
Structural studies and detection of nitroaromatics by luminescent 2D coordination polymers with angular dicarboxylate ligands†
Yadagiri
Rachuri
ab,
Bhavesh
Parmar
ab,
Kamal Kumar
Bisht
 abc and
Eringathodi
Suresh
*ab
abc and
Eringathodi
Suresh
*ab
aAnalytical Discipline and Centralized Instrument Facility, CSIR–Central Salt and Marine Chemicals Research Institute, Council of Scientific and Industrial Research, G. B. Marg, Bhavnagar-364 002, Gujarat, India. E-mail: esuresh@csmcri.org; sureshe123@rediffmail.com
bAcademy of Scientific and Innovative Research (AcSIR), CSIR–Central Salt and Marine Chemicals Research Institute, Council of Scientific and Industrial Research, G. B. Marg, Bhavnagar-364 002, Gujarat, India
cDepartment of Chemistry, University of Petroleum and Energy Studies (UPES), P.O. Bidholi via Premnagar, Dehradun 248007, Uttarakhand, India
First published on 14th January 2015
Abstract
Luminescent two dimensional coordination polymers (CPs) {[Cd(SDB)(H2O)]·3H2O}n1 and {[Zn3(μ-OH)2(SDB)2]·(PPZ)}n2, (where SDB = 4,4′-sulfonyldibenzoate; PPZ = piperazine) have been synthesized by solvothermal methods. The crystal structure of 1 revealed that the carboxylate moieties of the SDB ligands are involved in “paddle wheel” type coordination with the metal nodes to engender a double chain loop which is further connected by μ-2 type coordination from one of the carboxylate oxygens, generating a two dimensional coordination polymer. In the case of 2, the two dimensional framework is constructed by bridging the one dimensional tri-metallic (Zn3μ3-OH) strands with the SDB ligand with a cylindrical cavity occupied by a piperazine moiety. Comprehensive characterization of both pristine compounds 1 and 2 by various physico-chemical methods, structural analysis and photoluminescence properties of activated CPs 1′ and 2′ towards the detection of nitroaromatics have been investigated. Both activated compounds 1′ and 2′ showed sensing of nitro explosive TNP (2,4,6-trinitrophenol) compared to other nitro analytes by fluorescence quenching properties.
Introduction
The design and synthesis of coordination polymers (CPs) have progressed rapidly in the last decade due to their intriguing architectures and functional properties in gas storage/separation,1 heterogeneous catalysis,2 molecular magnetism,3 luminescence4 and drug delivery.5 Varieties of CPs have been assembled by various approaches, such as by the judicious choice of organic ligands bridging different metal nodes, changing the functional groups or tether length of the organic linker, incorporating different anions in the cationic framework and utilizing dual ligands as linkers in multi-dimensional networks. The structural outcome of CPs is influenced by many factors such as the solvent system, pH value, temperature and templates added during the synthetic process in addition to the coordination geometry around the metal center and the bridging mode of the multitopic ligand.6 Subtle changes in the above-mentioned factors can alter the structural diversity such as the topology and dimensionality of the network in CPs. Organic polycarboxylate ligands, alone or in conjunction with N-donor ligands, have been extensively employed in the preparation of CPs with multi-dimensional networks and interesting properties.7 Recently, more attention has been paid to flexible polycarboxylate ligands, however, CPs with V-shaped carboxylates such as meta-benzenedicarboxylate and 4,4′-oxydibenzoate, alone or in combination with different N-donor ligands, have been reported in the literature for generating multi-dimensional CPs with different metal centers.8 Angular dicarboxylates such as H2SDB (SDB = 4,4′-sulfonyldibenzoate) and H2IPA (IPA = isophthalic acid), owing to their diverse coordination capabilities and topological-directional preferences, have been used as notable organic spacers for the construction of coordination polymers in conjunction with various N-donor ligands.9 Properties such as the luminescence and porosity of CPs have been of particular interest because of their potential applications as chemical sensors. The luminescence quenching properties of CPs have been exploited for the sensing of aromatic nitro compounds/explosives and proven to be a sensitive and versatile method.10 In addition to the ongoing research with CPs/MOFs for the detection of nitroaromatics and nitro explosives, new materials such as nanocrystals, nanoflowers and gold nanoparticles by upconversion luminescence have been reported by Leyu Wang et al.11 At this juncture, it is also worth mentioning recent developments such as bioinspired phage-based colorimetric sensors and Ag nanocluster/DNA hybrid materials for the detection of nitroaromatic explosives.12We have been involved in the design and synthesis of new multi-dimensional CPs and investigation of their structural and functional aspects. Recently, we reported some mixed ligand CPs derived from angular dicarboxylic acid 4,4′-sulfonyldibenzoic acid (H2SDB) and N-donor ligands.13 Structural investigation of the above revealed that besides other factors, the hydrogen bonding ability of SDB through the sulfone functionality plays an important role in the supramolecular assembly of chiral and achiral CPs.13a We have also reported some ternary luminescent CPs and their application towards aromatic nitro detection by espousing the quenching phenomenon.14 In continuation of our quest for new CPs, we have chosen angular dicarboxylic acid 4,4′-sulfonyldibenzoic acid (H2SDB) as the spacer, considering the sulfone functionality to be involved in secondary interactions which can promote varied structural and topological features with d10 metal nodes (Cd+2/Zn+2) as well as application as a photoluminescent material. In the current work, we describe the synthesis, characterisation and crystal structures of new 2D CPs {[Cd(SDB)(H2O)]·3H2O}n1 and {[Zn3(μ-OH)2(SDB)2]·(PPZ)}n (PPZ = piperazine) 2, based on angular dicarboxylate SDB and study their sensing of nitroaromatics by exploiting their luminescence quenching properties.
Experimental section
Caution! Nitrophenol (TNP, 2,4-DNP, o-NP and p-NP) vapour is toxic and hazardous to human health. TNP is potentially explosive in nature. Proper precautions have been taken during the handling of these analytes throughout all of the experiments.Materials and method
4,4′-Sulfonyldibenzoic acid (H2SDB) and reagents for the synthesis of 1,4-bis(pyridylmethyl)piperazine (4-BPMP) were purchased from Sigma-Aldrich. Nitro analytes TNP (2,4,6-trinitrophenol or picric acid), 2,4-DNP (2,4-dinitrophenol), o-NP (ortho-nitrophenol) and p-NP (para-nitrophenol) were also purchased from Sigma-Aldrich. Metal salts and organic solvents were obtained from RANBAXY and SD Fine Chemicals, India. Distilled water was used for synthetic manipulations. All of the reagents and solvents were used as received without any further purification. Ligand 4-BPMP was synthesized by following a reported procedure.15 CHNS analyses were carried out using a Perkin-Elmer 2400 CHNS/O analyzer. FTIR spectra were recorded using KBr pellets on a Perkin-Elmer GX FTIR spectrometer. For each FTIR spectrum 10 scans were recorded at 4 cm−1 resolution. TGA analysis was carried out using Mettler Toledo Star SW 8.10. Solid state UV-vis spectra and luminescence spectra were recorded using a Shimadzu UV-3101PC spectrometer and Fluorolog Horiba Jobin Yvon spectrophotometer with stirring attachment, respectively. PXRD data were collected using a Philips X-Pert MPD system with CuKα radiation. Single crystal structures were determined using a BRUKER SMART APEX (CCD) diffractometer.Synthesis of the compounds
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and then sealed in a 23 mL Teflon-lined autoclave, which was heated at 398 K for 4 days. After slow cooling to room temperature, colourless block-shaped crystals were obtained. Yield ca. 62%. Elemental analysis (%) C14H14O9SCd calc.: C, 35.72; H, 3.00; S, 6.81; found (%): C, 35.67; H, 2.96; S, 6.78. FTIR cm−1 (KBr): 3486 (br), 3079 (w), 2967 (m), 2865 (w), 2366 (w), 1951 (w), 1628 (s), 1564 (m), 1398 (s), 1296 (m), 1167 (s), 1100 (m), 1013 (w), 856 (m), 734 (s), 686 (w), 618 (m), 496 (m).
:1) and then sealed in a 23 mL Teflon-lined autoclave, which was heated at 398 K for 4 days. After slow cooling to room temperature, light yellow coloured needle-shaped crystals suitable for single crystal X-ray analysis were obtained. During the solvothermal reaction in situ cleavage of the 4-BPMP ligand took place to produce the piperazine molecule, which is encapsulated in the crystal lattice. Yield ca. 32%. Elemental analysis (%) C32H28N2O14S2Zn3 calc.: C, 41.56; H, 3.05; N, 3.03; S, 6.93; found: C, 39.7; H, 2.90; N, 3.00; S, 6.9. FTIR cm−1 (KBr): 3649 (w), 3440 (br), 3161 (s), 2969 (w), 2408 (w), 1948 (w), 1614 (s), 1561 (m), 1387 (s), 1294 (m), 1164 (s), 1103 (s), 1012 (m), 872 (s), 745 (s), 632 (m), 569 (m), 476 (w).
:1) and then sealed in a 23 mL Teflon-lined autoclave, which was heated at 398 K for 4 days. After slow cooling to room temperature, colourless block-shaped crystals were obtained. Yield ca. 62%. Elemental analysis (%) C14H14O9SCd calc.: C, 35.72; H, 3.00; S, 6.81; found (%): C, 35.67; H, 2.96; S, 6.78. FTIR cm−1 (KBr): 3486 (br), 3079 (w), 2967 (m), 2865 (w), 2366 (w), 1951 (w), 1628 (s), 1564 (m), 1398 (s), 1296 (m), 1167 (s), 1100 (m), 1013 (w), 856 (m), 734 (s), 686 (w), 618 (m), 496 (m).
:1) and then sealed in a 23 mL Teflon-lined autoclave, which was heated at 398 K for 4 days. After slow cooling to room temperature, light yellow coloured needle-shaped crystals suitable for single crystal X-ray analysis were obtained. During the solvothermal reaction in situ cleavage of the 4-BPMP ligand took place to produce the piperazine molecule, which is encapsulated in the crystal lattice. Yield ca. 32%. Elemental analysis (%) C32H28N2O14S2Zn3 calc.: C, 41.56; H, 3.05; N, 3.03; S, 6.93; found: C, 39.7; H, 2.90; N, 3.00; S, 6.9. FTIR cm−1 (KBr): 3649 (w), 3440 (br), 3161 (s), 2969 (w), 2408 (w), 1948 (w), 1614 (s), 1561 (m), 1387 (s), 1294 (m), 1164 (s), 1103 (s), 1012 (m), 872 (s), 745 (s), 632 (m), 569 (m), 476 (w).
General procedure for the fluorescence study
In the fluorescence experiments the activated compounds of both CPs 1 and 2 (designated as 1′ and 2′) were used after removing the encapsulated lattice molecules. The activated compounds 1′ and 2′ were prepared by heating 1 and 2 activated at 200 °C for 8 h in a temperature controlled oven. In a typical experimental setup, 2 mg of the activated CP 1′ or 2′ was weighed and added to a cuvette (path length of 1 cm) containing 2 mL of acetone under constant stirring to make an emulsion. The fluorescence response was measured in the range 300–600 nm upon excitation at 290 nm in situ after incremental addition (40 μL) of freshly prepared nitro analyte solutions in acetone (2 mM) and the corresponding fluorescence intensity was monitored at 402 and 404 nm for 1′ and 2′, respectively. During titration experiments the net concentration of analytes present in the cuvette ranged from 0.04 mM to 0.20 mM. To maintain the homogeneity, the emulsion was stirred at a constant rate using the stirring attachment in the florescence quenching experiments.X-ray crystallography
A summary of the crystallographic data and details of the data collection for 1 and 2 are given in Table S1.† Single crystals with suitable dimensions were chosen under an optical microscope after immersion in paratone oil and mounted on a glass fibre for data collection. Intensity data for both crystals were collected using MoKα (λ = 0.71073 Å) radiation on a Bruker SMART APEX diffractometer equipped with a CCD area detector at 150 K. The data integration and reduction were processed using SAINT software16 and an empirical absorption correction was applied to the collected reflections with SADABS.17 The structures were solved by direct methods using SHELXTL and were refined on F2 by the full-matrix least-squares technique using the program SHELXL-97.18 All non-hydrogen atoms were refined anisotropically until convergence was reached. Hydrogen atoms attached to the organic moieties present in both CPs are either located from the difference Fourier map or fixed stereochemically. After refining the coordination polymeric networks for 1, diffused scattered peaks with residual electron density ranging from ∼ 2.5 Å−3 to 3 Å−3 were observed in the difference Fourier map which can be attributed to lattice solvent molecules (three water molecules) present in the crystal lattice. Attempts to model these disordered peaks were unsuccessful since residual electron density of the peaks obtained was diffused and there was no obvious major site occupations for the lattice solvent molecules. PLATON/SQUEEZE19 was used to correct the diffraction data for the contribution from disordered lattice solvent molecules. Final cycles of least-squares refinements improved both the R-values and goodness of fit significantly for 1 with the modified data set after subtracting the contribution from the disordered solvent molecules using the SQUEEZE program. The accessible void volumes per unit cell estimated by PLATON/SOLV for 1 and 2 after the complete removal of the lattice water molecules in 1 and piperazine moiety from 2 are 18.4% (620.5 Å3) and 19.0% (311.6 Å3), respectively.Results and discussion
Crystal and molecular structure of {[Cd(SDB)(H2O)]·3H2O}n1
Compound 1 crystallises in a monoclinic system with C2/c space group. The asymmetric unit of 1 is composed of one crystallographically independent Cd(II) ion, one molecule of SDB and one coordinated water molecule along with 3 disordered water molecules (whose contribution is removed from the crystallographic data by using the PLATON/SQUEEZE program). Single crystal analysis revealed that the structure of compound 1 is an extended 2D network. The five symmetrically arranged SDB units are involved in bridging the metal centers through the dicarboxylate oxygens via a η1:η1:η1:η2 mode of coordination. Pairs of Cd(II) ions are joined by four symmetrically oriented SDB ligands in a syn-anti mode to generate a binuclear Cd(II) “paddle wheel” type secondary building unit (SBU) with a Cd⋯Cd distance of 3.389(7) Å within the paddle wheel motif. These paddle wheel SBUs are further bridged via the second terminal carboxylate groups of the symmetrical SDB dianions into 1D double chain loops with a Cd⋯Cd bridging distance of 13.12 Å. As depicted in Fig. 1a, these one dimensional [Cd(SDB)]n dimeric double chain loops oriented along the b-axis are further linked through μ3–η1:η2 coordination adopted by the O1 and O2 carboxylate oxygens of the symmetrically arranged SDB moiety along the a-axis, generating a two dimensional network. The Cd⋯Cd separation distance bridged by carboxylate oxygen O1 via μ3–η2 coordination between the [Cd(SDB)]n dimeric double chain loop is 3.60 Å. Interestingly, the O5 and O6 carboxylate oxygens of the SDB ligand are involved only in μ2–η1:η1 coordination in the paddle wheel formation with the Cd(II) centers. Cd2+ in 1 possesses a distorted octahedral geometry provided by the carboxylate oxygens from five different SDB ligands adopting μ2–η1:η1 and μ3–η1:η2 coordination along with one water molecule, O7 (Fig. 1b & S1†). The Cd⋯O distance coordinated via the carboxylate oxygens ranges from 2.189(3) to 2.502(3) Å whereas the Cd⋯O7 distance is 2.286(4) Å. The mean plane angle between the phenyl rings of the SDB ligand in the double chain is 77.8°. The packing diagram clearly shows a cylindrical cavity down the a-axis with the approximate window size of the channel (13.18 × 11.42 Å); probably the disordered water molecules are encapsulated within the cylindrical channels.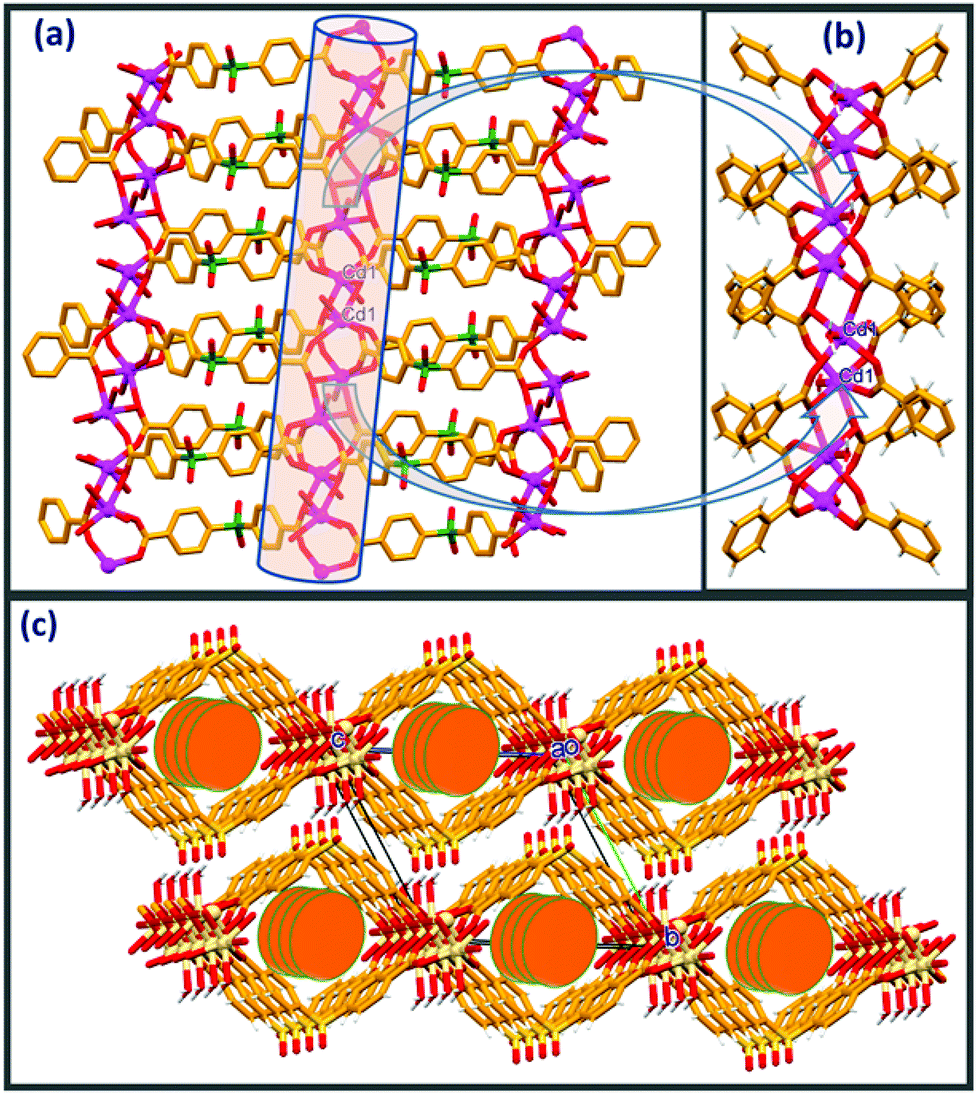 | ||
| Fig. 1 (a) 2D network in 1 comprising [Cd(SDB)]n paddle wheel loops, (b) close-up view depicting the coordination environment in 1, (c) packing diagram depicting the staggering of adjacent 2D sheets and the thorough cylindrical channels down the a-axis. | ||
As depicted in Fig. 1c, the packing diagram viewed down the a-axis clearly exposes a displacement of the adjacent 2D nets alternating along the b axis (by b/2 Å). Analysis of packing and hydrogen bonding clearly shows that adjacent layers of the 2D nets are drawn in via strong intermolecular O–H⋯O. Consequently, both coordinated water hydrogens H7A and H7B act as donors and are involved in intermolecular O–H⋯O contacts with sulfone oxygens O3 and O4, respectively, with the displaced adjacent 2D layers generating a three dimensional hydrogen bonded network. In addition to this, sulfone oxygen O3 is involved in an additional intramolecular C–H⋯O contact with phenyl hydrogen H9. Details of these pertinent hydrogen bonding interactions and symmetry codes are O(7)–H(7A)⋯O(4): H(7A)⋯O(4) = 1.90(2) Å, O(7)⋯O(4) = 2.804 (5) Å, ∠O(7)–H(7A)⋯O(4) = 164(4)°, symmetry code: 1/2 − x, −1/2 + y, 1/2–z; O(7)–H(7B)⋯O(3): H(7B)⋯O(3) = 2.02(5)Å, O(7)⋯O(3) = 2.936(6)Å, ∠O(7)–H(7B)⋯O(3) = 165(4)°, symmetry code: 1/2 − x, 1/2 − y, 1 − z; C(9)–H(9)⋯O(3): H(9)⋯O(3) = 2.56(4) Å, C(9)⋯O(3) = 3.317(6) Å, ∠C(9)–H(9)⋯O(3) = 139(3)°, symmetry code: x, 1 − y, −1/2 + z). It is worth mentioning here the crystal structure of an interesting three dimensional CP [Cd(SDB)(H2O)] with a (3,6)-connected rutile network in which the sulfonate oxygen of the SDB ligand is coordinated to the metal center.20
Crystal and molecular structure of {[Zn3(μ-OH)2(SDB)2]·(PPZ)}n2
The asymmetric unit of compound 2 contains divalent zinc Zn1 occupying a special position (0.5, 0.5, 0.5) and Zn2 occupying a general position, one hydroxyl ion (OH−) and one SDB ligand along with half a molecule of piperazine in the crystal lattice. Compound 2 can be described as a two dimensional coordination polymer (Fig. 2a) and crystallizes in a monoclinic system with P21/n space group.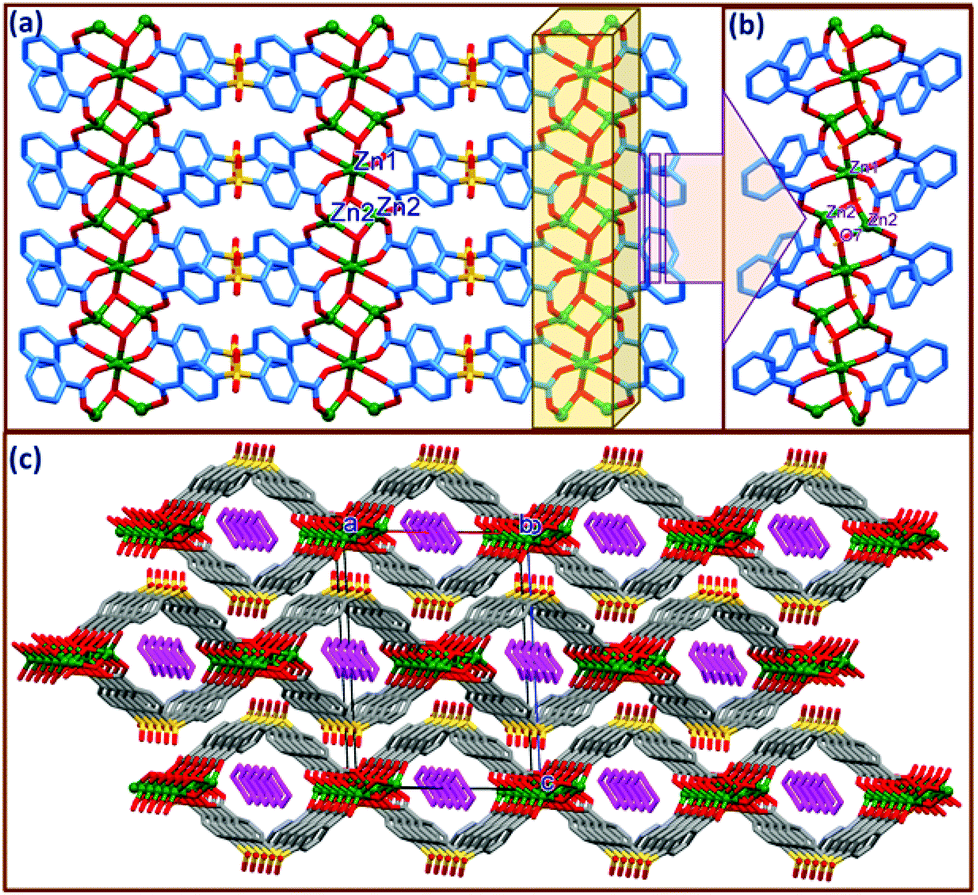 | ||
| Fig. 2 (a) Two dimensional coordination polymeric network of {[Zn3(μ-OH)2(SDB)2]n in 2, (b) close-up view depicting the coordination environment in 2, (c) packing diagram depicting the staggering of adjacent 2D sheets and through cylindrical channels down the b-axis with the encapsulated piperazine molecule. | ||
A close-up view depicting the coordination around the metal center is shown in Fig. 2b & S2.† As shown in Fig. 2b, the Zn1 and Zn2 metal units are linked via μ3-OH generating a trimeric (Zn3μ3-OH) cluster strand along the a-axis. The Zn2⋯Zn2 and Zn1⋯Zn2 distances within this strand are 2.891 and 2.934 Å, respectively. It is interesting to mention here that a similar type of μ3-OH bridging between the tri-metallic Zn cluster is observed in the case of MOF-69A ({[Zn3(OH)2(bpdc)2]·4DEF·2H2O}n) and MOF-69B ({[Zn3(OH)2(ndc)2]·4DEF·2H2O}n) reported by Yaghi et al. with rigid dicarboxylate moieties such as 4,4′-biphenyldicarboxylate (bpdc) and 2,6-naphthalenedicarboxylate (ndc), respectively.21 Three crystallographically unique Zn atoms within the strand connected by μ3-OH are further linked by four carboxylate groups in syn-anti fashion from different SDB ligands, generating a two dimensional coordination polymeric network with Zn1 and Zn2 possessing octahedral and tetrahedral coordination geometries, respectively. Thus, Zn1 has an octahedral coordination provided by four symmetrically oriented carboxylate oxygens (O1 and O5) as well as two axially coordinated oxygens from OH groups. The Zn1⋯O distances involving the carboxylate oxygens are Zn1⋯O = 2.076(4) Å, Zn1⋯O2 = 2.169(4) Å and with the hydroxyl oxygen O7 it is 2.030(5) Å. The cis angles involving the coordinating atoms range from 84.95 to 95.05° while the trans angles are almost 180° in the distorted octahedral coordination around Zn1. The tetrahedral coordination of Zn2 is provided by carboxylate oxygens O2 and O6 as well as symmetrically oriented hydroxyl oxygen O7 with Zn⋯O distances ranging from 1.914(4) Å to 1.965(4) Å and the tetrahedral angle ranges from 108.5(2)° to 113.8(2)°. The bond distances and angles observed in the present case are well within the range of similar Zn complexes reported by Yaghi et al.21 Details of selected bond distances and angles are given in the ESI Table S2.† μ3-OH connected trimeric Zn cluster strands are further connected by SDB ligands through carboxylate oxygens in a syn-anti mode via μ2–η1:η1 coordination making double chain loops with cylindrical channels down the b-axis. The piperazine molecule generated by the in situ cleavage of the 4-BPMP ligand during the solvothermal reaction is anchored within the cylindrical channels. To the best of our knowledge we are unaware of any report on the decomposition of the 4-BPMP ligand to give piperazine in the solvothermal synthesis of CPs, indicating that this is an unprecedented transformation. It is interesting to note that no obvious classical H-bonding interactions are observed between the lattice PPZ and the host framework. However, very weak C–H⋯π interactions involving the methyl hydrogens and phenyl rings of the SDB ligands are noticed. Thus, H16A and H16B from the centre of the symmetric piperazine moiety are making C–H⋯π contacts with the centroids of the phenyl rings Cg1 and Cg2, with H16A⋯Cg1 and H16B⋯Cg2 distances of 3.41 and 3.57 Å and ∠C12–H12⋯Cg1 and ∠C12–H12⋯Cg2 angles of 160.8° and 146.9°, respectively, where Cg1 and Cg2 are the centroids of the SDB rings C15–21 and C22–C28 (Fig. S3†). As depicted in Fig. 2c, the packing diagram of 2 viewed down the b-axis clearly exposes a displacement of the adjacent 2D nets alternating along the a-axis (by a/2 Å). Analysis of the supramolecular interactions revealed that adjacent offset 2D layers form strong intermolecular O–H⋯O hydrogen bonds between the H7 from the hydroxyl group and the sulfone oxygen O4 which stabilise the hydrogen bonded three dimensional nets. Details of these pertinent hydrogen bonding interactions are O(7)–H(7C)⋯O(4): H(7C)⋯O(4) = 2.24(1), O7⋯O(4) = 2.825(6), ∠O(7)–H(7C)⋯O(4) = 141(11)°, symmetry code: −1/2 + x, 3/2 − y, 1/2 + z.
FTIR, TGA and PXRD analyses
The lattice guests and compositions of 1 and 2 were ascertained by TGA, elemental and single crystal analyses. FTIR spectra can expose valuable information about the coordination mode of the SDB moiety. The difference between the asymmetric and symmetric carbonyl stretching frequencies (Δν = νasym − νsym) was used to obtain information about the metal–carboxylate binding modes. Distinguishable differences in the asymmetric and symmetric carbonyl stretching frequencies (Δν = νasym − νsym) were observed for both compounds and the Δν values apparently indicate the versatile coordination modes of the carboxylate moiety. Coordination polymers 1 and 2 showed νasym and νsym carbonyl frequencies centred at 1628, 1398 cm−1 (Δν = 230 cm−1) and 1614, 1387 cm−1 (Δν = 227 cm−1) respectively, inferring the μ2-bidentate mode of coordination by the carboxylate group in SDB. A broad band at 3486 cm−1 can be assigned to the lattice and coordinated water molecules for 1 (Fig. S4†) and the broad band at 3440 cm−1 in 2 is proposed to be due to the OH stretching frequency. A sharp band at 3161 cm−1 can be ascribed to the N–H stretching frequency of the piperazine molecule present in the lattice of 2 (Fig. S5†). Compound 1 loses 11.52% of its initial weight in the temperature range 90–200 °C which corroborates the expulsion of three lattice water molecules (calc. 11.5%). The dehydrated framework showed stability up to almost 420 °C (Fig. S6†).In the case of 2, expulsion of the lattice piperazine molecule starts in the wide temperature range of 190–280 °C and the weight loss corresponding to 9.0% supports well the eviction of the piperazine moiety (calc. 9.3%). The guest free framework was found to be stable up to 400 °C (Fig. S7†). Experimental powder XRD (PXRD) patterns of bulk samples of 1 and 2 concur with the simulated ones from respective single crystal data, confirming the phase purity of the bulk samples (Fig. 3 and Fig. S8†). For the photoluminescence experiments CPs 1 and 2 were activated at 200 °C for 8 hours and the activated samples are designated as 1′ and 2′. PXRD patterns of the activated compounds 1′ and 2′ (after the removal of the encapsulated lattice molecules from 1 and 2) corroborate well with the simulated SCXRD data of the pristine samples (with variation in peak intensities) indicating the robustness and stability of the framework topology of the activated compounds (Fig. 3 and Fig. S8†). Framework stabilities of up to 420 and 400 °C for the respective activated compounds 1′ and 2′ were also confirmed by TGA experiments (Fig. S9†).
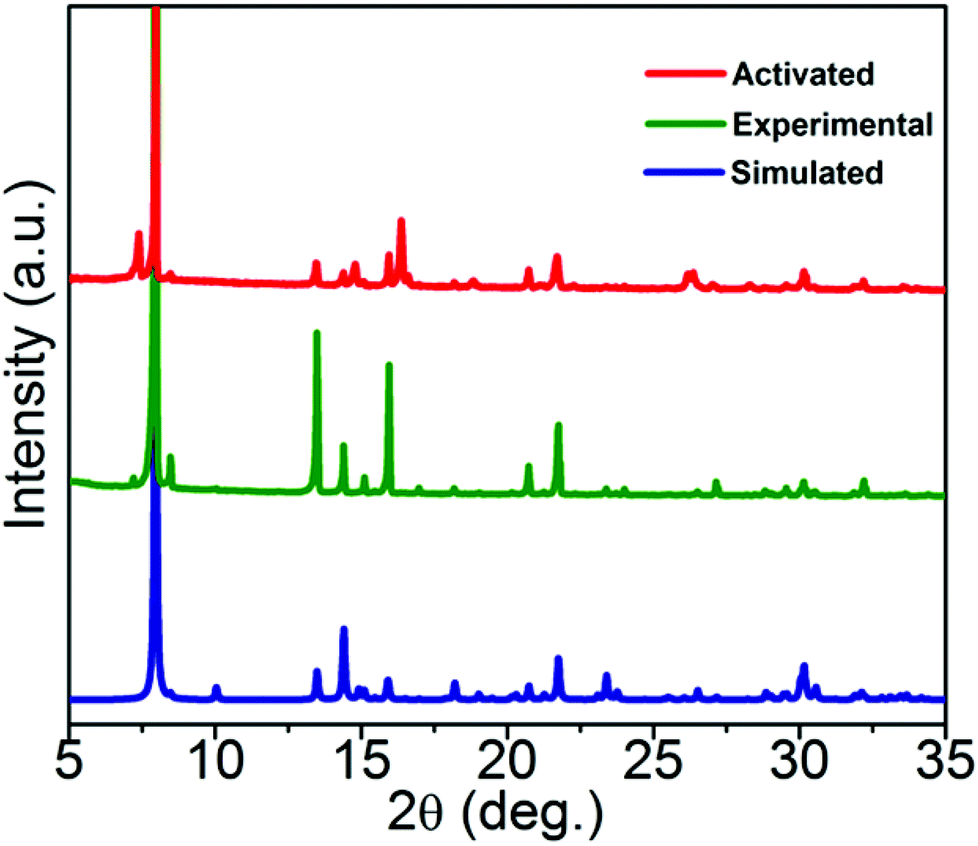 | ||
| Fig. 3 PXRD patterns of 1; comparison of experimental (bulk) and activated (1′) samples with the simulated PXRD of 1. | ||
Luminescence behaviour and sensing properties of 1′ and 2′
Coordination polymers designed and constructed from d10 metal nodes (Cd+2 and Zn+2) and conjugated organic linkers are well known for their photoluminescence properties and can be used as potential photoactive materials.22 Hence, solid state photoluminescence (PL) spectra of the H2SDB ligand and guest free activated CPs 1′ and 2′ were recorded at room temperature. Absorption maximum for H2SDB was observed at 270 nm; however, 1′ and 2′ showed major absorption bands at ca. 290 and 292 nm, respectively (Fig. S10†). Upon excitation at 270 nm, H2SDB showed emission at 333 nm. The emission band of the free ligands can be attributed to the π*→n or π*→π transitions. Strong fluorescence emission occurred at 451 and 447 nm, respectively, for 1′ and 2′ when they were excited at 330 nm (Fig. S11†). The red shift (ca. 117 nm) in 1′ and 2′ with respect to the emission wavelength of H2SDB may be considered as ligand to metal charge transfer (LMCT).23 The observed red shift in the emission bands from H2SDB to the CPs is attributed to the chelation of the ligand to the metal centers. The photoluminescence spectra of activated compounds 1′ and 2′ displayed strong emission bands at 451 nm and 447 nm, respectively, in the solid state at ambient temperature upon excitation at 330 nm, which prompted us to investigate their application for the detection of electron-withdrawing nitroaromatic compounds. The quenching phenomenon is explained by electrostatic interactions between the analytes and CPs. Usually, the analyte (electron deficient) accepts excited electrons from the LUMO of electron rich CPs which causes the quenching of emission intensity. Varying degrees of quenching for different analytes and the shifting of emission maxima may be attributed to the structural aspects of the CP cavity and analytes, electrostatic and weak intermolecular interactions. We have chosen four different analytes; TNP (2,4,6-trinitrophenol or picric acid), 2,4-DNP (2,4,-dinitrophenol), o-NP (ortho-nitrophenol) and p-NP (para-nitrophenol) for the sensing experiments. Thus, fluorescence quenching titrations were performed by the incremental addition of nitroaromatics in acetone solutions (from 40 μL to 200 μL) to emulsions of 1′ and 2′ in acetone. In all of the titration experiments after the addition of the nitroaromatics the emulsion was allowed to stir continuously for 15 minutes in order to obtain a homogenous mixture before the emission quenching measurements. A control reaction was also performed by photo-irradiating an emulsion mixture of acetone (2 mL) in the presence of 1′ and 2′ (2 mg) with constant stirring for 5 minutes before commencing the fluorescence quenching titrations with different nitroaromatics. The observed emission peaks for the blank emulsions of 1′ and 2′ are 402 and 404 nm upon excitation with 290 and 292 nm, respectively. The difference in emission peak wavelength (∼50 nm) for the acetone emulsions of 1′ and 2′ in the control reaction with respect to the solid state emission may be attributed to the solvent effect.Fig. 4 depicts the quenching of luminescence intensity upon the incremental addition of acetone solutions of TNP (40–200 μL) along with 1′. The maximum fluorescence intensity of 1′ was reduced by 67% upon addition of 200 μL TNP solution in acetone. The quenching percentage is calculated using the formula (I0 − I)/I0 × 100%, where I0 and I are the fluorescence intensities of 1′ before and after addition of the nitroaromatic. Quenching of the luminescence intensity and slight shifts in emission wavelength upon the incremental addition of TNP may be due to the supramolecular interactions of the nitroaromatic compound with the interior and surface sites of the CP.24
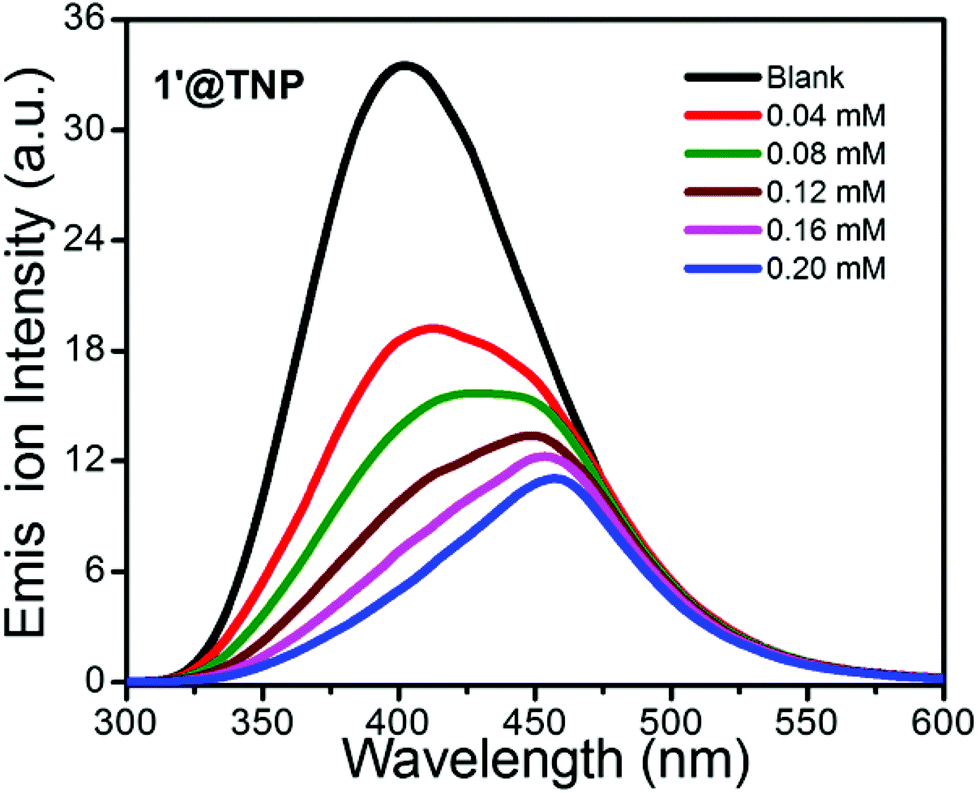 | ||
| Fig. 4 Emission spectra of 1′ (2 mg per 2 mL) with the incremental addition of 2 mM solution of TNP in acetone. Emission wavelength of 402 nm was obtained upon excitation at 290 nm. | ||
Compared to TNP, all other aromatic nitro compounds (2,4-DNP, o-NP and p-NP) showed poor fluorescence quenching with respect to 1′ (Fig. S12–S14†). 2,4-DNP, o-NP and p-NP quench the fluorescence intensity of 1′ by only 7.1%, 21% and 5% upon addition of 200 μL of the respective acetone solutions (Fig. 5). An almost similar trend was observed for the nitro detection studies with 2′ with the same series of analytes (Fig. S15†). The quenching efficiencies for 200 μL of TNP, 2,4-DNP, o-NP and p-NP for the fluorescence intensity of 2′ are 56%, 15%, 15%, and 13%, respectively (Fig. S16–19†). For better clarity, the quenching efficiency of the analytes has been provided by Stern–Volmer (SV) plots in the ESI (Fig. S24–25†). From the above fluorescence quenching studies it can be inferred that both 1′ and 2′ are good candidates for the detection of TNP from the small pool of analytes investigated and the fluorescence quenching efficiency of 1′ is slightly better than that of 2′. The efficient quenching by 1′ and 2′ towards TNP can be attributed to photo induced electron transfer from the excited CP framework (as an electron donor) to the highly electron deficient TNP (electron acceptor). We also checked the recyclability and reusability of 1′ and 2′ by centrifugation of the emulsion after use and washing several times with acetone.
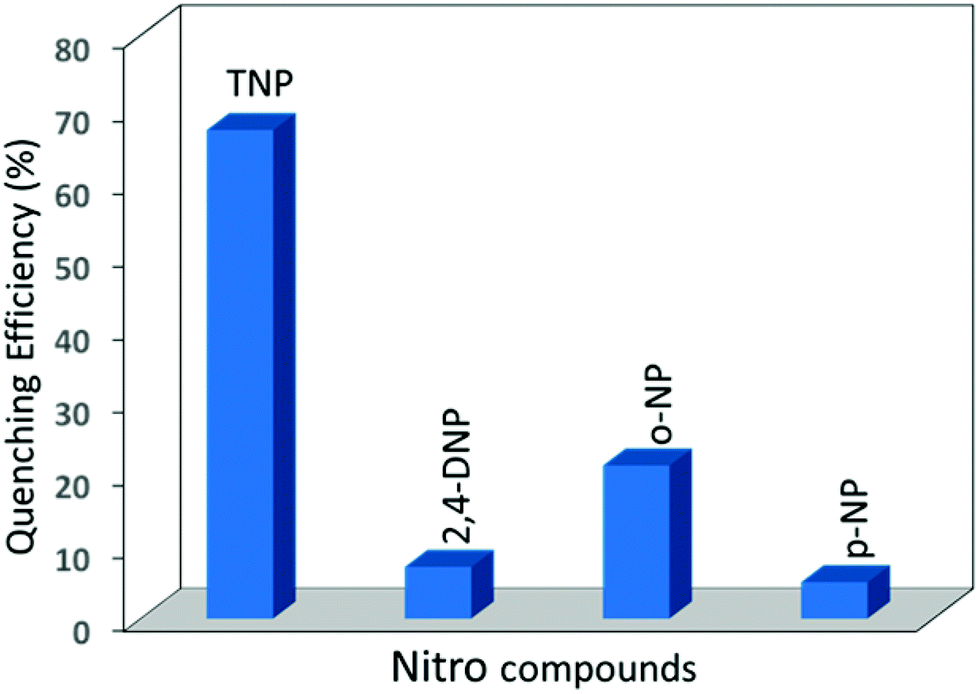 | ||
| Fig. 5 Percentage of fluorescence quenching efficiency of 1′ by different nitroaromatic compounds. | ||
As depicted in Fig. 6, the quenching efficiency was tested for 3 cycles and all of the cycles showed almost the same results (∼ 67% and 56% for 1′ and 2′) via screening the emission spectra of 1′ and 2′ (Fig. S20†) dispersed in acetone solutions of the nitroaromatics. The phase purity of the recovered materials 1′ and 2′ from each experimental cycle was also confirmed by PXRD data which demonstrated the crystallinity and stability of the framework (Fig. S21†). In the case of TNP, both 1′ and 2′ showed considerable photoluminescence quenching (67% and 56%) of the emission peak intensity and these d10 luminescent compounds could be regarded as potential materials for the detection of TNP compared to the other three nitro analytes.
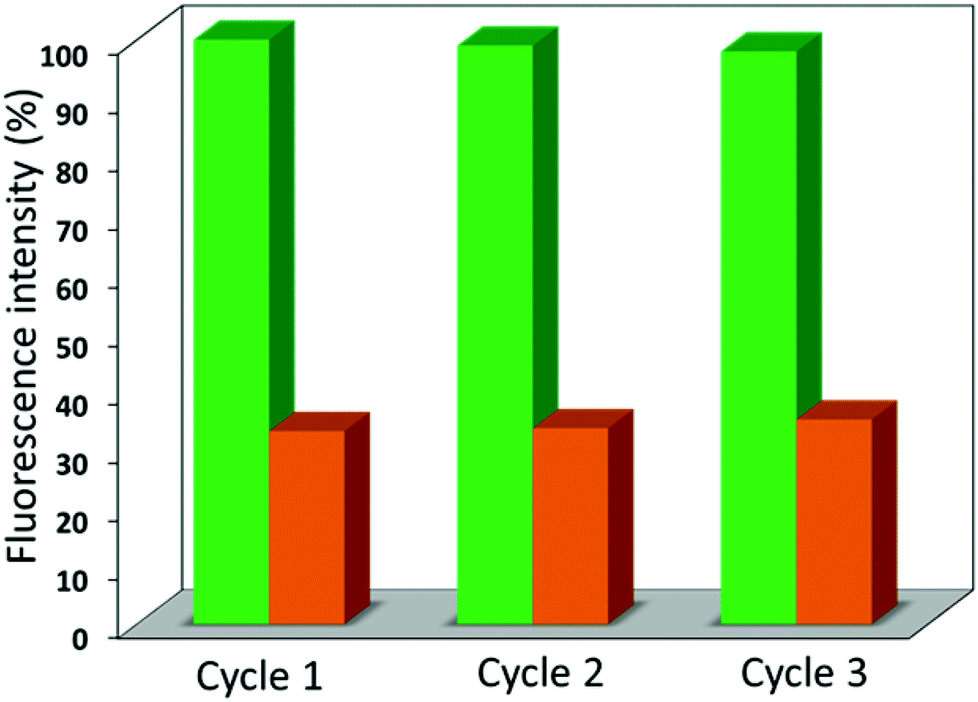 | ||
| Fig. 6 Reproducibility of the quenching capacity of 1′ dispersed in acetone with titration of TNP in acetone. The material was recovered by centrifuging after each experimental cycle and washing several times with acetone. The fluorescence quenching efficiency of 1′ was effective after 3 cycles [green bars = initial intensity, orange bars = after addition of 200 μL (2 mM) TNP solution]. | ||
Conclusions
Luminescent two dimensional coordination polymers {[Cd(SDB)(H2O)]·3(H2O)2}n1 and {[Zn3(μ-OH)2(SDB)2]n·(PPZ)}n2, (where SDB = 4,4′-sulfonyldibenzoate; PPZ = piperazine) have been synthesized by solvothermal methods and characterised by various physico-chemical methods including SCXRD. SCXRD analysis revealed the presence of water molecules and in situ generated piperazine moieties (under solvothermal conditions from the 4-BPMP ligand) in the cylindrical channels of pristine CPs 1 and 2, respectively. The pristine compounds 1 and 2 were activated at 200 °C for 8 hours and the activated samples (1′ and 2′) showed robustness and stability of the framework topology up to 400 and 420 °C. This is further confirmed by TGA and PXRD experiments. The photoluminescence properties of the activated compounds 1′ and 2′ were investigated for sensing electron withdrawing nitroaromatic compounds by fluorescence quenching phenomena. A series of nitroaromatic compounds; TNP (2,4,6-trinitrophenol or picric acid), 2,4-DNP (2,4,-dinitrophenol), o-NP (ortho-nitrophenol) and p-NP (para-nitrophenol) have been studied in the sensing experiments. Both 1′ and 2′ showed considerable photoluminescence quenching for TNP (67% and 56%) while the other analytes showed comparatively poor quenching efficiencies (up to 21%). Thus, the present study enriches research insight into the design and synthesis of luminescent CPs towards the sensing of hazardous nitroaromatic compounds.Acknowledgements
The authors thank CSIR, New Delhi for generous support towards infrastructure and core competency development. We acknowledge Ms. Riddhi Laiya for the PXRD data, Mrs Monika Gupta for the TGA data, Mr Viral Vakani for the FTIR data, Mr Satyaveer Gothwal for the CHNS analysis and Dr P. Paul for all round analytical support. KKB & BP acknowledge CSIR (India) and YR acknowledges UGC (India) for research fellowships. Publication Registration Number: CSIR-CSMCRI–178/2014.Notes and references
- (a) J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC; (b) O. K. Farha, A. Ö. Yazaydın, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944 CrossRef CAS PubMed; (c) M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 6782 CrossRef PubMed; (d) J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869 CrossRef CAS PubMed; (e) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed; (f) P. D. C. Dietzel, V. Besikiotis and R. Blom, J. Mater. Chem., 2009, 19, 7362 RSC; (g) S. Ma and H.-C. Zhou, Chem. Commun., 2010, 46, 44 RSC.
- (a) A. Corma, H. García, F. X. Llabres and I. Xaména, Chem. Rev., 2010, 110, 4606 CrossRef CAS PubMed; (b) Y. Liu, W. Xuan and Y. Cui, Adv. Mater., 2010, 22, 4112 CrossRef CAS PubMed; (c) L. Ma, C. Abneya and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC; (d) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC; (e) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196 CrossRef CAS PubMed; (f) C.-D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940 CrossRef CAS PubMed; (g) C.-D. Wu and W. Lin, Angew. Chem., Int. Ed., 2007, 119, 1093 CrossRef; (h) S. Parshamoni, S. Sanda, H. S. Jena and S. Konar, Dalton Trans., 2014, 43, 7191 RSC.
- (a) P. Dechambenoit and J. R. Long, Chem. Soc. Rev., 2011, 40, 3249 RSC; (b) D. F. Weng, Z. M. Wang and S. Gao, Chem. Soc. Rev., 2011, 40, 3157 RSC; (c) M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353 RSC; (d) C. N. R. Rao, A. K. Cheetham and A. Thirumurugan, J. Phys.: Condens. Matter, 2008, 20, 083202 CrossRef; (e) A. Chakraborty, B. K. Ghosh, J. B. Arino, J. Ribas and T. K. Maji, Inorg. Chem., 2012, 51, 6440 CrossRef CAS PubMed; (f) C. Dey, R. Das, B. K. Saha, P. Poddar and R. Banerjee, Chem. Commun., 2011, 47, 11008 RSC; (g) B. K. Tripuramallu, P. Manna and S. K. Das, CrystEngComm, 2014, 16, 4816 RSC; (h) S. Sanda, S. Goswami, H. S. Jena, S. Parshamoni and S. Konar, CrystEngComm, 2014, 16, 4742 RSC.
- (a) M. D. Allendorf, C. A. Bauer, R. K. Bhaktaa and R. J. T. Houka, Chem. Soc. Rev., 2009, 38, 1330 RSC; (b) Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed; (c) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC; (d) D. Banerjee, Z. Hua and J. Li, Dalton Trans., 2014, 43, 10668 RSC.
- (a) P. Horcajada, C. Serre, M. Vallet-Reg, M. Sebban, F. Taulelle and G. Férey, Angew. Chem., Int. Ed., 2006, 118, 6120 CrossRef; (b) P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172 CrossRef CAS PubMed; (c) J. D. Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957 CrossRef PubMed; (d) A. C. McKinlay, R. E. Morris, P. Horcajada, G. Frey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260 CrossRef CAS PubMed; (e) F. Ke, Y.-P. Yuan, L.-G. Qiu, Y.-H. Shen, A.-J. Xie, J.-F. Zhu, X.-Y. Tian and L.-D. Zhang, J. Mater. Chem., 2011, 21, 3843 RSC; (f) P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232 CrossRef CAS PubMed.
- (a) G.-P. Yang, L. Hou, L.-F. Ma and Y.-Y. Wang, CrystEngComm, 2013, 15, 2561 RSC; (b) Y. B. Go, X. Wang, E. V. Anokhina and A. J. Jacobson, Inorg. Chem., 2005, 44, 8265 CrossRef CAS PubMed; (c) L. Luo, K. Chen, Q. Liu, Y. Lu, T. Okamura, G.-C. Lv, Y. Zhao and W.-Y. Sun, Cryst. Growth Des., 2013, 13, 2312 CrossRef CAS; (d) Y.-X. Sun and W.-Y. Sun, Chin. Chem. Lett., 2014, 25, 823 CrossRef CAS PubMed; (e) A. D. Burrows, K. Cassar, R. M. W. Friend, M. F. Mahon, S. P. Rigby and J. E. Warren, CrystEngComm, 2005, 7, 548 RSC; (f) L.-S. Long, CrystEngComm, 2010, 12, 1354 RSC; (g) F. Yuan, J. Xie, H.-M. Hu, C.-M. Yuan, B. Xu, M.-L. Yang, F.-X. Dong and G.-L. Xue, CrystEngComm, 2013, 15, 1460 RSC.
- (a) S. Zhang, W. Shi, L. Li, E. Duan and P. Cheng, Inorg. Chem., 2014, 53, 10340 CrossRef CAS PubMed; (b) R. Grünker, V. Bon, P. Müller, U. Stoeck, S. Krause, U. Mueller, I. Senkovska and S. Kaske, Chem. Commun., 2014, 50, 3450 RSC; (c) N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933 CrossRef CAS PubMed; (d) H.-T. Ye, C.-Y. Ren, G.-F. Hou, Y.-H. Yu, X. Xu, J.-S. Gao, P.-F. Yan and S.-W. Ng, Cryst. Growth Des., 2014, 14, 3309 CrossRef CAS; (e) J. E. Warren, C. G. Perkins, K. E. Jelfs, P. Boldrin, P. A. Chater, G. J. Miller, T. D. Manning, M. E. Briggs, K. C. Stylianou, J. B. Claridge and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2014, 53, 4592 CrossRef CAS PubMed; (f) Z.-X. Zhang, N.-N. Ding, W.-H. Zhang, J.-X. Chen, D. J. Young and T. S. Andy Hor, Angew. Chem., Int. Ed., 2014, 53, 4628 CrossRef CAS PubMed; (g) Y. Rachuri, K. K. Bisht and E. Suresh, Cryst. Growth Des., 2014, 14, 3300 CrossRef CAS.
- (a) W.-X. Chen, G.-L. Zhuang, H.-X. Zhao, L.-S. Long, R.-B. Huang and L.-S. Zheng, Dalton Trans., 2011, 40, 10237 RSC; (b) D.-X. Xue, Y.-Y. Lin, X.-N. Cheng and X.-M. Chen, Cryst. Growth Des., 2007, 7, 1332 CrossRef CAS; (c) S. Horike, M. Sugimoto, K. Kongpatpanich, Y. Hijikata, M. Inukai, D. Umeyama, S. Kitao, M. Setod and S. Kitagawa, J. Mater. Chem. A, 2013, 1, 3675 RSC; (d) K.-J. Chen, R.-B. Lin, P.-Q. Liao, C.-T. He, J.-B. Lin, W. Xue, Y.-B. Zhang, J.-P. Zhang and X.-M. Chen, Cryst. Growth Des., 2013, 13, 2118 CrossRef CAS; (e) Z. Fu, Y. Chen, J. Zhang and S. Liao, J. Mater. Chem., 2011, 21, 7895 RSC; (f) E. Suresh, K. Boopalan, R. V. Jasra and M. M. Bhadbhade, Inorg. Chem., 2001, 40, 4078 CrossRef CAS; (g) Y. Hijikata, S. Horike, D. Tanaka, J. Groll, M. Mizuno, J. Kim, M. Takatade and S. Kitagawa, Chem. Commun., 2011, 47, 7632 RSC; (h) P. Mahata, C.-M. Draznieks, P. Roy and S. Natarajan, Cryst. Growth Des., 2013, 13, 155 CrossRef CAS; (i) X.-L. Chen, B. Zhang, H.-M. Hu, F. Fu, X.-L. Wu, T. Qin, M.-L. Yang, G.-L. Xue and J.-W. Wang, Cryst. Growth Des., 2008, 8, 3706 CrossRef CAS; (j) M. Kondo, Y. Irie, Y. Shimizu, M. Miyazawa, H. Kawaguchi, A. Nakamura, T. Naito, K. Maeda and F. Uchida, Inorg. Chem., 2004, 43, 6139 CrossRef CAS PubMed; (k) I.-H. Park, S. S. Lee and J. J. Vittal, Chem. – Eur. J., 2013, 19, 2695 CrossRef CAS PubMed; (l) L. Qin, J.-S. Hu, M.-D. Zhang, Z.-J. Guo and H.-G. Zheng, Chem. Commun., 2012, 48, 10757 RSC; (m) S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153 CrossRef CAS PubMed; (n) P. Mahata, S. Natarajan, P. Panissod and M. Drillon, J. Am. Chem. Soc., 2009, 131, 10140 CrossRef CAS PubMed.
- (a) F.-Y. Lian, F.-L. Jiang, D.-Q. Yuan, J.-T. Chen, M.-Y. Wu and M.-C. Hong, CrystEngComm, 2008, 10, 905 RSC; (b) Y. Hijikata, S. Horike, M. Sugimoto, M. Inukai, T. Fukushima and S. Kitagawa, Inorg. Chem., 2013, 52, 3634 CrossRef CAS PubMed; (c) X.-L. Chen, L. Gou, H.-M. Hu, F. Fu, Z.-X. Han, H.-M. Shu, M.-L. Yang, G.-L. Xue and C.-Q. Du, Eur. J. Inorg. Chem., 2008, 239 CrossRef CAS.
- (a) S. S. Nagarkar, B. Joarder, A. K. Chaudhari, S. Mukherjee and S. K. Ghosh, Angew. Chem., Int. Ed., 2013, 52, 2881 CrossRef CAS PubMed; (b) S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153–4155 CrossRef CAS PubMed; (c) A. Lan, K. Li, H. Wu, D. H. Olson, T. J. Emge, W. Ki, M. Hong and J. Li, Angew. Chem., Int. Ed., 2009, 48, 2334–2338 CrossRef CAS PubMed; (d) H. Xu, F. Liu, Y. Cui, B. Chen and G. Qian, Chem. Commun., 2011, 47, 3153 RSC.
- (a) Y. Ma, S. Huang, M. Deng and L. Wang, Appl. Mater. Interfaces, 2014, 6, 7790 CrossRef CAS PubMed; (b) N. Tu and L. Wang, Chem. Commun., 2013, 49, 6319 RSC.
- (a) N. Enkin, E. Sharon, E. Golub and I. Willner, Nano Lett., 2014, 14, 4918 CrossRef CAS PubMed; (b) J.-W. Oh, W.-J. Chung, K. Heo, H.-E. Jin, B. Y. Lee, E. Wang, C. Zueger, W. Wong, J. Meyer, C. Kim, S.-Y. Lee, W.-G. Kim, M. Zemla, M. Auer, A. Hexemer and S.-W. Lee, Nat. Commun., 2014, 5 DOI:10.1038/ncomms4043.
- (a) K. K. Bisht and E. Suresh, Inorg. Chem., 2012, 51, 9577 CrossRef CAS PubMed; (b) K. K. Bisht and E. Suresh, Cryst. Growth Des., 2013, 13, 664 CrossRef CAS; (c) Y. Rachuri, K. K. Bisht, B. Parmar and E. Suresh, J. Solid State Chem., 2014, 213, 43 CrossRef PubMed.
- Y. Rachuri, K. K. Bisht, B. Parmar and E. Suresh, J. Solid State Chem., 2014 DOI:10.1016/j.jssc.2014.05.012.
- D. Pocic;, J.-M. Planeix, N. Kyritsakas, A. Jouaiti and M. W. Hosseini, CrystEngComm, 2005, 7, 624 RSC.
- G. M. Sheldrick, SAINT 5.1 ed, Siemens Industrial Automation Inc., Madison, WI, 1995 Search PubMed.
- SADABS, Empirical Absorption Correction Program, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- (a) G. M. Sheldrick, SHELXTL Reference Manual: Version 5.1, Bruker AXS, Madison, WI, 1997 Search PubMed; (b) G. M. Sheldrick, SHELXL–97 Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, C34 Search PubMed.
- D. Xiao, H. Chen, D. Suna, G. Zhang, J. He, R. Yuan and E. Wang, Solid State Sci., 2011, 13, 1573 CrossRef CAS PubMed.
- N. L. Rosi, M. Eddaoudi, J. Kim, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2002, 41, 284–287 CrossRef CAS.
- (a) S. Pramanik, Z. Hu, X. Zhang, C. Zheng, S. Kelly and J. Li, Chem. – Eur. J., 2013, 19, 15964 CrossRef CAS PubMed; (b) B. Gole, A. K. Bar and P. S. Mukherjee, Chem. Commun., 2011, 47, 12137 RSC; (c) M. Guo and Z.-M. Sun, J. Mater. Chem., 2012, 22, 15939 RSC; (d) D. Ma, B. Li, X. Zhou, Q. Zhou, K. Liu, G. Zeng, G. Li, Z. Shi and S. Feng, Chem. Commun., 2013, 49, 8964 RSC.
- (a) J.-C. Dai, X.-T. Wu, Z.-Y. Fu, S.-M. Hu, W.-X. Du, C.-P. Cui, L.-M. Wu, H.-H. Zhang and R.-Q. Sun, Chem. Commun., 2002, 12 RSC; (b) X. L. Wang, C. Qin, E. B. Wang, Y. G. Li, N. Hao, C. W. Hu and A. L. Xu, Inorg. Chem., 2004, 43, 1850 CrossRef CAS PubMed; (c) S. Parshamoni, H. S. Jena, S. Sanda and S. Konar, Inorg. Chem. Front., 2014, 1, 611 RSC; (d) L. Wang, M. Yang, G. H. Li, Z. Shi and S. H. Feng, Inorg. Chem., 2006, 45, 2474 CrossRef CAS PubMed.
- (a) S. S. Nagarkar, A. V. Desai and S. K. Ghosh, Chem. Commun., 2014, 50, 8915 RSC; (b) Y.-N. Gong, Y.-L. Huang, L. Jiang and T.-B. Lu, Inorg. Chem., 2014, 53, 9457 CrossRef CAS PubMed; (c) L. B. Sun, H. Z. Xing, J. Xu, Z. Q. Liang, J. H. Yu and R. R. Xu, Dalton Trans., 2013, 42, 5508 RSC; (d) Y. S. Xue, Y. B. He, L. Zhou, F. J. Chen, Y. Xu, H. B. Du, X. Z. You and B. L. Chen, J. Mater. Chem. A, 2013, 1, 4525 RSC; (e) J.-D. Xiao, L.-G. Qiu, F. Ke, Y.-P. Yuan, G.-S. Xu, Y.-M. Wanga and X. Jiang, J. Mater. Chem. A, 2013, 1, 8745 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: ORTEP diagram, FTIR, TGA, PXRD, UV-Vis, PL spectra and selected bond lengths and angles. CCDC 1024317 and 1024318. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00175c |
| This journal is © the Partner Organisations 2015 |