
Catalytic dehydrogenative borylation of terminal alkynes by POCOP-supported palladium complexes†
Christopher J.
Pell
and
Oleg V.
Ozerov
*
Department of Chemistry, Texas A&M University, College Station, TX 77842, USA
First published on 12th June 2015
Abstract
A (POCOP)-supported palladium complex was used to catalyse the dehydrogenative borylation of terminal alkynes to form alkynylboronates. Competing hydrogenation reactions could be mitigated through the use of additives such as phosphines or elemental mercury.
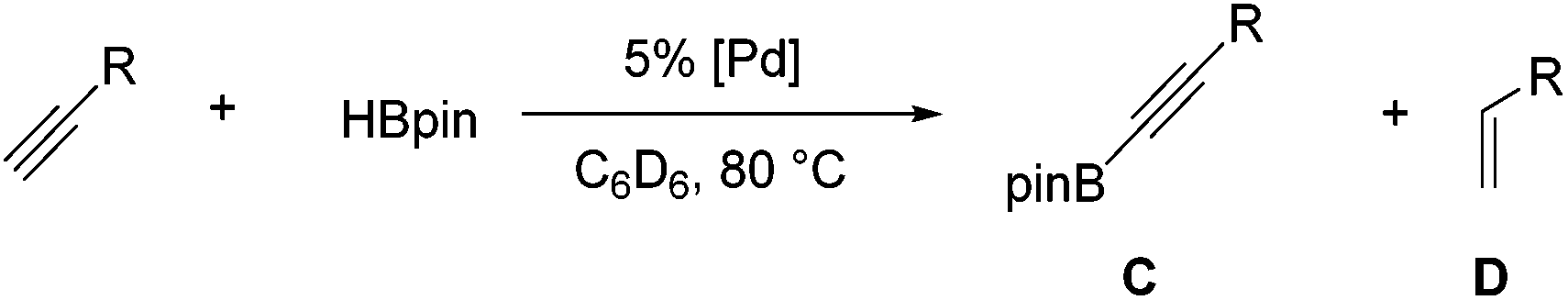
The dehydrogenative formation of carbon–boron bonds directly from carbon–hydrogen bonds is one of the most attractive methods of transition metal-catalysed C–H functionalization due to the utility of the resulting organoboronate reagent.1 Significant progress has been achieved with iridium and rhodium catalysts, especially in borylation of aromatic C–H bonds.2 Group 10 metal catalysts have featured less prominently in the studies of C–H borylation. Palladium is responsible for most of these examples such as the borylation of olefins,3 isoindoles,4 phenylpyridines,5 and benzylic arms of alkylbenzenes.6 Nickel has recently made its first appearance as a dehydrogenative C–H borylation catalyst with the borylation of arenes and indoles.7 Platinum is rarely mentioned in the field of dehydrogenative borylation, but has been shown to catalyse the dehydrogenative borylation of olefins with borane clusters.3b
Our group recently introduced dehydrogenative borylation of terminal alkynes (DHBTA), a new type of a C–H borylation reaction that produces alkynylboronates (Scheme 1). Alkynylboronates are widely applicable in organic synthesis and their use has recently been reviewed.8 In the original report, we utilized an iridium catalyst supported by a SiNN-type ligand,9 and later improved on it through the use of other pincer ligands.10 A slower DHBTA catalysis with a similar scope was reported by Tsuchimoto et al. using Zn(OTf)2/pyridine.11 Interestingly, SiNN complexes of Rh and the non-pincer Ir catalysts that excel at aromatic C–H borylation do not catalyse DHBTA. In the search for alternative DHBTA catalysts outside of group 9, we sought to examine pincer complexes of group 10 metals. We surmised that pincer-ligated group 10 metal hydrides should be appropriate for entry into potential DHBTA catalysis, especially since they have been shown to dehydrogenatively form metal alkynyl and metal boryl species, plausible catalytic intermediates.12–15 We chose to focus on the aryl/bisphosphinite ligand family16 because of its simple synthesis and the relative ease with which we can vary the size of the phosphine substituents.
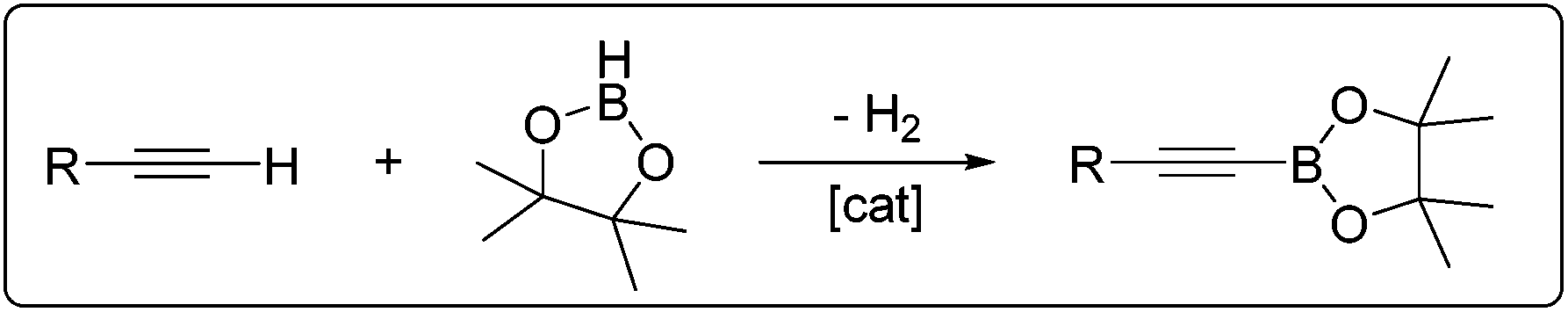 | ||
| Scheme 1 Dehydrogenative borylation of terminal alkynes. | ||
Compound 1a was synthesized by a literature procedure17 while 2a and 3a were synthesized by treating 2 and 3 with NaHBEt3 (Scheme 2). To test the catalytic potential of the (POCOP) group 10 metal hydrides, 5 mol% of 1a, 2a, and 3a were dissolved in C6D6 and treated with 4-ethynyltoluene and pinacolborane (HBpin) at 80 °C for 1 day (Table 1). The reaction with 1a (entry 1) gave a complex catalytic mixture of products containing olefinic signals as seen by 1H NMR spectroscopy due to the ability of terminal alkynes to insert into the nickel–hydride bond,18 while 3a yielded about 5% of the hydroboration product, (E)-(4-methylstyryl)Bpin (entry 2). 2a did act as a DHBTA catalyst, converting 40% of the alkyne into the alkynylboronate (A). However there was an almost equal amount of 4-methylstyrene produced from the semihydrogenation of 4-ethynyltoluene (entry 3).
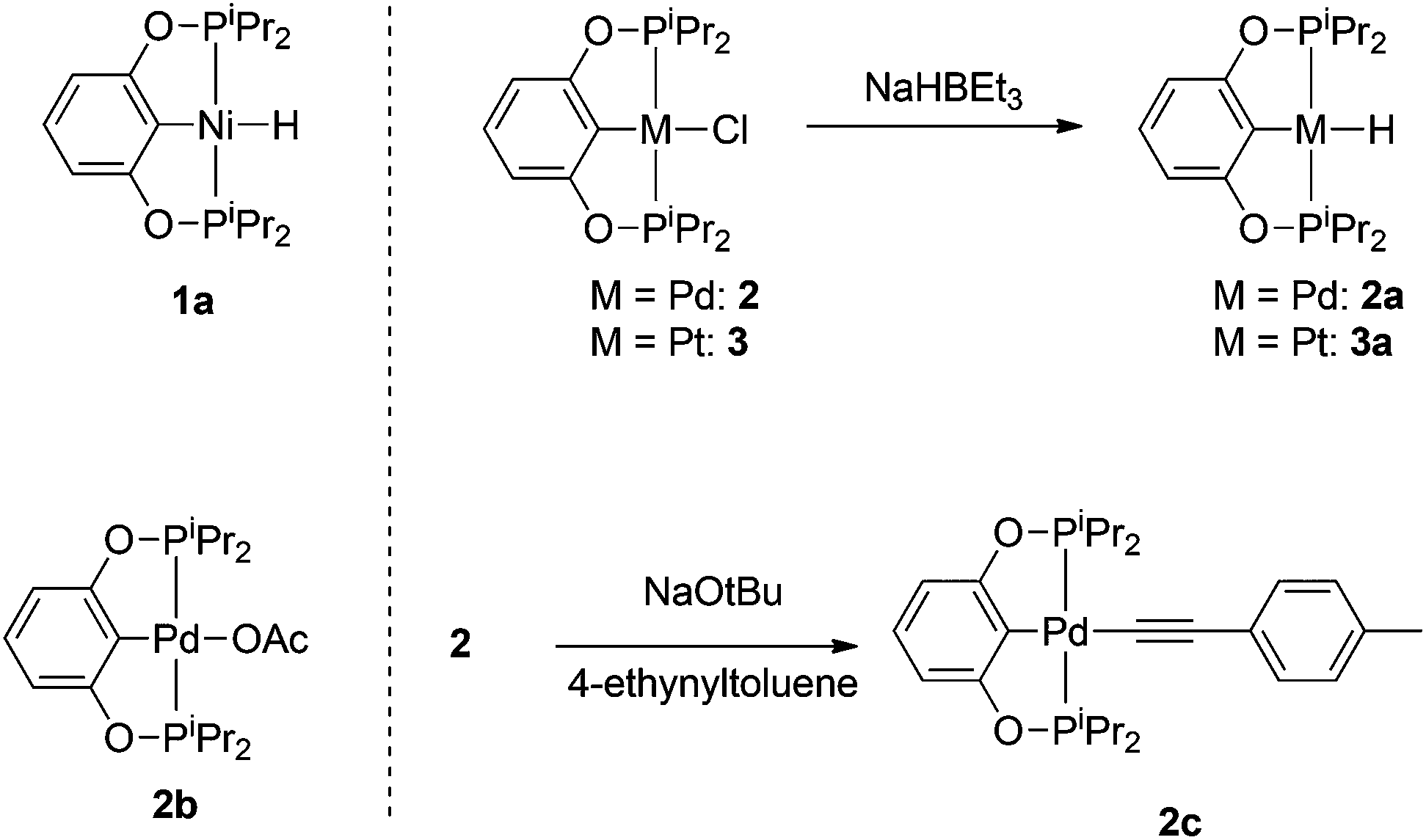 | ||
| Scheme 2 POCOP-supported Ni, Pd, and Pt precatalysts. | ||
|
|
|||||
|---|---|---|---|---|---|
| # | [Pd] | A | B , | Time | Additives |
| a All reactions performed at 80 °C in C6D6 with 0.34 mmol alkyne, 0.34 mmol HBpin, and 5% catalyst loading. b Yields were determined by 1H NMR integration versus an internal standard (1,4-dioxane). c Reactions that produced significant amounts of B also showed trace production of 4-ethyltoluene. d Many intractable products observed containing olefinic resonances. e 5% (E)-(4-methylstyryl)Bpin was observed by 1H NMR spectroscopy. f 1.7 μmol of 5b used. | |||||
| 1 | 1a | <5% | 7% | 1 d | X |
| 2 | 3a | 0% | <5% | 1 d | X |
| 3 | 2a | 40% | 39% | 1 d | X |
| 4 | 2b | 44% | 49% | 1 d | X |
| 5 | 2c | 45% | 42% | 1 d | X |
| 6 | 2a | >95% | <5% | 6 d | 0.45 mmol Hg |
| 7 | 2b | 53% | 43% | 1 d | 1.12 mmol Ga |
| 8 | 2b | 49% | 51% | 1 d | 5.45 mmol Ga |
| 9 | 2a | 63% | 35% | 2 d | 0.017 mmol PPh3 |
| 10 | 2a | 87% | <5% | 3 d | 0.034 mmol PPh3 |
| 11 | 2b | 85% | <5% | 3 d | 0.034 mmol PPh3 |
| 12 | 2b | 88% | 6% | 3 d | 0.034 mmol PMe3 |
| 13 | 2b | 46% | <5% | 4 d | 0.034 mmol P(OMe)3 |
| 14 | 2b | 45% | 48% | 1 d | 0.034 mmol SiPr2 |
| 15 | 2b | 44% | 43% | 1 d | 150 eq. PVPy |
| 16 | 4a | 2% | 10% | 3 d | X |
| 17 | 5b | 40% | 37% | 2 h | X |
| 18 | 5c | 36% | 27% | 2 h | X |
| 19 | 5b | 91% | <5% | 5 h | 0.45 mmol Hg |
| 20 | 5b | 85% | <5% | 1 d | 0.034 mmol PPh3 |
| 21 | 5b | 64% (32%) | 8% | 1 d | 0.034 mmol PMe3 |
| 22 | 5b | <5% | — | 1 d | 0.034 mmol P(OMe)3 |
| 23 | 6b | 7% | 13% | 1 d | X |
| 24 | 7b | 49% | 43% | 1 d | X |
| 25 | 8b | 43% | 48% | 3 d | X |
| 26 | 5b | 38% | <5% | 1 d | 0.45 mmol Hg |
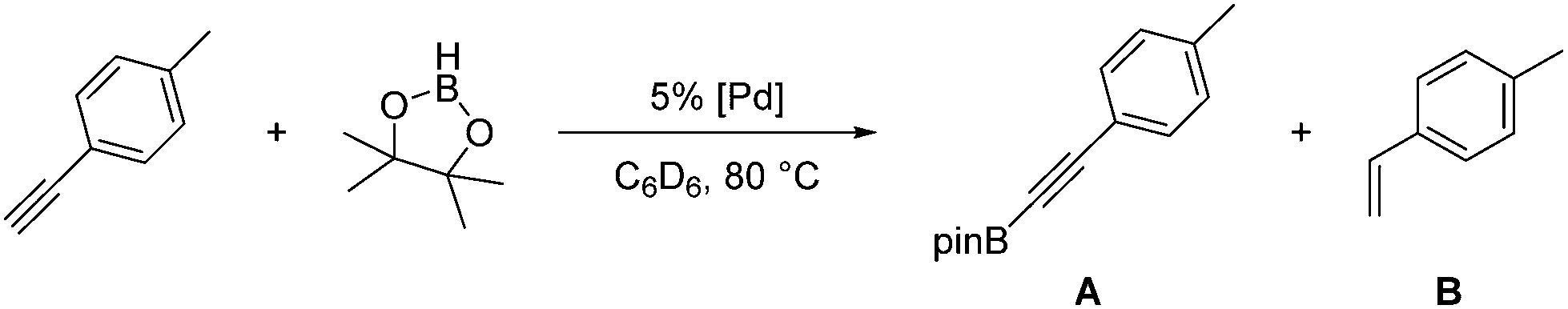
Known synthetic procedures for isolating pure 2a are low-yielding and not fully reproducible.15 However, it was found that the easily synthesized 2b19 could exchange an acetate group for the hydride of HBpin to form 2ain situ within seconds. 2c was synthesized by treating 2 with 2 eq. of NaOtBu in toluene in the presence of 4-ethynyltoluene (Scheme 1). 2c was also observed when 2b was treated with 1 eq. of 4-ethynyltoluene, which resulted in the formation of acetic acid and a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 equilibrium mixture of compounds 2b:2c. When this equilibrium mixture was treated with 1 eq. HBpin, acetic acid was converted to AcOBpin and 2c was the resulting product. The use of 2a, 2b, or 2c as precatalysts was observed to lead to essentially the same results in the catalytic borylation of 4-ethynyltoluene (entries 3–5), indicating that all three allowed equal access to the catalytically active species. Since AcOBpin was not detrimental to catalysis and because 2b was easiest to make and store, it is the most practical choice of precatalyst.
:2 equilibrium mixture of compounds 2b:2c. When this equilibrium mixture was treated with 1 eq. HBpin, acetic acid was converted to AcOBpin and 2c was the resulting product. The use of 2a, 2b, or 2c as precatalysts was observed to lead to essentially the same results in the catalytic borylation of 4-ethynyltoluene (entries 3–5), indicating that all three allowed equal access to the catalytically active species. Since AcOBpin was not detrimental to catalysis and because 2b was easiest to make and store, it is the most practical choice of precatalyst.
Scheme 3 shows the catalytic cycle we propose for (POCOP)Pd catalysed DHBTA. It entails the reaction of 2a with the terminal alkyne to evolve H2 and produce 2c, which would then react with HBpin to release the alkynylboronate product and regenerate 2a. 2c was the sole product detectable by 31P{1H} NMR spectroscopy until the end of the catalytic reactions using compounds 2a, 2b, and 2c; and it appears to be the resting state of the catalyst. Stoichiometric experiments are also consistent with the mechanistic proposal. Reaction of 2a with 1 eq. of 4-ethynyltoluene led to a clean conversion to 2c within minutes.20 Treatment of 2c with 5 eq. of pinacolborane resulted in 50% conversion to 2a after 5 h at 80 °C, with concomitant formation of A.21 This reaction is reversible; when 2a was thermolysed with 1 eq. of A at 80 °C for 3 h, analysis by 31P{1H} NMR spectroscopy showed 16% conversion to 2c. An alternative mechanistic pathway might involve intermediacy of (POCOP)Pd-Bpin, however, we have no evidence of its formation in catalytic reactions or in stoichiometric reactions of 2a or 2c with HBpin.
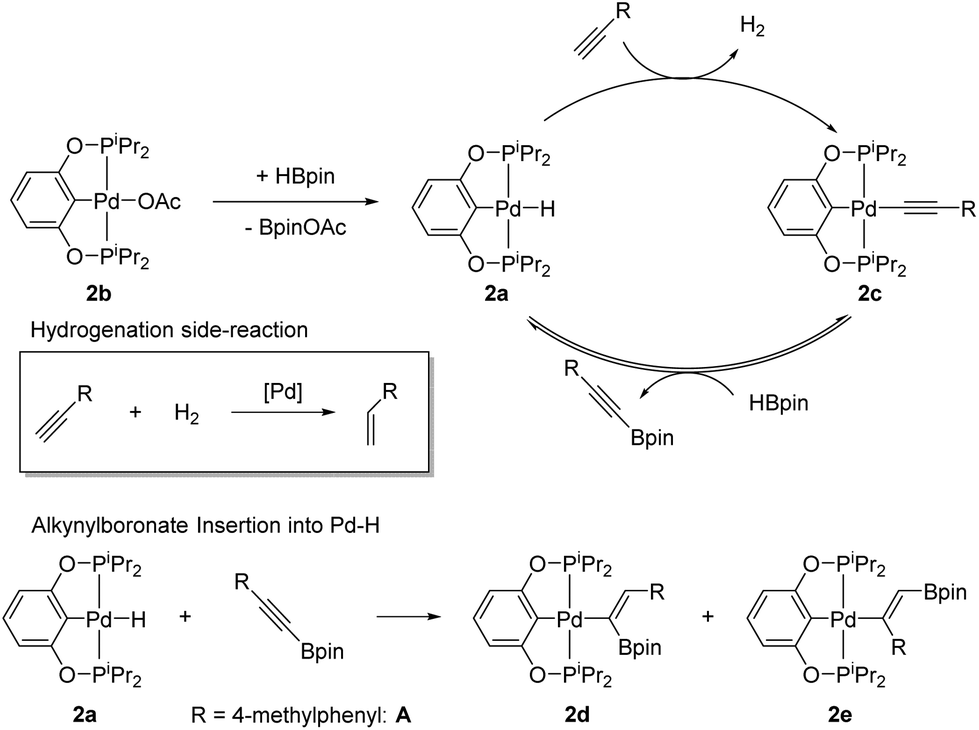 | ||
| Scheme 3 Proposed mechanism for (POCOP)Pd catalysed DHBTA and competing side reactions. | ||
The equilibrium between 2c/HBpin and 2a/A is eventually disturbed by the irreversible insertion of the alkynylboronate into the palladium hydride bond. When 2a was treated with 1 eq. of A and heated for 4 days at 80 °C, 2d and 2e were formed and accounted for 71% of the reaction mixture as judged by 31P{1H} NMR integration (Scheme 3). Treating this mixture with concentrated HCl converted all of the (POCOP)Pd compounds to 2, and (Z)-(4-methylstyryl)Bpin was observed by 1H NMR spectroscopy, consistent with the proposed isomeric structures for 2d and 2e. Under catalytic conditions, the insertion of the alkynylboronate is much slower than the rate of DHBTA and we have not observed formation of any alkenyl products in catalytic reactions with 2a,b,c until the reaction is nearly complete and the concentration of alkyne in solution is low enough to allow 2a to start accumulating. However, insertion of alkynes into the Pd–H bond may be responsible for the ultimate demise of the catalyst.
Catalytic 4-ethynyltoluene DHBTA reactions with 2a, 2b, or 2c precatatalysts inevitably produce comparable quantities of the semihydrogenation product (4-methylstyrene), presumably by using the H2 formed as a byproduct of DHBTA. The Guan group reported that 2a functions as an alkyne semihydrogenation catalyst, but ostensibly through generation of Pd(0) nanoparticles as the hydrogenation catalysis was inhibited by elemental mercury.15 Many pincer-ligated palladium complexes have been shown to leach soluble Pd(0) into solution.22 Over the course of the reaction, (POCOP)Pd-catalyzed DHBTA reaction mixtures developed a red colour associated with palladium nanoparticles in solution.23 Introduction of elemental mercury inhibited the hydrogenation of the terminal alkynes in DHBTA mixtures, and >95% of the terminal alkyne was converted to A (entry 6). It was also determined that the rate of DHBTA was not affected by the presence of mercury. After 1 day, parallel reactions catalysed by 2a with and without mercury (entries 6 and 3, respectively) had both converted about 40% of the terminal alkyne to A. We were intrigued by the possible use of gallium as an alternative, less toxic liquid metal trap for palladium nanoparticles, but it proved completely ineffective at stopping hydrogenation (entries 7 and 8).
Hydrogenation of the alkyne could also be mitigated by the addition of PMe3, PPh3, or P(OMe)3 (entries 9–13). PPh3 and PMe3 were similarly effective at 10% loading. 10% P(OMe)3 also inhibited hydrogenation, but supressed the formation of A, as well (entry 13). Addition of 10% SiPr2 (entry 14) or of a large excess of poly(4-vinylpyridine)23a (entry 15) had no effect on the reaction.
We prepared Pd complexes supported by several other (PCP)- and POCOP-type pincer ligands (Scheme 4) to test as catalysts and to compare them to 2b. Complexes 4a, 5b, 6b, 7b, 8b were synthesized according to literature procedures and precedent and were used as precatalysts (Scheme 4).15,19,245c was synthesized through the same method as complex 2c. Our catalogue of different palladium catalysts were tested for DHBTA at 5 mol% loading with 4-ethynyltoluene and pinacolborane. It was discovered that the relative rate of alkyne consumption is greatly dependent on the size of the substituents on the phosphine arms. The bulkier 4a only produced trace amounts of alkynylboronate (entry 16), while the less sterically imposing 5b was capable of consuming all of the alkyne substrate within 2 h (entry 17), and 5c was also found to be an analogous precatalyst (entry 18). Similar to 2b, phosphine additives and mercury were able to inhibit the semihydrogenation reaction with 5b (entries 19–22). Higher concentrations of HBpin were also examined using 5b, but did not prove to be any more beneficial than 1 equivalent.256b did not perform nearly as well as the resorcinol-based pincer ligands (entry 23), and 7b performed similarly to the unsubstituted 2b (entry 24). 8b gave a nearly identical distribution of A and semihydrogenated product, but required a much longer reaction time of three days (entry 25) possibly resulting from the slightly bulkier nature of the ligand compared to POCOP.26 The longevity of 5b was investigated using mercury as a hydrogenation inhibitor by using 0.5 mol% catalyst loading for the borylation of 4-ethynyltoluene (entry 26). The catalysis ceased after 1 day and 38% conversion to A, which accounted for about 75 turnovers. Control reactions using Pd2(DBA)3 and Pd(COD)Cl2 as catalysts did not show any formation of alkynylboronate.27
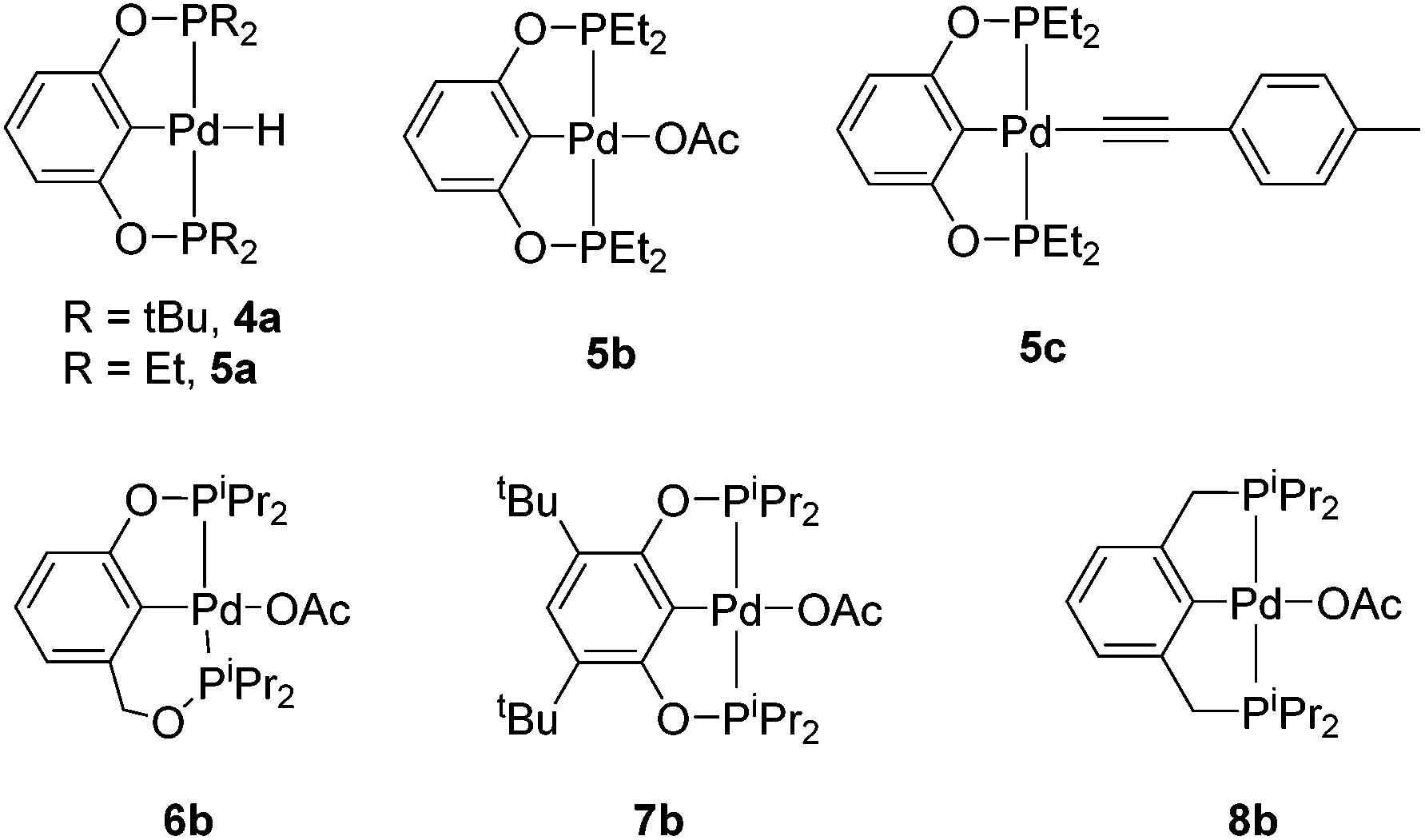 | ||
| Scheme 4 Palladium DHBTA precatalysts. | ||
5a could not be isolated due to its inherent instability. Attempts to synthesize it through the same means of obtaining 2a did not prove successful, and attempts to treat 5b with HBpin resulted in the immediate formation of dihydrogen and a rapid colour change from a clear solution to dark brown signifying decomposition of the palladium complex.27 However, analysis of catalytic mixtures with 5b by 31P{1H} NMR spectroscopy showed the presence of 5c under catalytic conditions, and stoichiometric reactions treating 5c with HBpin did lead to the production of A.
The substrate scope of our system was briefly examined using 2b and 5b as catalysts and is outlined in Table 2. 5b gave better results than 2b in DHBTA of 4-ethynyltoluene (entries 11 and 20) and Me3SiCCH (entries 27 and 28), but fared less well in DHBTA of 1-hexyne (entries 29 and 30).
|
|
|||||
|---|---|---|---|---|---|
| # | [Pd] | R | C | D | Other |
| a All reactions performed for 1 day at 80 °C in C6D6 with 0.34 mmol alkyne, 0.34 mmol HBpin, 0.034 mmol PPh3, 5% catalyst loading. b Yields were determined by 1H NMR integration versus an internal standard (1,4-dioxane). c SM = starting material. | |||||
| 27 | 5b | Me3Si | 80% | 4% | 16% SMc |
| 28 | 2b | Me3Si | 11% | — | 85% SM |
| 29 | 5b | n Bu | <5% | 11% | 85% SM |
| 30 | 2b | n Bu | 84% (58%) | <5% | 12% SM |
| 31 | 2b | Me3SiOCH2 | 50% | <5% | 8% Me3SiOBpin |
| 32 | 5b | Me3SiOCH2 | — | — | 90% Me3SiOBpin |
| 33 | 2b | PhOCH2 | 20% | 19% | 15% PhOBpin |
| 34 | 5b | PhOCH2 | — | — | 95% PhOBpin |
| 35 | 2b | PhSCH2 | 5% | — | 77% SM |
| 36 | 5b | PhSCH2 | — | — | 80% SM |
Propargyl-functionalized alkynes had been problematic with the original iridium system9 and also proved to be difficult for our palladium systems. 5b was not effective at DHBTA with propargyl substrates at all (entries 32, 34, and 36), while 2b resulted in some formation of DHBTA products (entries 31, 33, and 35). However, side reactions dominated in all cases. Phenyl propargyl ether was susceptible to hydrogenation and C–O bond cleavage to form PhOBpin, while trimethyl silyl propargyl ether was mostly susceptible to C–O bond cleavage which resulted in the formation of Me3Si–O–Bpin. Propyne and propylene were also detected by 1H NMR spectroscopy in reaction mixtures.
In conclusion, we have shown that palladium can perform dehydrogenative borylation of terminal alkynes. This expands the realm of substrates that palladium can borylate and also shows that DHBTA can be performed by other pincer-ligated metals that behave differently from the original (SiNN)Ir catalyst.
This palladium system is limited by the propensity for pincer-ligated palladium complexes to leach palladium(0) into solution, which can then perform hydrogenation of the terminal alkyne. However, hydrogenation can be prevented with the addition of phosphines to the reaction mixture. This work also shows the ability of pincer-ligated Pd–X compounds to perform metathesis reactions to couple it's X-substituent with another reagent.
Acknowledgement is made to the US National Science Foundation (grant CHE-1300299 to O. V. O.) and the Welch Foundation (grant A-1717 to O. V. O.).
Notes and references
- Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine, ed. D. G. Hall, Wiley-VCH, Weinheim, Germany, 2nd edn, 2005 Search PubMed.
- (a) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed; (b) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864–873 CrossRef CAS PubMed; (c) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS PubMed; (d) S. M. Preshlock, B. Ghaffari, P. E. Maligres, S. W. Krska, R. E. Maleczka Jr. and M. R. Smith III, J. Am. Chem. Soc., 2013, 135, 7572–7582 CrossRef CAS PubMed; (e) J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka Jr. and M. R. Smith III, Science, 2002, 295, 305–308 CrossRef CAS PubMed.
- (a) T. Davan, E. W. Corcoran Jr. and L. G. Sneddon, Organometallics, 1983, 2, 1693–1694 CrossRef CAS; (b) D. E. Kadlecek, P. J. Carroll and L. G. Sneddon, J. Am. Chem. Soc., 2000, 122, 10868–10877 CrossRef CAS; (c) J. Takaya, N. Kirai and N. Iwasawa, J. Am. Chem. Soc., 2011, 133, 12980–12983 CrossRef CAS PubMed.
- T. Ohmura, A. Kijima and M. Suginome, J. Am. Chem. Soc., 2009, 131, 6070–6071 CrossRef CAS PubMed.
- Y. Kuninobu, T. Iwanaga, T. Omura and K. Takai, Angew. Chem., Int. Ed., 2013, 52, 4431–4434 CrossRef CAS PubMed.
- T. Ishiyama, K. Ishida, J. Takagi and N. Miyaura, Chem. Lett., 2001, 1082–1083 CrossRef CAS.
- T. Furukawa, M. Tobisu and N. Chatani, Chem. Commun., 2015, 51, 6508–6511 RSC.
- J. Jiao and Y. Nishihara, J. Organomet. Chem., 2012, 721, 3–16 CrossRef PubMed.
- C.-I. Lee, J. Zhou and O. V. Ozerov, J. Am. Chem. Soc., 2013, 135, 3560–3566 CrossRef CAS PubMed.
- C.-I. Lee, J. Zhou, J. C. DeMott, N. Bhuvanesh, A. Christopher and O. V. Ozerov, submitted.
- T. Tsuchimoto, H. Utsugi, T. Sugiura and S. Horio, Adv. Synth. Catal., 2015, 357, 77–82 CrossRef CAS PubMed.
- R. Johansson and O. F. Wendt, Organometallics, 2007, 26, 2426–2430 CrossRef CAS.
- D. Adhikari, J. C. Huffman and D. J. Mindiola, Chem. Commun., 2007, 4489–4491 RSC.
- Y. Zhu, C.-H. Chen, C. M. Fafard, B. M. Foxman and O. V. Ozerov, Inorg. Chem., 2011, 50, 7980–7987 CrossRef CAS PubMed.
- A. Adhikary, J. R. Schwartz, L. M. Meadows, J. A. Krause and H. Guan, Inorg. Chem. Front., 2014, 1, 71–82 RSC.
- (a) M. E. van der Bloom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef PubMed; (b) The Chemistry of Pincer Compounds, ed. D. Morales-Morales and C. M. Jensen, Elsevier, Amsterdam, 2007, pp. 151–179 Search PubMed.
- S. Chakraborty, J. A. Krause and H. Guan, Organometallics, 2009, 28, 582–586 CrossRef CAS.
- G. L. O. Wilson, M. Abraha, J. A. Krause and H. Guan, Dalton Trans., 2015, 44 10.1039/C5DT00161G , in press.
- Z. Wang, M. R. Eberhard, C. M. Jensen, S. Matsukawa and Y. Yamamoto, J. Organomet. Chem., 2003, 681, 189–195 CrossRef CAS.
- The Guan laboratory recently reported (ref. 15) that the addition of 1 eq. of phenylacetylene to 2a led to 7% alkyne insertion products in addition to formation of the palladium alkynyl as quantified by 31P{1H} NMR integration. In our hands, 4-ethynyltoluene was not seen to insert into the Pd–H under stoichiometric conditions at room temperature.
- Alkynylboronate insertion products (2d and 2e) were formed upon further heating of this mixture.
- (a) M. R. Eberhard, Org. Lett., 2004, 6, 2125–2128 CrossRef CAS PubMed; (b) K. Yu, W. Sommer, J. M. Richardson, M. Weck and C. W. Jones, Adv. Synth. Catal., 2005, 347, 161–171 CrossRef CAS PubMed; (c) J. G. de Vries, Dalton Trans., 2006, 421–429 RSC; (d) J. L. Bolliger, O. Blacque and C. M. Frech, Chem. – Eur. J., 2008, 14, 7969–7977 CrossRef CAS PubMed; (e) C. Melero, L. M. Martínez-Prieto, P. Palma, D. del Rio, E. Álvarez and J. Cámpora, Chem. Commun., 2010, 46, 8851–8853 RSC.
- (a) K. Yu, W. Sommer, J. M. Richardson, M. Weck and C. W. Jones, Adv. Synth. Catal., 2005, 347, 161–171 CrossRef CAS PubMed; (b) D. Pun, T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 8213–8221 CrossRef CAS PubMed.
- L. M. Martínez-Prieto, C. Melero, D. del Río, P. Palma, J. Cámpora and E. Álvarez, Organometallics, 2012, 31, 1425–1438 CrossRef.
- See ESI† for further discussion.
- J. Choi, A. H. Roy MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed.
- See ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectroscopic characterization. See DOI: 10.1039/c5qi00074b |
| This journal is © the Partner Organisations 2015 |