
Compound Q is finally deciphered
Ambika
Bhagi-Damodaran
and
Yi
Lu
*
Department of Chemistry, University of Illinois, Urbana-Champaign, Urbana, Illinois 61801, USA. E-mail: yi-lu@illinois.edu
First published on 28th July 2015
Abstract
Methane monooxygenases (MMOs) activate the high energy C–H bond of methane and convert it to methanol with high selectivity and under physiological conditions. Despite decades of efforts focusing on elucidating the structure, function and mechanism of soluble MMOs, the structure of a key intermediate (called compound Q) remains unknown. This article highlights a recent report by Banerjee et al. which not only firmly establishes the core-structure of Q, but also provides significant insight into its formation, reaction with methane and eventual decay.
Methane is the primary component of natural gas; it is relatively abundant and could potentially serve as a practical substitute for petroleum-based fuels. However, its gaseous form makes its storage and transportation a challenging task. Moreover, the gas may leak from wells or through pipelines and act as a potent greenhouse gas. One solution to these challenges is to convert methane to liquid methanol, which is not only easier to store/transport but also serves as a raw material for several chemicals and polymers. The commercial process for methanol production involves industrial cracking of syn gas, which is a multi-step energy-consuming process.1 In contrast, the methane monooxygenase (MMO) enzyme from methanotrophic bacteria utilizes inexpensive and readily available oxygen to oxidize the high energy C–H bond (104 kcal mol−1) of methane to produce methanol under ambient conditions without producing over-oxidized products such as formaldehyde or CO2. Two types of MMOs have evolved to perform this reaction: a membrane-bound particulate MMO present in most methanotrophs and a soluble MMO expressed in some methanotrophs under copper-limiting conditions.2 Although there is some debate regarding the catalytic active site of the particulate MMO as to whether it is a mono-, di- or tri-nuclear copper center, the catalytic centre in the hydroxylase subunit of soluble MMO (called MMOH) is unquestionably characterized as a carboxylate-bridged diiron unit (Fig. 1). The oxidative chemistry taking place at these catalytic sites, especially the latter, has been investigated extensively over the last 20 years.3 Nevertheless, the chemical structure of the key oxidizing species that reacts with methane and cleaves the high energy C–H bond, called compound Q for MMOH, has remained unclear. A recent report by Banerjee et al. solved this missing piece in the puzzle of the MMOH mechanism by unequivocally establishing the structure of Q as a diamond-core bis-μ-oxo FeIV FeIV cluster.4
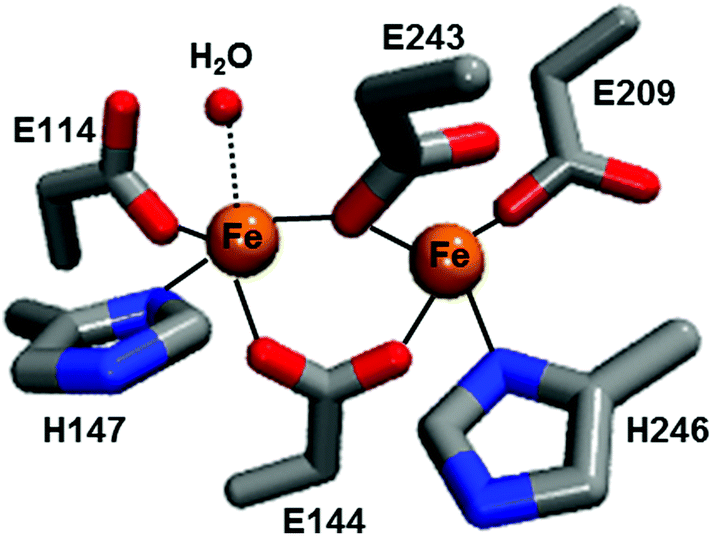 | ||
| Fig. 1 Diiron active site structure of reduced Mc MMOH (PDB: 1FYZ). The amino acid residues are shown in stick representation while iron and water molecules are shown as spheres. | ||
The compound Q was first detected by transient kinetics in a single-turnover reaction between di-ferrous MMOH and oxygen5 (Fig. 2). The experiment revealed the formation of three intermediates named P, Q and T. While many spectroscopic studies have identified the intermediates P and T as μ-peroxodiiron(III) and μ-oxodiiron(III) respectively, the structure of compound Q, which displays broad absorbance bands around 330 nm and 430 nm, has been very difficult to pinpoint, because it does not exhibit any EPR signal, attributable to exchange coupled high-valent FeIVFeIV form (as confirmed later by Mössbauer spectroscopy6). Additionally, the decay rate of compound Q increased linearly with the methane concentration, suggesting it to be an activated form of MMOH that directly catalysed methane oxidation.5 Thus, it was necessary to elucidate the exact structure of Q in order to understand how MMOH catalyses methane oxidation and to design more robust and less expensive artificial catalysts to carry out the same reaction. Even though the oxidation and spin states of compound Q were fully characterized via UV-Vis, EPR and Mössbauer spectroscopic methods, its core structure remained ambiguous. To address this issue, an extended X-ray absorption fine structure (EXAFS) study on Q revealed a short Fe–Fe separation of 2.47 Å and two short Fe–O bonds of slightly different lengths. It was proposed that these structural characteristics could best be explained by the presence of two asymmetric single-atom oxygen bridges between the irons forming a diamond-shaped core.7 This proposal was further supported by comparison with spectroscopic data of structurally characterized synthetic model complexes as well as the oxygen evolving complex of photosystem II, which exhibited an M2(μ-O)2 geometry and metal distances ranging between 2.5 and 2.9 Å. However, because the EXAFS data only definitively reflected the presence of two short Fe–O bonds, other core structures for compound Q were also conceivable. As a result of this uncertainty, the ‘diamond-core’ hypothesis came under scrutiny by several experimental and computational observations. For example, high-level density functional theory (DFT) calculations could not reproduce the short Fe–Fe distance of compound Q as predicted by EXAFS studies.8 Furthermore, a synthetic model, possessing a [FeIV2(μ-O)2] diamond core and anti-ferromagnetic coupling similar to compound Q, exhibited an Fe–Fe distance of 2.73 Å. Even though the irons in the synthetic model differed in spin state and ligation from Q, the Fe–Fe distance was clearly longer.9 Finally, the conversion of a synthetic complex with a valence-delocalized [Fe3.5(μ-O)2Fe3.5]3+ diamond-core structure into a complex with a valence-localized [HO–FeIII–O–FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O]2+ open core resulted in over a million-fold faster aliphatic C–H bond cleavage. This activity enhancement, presumed to be resulting from the formation of a terminal oxo-iron(IV) moiety with a high oxidizing capability and conversion of the low-spin (S = 1) centre to a high-spin (S = 2) centre, further raised the question of the viability of a diamond core as the structure of the key oxidizing species of compound Q.10
O]2+ open core resulted in over a million-fold faster aliphatic C–H bond cleavage. This activity enhancement, presumed to be resulting from the formation of a terminal oxo-iron(IV) moiety with a high oxidizing capability and conversion of the low-spin (S = 1) centre to a high-spin (S = 2) centre, further raised the question of the viability of a diamond core as the structure of the key oxidizing species of compound Q.10
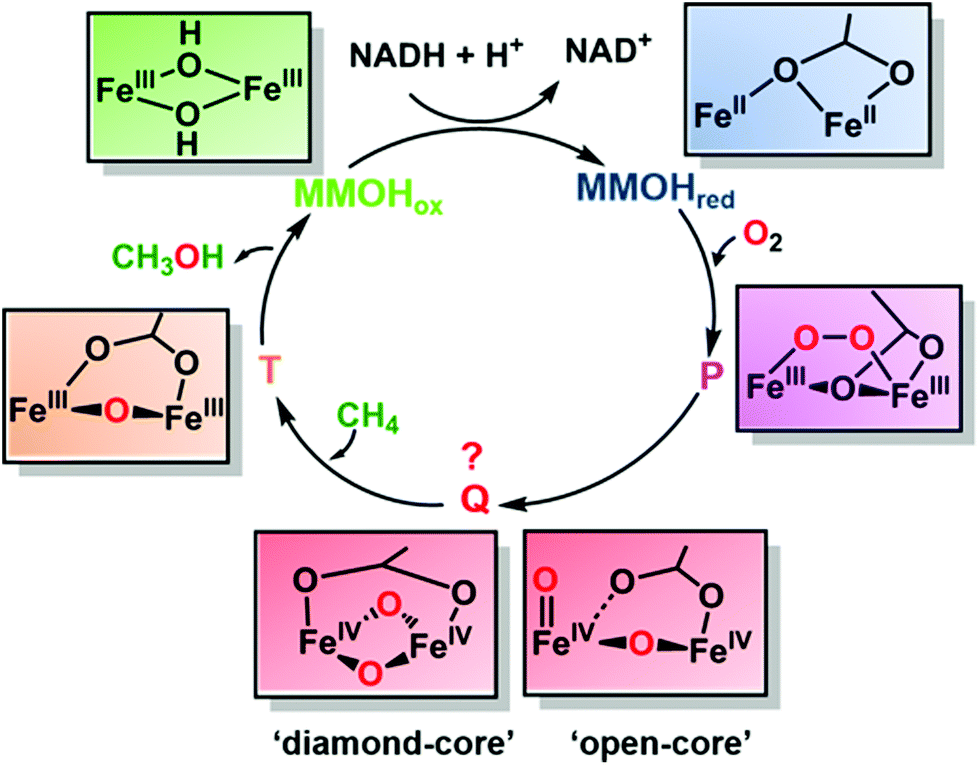 | ||
| Fig. 2 Simplified mechanism of methane oxidation in MMOH. The reduced diiron center (MMOHred) binds an oxygen molecule (in red) to form a peroxy intermediate (P), which then cleaves the O–O bond to form Q. Compound Q is the key oxidizing species in MMOH and could possess either closed ‘diamond-core’ or ‘open-core’ structure. It reacts with methane to oxidize the high energy C–H bond to form methanol converting itself to intermediate T, which finally decomposes to the oxidized form of the enzyme (MMOHox). | ||
As a result, the structure of compound Q had remained a mystery for many years. The definite characterization of the core structure of compound Q in MMOH, in principle, could be possible using resonance Raman (RR) spectroscopy, as it can detect molecular vibrations from the stretching/bending of Fe–O bonds providing clues to the Fe–O structure and bonding. However, there were several challenges associated with such an experiment, for example (1) relatively weak Raman enhancement of signals demanded a high sample concentration and (2) photolysis of compound Q resulted in the destruction of the intermediate before Raman spectra could be collected. Banerjee et al. overcame these obstacles using a custom-designed time-resolved RR instrument11 that continuously mixed the reduced MMOH with oxygen and a substrate (methane or furan) in a flowing system. By recording at a specific time after mixing in the flowing system, the RR spectrum of the short-lived compound Q could be averaged for hours at high concentration and without photolysis. By comparing the spectra generated using oxygen-16 isotope (16O2) with that generated using oxygen-18 isotope (18O2), they isolated vibrations due to Fe–O bonds in MMOH from vibrations due to the protein backbone or the solvent. On mixing di-ferrous MMOH with oxygen in this experimental set-up, they observed a vibration at 690 cm−1 which disappeared on the addition of substrates methane/furan and exhibited a large deuterium kinetic isotope effect when CD4 was used as the substrate, suggesting the vibration to be due to compound Q. Since the frequency of the compound Q vibration was much lower than that typically observed for Fe(IV)O species (850 cm−1), but much higher than that reported for Fe(IV)–OH species (565 cm−1), this finding ruled out the possibility of an ‘open’ structure. Furthermore, the compound Q vibration at 690 cm−1 was similar to that of other structurally characterized synthetic models that bear diamond-core (FeIVμ-O)2 structures (666–674 cm−1), making it possible to define the structure of compound Q as a ‘closed-diamond’ core (Fig. 2). In order to probe the origin of the two oxygen atoms in compound Q, which could be from either an activated oxygen molecule or the solvent water molecule, the authors used a mixed isotopic form of dioxygen (16O18O) and observed a new frequency in the spectrum at 673 cm−1. The exact positioning of this feature, midway between those of 16O2 (690 cm−1) and 18O2 (654 cm−1) incorporated Q, could only be explained as a tetratomic vibration arising from a diamond-core structure into which both 16O and 18O atoms of the dioxygen (16O18O) molecule have been incorporated. The experiment suggests that the O–O bond in the peroxo- (intermediate P) cleaves to form compound Q. However, the mechanism of O–O bond cleavage, whether homolytic or heterolytic, remains uncertain. The energy constraints of the system, in fact, favor the homolytic cleavage of the O–O bond for MMOH, in stark contrast to heme-containing cytochrome P450s which also activates oxygen to oxidize aliphatic C–H bonds but cleaves the O–O bond heterolytically to form the key oxidizing species heme compound I.12
Overall, the study makes significant progress towards not only elucidating the mechanism of methane oxidation but also understanding the structure–function relationship of other oxygen activating diiron enzymes, such as ribonucleotide reductase which generates an intermediate ‘X’ possessing a relatively short FeIII FeIV bond length similar to the intermediate ‘Q’.13 The study also opens up several questions in the field of bioinorganic chemistry regarding the reactivity and formation of compound Q. For example, why are biomimetic models exhibiting a diamond-core structure like compound Q not reactive enough to oxidize methane? Potential causes may include factors such as the low-spin state of synthetic models9,10 or the lack of hydrogen bonding provided by glutamate in synthetic models.14 Furthermore, what properties of MMOH cause the peroxo-species (intermediate P) to spontaneously convert to compound Q whereas the peroxodiiron(III) units of most other diiron enzymes do not appear to access such high-valent intermediates?9 Potential causes may include water molecules in the active site of MMOH which may help to polarize and activate the bound peroxide.15
Like many important discoveries in science and engineering, Banerjee et al. were able to achieve a milestone in methane oxidation chemistry by working extremely hard (the experiments required a whopping 30 g of highly purified MMOH), developing and optimizing a novel experimental setup (that of continuously mixing and flowing time-resolved RR spectroscopy) and meticulously performing and analysing the data. The work addresses a key issue that puzzled researchers in the MMO field for many years and provides a strong basis for understanding nonheme diiron enzymes and making efficient catalysts for applications in C–H bond oxidation.
Acknowledgements
The authors acknowledge and thank Professors J. D. Lipscomb and D. Proshlyakov, Shiliang Tian and Braddock A. Sandoval for critical reading of the manuscript and important inputs. The work in the Lu group has been financially supported by the US National Institutes of Health (GM062211).References
- C. A. Haynes and R. Gonzalez, Nat. Chem. Biol., 2014, 10, 331 CrossRef CAS PubMed.
- S. J. Lippard and C. E. Tinberg, Acc. Chem. Res., 2011, 44, 280 CrossRef PubMed.
- S. Sirajuddin and A. C. Rosenzweig, Biochemistry, 2015, 54, 2283 CrossRef CAS PubMed.
- R. Banerjee, Y. Proshlyakov, J. D. Lipscomb and D. Proshlyakov, Nature, 2015, 518, 433 CrossRef PubMed.
- S.-K. Lee, J. Nesheim and J. D. Lipscomb, J. Biol. Chem., 1993, 268, 21569 CAS.
- S.-K. Lee, B. G. Fox, W. A. Froland, J. D. Lipscomb and E. Münck, J. Am. Chem. Soc., 1993, 115, 6450 CrossRef CAS.
- L. Shu, J. D. Lipscomb and L. Que Jr., Science, 1997, 275, 515 CrossRef CAS.
- B. F. Gherman, M.-H. Baik, S. J. Lippard and R. A. Friesner, J. Am. Chem. Soc., 2004, 126, 2978 CrossRef CAS PubMed.
- G. Xue, D. Wang, R. De Hont, A. T. Fiedler, X. Shan, E. Münck and L. Que Jr., Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20713 CrossRef CAS PubMed.
- G. Xue, E. Münck and L. Que Jr., Nat. Chem., 2010, 2, 400 CrossRef CAS PubMed.
- P. K. Grzyska, E. H. Appelman, R. P. Hausinger and D. A. Proshlyakov, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3982 CrossRef CAS PubMed.
- J. Rittle and M. T. Green, Science, 2010, 330, 6006 CrossRef PubMed.
- N. Mitic, M. D. Clay, L. Saleh, J. M. Bollinger Jr. and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 9049 CrossRef CAS PubMed.
- L. H. Do, T. Hayashi, P. Moënnoz-Loccoz and S. J. Lippard, J. Am. Chem. Soc., 2010, 132, 1273 CrossRef CAS PubMed.
- D. M. York and S.-T. Lee, Multi-scale Quantum Models for Biocatalysis, Springer, 2009 Search PubMed.
| This journal is © the Partner Organisations 2015 |