
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuneable pH-regulated supramolecular copolymerisation by mixing mismatched dendritic peptide comonomers†‡
P.
Ahlers§
ab,
H.
Frisch§
ab and
P.
Besenius
 *a
*a
aInstitut für Organische Chemie, Johannes Gutenberg-Universität Mainz, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: besenius@uni-mainz.de; Tel: +49 6131 39 22355
bOrganisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstrasse 40, 48149 Münster, Germany
First published on 18th August 2015
Abstract
Charged phenylalanine-rich dendritic peptides form highly stable and pH-switchable rod-like supramolecular copolymers, when co-assembled with a matching oppositely charged dendritic comonomer. Here, we demonstrate that by mismatching a strong with a weak β-sheet encoded comonomer, both the stability and the pH-triggered disassembly of the copolymers shifts drastically from pH 4.2 to biologically relevant pH 5.8.
Research activities have intensified in using oligopeptide building blocks as supramolecular monomers to form ordered supramolecular polymers and nanostructures in water, demonstrating that rules from the three-dimensional organisation of natural proteins can be translated into the design of peptide-based supramolecular functional materials.1,2 A large body of architectures are accessible including cages, spheres, tubes, fibre-like morphologies, tapes and sheet-like arrays. The building blocks are either based on biological derivatives such as collagens,3 elastins,4,5 β-sheet systems6–10 and α-helices/coiled-coils11–16 or are based on new design motifs, e.g. cyclic,17,18 aliphatic19–21 or aromatic amphiphiles,22,23 and dendritic oligopeptides.24,25 The most exciting features for the development of functional soft matter rely on their robust, yet stimuli-responsive properties,26–29 emergence of molecular networks and complex behaviour,30,31 and biomedical applications in tissue engineering and regenerative medicine.32,33
We have previously reported dendritic anionic and cationic amphiphiles encoded with β-sheet peptide motifs.25,34 These form supramolecular alternating copolymers when mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 feed ratio of the comonomers, and are able to respond to pH triggers: the self-assembly is only turned on at a neutral pH when both comonomers are charged. By protonating the anionic glutamic acid (E) comonomer at low pH, or deprotonating the cationic lysine (K) complementary comonomer at high pH, attractive Coulomb interactions are screened. This, in turn, induces the disassembly of the copolymers into the monomers, when using alanine (A) and leucine (L) containing peptides.25 The use of aromatic phenylalanine (F) alternated with charged amino acids, does not lead to a simple disassembly of the copolymers, but a pH-triggered copolymer to homopolymer transition, both at low and at high pH.34 Herein we show that long phenylalanine rich dendritic peptides form nanorod-like supramolecular copolymers, that are extremely stable towards changes in pH and ionic strength. Shortening the peptide sequence via the removal of just one aromatic amino acid reduces the stability, due to reduced hydrophobic shielding of hydrogen bonding and Coulomb attractive interactions. Crucially, we demonstrate that by co-assembling a stronger β-sheet comonomer, with the second weaker β-sheet one, the stability of the copolymers is reduced and the pH-triggered disassembly for the copolymers is shifted from pH 4.2 to biologically relevant pH 5.8. A change in the pH value as the stimulus for a programmed material response35 opens exciting avenues in biomedical applications.36 In living cells and tissues, intra- and extracellular pH values are tightly regulated, but can deviate from pH neutral and drop to pH 5.5–6, as observed for example in tumorous, inflammatory sites, in endocytic pathways and specific cellular compartments.37,38 We refer to the reported tuneable pH-regulated polymerisation as “comonomer mismatch”. We expect it to have an important impact on the design of oligopeptide based delivery vehicles, where the triggered disassembly of surface functionalised polycationic scaffolds into monomeric building blocks is an exciting strategy to release oligonucleotide after the loss of multivalent points of interaction.39–42 This phenomenon is somewhat similar to native protein cages, whereby the release of cargo material in intracellular compartments occurs via a delicate balance of decrease in pH and osmotic swelling.
:1 feed ratio of the comonomers, and are able to respond to pH triggers: the self-assembly is only turned on at a neutral pH when both comonomers are charged. By protonating the anionic glutamic acid (E) comonomer at low pH, or deprotonating the cationic lysine (K) complementary comonomer at high pH, attractive Coulomb interactions are screened. This, in turn, induces the disassembly of the copolymers into the monomers, when using alanine (A) and leucine (L) containing peptides.25 The use of aromatic phenylalanine (F) alternated with charged amino acids, does not lead to a simple disassembly of the copolymers, but a pH-triggered copolymer to homopolymer transition, both at low and at high pH.34 Herein we show that long phenylalanine rich dendritic peptides form nanorod-like supramolecular copolymers, that are extremely stable towards changes in pH and ionic strength. Shortening the peptide sequence via the removal of just one aromatic amino acid reduces the stability, due to reduced hydrophobic shielding of hydrogen bonding and Coulomb attractive interactions. Crucially, we demonstrate that by co-assembling a stronger β-sheet comonomer, with the second weaker β-sheet one, the stability of the copolymers is reduced and the pH-triggered disassembly for the copolymers is shifted from pH 4.2 to biologically relevant pH 5.8. A change in the pH value as the stimulus for a programmed material response35 opens exciting avenues in biomedical applications.36 In living cells and tissues, intra- and extracellular pH values are tightly regulated, but can deviate from pH neutral and drop to pH 5.5–6, as observed for example in tumorous, inflammatory sites, in endocytic pathways and specific cellular compartments.37,38 We refer to the reported tuneable pH-regulated polymerisation as “comonomer mismatch”. We expect it to have an important impact on the design of oligopeptide based delivery vehicles, where the triggered disassembly of surface functionalised polycationic scaffolds into monomeric building blocks is an exciting strategy to release oligonucleotide after the loss of multivalent points of interaction.39–42 This phenomenon is somewhat similar to native protein cages, whereby the release of cargo material in intracellular compartments occurs via a delicate balance of decrease in pH and osmotic swelling.
All four C3-symmetrical dendritic peptide comonomers 1a, 1b and 2a, 2b were synthesised using a convergent synthetic approach.34 We have incorporated hydrophobic FE and FK based alternating amino acid sequences in each side arm of the C3-symmetrical comonomers, including an apolar hexyl-spacer separating the peptide block from a water solubilising tetraethylene glycol peripheral dendron (Fig. 1). The latter were introduced as reported previously in order to increase the solubility of the materials.25,34 Note that in the cationic comonomer 1b, we incorporated a C-terminal p-cyanophenylalanine (Cnf), instead of phenylalanine on the complementary anionic comonomer 1a, as an optical probe. This leads to an unambiguous differentiation of homo- and copolymers via circular dichroism (CD) spectroscopy. The detailed synthesis and characterisation of the amphiphilic anionic and cationic peptide comonomers 1a (using a GFEFEF peptide sequence), 1b (GFKFKCnf), 2a (GFEFE) and 2b (GFKFK) can be found in the ESI.‡
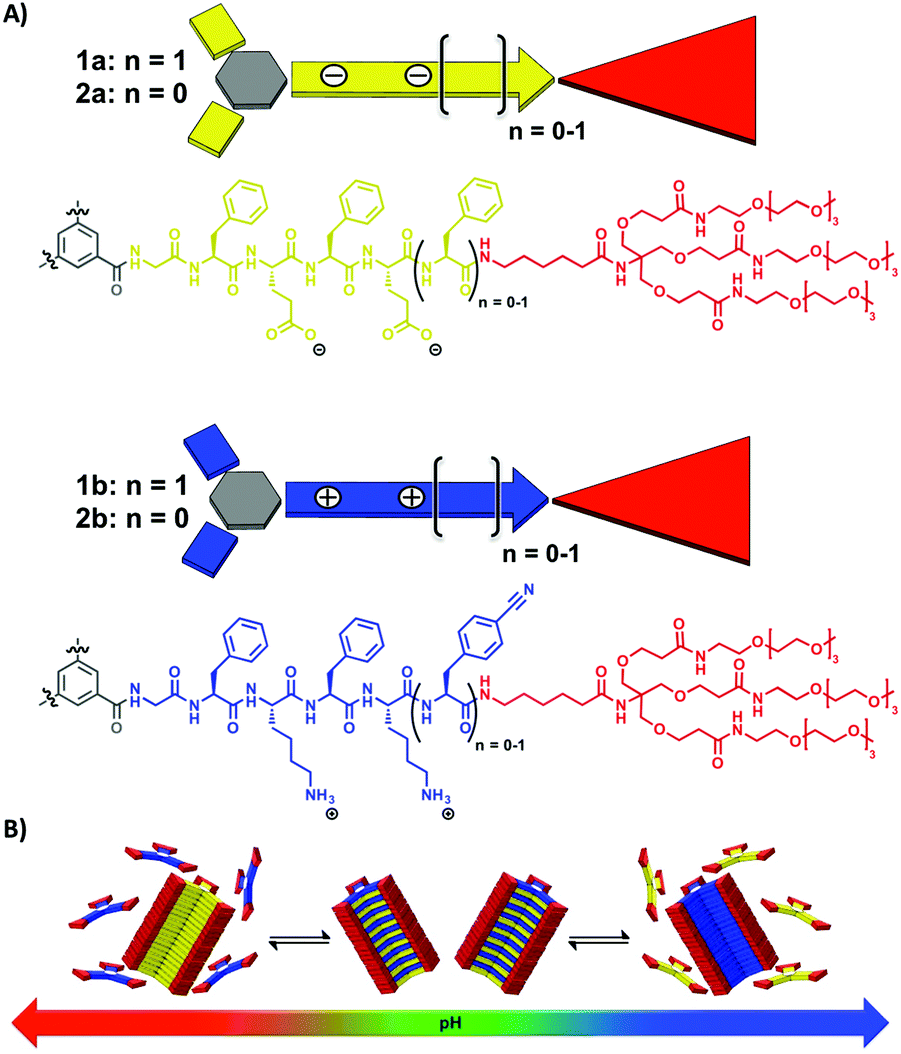 | ||
| Fig. 1 Chemical structures of the C3-symmetrical dendritic peptide comonomers 1a, 1b and 2a, 2b (A) and their pH-regulated supramolecular polymerisation into copolymers at neutral pH, and homopolymers at high and low pH respectively (B). | ||
CD spectroscopy experiments using the isolated solutions of comonomers at neutral pH show weak positive CD bands at λ = 220 nm for 1a and 1b (Fig. 2A and B). There is no indication of self-assembly into homopolymers, as expected due to Coulomb repulsion between monomers of the same charge. However by changing the pH, we observe the triggered self-assembly into homopolymers for 1a at pH 5.2 and 1b at pH 10.3. The changes in CD bands are significant with the appearance of a strong negative band at λ = 212 nm (Fig. 2A and B). We observe a shift in the apparent pKa values for the dendritic peptides compared to the isolated amino acids of 4.3 for glutamic acid and 10.5 for lysine, most likely driven by the aggregation process. We were furthermore able to perform ionic strength dependent experiments for monomer 1a. Increasing the concentration of NaCl to >500 mM shifts the monomer to homopolymer transition to slightly higher pH values, due to the increased thermodynamic driving force for supramolecular polymerisation of the very hydrophobic phenylalanine containing building blocks (Fig. S1‡). This finding is in agreement with our previous investigations on frustrated self-assemblies, whereby due to the electrostatic screening of the negative charges of carboxylate side-chains, the supramolecular polymerisation becomes more favourable at higher ionic strength and neutral pH.43–45 Mixing both monomeric building blocks in a 1:1 ratio at pH = 7.4 instantly leads to a strong negative CD band at λ = 212 nm characteristic for β-sheet formation, which is a strong indication for self-assembly into supramolecular alternating copolymers (Fig. 3A). In the past we have successfully correlated CD spectral bands with photoluminescence experiments using minimalistic FRET pairs, as well as detailed transmission electron microscopy (TEM) characterisation of the monomers, their homopolymers and hetero-copolymers.34
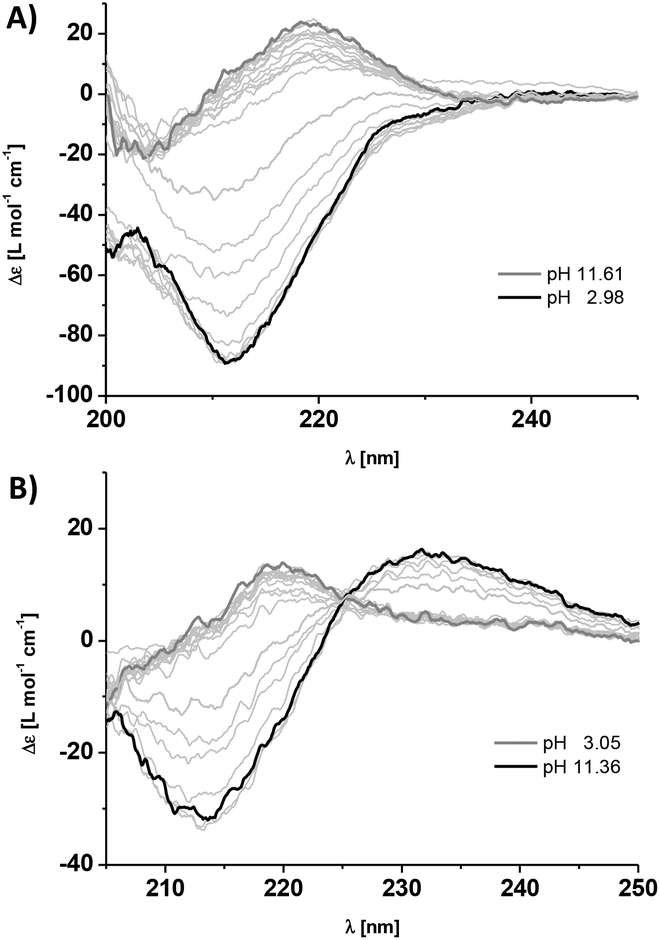 | ||
| Fig. 2 Titration experiments and CD spectra of a 60 μM aqueous solution of monomer 1a (A) and monomer 1b (B) at different pH values in 10 mM phosphate buffer at 293 K. The black spectra represent the self-assembled state into homopolymers (pH 2.98 for 1a and pH 11.61 for 1b), and the dark grey spectra (thick lines) the monomeric state (pH 11.36 for 1a and pH 3.05 for 1b). | ||
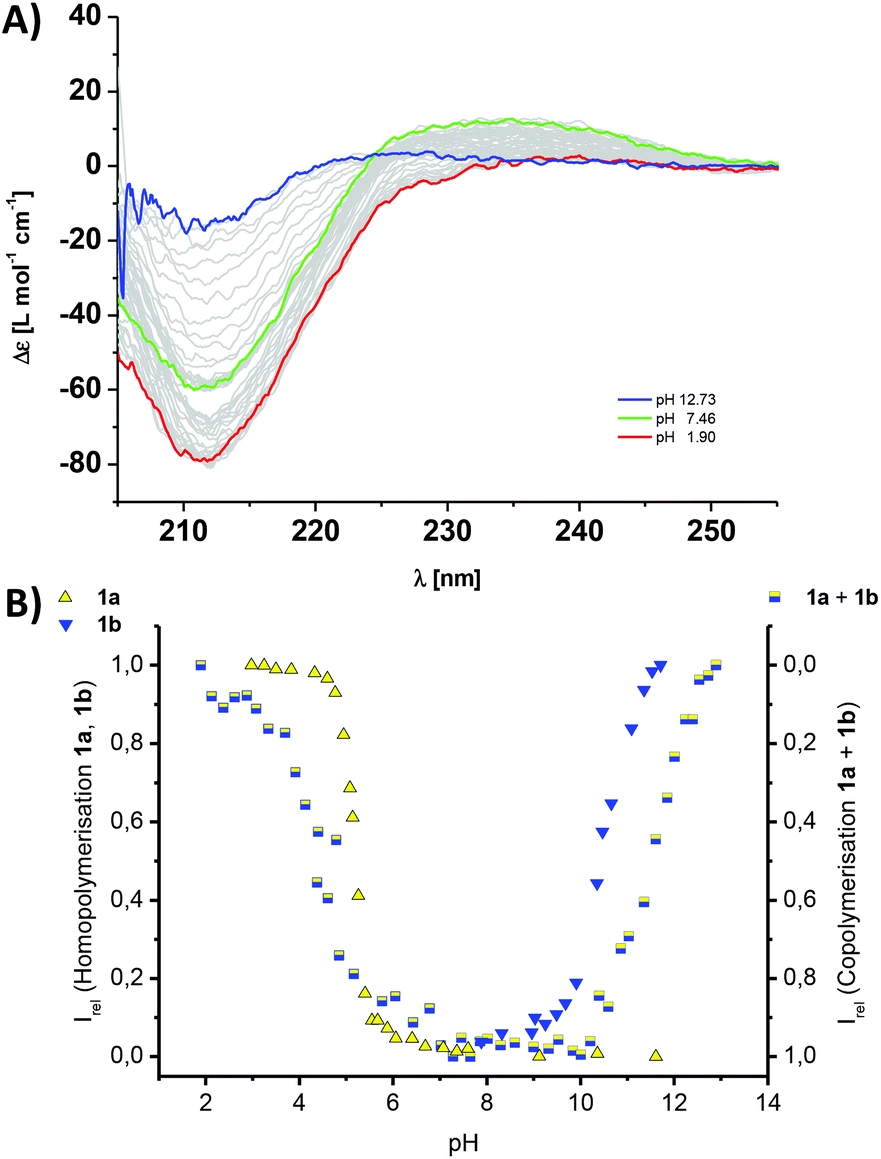 | ||
| Fig. 3 (A) Titration experiments and CD spectra of a 60 μM aqueous solution containing a 1:1 mixture of comonomers 1a and 1b at different pH values in 10 mM phosphate buffer; the black spectrum represents the co-assembled state into copolymers (pH 7.46), the yellow spectrum the mixture of homopolymers of 1a and monomeric 1b (pH 1.9) and the blue spectrum for monomeric 1a and homopolymers of 1b (12.73). (B) The titration curves and normalised CD data for the isolated homopolymerisations (left y-axis) of 1a (yellow triangles, Fig. 2A) or 1b (blue triangles, Fig. 2B) and the 1a–1b copolymers (mixed yellow-blue squares, right y-axis) following the intensity of the CD band at λ = 216 nm (A); Irel = 1 is set for the polymerised state as ‘on’ and Irel = 0 is set for the polymerised state as switched ‘off’. | ||
To further elucidate the stability of the 1a–1b copolymers towards changes in the pH and ionic strength, we performed titration experiments using CD spectroscopy. To our surprise the copolymers were extremely stable. Upon acidifying the phosphate buffer, the intensity of the CD band at λ = 212 nm only changed at pH 4.2, and by adding base at pH 11.5 (Fig. 3). This is a strong indication that the pH-triggered transition from co- to homopolymers and simultaneous release of the complementary comonomer is shifted by one pH unit compared to the values expected from the homopolymerisation of the isolated monomer solutions of 1a and 1b (Fig. 3B). To confirm that the pH-stability window of the copolymers is increased, we performed NaCl titrations in neutral phosphate buffer. Remarkably no changes in the characteristic β-sheet band was observed up to >1 M NaCl (Fig. S2‡). Phenylalanine rich peptide sequences are the main source of stabilisation for the hydrogen bonded and electrostatics driven β-sheet formation. In contrast, the previously reported alanine derivates using GAEAE and GAKAK encoded building blocks, disassemble already at <50 mM NaCl in phosphate buffer.25 We point out that Matile and coworkers have exploited a similar strategy in the β-sheet directed self-assembly of p-octaphenyl β-barrel pores embedded in lipid bilayers.46–48 Due to the spacial proximity and electrostatic repulsion of the charged basic or acidic peptide side chains in these synthetic pores, the intrinsic pKa of the base/acid is shifted, an observation which was summarized in an ‘intermediate internal charge repulsion’ (ICR) model.47,49 Here, the balance of attractive and repulsive contributions in the barrel-stave supramolecules is not too dissimlar to our pH-regulated supramolecular co- and homopolymerisation of oppositely charged comonomers: lack of ICR has been suggested to account for ‘implosion’, low ICR for contraction, high ICR for expansion, and excess ICR for ‘explosion’ of the β-barrels.
To correlate the spectroscopic findings with morphological investigations we performed negative stain TEM experiments (Fig. 4 and S5–S14‡). By depositing a solution of either building block 1a at pH 2.0, or 1b at pH 12.0, anisotropic nanorod-like structures are obtained (Fig. 4A and B and S5–S8‡). The thickness of the rods 9 nm is in good agreement with the diameter of the molecular building blocks, which corresponds to an estimated 7.2 nm for the extended hydrophobic core and 12.5 nm including the stretched out hydrophilic dendron. If both comonomers are pre-mixed in a 1:1 ratio at pH 7.4, well-defined anisotropic structures are observed (Fig. 4C and S9–S10‡). Their thickness of 8 nm is again in good agreement with the diameter of the molecular buildings blocks.
 | ||
| Fig. 4 (A) TEM images of the homopolymer of 1a at pH 2.0, (B) the 1a–1b copolymer at pH 7.4 and (C) the homopolymer of 1b at pH 12.0, in Tris buffer (10 mM); scale bars are 200 nm (A), (C) and 100 nm (B). | ||
Due to our interest in using ampholytic supramolecular copolymers as pH-switchable delivery vectors for biomedical applications, we decided to utilise a further pair of the comonomer building blocks 2a (GFEFE) and 2b (GFKFK). These lack the final C-terminal aromatic phenylalanine and p-cyanophenylalanine compared to 1a and 1b. The pH-induced monomer to homopolymer transitions for 2a at pH 5.8 and 2b at pH 9.2 were only marginally influenced by the decreased hydrophobicity of the peptide side arms (Fig. 4).¶ However the pH titration curves clearly show that the transitions are less sharp. Note that in temperature or concentration dependent supramolecular polymerisations, a more shallow transition would suggest a less cooperative growth mechanism.50–54
Our main goal herein was to co-assemble the mismatched comonomer pairs, between a longer and a shorter β-sheet comonomer and mix 1a with 2b, or 1b with 2a. Intriguingly the CD spectroscopic investigations show that both mismatched pairs co-assemble into supramolecular copolymers (Fig. S3‡) (Fig. 5).
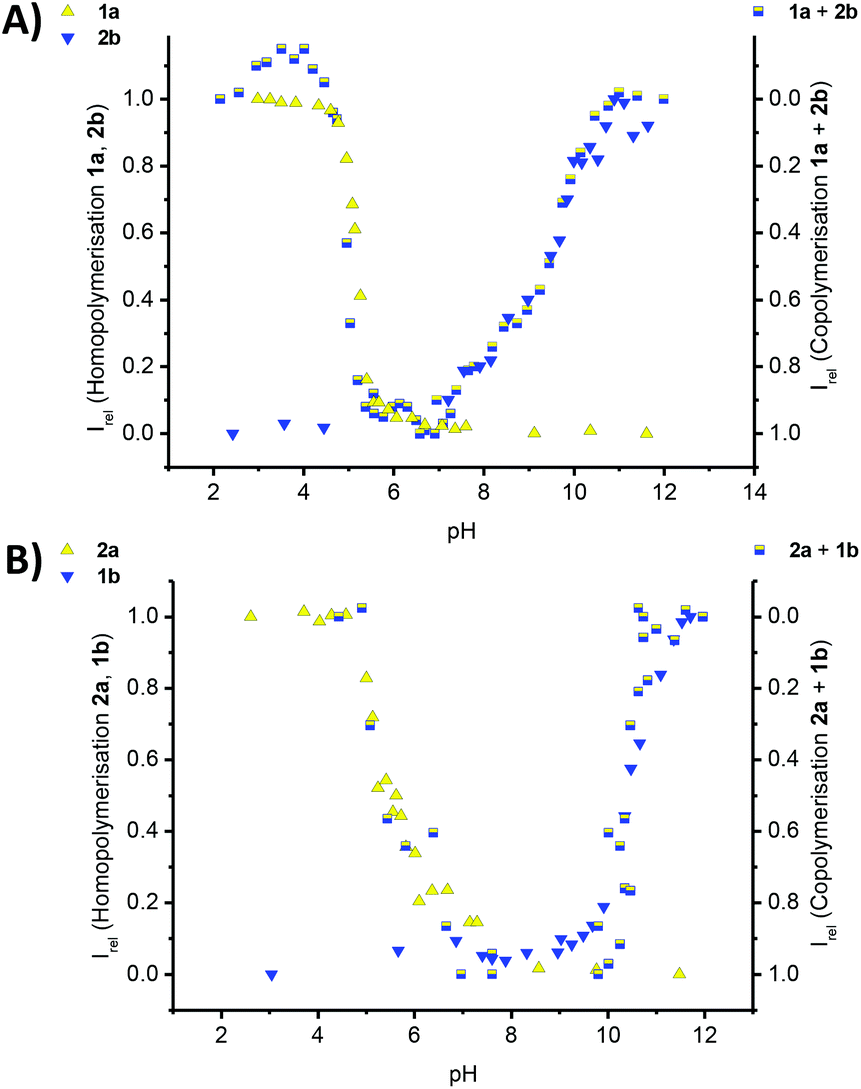 | ||
| Fig. 5 Titration experiments of 60 μM aqueous solutions of monomers in 10 mM phosphate buffer at 293 K; (A) the titration curves for the homopolymerisation of an isolated solution of 1a (yellow triangles, Fig. 2A) and 2b (blue triangles, Fig. S4B‡) and the 1a–2b copolymers (mixed yellow-blue squares, Fig. S3A‡); (B) the titration curves and normalised CD data for the homopolymerisation (left y-axis) of an isolated solution of 2a (yellow triangles, Fig. S4A‡) and 1b (blue triangles, Fig. 2A) and the 1b–2a copolymers (mixed yellow-blue squares, right y-axis, Fig. S3B‡), following the intensity of the CD band at λ = 216 nm in all cases, except for the 1b–2a copolymer (λ = 237 nm); Irel = 1 is set for the polymerized state as ‘on’ and Irel = 0 is set for the polymerized state as switched ‘off’. | ||
The pH titrations for both 1a–2b and 1b–2a copolymers, lead to pH curves that reflect our predicted trend. Due to the omission of one hydrophobic amino acid, the thermodynamic stability of the copolymer formation is reduced, the copolymerisation thereby weakened and the pH-stability window of the copolymers narrowed. The copolymer to homopolymer transitions and release of complementary comonomer overlap with the pH value expected for the homopolymer formation in a solution of the isolated monomer. Furthermore the disassembly of the copolymer is very sharp at low pH for 1a–2b, and at high pH for 1b–2a. In the contrary, the transition is shallow at high pH for 1a–2b, and at low pH for 1b–2a. This result is significant for two reasons: first we were able to shift the triggered disassembly from pH 4.2 for 1a–1b, to pH 5.2 for 1a–2b and to pH 5.8 for 2a–1b. Secondly, the transitions at pH 4.2 and at pH 5.2 are sharp when using the strongly aggregating co-monomer 1a, but more shallow for the transition at pH 5.8 in the case of the weakly aggregating comonomer 2a. Clearly, the choice and design of comonomers dictates the exact pH at which the disassembly occurs, but also the cooperative response to the external trigger. In case a controlled release of cargo is preferred over burst-type release often observed for conventional micellar assemblies, our design features will have important implications for the structural design parameters of biomedical delivery vehicles.
Conclusions
In summary, we have synthesised two different pairs of oppositely charged amphiphilic dendritic peptides. These co-assemble into pH-switchable nanorod-like supramolecular copolymers in neutral buffer. In the first set of pairs we have used long phenylalanine rich dendritic peptides that are extremely stable towards changes in pH and ionic strength. By shortening the peptide sequence in the second comonomer pair the removal of a single aromatic amino acid reduces the stability significantly, due to reduced hydrophobic shielding. Crucially we demonstrate that by co-assembling a longer and stronger β-sheet comonomer, with a second weaker β-sheet design, the stability of the copolymers is tuneable. The pH-triggered disassembly for the copolymers is shifted from pH 4.2 for the two stronger β-sheet monomers to biologically relevant pH 5.8 for the mismatched pair between a strong cationic and a weak anionic β-sheet synthon. We refer to this tuneable pH-regulated polymerisation as comonomer mismatch strategy, and expect it to have an impact on the design of peptidic delivery vehicles for biomedical applications, since the release of cargo material in intracellular compartments is a balance between decrease of the pH and osmotic swelling.Acknowledgements
We thank Daniel Spitzer (Institut für Organische Chemie, JGU Mainz) Frank Depoix (Institut für Zoologie, JGU Mainz) for help with the TEM experiments, the ‘Fonds der Chemischen Industrie’ (FCI) for financial support and COST Action CM1005 (Supramolecular Chemistry in Water).Notes and references
- T. O. Yeates and J. E. Padilla, Curr. Opin. Struct. Biol., 2002, 12, 464–470 CrossRef CAS.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- J. A. Fallas, L. E. R. O'Leary and J. D. Hartgerink, Chem. Soc. Rev., 2010, 39, 3510–3527 RSC.
- J. F. Almine, D. V. Bax, S. M. Mithieux, L. Nivison-Smith, J. Rnjak, A. Waterhouse, S. G. Wise and A. S. Weiss, Chem. Soc. Rev., 2010, 39, 3371–3379 RSC.
- S. R. MacEwan and A. Chilkoti, Biopolymers, 2010, 94, 60–77 CrossRef CAS PubMed.
- M. Krejchi, E. Atkins, A. Waddon, M. Fournier, T. Mason and D. Tirrell, Science, 1994, 265, 1427–1432 CAS.
- M. W. West, W. Wang, J. Patterson, J. D. Mancias, J. R. Beasley and M. H. Hecht, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11211–11216 CrossRef CAS.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- I. W. Hamley, Angew. Chem., Int. Ed., 2007, 46, 8128–8147 CrossRef CAS PubMed.
- A. Saiani, A. Mohammed, H. Frielinghaus, R. Collins, N. Hodson, C. M. Kielty, M. J. Sherratt and A. F. Miller, Soft Matter, 2009, 5, 193–202 RSC.
- C. Cohen and D. A. D. Parry, Proteins: Struct., Funct., Genet., 1990, 7, 1–15 CrossRef CAS PubMed.
- T. Alber, Curr. Opin. Genet. Dev., 1992, 2, 205–210 CrossRef CAS.
- R. B. Hill, D. P. Raleigh, A. Lombardi and W. F. DeGrado, Acc. Chem. Res., 2000, 33, 745–754 CrossRef CAS PubMed.
- B. Apostolovic, M. Danial and H.-A. Klok, Chem. Soc. Rev., 2010, 39, 3541–3575 RSC.
- D. N. Woolfson and Z. N. Mahmoud, Chem. Soc. Rev., 2010, 39, 3464–3479 RSC.
- N. R. Zaccai, B. Chi, A. R. Thomson, A. L. Boyle, G. J. Bartlett, M. Bruning, N. Linden, R. B. Sessions, P. J. Booth, R. L. Brady and D. N. Woolfson, Nat. Chem. Biol., 2011, 7, 935–941 CrossRef CAS PubMed.
- R. Chapman, M. Danial, M. L. Koh, K. A. Jolliffe and S. Perrier, Chem. Soc. Rev., 2012, 41, 6023–6041 RSC.
- J. Montenegro, M. R. Ghadiri and J. R. Granja, Acc. Chem. Res., 2013, 46, 2955–2965 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- J. D. Hartgerink, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5133–5138 CrossRef CAS PubMed.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS PubMed.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS PubMed.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- V. Percec, A. E. Dulcey, M. Peterca, M. Ilies, M. J. Sienkowska and P. A. Heiney, J. Am. Chem. Soc., 2005, 127, 17902–17909 CrossRef CAS PubMed.
- H. Frisch, J. P. Unsleber, D. Lüdeker, M. Peterlechner, G. Brunklaus, M. Waller and P. Besenius, Angew. Chem., Int. Ed., 2013, 52, 10097–10101 CrossRef CAS PubMed.
- D. W. P. M. Löwik, E. H. P. Leunissen, M. van den Heuvel, M. B. Hansen and J. C. M. van Hest, Chem. Soc. Rev., 2010, 39, 3394–3412 RSC.
- J. M. Zayed, N. Nouvel, U. Rauwald and O. A. Scherman, Chem. Soc. Rev., 2010, 39, 2806–2816 RSC.
- E. Krieg and B. Rybtchinski, Chem. – Eur. J., 2011, 17, 9016–9026 CrossRef CAS PubMed.
- X. J. Loh, J. del Barrio, T.-C. Lee and O. A. Scherman, Chem. Commun., 2014, 50, 3033–3035 RSC.
- R. F. Ludlow and S. Otto, Chem. Soc. Rev., 2008, 37, 101–108 RSC.
- E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111–119 CrossRef CAS PubMed.
- H. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1–18 CrossRef CAS PubMed.
- X. Zhao, F. Pan, H. Xu, M. Yaseen, H. Shan, C. A. E. Hauser, S. Zhang and J. R. Lu, Chem. Soc. Rev., 2010, 39, 3480–3498 RSC.
- H. Frisch, Y. Nie, S. Raunser and P. Besenius, Chem. – Eur. J., 2015, 21, 3304–3309 CrossRef CAS PubMed.
- H. Frisch and P. Besenius, Macromol. Rapid Commun., 2015, 36, 346–363 CrossRef CAS PubMed.
- H. Cabral and K. Kataoka, J. Controlled Release, 2014, 190, 465–476 CrossRef CAS PubMed.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS PubMed.
- J. R. Casey, S. Grinstein and J. Orlowski, Nat. Rev. Mol. Cell Biol., 2010, 11, 50–61 CrossRef CAS PubMed.
- A. Barnard and D. K. Smith, Angew. Chem., Int. Ed., 2012, 51, 6572–6581 CrossRef CAS PubMed.
- S. K. M. Nalluri, J. Voskuhl, J. B. Bultema, E. J. Boekema and B. J. Ravoo, Angew. Chem., Int. Ed., 2011, 50, 9747–9751 CrossRef CAS PubMed.
- S. P. Jones, N. P. Gabrielson, D. W. Pack and D. K. Smith, Chem. Commun., 2008, 4700–4702 RSC.
- D. Joester, M. Losson, R. Pugin, H. Heinzelmann, E. Walter, H. P. Merkle and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1486–1490 CrossRef CAS PubMed.
- M. von Gröning, I. de Feijter, M. C. A. Stuart, I. K. Voets and P. Besenius, J. Mater. Chem. B, 2013, 1, 2008–2012 RSC.
- P. Ahlers, H. Frisch, D. Spitzer, Z. Vobecka, F. Vilela and P. Besenius, Chem. – Asian J., 2014, 9, 2052–2057 CrossRef CAS PubMed.
- R. Appel, S. Tacke, J. Klingauf and P. Besenius, Org. Biomol. Chem., 2015, 13, 1030–1039 CAS.
- S. Matile, Chem. Soc. Rev., 2001, 30, 158–167 RSC.
- B. Baumeister, A. Som, G. Das, N. Sakai, F. Vilbois, D. Gerard, S. P. Shahi and S. Matile, Helv. Chim. Acta, 2002, 85, 2740–2753 CrossRef CAS.
- N. Sakai, J. Mareda and S. Matile, Acc. Chem. Res., 2008, 41, 1354–1365 CrossRef CAS PubMed.
- A. Som and S. Matile, Chem. Biodiversity, 2005, 2, 717–729 CAS.
- D. Zhao and J. S. Moore, Org. Biomol. Chem., 2003, 1, 3471–3491 CAS.
- T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- Z. Chen, A. Lohr, C. R. Saha-Möller and F. Würthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC.
- M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. d. Greef, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Chem. – Eur. J., 2010, 16, 362–367 CrossRef CAS PubMed.
- C. Rest, R. Kandanelli and G. Fernandez, Chem. Soc. Rev., 2015, 44, 2543–2572 RSC.
Footnotes |
| † We dedicate this paper to Professor David C. Sherrington, an inspiring supervisor and mentor, as well as a true gentleman. |
| ‡ Electronic supplementary information (ESI) available: Additional Fig. S1–S14, detailed experimental section: materials, synthesis, characterisation and analysis. See DOI: 10.1039/c5py01241d |
| § These authors contributed equally to this work. |
| ¶ For TEM experiments of the self-assembled 2a and 2b, see Fig. S11–S14. |
| This journal is © The Royal Society of Chemistry 2015 |