hands (we observed hydrogenation of the pyridine ring).43 The reaction with iron powder in concentrated hydrochloric acid solution failed too; however the procedure developed by Kundu and co-workers, involving tin(II) chloride, gave the desired 2-(2′-aminophenyl) substituted imidazo[1,2-a]pyridines 13–16 in good yields.44 Even better results could be obtained using indium powder in hydrochloric acid which seems to be the best but a bit expensive reductant (Table 2).
| a Reaction conditions: Reaction conditions: Nitroimidazopyridine (10 mmol), SnCl2 (40 mmol), EtOH, 100 °C, 1.5 h. bNitroimidazopyridine (1 mmol), indium powder (4 mmol), THF, 3 h, rt. cAmine (1 mmol), TsCl (2 mmol), Et3N (2 mmol), AcOEt, 50 °C, 16 h. |
|---|
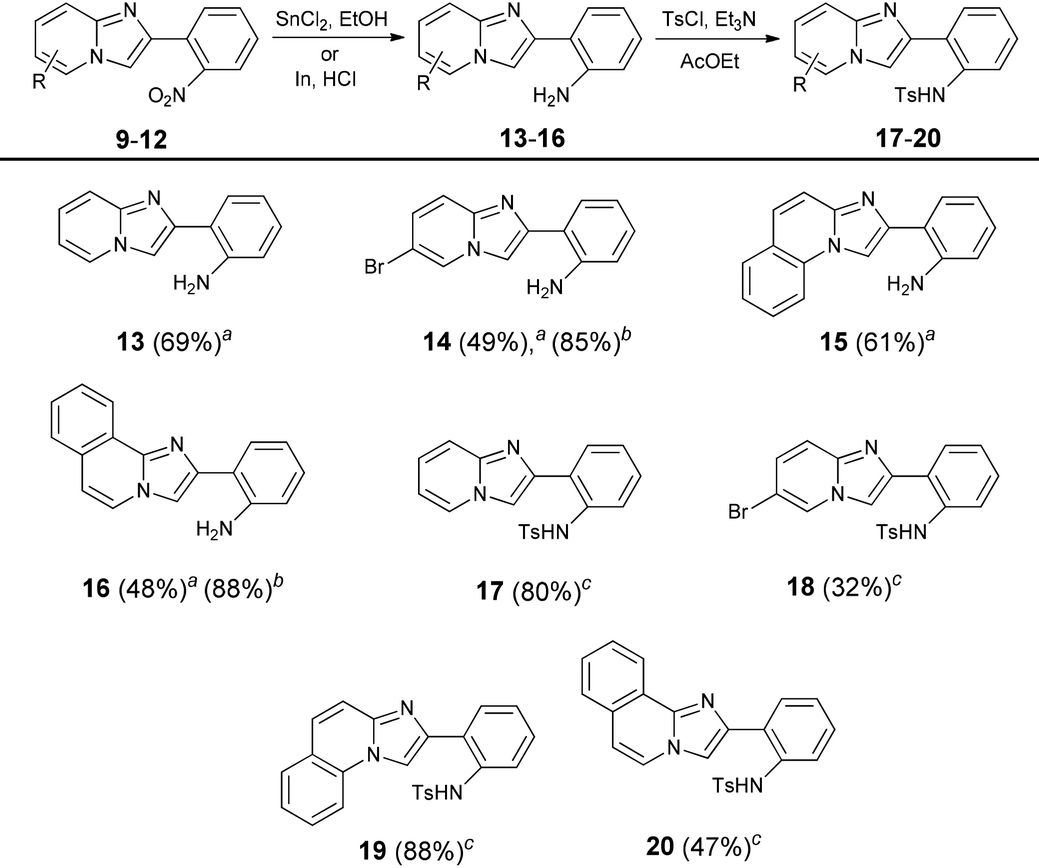
|
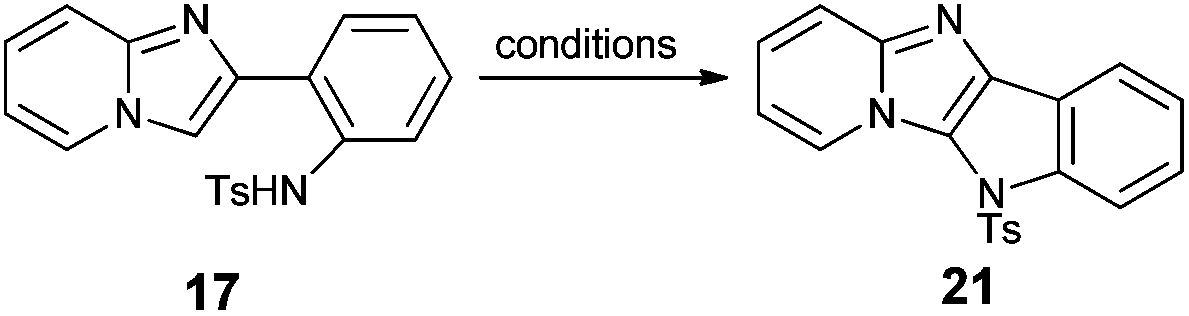
Having 2-(2′-aminophenyl)imidazo[1,2-a]pyridine (13) as a key substrate, we started to search for suitable conditions for oxidative C–H amination. Literature data revealed that N-substituted 2-aryl-1-aminobenzenes possessing various electron-withdrawing groups on the nitrogen atom have been transformed into corresponding carbazoles.45–48 In the preliminary phase of our studies it was found that amination reactions of acetyl- and trifluoromethanesulfonyl-protected amine 13 were sluggish regardless of the conditions, occasionally giving the expected product in very low yields. Given that subsequently we discovered that the tosyl protecting group is the most effective in C–H bond amination we transformed all amines into tosylamides 17–20.
We decided to test four procedures published for carbazoles45–48 in oxidative coupling of amide 17 as a model compound. Buchwald's methodology using palladium(II) acetate as a catalyst and copper(II) acetate/oxygen as a reoxidant did not lead to the expected product (Table 3, entry 1).45 Thus, we adopted the Youn procedure which requires oxone or (diacetoxyiodo)benzene (PIDA) as a reoxidant of palladium(II) acetate (entries 2–5).46 The latter reoxidant allowed us to isolate the desired tetracyclic compound 21; however, the yield was still not satisfactory (entry 4). Finally, we decided to apply Chang's method, which uses copper(II) triflate in combination with PIDA in ethylene chloride.47 This allowed us to isolate 21 in excellent yield and purity (entry 7). There are some reports on an analogous transformation that could be achieved without the use of any metal catalyst source (intramolecular organocatalytic C–H bond amination). Antonchick and co-workers found that N-substituted carbazoles can be obtained by treatment of N-protected diphenylamines with PIDA.48 They also observed the positive effect of a fluorinated co-solvent. As this approach offers a number of advantages over a transition-metal-catalyzed amination, we tried to apply these conditions for oxidation of tosylamide 17 which afforded fused compound 21 in fairly good yield (62%, entry 6). Finally, it was found that the second procedure published by Chang and co-workers,47i.e. (bis-(trifluoroacetoxy)iodo)benzene (PIFA) in the presence of trifluoroacetic acid (TFA), could also be applied, albeit the yield of compound 21 was slightly lower (77%, entry 8).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Reoxidant | Additives | Solvent | Time (h) | Yielda (%) |
| a Isolated yields. b 120 °C. c Room temperature. d 50 °C. | ||||||
| 1 | Pd(OAc)2 | Cu(OAc)2/O2 | Mol. sieves | Toluene | 12b | 0 |
| 2 | Pd(OAc)2 | Oxone | PivOH–DMF | 12c | 0 | |
| 3 | Pd(OAc)2 | Oxone | TsOH | PivOH–DMF | 12c | 0 |
| 4 | Pd(OAc)2 | PIDA | AcOH–DMF | 12c | 40 | |
| 5 | Pd(OAc)2 | PIDA | TsOH | AcOH–DMF | 12c | 32 |
| 6 | PIDA | HFIP–DCM | 16c | 62 | ||
| 7 | Cu(OTf)2 | PIDA | TFA | DCE | 0.5 | 93 |
| 8 | — | PIFA | TFA | DCE | 0.5d | 77 |
Following the well-supported Chang's proposition47 we think that the copper species works as a Lewis acid to activate PIFA, which is the main oxidant inducing the formation of radical intermediates. Having optimized the C–H bond amination, we tried to transform the other sulfonamides. Although bromoderivative 22 smoothly undergoes this reaction, in the case of benzo-fused analogues 19 and 20 we observed the formation of many by-products (Table 4). Therefore we changed the catalytic system to a copper-free PIFA–TFA mixture which gave a much cleaner conversion and higher yields of compounds 23 and 24. The presence of a bromine atom in compound 22 gives an opportunity for further extension of the π-system using palladium catalyzed Sonogashira coupling. This reaction proceeded smoothly, resulting in the formation of compound 25.
| a Reaction conditions: Reaction conditions: Tosylamide (0.2 mmol), PIDA (0.3 mmol), Cu(OTf)2 (0.01 mmol), TFA (0.6 mmol), DCE, 50 °C, 0.5 h. bTosylamide (0.2 mmol), PIFA (0.3 mmol), TFA (0.6 mmol), DCE, 50 °C, 0.5 h. c22 (0.27 mmol), 4-ethynyl-N,N-dimethylaniline (0.69 mmol), Cs2CO3 (0.76 mmol), Pd(OAc)2 (0.035 mmol), PPh3 (0.14 mmol), DMSO, 80 °C, 24 h. |
|---|
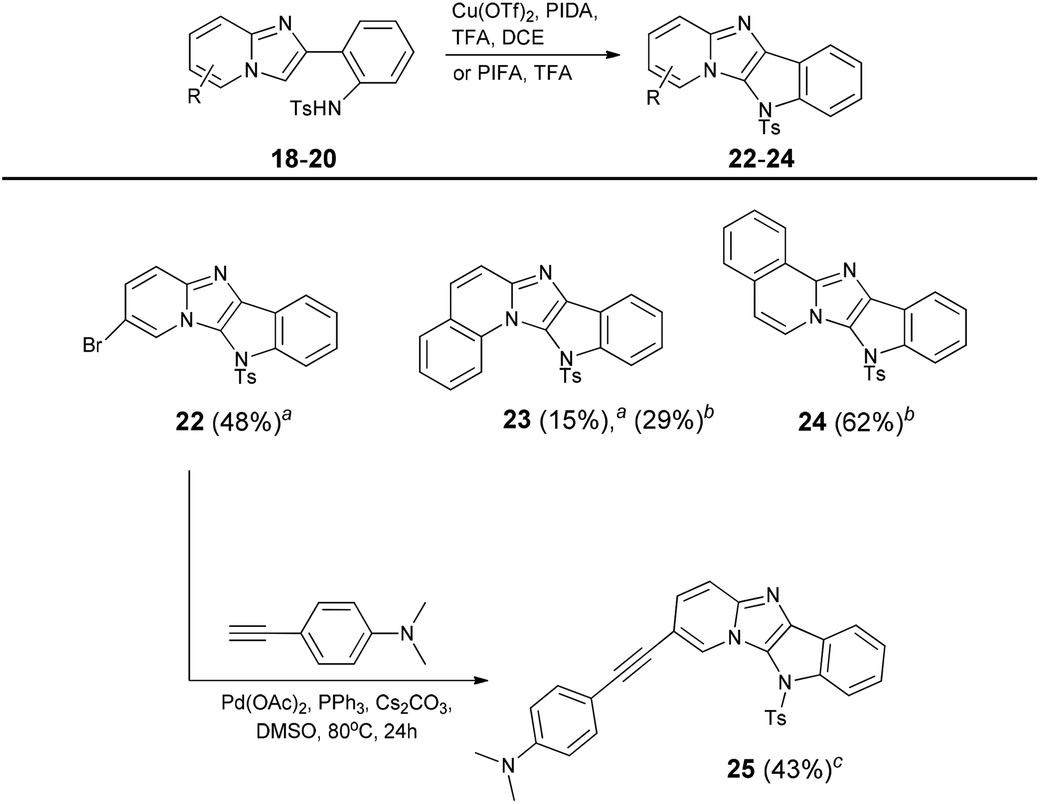
|
The successful synthesis of a small library of 5H-pyrido[2′,1′:2,3]imidazo[4,5-b]indoles and their π-expanded analogs gave us an excellent opportunity for measuring their photophysical properties for the first time. In the case of ladder-type fused heterocycles it is well-known that extending the π-system alters some of the photophysical properties but usually it does not lead to a bathochromic shift of absorption.29,30 The spectroscopic data collected for compounds 21–25 are presented in Table 5. The electronic spectra of indole-fused imidazopyridines typically consist of two strong bands in the 254–355 nm region, whereas a single band was observed in their fluorescence spectra (Fig. 2). Comparison of the properties of compounds 21 and 22 with unsubstituted imidazo[1,2-a]pyridine IP led to a conclusion that fusion with an indole scaffold results in 10–30 nm and 58–68 nm bathochromic shift of absorption and emission maxima, respectively. Due to the angular type of fusion of an additional benzene ring in derivatives 23 and 24, their optical properties strongly resemble those of compound 21. On the other hand, N,N-dimethylphenylethynyl substituted imidazopyridine 25 exhibits well pronounced red-shifted absorption and emission maxima at 355 and 461 nm, respectively. As expected, this compound displays the most intense absorption bands (with ε reaching 130![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) as a result of high π-conjugation and strong dipolar character. The largest Stokes shift was observed for compound 21, which was almost two times higher than that calculated for unsubstituted imidazopyridine. In contrast to imidazo[1,2-a]pyridines compounds 21–25 display very weak fluorescence. Interestingly, the highest fluorescence quantum yield was measured for compound 21. Substitution with a heavy atom (bromo-derivative 22), according to expectations, led to a further decrease in fluorescence intensity. Extension of the π-system probably opens the non-radiative deactivation pathways, which resulted in the lower fluorescence quantum yield observed for compounds 21–25.
000 M−1 cm−1) as a result of high π-conjugation and strong dipolar character. The largest Stokes shift was observed for compound 21, which was almost two times higher than that calculated for unsubstituted imidazopyridine. In contrast to imidazo[1,2-a]pyridines compounds 21–25 display very weak fluorescence. Interestingly, the highest fluorescence quantum yield was measured for compound 21. Substitution with a heavy atom (bromo-derivative 22), according to expectations, led to a further decrease in fluorescence intensity. Extension of the π-system probably opens the non-radiative deactivation pathways, which resulted in the lower fluorescence quantum yield observed for compounds 21–25.
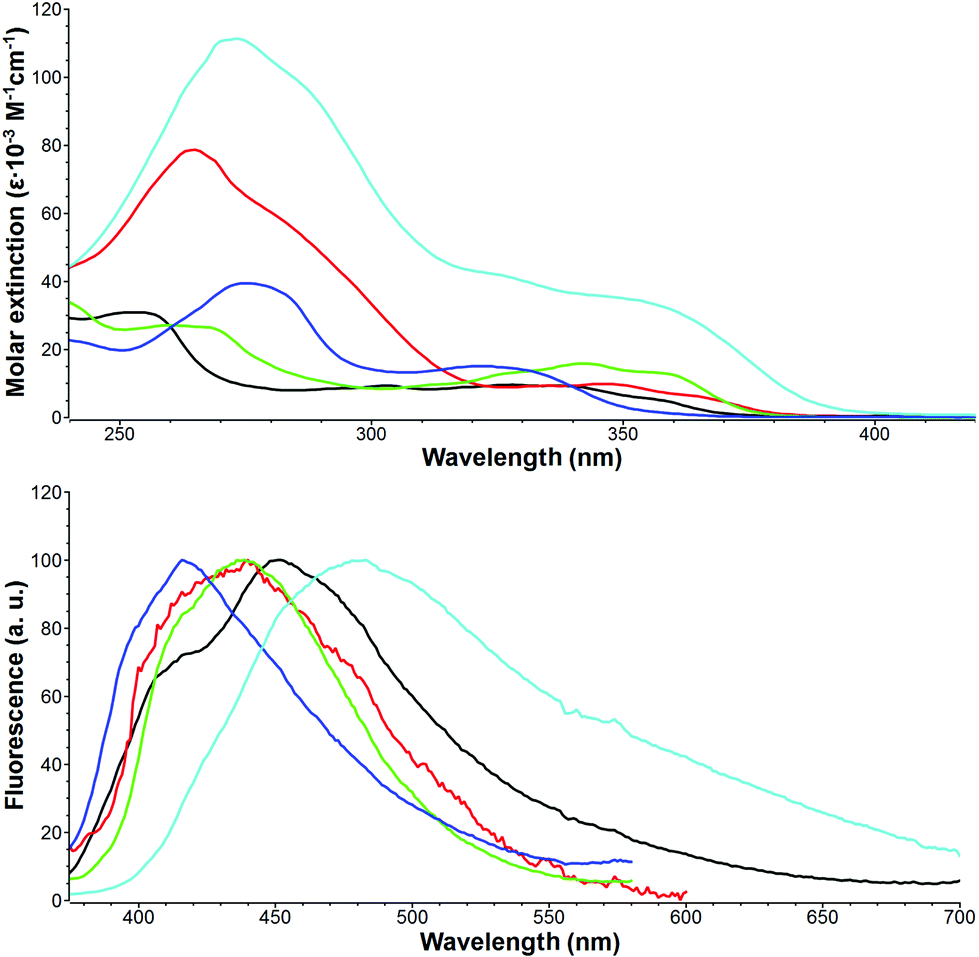 | ||
| Fig. 2 Absorption (top) and normalized fluorescence (bottom) spectra of 21 (black), 22 (red), 23 (green), 24 (blue) and 25 (cyan) measured in DCM. | ||
| Compd | Absmax (nm) | ε (×10−3 M−1 cm−1) | Emissionmax (nm) | Stokes shift (cm−1) | Φ fl |
|---|---|---|---|---|---|
| a Measured in DCM. b IP = unsubstituted imidazo[1,2-a]pyridine (data taken from ref. 49). c Measured with quinine sulphate as a standard. d Excited at 360 nm. e Excited at 310 nm. | |||||
| IP | 318 | 376 | 4800 | ||
| 21 | 254 | 31.1 | 444 | 8000 | 0.034d |
| 303 | 9.28 | ||||
| 328 | 9.51 | ||||
| 22 | 265 | 78.9 | 434 | 5700 | 0.025d |
| 348 | 9.76 | ||||
| 23 | 260 | 27.2 | 432 | 6100 | 0.006e |
| 342 | 15.8 | ||||
| 24 | 275 | 39.7 | 415 | 7000 | 0.008e |
| 322 | 15.1 | ||||
| 25 | 274 | 134 | 461 | 6500 | 0.009d |
| 355 | 33.2 | ||||
Conclusions
It has been proven that intramolecular oxidative C–H amination can also occur with heterocycles bearing a basic nitrogen atom. A new, straightforward, four-step synthesis of 5H-pyrido[2′,1′:2,3]imidazo[4,5-b]indoles was developed. Two sets of suitable reaction conditions were found for the key step, i.e. intramolecular C–H bond amination: Cu(OTf)2–PIDA–TFA and organocatalytic PIFA–TFA combinations. In contrast to many analogous structures, the fluorescence quantum yields of the prepared blue-emitting π-expanded imidazo[1,2-a]pyridine analogs turned out to be low.Experimental section
General information
All the reported NMR spectra (1H NMR and 13C NMR) were recorded on a Varian 500 spectrometer. Chemical shifts (δ ppm) were determined with TMS as the internal reference; J values are given in Hz. High resolution mass spectra (HRMS) were obtained via electron ions (EI). IR spectra were recorded on a JASCO FT/IR-6200 Spectrometer. UV-Vis absorption spectra were recorded on a PerkinElmer Lambda 25 Spectrometer. Fluorescence spectra were recorded on a HITACHI F-7000 Fluorescence Spectrophotometer. Chromatography was performed on silica gel 60 (230–400 mesh) and thin layer chromatography was performed on TLC plates (Merck, silica gel 60 F254). Compound 13 was prepared according to a previously reported procedure.44General procedure for the synthesis of 2-(2-nitrophenyl)imidazo[1,2-a]pyridines
A mixture of 1-(2′-nitrophenyl)ethanone (10 mmol), 2-aminopyridine (20 mmol) and iodine (10 mmol) was heated at 110 °C. After 4 h the temperature of the oil bath was reduced to 70 °C and stirring was continued for an additional 12 h. The resulting waxy solid was dissolved in distilled water and an excess of conc. aqueous sodium hydroxide (45%) was added. Then the reaction mixture was stirred at 100 °C for 1 h, cooled and diluted with CH2Cl2. The pH of the resulting mixture was adjusted to neutral using aqueous HCl (10%) and layers were separated. The aqueous layer was extracted with CH2Cl2, and the combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated under reduced pressure. Column chromatography (silica, CH2Cl2–AcOEt 95:5, gradually increasing the amount of AcOEt) followed by crystallization from AcOEt–hexanes afforded products of analytical purity.
General procedure for the reduction of nitro-imidazo[1,2-a]pyridines
Method A: A solution of nitroimidazopyridine (10 mmol) and SnCl2 (40 mmol) in ethanol was stirred at 100 °C for 1.5 h under argon. Then the reaction mixture was cooled to rt and 5% aqueous NaHCO3 was added until the pH was made slightly basic (pH 8). The resulting slurry was then filtered through a pad of Celite, washed with a small amount of ethanol and concentrated. The aqueous solution was extracted with AcOEt and the combined organic layers were washed with water and brine, dried over Na2SO4 and concentrated under reduced pressure. Purification by column chromatography (silica, CH2Cl2–AcOEt 3:1) and crystallization from AcOEt–hexanes gave the expected product of analytical purity.
Method B: To a solution of nitroimidazopyridine (0.56 mmol) in aq. THF (3 mL), indium powder (260 mg, 2.26 mmol) was added, followed by concentrated HCl (300 μL). After stirring for 3 h at r.t., 5% aqueous NaHCO3 was added until the pH was neutral. After evaporation the crude product was used in the next step without any further purification.
Typical procedure for the preparation of sulfonamides
A solution of toluenesulphonyl chloride (2 mmol) in AcOEt (15 mL) was slowly added to the mixture of aminoimidazo[1,2-α]pyridines (1 mmol) and dry Et3N (2 mmol) in AcOEt (15 mL). The reaction mixture was stirred at 50 °C for 12 h under argon. The resulting suspension was diluted with AcOEt and extracted with H2O. The organic layer was subsequently washed with brine, dried over Na2SO4 and concentrated under reduced pressure. Crystallization from MeOH afforded pure products.Typical procedure for the oxidative coupling of tosylamides
Method A: CF3COOH (46 μL, 0.6 mmol) was slowly added to the mixture of tosylamides 17 or 18 (0.2 mmol), Cu(OTf)2 (0.01 mmol) and PhI(OAc)2 (0.3 mmol) in 1,2-dichloroethane (2 mL) and the whole reaction mixture was stirred at 50 °C for 30 minutes under argon. The resulting solution was cooled, filtered through a pad of Celite and washed with AcOEt. Purification by column chromatography (silica, AcOEt–hexanes 6:4) followed by crystallization from AcOEt–hexanes afforded pure products.
Method B: CF3COOH (46 μL, 0.6 mmol) was slowly added to the mixture of tosylamide 19 or 20 (0.2 mmol) and PhI(CF3COO)2 (0.3 mmol) in 1,2-dichloroethane (2 mL) and the whole reaction mixture was stirred at 50 °C for 30 minutes under argon. The product was purified according to the procedure described in Method A.
:3) followed by crystallization from AcOEt–hexanes afforded 25 as yellow crystals (0.059 g, 43% yield). M.p. 229–232 °C. 1H NMR (DMSO, 500 MHz): δ 9.21–9.19 (m, 1H), 8.19 (d, J = 8.2 Hz, 1H), 7.79 (d, J = 7.7 Hz, 1H), 7.76 (dd, J = 9.5, 0.8 Hz, 1H), 7.54–7.48 (m, 2H), 7.46–7.41 (m, 5H), 7.19 (d, J = 8.2 Hz, 2H), 6.75 (d, J = 9.0 Hz, 2H), 2.89 (s, 6H), 2.21 (s, 3H). 13C NMR (DMSO, 125 MHz): δ 21.4, 84.2, 92.8, 108.2, 109.9, 112.3, 117.6, 118.5, 120.0, 123.4, 126.3, 126.5, 127.1, 127.3, 127.4, 128.0, 130.5, 130.8, 133.0, 139.5, 141.6, 146.5, 147.0, 150.8. IR (KBr, cm−1): 539, 574, 673, 815, 931, 1172, 1371, 1536, 1604, 2211, 3037, 3131. HRMS (EI): m/z calculated for C30H24N4O2S [M˙+] = 504.1620; found: 504.1622.
Acknowledgements
Authors would like to kindly acknowledge financial support from the National Science Centre, Poland (the grant MAESTRO).Notes and references
- S. Z. Langer, S. Arbilla and J. Benavides, Adv. Biochem. Psychopharmacol., 1990, 46, 61 CAS.
- T. S. Harrison and G. M. Keating, CNS Drugs, 2005, 19, 65 CrossRef CAS PubMed.
- A. Elhakmaoui, A. Gueiffier, J.-C. Milhavet, Y. Blache, J.-P. Chapat, O. Chavignon, J.-C. Teulade, R. Snoeck, G. Andrei and E. De Clercq, Bioorg. Med. Chem. Lett., 1994, 4, 1937 CrossRef.
- L. B. Townsend and J. C. Drach, WO9727205, 2009 Search PubMed.
- S. Mavel, J. L. Renou, C. Galtier, R. Snoeck, G. Andrei, J. Balzarini, E. De Clercq and A. Gueiffier, Arzneim.-Forsch., 2001, 51, 304 CAS.
- T. Biftu, D. Feng, M. Fisher, G.-B. Liang, X. Qian, A. Scribner, R. Dennis, S. Lee, P. A. Liberator, C. Brown, A. Gurnett, P. S. Leavitt, D. Thompson, J. Mathew, A. Misura, S. Samaras, T. Tamas, J. F. Sina, K. A. McNulty, C. G. McKnight, D. M. Schmatz and M. Wyvratta, Bioorg. Med. Chem. Lett., 2006, 16, 2479 CrossRef CAS PubMed.
- K. F. Byth, J. D. Culshaw, S. Green, S. E. Oakes and A. P. Thomaset, Bioorg. Med. Chem. Lett., 2004, 14, 2245 CrossRef CAS PubMed.
- R. B. Lacerda, C. K. De Lima and L. L. Da Silva, Bioorg. Med. Chem. Lett., 2009, 17, 74 CrossRef CAS PubMed.
- E. Abignente, F. Arena, E. Luraschi, C. Saturnino, F. Rossi, E. Lampa, M. Cazzola, E. Brandinelli, R. Marrazzo and E. Marmo, Rend. Atti Accad. Sci. Med. Chir., 1985, 139, 313 CAS.
- L. Almirante, L. Polo, A. Mugnaini, E. Provinciali, P. Rugarli, A. Biancotti, A. Gamba and W. Murmann, J. Med. Chem., 1965, 8, 305 CrossRef CAS.
- K. Fuchs, M. Romig, K. Mendla, H. Briem and K. Fechteler, WO2002014313 Search PubMed; Chem. Abstr. 2002 136 183824r Search PubMed.
- J. Lee, J. Chung, S. M. Byun, B. M. Kim and C. Lee, Tetrahedron, 2013, 69, 5660 CrossRef CAS PubMed.
- A. K. Bagdi, M. Rahman, S. Santra, A. Majee and A. Hajra, Adv. Synth. Catal., 2013, 355, 1741 CrossRef CAS.
- K. Monir, A. K. Bagdi, S. Mishra, A. Majee and A. Hajra, Adv. Synth. Catal., 2014, 356, 1105 CrossRef CAS.
- H. Yan, Y. Wang, C. Pan, H. Zhang, S. Yang, X. Ren, J. Li and G. Huang, Eur. J. Org. Chem., 2014, 2754 CrossRef CAS.
- C. Ravi, D. C. Mohan and S. Adimurthy, Org. Lett., 2014, 16, 2978 CrossRef CAS PubMed.
- L. Zhao, H. Zhan, J. Liao, J. Huang, Q. Chen, H. Qiu and H. Cao, Catal. Commun., 2014, 56, 65 CrossRef CAS PubMed.
- S. Santra, S. Mitra, A. K. Bagdi, A. Majee and A. Hajra, Tetrahedron Lett., 2014, 55, 5151 CrossRef CAS PubMed.
- K. Monir, A. K. Bagdi, M. Ghosh and A. Hajra, Org. Lett., 2014, 16, 4630 CrossRef CAS PubMed.
- H. Cao, X. Liu, L. Zhao, J. Cen, J. Lin, Q. Zhu and M. Fu, Org. Lett., 2014, 16, 146 CrossRef CAS PubMed.
- S. Santra, A. K. Bagdi, A. Majee and A. Hajra, Adv. Synth. Catal., 2013, 355, 1065 CrossRef CAS.
- A. Douhal, F. Amat-Guerri and A. U. Acuña, J. Phys. Chem., 1995, 99, 76 CrossRef CAS.
- A. Douhal, F. Amat-Guerri and A. U. Acuña, Angew. Chem., Int. Ed. Engl., 1997, 36, 1514 CrossRef CAS.
- A. Douhal, Ber. Bunsen-Ges. Phys. Chem., 1998, 102, 448 CrossRef CAS.
- A. J. Stasyuk, M. Banasiewicz, M. K. Cyrański and D. T. Gryko, J. Org. Chem., 2012, 77, 5552 CrossRef CAS PubMed.
- D. Firmansyah, A. I. Ciuciu, V. Hugues, M. Blanchard-Desce, L. Flamigni and D. T. Gryko, Chem. – Asian J., 2013, 8, 1279 CrossRef CAS PubMed.
- A. J. Stasyuk, M. Banasiewicz, B. Ventura, M. K. Cyrański and D. T. Gryko, New J. Chem., 2014, 38, 189 RSC.
- D. Firmansyah, M. Banasiewicz, I. Deperasińska, A. Makarewicz, B. Kozankiewicz and D. T. Gryko, Chem. – Asian J., 2014, 9, 2483 CrossRef CAS PubMed.
- A. Fukazawa and S. Yamaguchi, Chem. – Asian J., 2009, 4, 1386 CrossRef CAS PubMed.
- (a) K. Mouri, A. Wakamiya, H. Yamada, T. Kajiwara and S. Yamaguchi, Org. Lett., 2007, 9, 93–96 CrossRef CAS PubMed; (b) R. Gu, S. Van Snick, K. Robeyns, L. Van Meervelt and W. Dehaen, Org. Biomol. Chem., 2009, 7, 380–385 RSC; (c) M. T. Levick, I. Grace, S.-Y. Dai, N. Kasch, C. Muryn, C. Lambert, M. L. Turner and D. J. Procter, Org. Lett., 2014, 16, 2292–2295 CrossRef CAS PubMed; (d) C. Wetzel, A. Mishra, E. Mena-Osteritz, A. Liess, M. Stolte, F. Würthner and P. Bäuerle, Org. Lett., 2014, 16, 362–365 CrossRef CAS PubMed; (e) A. Janiga, E. Głodkowska-Mrówka, T. Stokłosa and D. T. Gryko, Asian J. Org. Chem., 2013, 411 CrossRef CAS; (f) T. Brock-Nannestad, C. B. Nielsen, M. Schau-Magnussen, P. Hammershøj, T. K. Reenberg, A. B. Petersen, D. Trpcevski and M. Pittelkow, Eur. J. Org. Chem., 2011, 6320–6325 CrossRef CAS.
- P. Golubev, Chem. Ber., 1884, 17c, 581 Search PubMed.
- P. Kaszynski and D. A. Dougherty, J. Org. Chem., 1993, 58, 5209 CrossRef CAS.
- Z. Sui, X. Zhang and X. Li, Be. Patent WO047017 A1, 2006 Search PubMed.
- K. W. Bair, U.K. Patent EP0447703 A1, 1991 Search PubMed.
- L. Qiu, X. Zhuang, N. Zhao, X. Wang, Z. An, Z. Lan and X. Wan, Chem. Commun., 2014, 50, 3324 RSC.
- L. Qiu, C. Yu, N. Zhao, W. Chen, Y. Guo, X. Wan, R. Yang and Y. Liu, Chem. Commun., 2012, 48, 12225 RSC.
- W. D. Ollis and S. P. Stanforth, J. Chem. Soc., Perkin Trans. 1, 1989, 961 RSC.
- P. K. Adhikary, S. K. Das and B. A. Hess Jr., J. Med. Chem., 1976, 19, 1352 CrossRef CAS.
- J.-C. Teulade, A. Gueiffier, H. Viols, J.-P. Chapat, G. Grassy, B. Perly and G. Dauphin, J. Chem. Soc., Perkin Trans. 1, 1989, 1895 RSC.
- A. El Akkaoui, M.-A. Hiebel, A. Mouaddib, A. Berteina-Raboin and G. Guillaumet, Tetrahedron, 2012, 68, 9131 CrossRef CAS PubMed.
- V. Tyagi, S. Khan, V. Bajpai, H. M. Gauniyal, B. Kumar and P. M. S. Chauhan, J. Org. Chem., 2012, 77, 1414 CrossRef CAS PubMed.
- I. R. Siddiqui, S. S. Shamim, M. A. Waseem, A. A. H. Abumhdi, A. Srivastava and A. Srivastava, Tetrahedron Lett., 2013, 54, 5083 CrossRef CAS PubMed.
- J. J. Nunes, J. Milne, J. Bemis, R. Xie, C. B. Vu, P. Y. Ng and J. S. Disch, WO2007019416 Search PubMed.
- S. Sharma, B. Saha, D. Sawant and B. Kundu, J. Comb. Chem., 2007, 9, 783 CrossRef CAS PubMed.
- W. C. P. Tsang, R. H. Munday, G. Brasche, N. Zheng and S. L. Buchwald, J. Org. Chem., 2008, 73, 7603 CrossRef CAS PubMed.
- S. W. Youn, J. H. Bihn and B. S. Kim, Org. Lett., 2011, 13, 3738 CrossRef CAS PubMed.
- S. H. Cho, J. Yoon and S. Chang, J. Am. Chem. Soc., 2011, 133, 5996 CrossRef CAS PubMed.
- A. P. Antonchick, R. Samanta, K. Kulikov and J. Lategahn, Angew. Chem., Int. Ed., 2011, 50, 8605 CrossRef CAS PubMed.
- S. Velázquez-Olvera, H. Salgado-Zamora, M. Velázquez-Ponce, E. Campos-Aldrete, A. Reyes-Arellano and C. Pérez-González, Chem. Cent. J., 2012, 6, 83 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra. See DOI: 10.1039/c4qo00248b |
| This journal is © the Partner Organisations 2015 |