
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-step grafting of polymers to graphene oxide†
Helen R.
Thomas
a,
Daniel J.
Phillips
a,
Neil R.
Wilson
b,
Matthew I.
Gibson
 *a and
Jonathan P.
Rourke
*a
*a and
Jonathan P.
Rourke
*a
aDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: j.p.rourke@warwick.ac.uk; m.i.gibson@warwick.ac.uk
bDepartment of Physics, University of Warwick, Coventry CV4 7AL, UK
First published on 6th October 2015
Abstract
The direct grafting of poly(N-isopropylacrylamide) to the basal plane of graphene oxide has been achieved in a single step: cleavage of the terminal thiocarbonylthio group on RAFT grown poly(N-isopropylacrylamide) reveals a reactive thiol that attacks the epoxides present across the surface of graphene oxide. The new composite material was characterised by a combination of SSNMR, FTIR, Raman, EDX, XPS, TGA and contact angle measurement; it shows enhanced thermal stability and solubility in water.
Introduction
There is currently much interest in exploiting graphene1 for a variety of applications given its many superlative physical properties: it is exceptionally electrically2 and thermally3 conductive, transparent,4 elastic,5 highly impermeable,6 extremely strong5b and yet is the thinnest6a 2D material ever obtained. However, rather than using pure graphene, which is only available in small quantities via mechanical exfoliation or chemical vapour deposition (CVD) growth, graphene oxide (GO) has become the graphene-like material of choice. Though not as strong or as conductive as graphene, GO is arguably more commercially applicable owing to its cheaper synthesis and potential for routine, large-scale production; it can be manipulated chemically7 and it is possible to reduce graphene oxide back to a more graphene-like material by thermal8 or chemical9 means. Moreover, the versatility of GO may be extended by combination with other species as part of a composite material. Considering this, polymers provide an obvious partner given the plethora of interesting properties (solubility, switchable characteristics, stimuli responsiveness,10 or impacting ‘stealth’ properties for in vivo application11) they can possess and which have been applied to controlling the interfacial properties of nanoparticles. Most previous attempts at combining GO with polymers have focussed on a simple mixing procedure,12 though this approach can be hampered by phase separation and aggregation issues.An alternative approach is to covalently functionalise a graphene-like structure with polymer chains following a traditional “grafting-from” or “grafting-to” like methodology13 as has been used for the functionalization of solid substrates and particles. Controlled-radical polymerisation technologies now enable the routine formation of materials with increasing levels of control over the molecular weights and structures.14 However, to apply the likes of Nitroxide-Mediated Polymerisation (NMP),15 copper-mediated polymerisations16 or Reversible Addition–Fragmentation Chain Transfer (RAFT)17 polymerisation using the grafting-from approach would require the functionalisation of the GO sheet with initiating or chain-transfer type species, which prevents characterization of the polymer chains and introduces batch-batch variability. A simpler alternative, which we demonstrate in this work, is therefore to covalently add pre-formed polymer chains of controlled sizes to the GO surface. Previous approaches to this grafting-to methodology have relied on multi-step synthetic routes that typically target the carboxyl groups found at the edges of the GO flakes.18
Recently, we have demonstrated a way of functionalising GO with thiol functional groups. This was achieved by ring-opening the epoxides present on the surface of GO with nucleophilic thioacetate anions followed by a simple hydrolysis procedure.19 Here, we extend this concept to directly attach a thiol-terminated polymer to GO in one reaction step. This is achieved by exploiting the thiocarbonylthio moiety at the ω-terminus of a RAFT-derived polymer chain which can be readily converted to a thiol group upon the addition of a nucleophile such as a primary amine. We demonstrate not only the successful attachment of polymer chains to a GO surface in this fashion, but also its value as a tool with which to significantly change the properties of the system.
Results and discussion
A series of well-defined water-soluble poly(N-isopropylacrylamide), pNIPAM, polymers with differing chain lengths (i.e. 30, 200 and 440 repeat units) was prepared using the RAFT methodology, giving a thiocarbonylthio moiety at the ω-chain-end (Scheme 1 and ESI, Table S1†). Each polymer was then treated with methyl amine to cleave the thiocarbonylthio group and added directly to a suspension of base-washed GO (bwGO) – used here to avoid any issues relating to reactions with oxidative debris.20 The resulting thiol-terminated polymer chains reacted in situ with the bwGO via an epoxide ring-opening reaction, which can proceed by both direct attack at the epoxide (an SN2 reaction) or by an attack on the double bond adjacent to the epoxide (an SN2′ reaction) to produce final products with polymer chains grafted randomly across both faces of the graphene-like sheet (Scheme 1).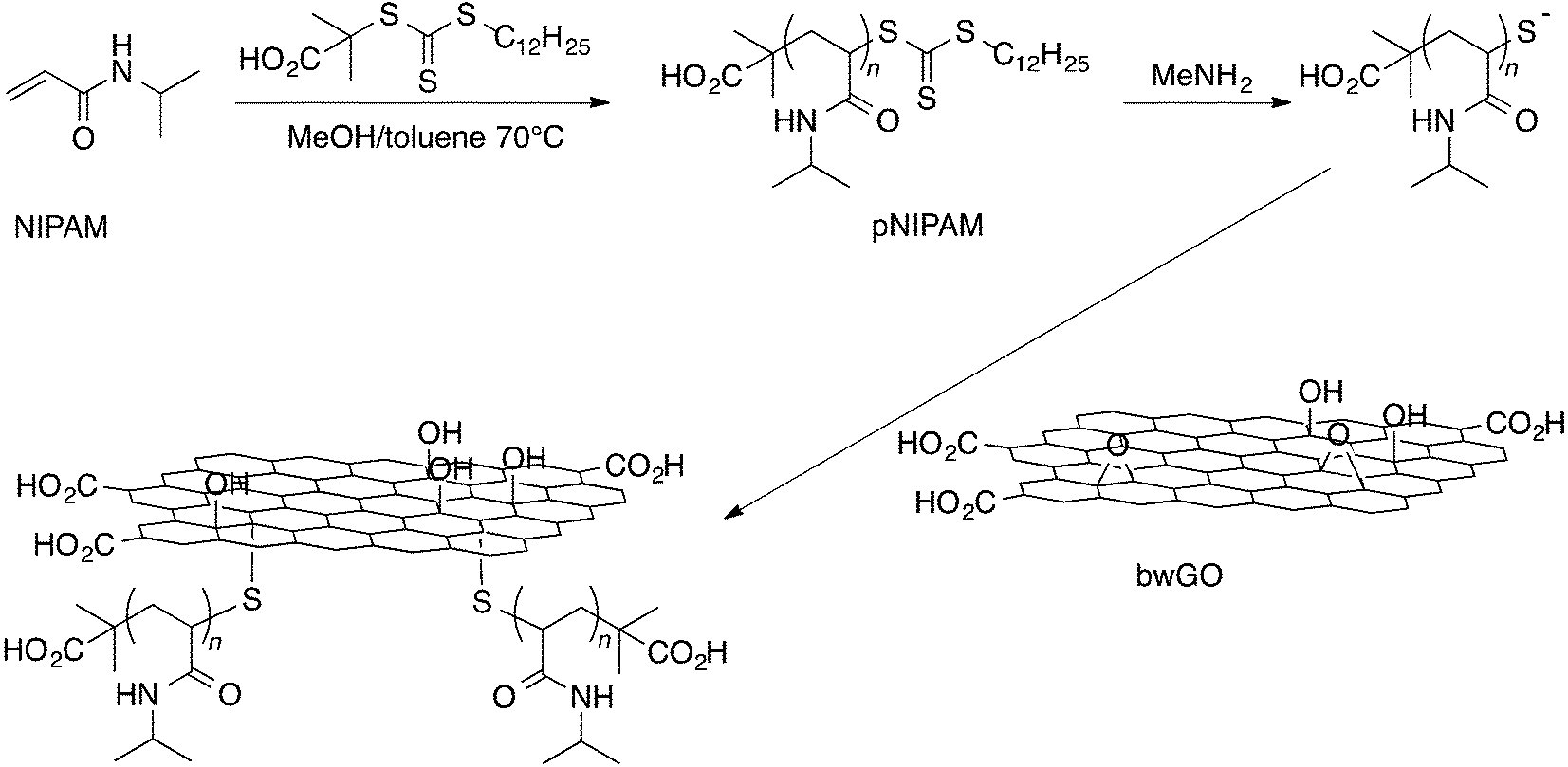 | ||
| Scheme 1 Synthetic route used to the GO-pNIPAMn samples. | ||
For each polymer sample, a single graphene containing material, free of unbound polymer was isolated following extensive washing with a variety of solvents. Five spectroscopic techniques (ssNMR, FTIR, EDX, XPS and Raman) were then used to confirm the presence of the polymer chains on the bwGO, whilst TGA and WCA measurements indicated changes in property following the chemical modification.
Sample characterisation
The solid-state 13C NMR spectra of the polymer/GO hybrids clearly show the expected peaks for the GO (broad peak at around 126 ppm for the sp2 areas of the GO sheets and the epoxide and hydroxide functionlised carbons at 61 and 72 ppm, respectively) and for those of the polymer side groups (sp3 aliphatic carbons at 30 and 35 ppm and the amide carbon at around 172 ppm) and the main polymer chain (shoulder at 40 ppm). Fig. 1 shows the spectrum for the GO-pNIPAM440 material, but other samples showed similar spectra. Alongside the new peaks in the spectrum, there is a decrease in the intensity of the epoxide carbon in the GO, consistent with reaction taking place at this site. An approximate integral shows that the total area represented by the polymer peaks corresponds to around 20% of the total signal area. | ||
| Fig. 1 The direct excitation 13C MAS ssNMR spectra of bwGO and GO-pNIPAM440. | ||
FTIR spectra of the polymer-functionalised GO (Fig. ESI.2†) reveal peaks at 2880 and 2900 cm−1 due to C–H stretches from the alkyl component of the polymer, and a reduction in the intensity of peaks from epoxy groups at 1240 cm−1.21 The expected C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch of an amide to falls between 1630 and 1670 cm−1, but in practice it cannot be distinguished from the overlapping carboxyl CO and CC bands found in GO itself.
O stretch of an amide to falls between 1630 and 1670 cm−1, but in practice it cannot be distinguished from the overlapping carboxyl CO and CC bands found in GO itself.
EDX (ESI Table 2†) shows nitrogen incorporation (approximately 2.7 ± 0.4 atomic% for GO-pNIPAM440) consistent with amide functionalities being introduced, but cannot be used to distinguish the chemical environments of the nitrogen. XPS measurements give a similar level of N incorporation (2.9 ± 0.4 atomic% nitrogen) and Fig. 2a shows the survey scan of GO-pNIPAM440 which gives the incorporation level. The XPS core level spectra were used to determine the chemical environment of the both the carbon and the nitrogen. The C 1s core level spectrum (Fig. 2b) shows the environments expected for a GO-based material with little apparent change upon polymer incorporation. However, since the polymer chain consists of a simple alkyl chain with amide functionality, it is not possible to identify peaks from the alkyl chain, as they will be obscured by the C–C bonded carbons in GO; likewise the amide carbons, which are expected to have a binding energy of 287.9–288.3 eV,22 will be obscured by the C–O (hydroxy and epoxy) and CO (keto, carboxy) peaks of GO itself. The N 1s core level spectrum (Fig. 2c) is much more illuminating and shows nitrogen in two different chemical environments. The principal contribution has a measured binding energy of 400 eV, consistent with amide functionalities;22 by area, this peak corresponds to 80% of the nitrogen present in GO-pNIPAM440. The smaller peak at 401.5 eV can be assigned to residual DMF solvent in this particular sample.
 | ||
| Fig. 2 XPS analysis of GO-NIPAM400, complete with survey scan (a), and core level spectra C 1s (b) and N 1s (c). The experimental data is shown as points with the underlying lines showing the fitted peaks (assignments as labelled). | ||
The Raman spectrum of the GO-pNIPAM samples (Fig. SI.6†) is as expected; showing strong D, G and G′ bands, consistent with a disordered monolayer structure. The D/G ratio is calculated to be 1.3, indicating that the addition of the polymer chain has caused no significant change (e.g. reduction) to the sheet structure of GO.
Using our elemental analysis data, it is possible to approximate the relative grafting density for each of the different polymers. Increasing the chain length from pNIPAM30 to pNIPAM200 gave a increase in nitrogen from 1.3 to 2.9%, consistent with a doubling of the mass of polymer being incorporated. However, this must be normalised by the relative length of the polymer chains, and therefore actually corresponds to a 3 fold decrease in grafting density (Table 2 ESI†). This is consistent with observations on flat surfaces (and RAFTed polymers onto gold particles) as the increased steric crowding of longer polymers lower the grafting density, even though more ‘mass’ is absorbed.23 pNIPAM440 had a similar nitrogen level to pNIPAM200, indicating another corresponding decrease in grafting density. A more detailed analysis of the EDX data showed that there was approximately 1 polymer chain per 1100 carbon atoms for pNIPAM30 (roughly equal to 1 chain per 150 nm2, which is a circle with radius 6.9 nm). As polymers can normally graft at densities up to 2 × Rg (radius of gyration), we can conclude that for small polymers, the grafting density was limited by the availability of the reactive sites (as we would expect an Rg < 1 nm), but as the chain length increases, sterics dominate as the longer polymers would be expected to have a radius of gyration >5 nm.24 Thus the composition (and any subsequent applications) of these nano-composites must take into account the balance between the grafted chain length and the achieved grafting densities. Any polymer derived from RAFT/Xanthate polymerization will have this terminal thiol group making this a versatile approach. However, it must be noted that issues such as steric hindrance from both the monomer class (acrylamide verses methacrylamide) and the monomer side chains will play a role in the final grafting efficiency, as would monomers with competing nucleophiles (e.g. thiols). Such considerations have previously been reviewed in detail in the context of RAFT end-group modification.13a,25
Properties
The new hybrid material has properties that reflect its composition: TGA measurements show a major decomposition temperature of the new material, at 550–600 °C, higher than that of both the isolated polymer, which decomposes sharply at around 380 °C (Fig. 3), and of that of the bwGO which loses the majority of its mass at around 460 °C. The decomposition temperature of the new material is comparable with that of other functionalised GO samples, where we see a stabilisation of the GO sheets which is attributable to the capping of the reactive surface functionalities which are believed to be the sites of initial decomposition.8,21,26 | ||
| Fig. 3 TGA profile of bwGO, pNIPAM440 and GO-pNIPAM440. | ||
This increase in thermal stability does not come at the cost of reduced solubility. Indeed, the GO–polymer composites show smaller contact angles (34°) than bwGO, suggesting an increase in hydrophilicity due to the hydrophilicity of the polymer chains (Fig. ESI.13†), and it could be dispersed in water. pNIPAM has a strong concentration (and co-operative) dependence on its LCST27 (cloud point) but the grafting densities employed here are low enough to prevent this transition at the temperatures employed, thus rendering this essentially a permanently hydrophilic polymer.
Conclusions
Here we have demonstrated a simple ‘grafting-to’ methodology and obtained graphene oxide/polymer hybrid materials. RAFT polymerization was used to generate well-defined polymers with a masked thiol at the chain end, and this was then exploited to conjugate the polymers directly to graphene oxide via the epoxide groups that are present across the entire basal plane. As all RAFT-derived polymers have this masked thiol present, this demonstrates a versatile, yet facile, route to polymer/GO composites. Here, hydrophilic poly(N-isopropyl acrylamide) was grafted onto the GO to give water-dispersible composites, but the methodology could equally well be used to graft hydrophobic polystyrene or bioactive polymers such as glycopolymers to the surface. Grafting was confirmed by high resolution TEM, XPS, TGA and solid state NMR. These revealed that, for shorter polymers, the grafting density was limited by the number of reactive sites, but for longer polymers by steric hindrance. This new and accessible synthetic route is highly scalable, using GO in place of graphene and a 1-step conjugation method and offers opportunities to precisely control the interfacial properties of GO in a wide range of applications.Acknowledgements
We thank EPSRC for a studentship award to HRT and the University of Warwick for a Scholarship to DJP. The TGA, XPS and some of the TEM equipment used in this research were purchased with support from Advantage West Midlands (part funded by the European Regional Development Fund). We thank Marc Walker for assistance with XPS measurements. Solid-state NMR spectra were obtained at the EPSRC UK National Solid-state NMR Service at Durham. MIG acknowledges the European Research Council for a Starter Grant (638661 CRYOMAT).References
- K. S. Novoselov, Angew. Chem., Int. Ed., 2011, 50, 6986–7002 CrossRef CAS PubMed.
- I. Meric, M. Y. Han, A. F. Young, B. Ozyilmaz, P. Kim and K. L. Shepard, Nat. Nanotechnol., 2008, 3, 654–659 CrossRef CAS PubMed.
- S. Ghosh, D. L. Nika, E. P. Pokatilov and A. A. Balandin, New J. Phys., 2009, 11 Search PubMed.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS PubMed.
- (a) A. Sakhaee-Pour, Solid State Commun., 2009, 149, 91–95 CrossRef CAS PubMed; (b) C. Lee, X. D. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- (a) V. Berry, Carbon, 2013, 62, 1–10 CrossRef CAS PubMed; (b) J. S. Bunch, S. S. Verbridge, J. S. Alden, A. M. van der Zande, J. M. Parpia, H. G. Craighead and P. L. McEuen, Nano Lett., 2008, 8, 2458–2462 CrossRef CAS PubMed.
- D. R. Dreyer, A. D. Todd and C. W. Bielawski, Chem. Soc. Rev., 2014, 43, 5288–5301 RSC.
- R. Larciprete, S. Fabris, T. Sun, P. Lacovig, A. Baraldi and S. Lizzit, J. Am. Chem. Soc., 2011, 133, 17315–17321 CrossRef CAS PubMed.
- P. Cui, J. Lee, E. Hwang and H. Lee, Chem. Commun., 2011, 47, 12370–12372 RSC.
- D. J. Phillips and M. I. Gibson, Polym. Chem., 2015, 6, 1033–1043 RSC.
- G. Pasut and F. M. Veronese, Prog. Polym. Sci., 2007, 32, 933–961 CrossRef CAS PubMed.
- C. Vallés, I. A. Kinloch, R. J. Young, N. R. Wilson and J. P. Rourke, Compos. Sci. Technol., 2013, 88, 158–164 CrossRef PubMed.
- (a) R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS PubMed; (b) H. J. Salavagione, G. Martinez and G. Ellis, Macromol. Rapid Commun., 2011, 32, 1771–1789 CrossRef CAS PubMed.
- K. Matyjaszewski and J. Spanswick, Mater. Today, 2005, 8, 26–33 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3668 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- (a) Y. Tan, L. Fang, J. Xiao, Y. Song and Q. Zheng, Polym. Chem., 2013, 4, 2939–2944 RSC; (b) Y. Cao, Z. Lai, J. Feng and P. Wu, J. Mater. Chem., 2011, 21, 9271–9278 RSC.
- H. R. Thomas, A. J. Marsden, M. Walker, N. R. Wilson and J. P. Rourke, Angew. Chem., Int. Ed., 2014, 53, 7613–7618 CrossRef CAS PubMed.
- (a) J. P. Rourke, P. A. Pandey, J. J. Moore, M. Bates, I. A. Kinloch, R. J. Young and N. R. Wilson, Angew. Chem., Int. Ed., 2011, 50, 3173–3177 CrossRef CAS PubMed; (b) H. R. Thomas, S. P. Day, W. E. Woodruff, C. Vallés, R. J. Young, I. A. Kinloch, G. W. Morley, J. V. Hanna, N. R. Wilson and J. P. Rourke, Chem. Mater., 2013, 25, 3580–3588 CrossRef CAS.
- A. Bagri, C. Mattevi, M. Acik, Y. J. Chabal, M. Chhowalla and V. B. Shenoy, Nat. Chem., 2010, 2, 581–587 CrossRef CAS PubMed.
- E. Jagst, Surface Functional Group Characterization Using Chemical Derivatization X-ray Photoelectron Spectroscopy (CD-XPS), BAM, 2010 Search PubMed.
- M. I. Gibson, D. Paripovic and H. A. Klok, Adv. Mater., 2010, 22, 4721–4725 CrossRef CAS PubMed.
- P. C. Hiemenz and T. P. Lodge, Polymer Chemistry, CRC Press, 2nd edn, 2007 Search PubMed.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- W. Gao, L. B. Alemany, L. J. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403–408 CrossRef CAS PubMed.
- D. J. Phillips and M. I. Gibson, Chem. Commun., 2012, 48, 1054–1056 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data. See DOI: 10.1039/c5py01358e |
| This journal is © The Royal Society of Chemistry 2015 |