
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Designed enzymatically degradable amphiphilic conetworks by radical ring-opening polymerization†
Yinfeng
Shi
,
Holger
Schmalz
and
Seema
Agarwal
*
Macromolecular Chemistry II and Bayreuth Center for Colloids and Interfaces, University of Bayreuth, Universitätsstraße 30, 95440 Bayreuth, Germany. E-mail: agarwal@uni-bayreuth.de
First published on 24th July 2015
Abstract
A new route for the preparation of enzymatically degradable amphiphilic conetworks (APCNs) based on unsaturated polyesters by radical ring-opening copolymerization of vinylcyclopropane (VCP) with cyclic ketene acetal (CKA) is presented in this article. In the first step, the unsaturated biodegradable polyesters with random distribution of cross-linkable double bonds and degradable ester units were prepared by radical ring-opening copolymerization of VCP and CKA such as 2-methylene-4-phenyl-1,3-dioxolane (MPDO). Very similar reactivity ratios (rVCP = 0.23 ± 0.08 and rMPDO = 0.18 ± 0.02), unimodal gel permeation chromatography (GPC) curves and 2D NMR technique (heteronuclear multiple bond correlation, HMBC) showed the formation of random copolymers with unsaturation and ester units. The unsaturated units were used for cross-linking by radical polymerization with a hydrophilic macromonomer (oligo(ethylene glycol) methacrylate, OEGMA) in a second step for the formation of enzymatically degradable amphiphilic conetworks (APCNs). Enzymatic degradability was studied using Lipase from Pseudomonas cepacia. Due to the hydrophilic (HI) and hydrophobic (HO) microphase separation, the APCNs showed swelling in both water and organic solvents with different optical properties. The method provides an interesting route for making functional biodegradable APCNs using radical chemistry in the future by choosing appropriate vinyl comonomers.
Introduction
Amphiphilic conetworks (APCNs), composed of covalently linked hydrophilic and hydrophobic (HI/HO) chain segments, have the capability of swelling in both water and organic solvents.1–5 Owing to the unique structures and properties, APCNs were developed for various applications.6–15 One of the applications of APCNs is in the stabilization and enhancement of enzymatic activity in organic solvents.14The most common method of making APCNs, in fact one of the first examples, was using radical copolymerization of polymerizable telechelics (low molar mass polymers with polymerizable chain-ends) with a suitable comonomer.16–18 Other methods used for the formation of APCNs were click chemistry,5 thiol–ene reaction,19 Diels–Alder reaction20etc.
Biodegradable APCNs would be highly interesting for many biomedical applications and have also been made using photo, thermal conventional/controlled radical polymerization of α,ω-acrylate chain-end functionalized common biodegradable polymers such as poly(L-lactide), polyglycolide and poly(ε-caprolactone) (PCL) with comonomers like 2-hydroxyethyl methacrylate and N,N-dimethylaminoethyl methacrylate.21–23 In all these examples aliphatic polyesters made by metal catalyzed ring-opening polymerization (ROP) of corresponding cyclic esters were used as degradable hydrophobic segment.24 Both chain-ends were modified with polymerizable double bonds, i.e. acrylate units, to make them suitable as cross-linker in radical copolymerization with hydrophilic comonomers in a second step. In addition, azide–alkyne coupling of diazide chain-end functionalised ABA triblock copolymers of PCL and PEO with tetrakis(2-propynyloxymethyl) methane (TMOP) was also applied.5
Polyesters, in general, can also be prepared by radical ring-opening polymerization (RROP) of cyclic ketene acetals (CKAs). This method provides opportunities of making functional polyesters in a simple but versatile way by copolymerization of CKAs with a large variety of vinyl comonomers available.25–38 The formation of polyester during copolymerization depends upon the reaction conditions and comonomer type as CKAs can also undergo vinyl polymerization without isomerization leading to polyacetals with ring-retained structures.
Vinylcyclopropane (VCP) and its derivatives are interesting vinyl monomers undergoing radical ring-opening polymerization for the formation of polymers with unsaturated units in the backbone39–47 and have been intensively studied for dental applications as low volume shrinkage monomers (Scheme 1A). The copolymerization of VCP with CKAs could provide new polymers with cross-linkable unsaturated units and biodegradable ester linkages randomly distributed on the polymer backbone (Scheme 1C), which could be useful for making APCNs. Unsaturated biodegradable amphiphilic hydrogels made by condensation polymerization of diol functionalised PCL and poly(ethylene glycol) with diacid functionalised poly(propylene fumarate), followed by cross-linking with polyacrylamide are known in the literature with main focus on swellability in water.48,49
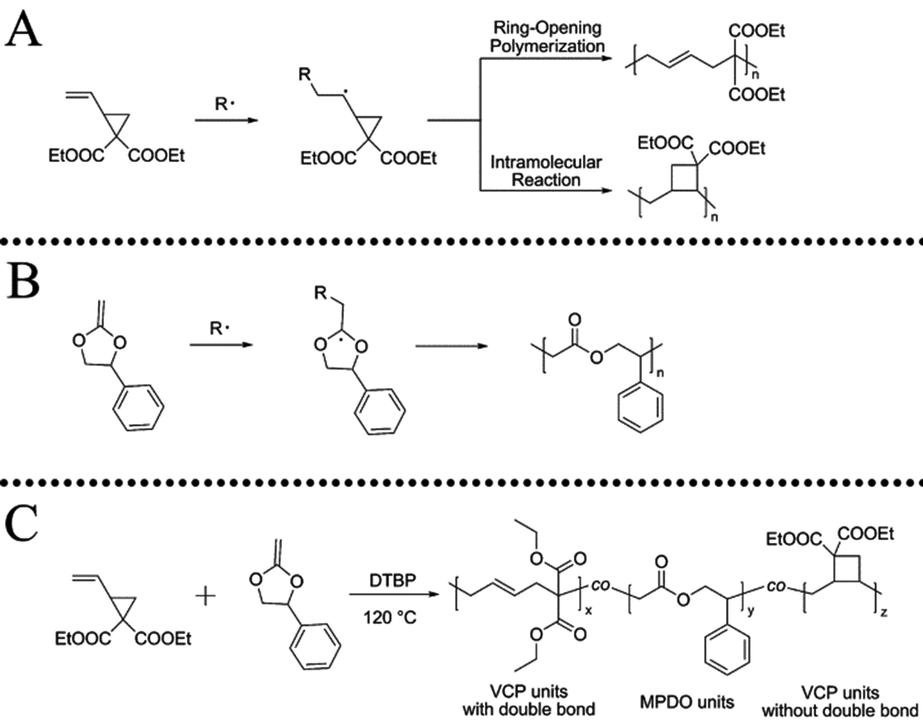 | ||
| Scheme 1 Schematic process of radical ring-opening polymerization (RROP) of (A) vinylcyclopropane (VCP),39 (B) 2-methylene-4-phenyl-1,3-dioxolane (MPDO),50 and (C) VCP and MPDO. | ||
We highlight the formation of APCNs, in which the hydrophobic part is made by radical ring-opening copolymerization of vinylcyclopropane (VCP) and 2-methylene-4-phenyl-1,3-dioxolane (MPDO), followed by cross-linking with a hydrophilic macromonomer oligo(ethylene glycol) methacrylate (OEGMA, Mn ∼ 500). The copolymerization of VCP and MPDO provides an unsaturated polyester with random distribution of carbon–carbon double bonds and ester units, as characterized by reactivity ratio determination. This is a new method of making unsaturated polyesters (UPs) by radical polymerization with the advantage of easy and versatile introduction of additional functional groups simply by choosing appropriate vinyl comonomers. The state-of-the art method of making UPs is by condensation polymerization using high temperatures that restricts the use of many functional monomers. The APCNs were characterized by swelling studies in water and DMF, and the enzymatic degradability of APCNs was studied using Lipase from Pseudomonas cepacia in pH = 7 PBS buffer solution. The unique swelling property and biodegradability of the prepared APCNs suggests the potential application in the biomaterial field.
Experimental section
Materials
1-Phenyl-1,2-ethanediol (Aldrich), potassium tert-butoxide (Acros), p-toluenesulfonic acid (Aldrich), 1,4-dibromo-2-butene (Aldrich), diethyl malonate (Aldrich), sodium (Aldrich), di-tert-butyl peroxide (DTBP, Aldrich) and Lipase (from Pseudomonas cepacia, ≥30 U mg−1, Aldrich) were used as received. All solvents were distilled before use. Oligo(ethylene glycol) methacrylate (OEGMA, Mn ∼ 500, Aldrich) was passed through a basic alumina column to remove the inhibitor.2-Methylene-4-phenyl-1,3-dioxolane (MPDO) was synthesized using the literature method of W. J. Bailey et al.50 with some modifications. 2-Vinylcyclopropane-1,1-dicarboxylate (VCP) was synthesized using the literature method of T. Endo et al.39
Instrumentation
1H (300 MHz), 13C (75 MHz) NMR spectra were recorded on a Bruker Ultrashied-300 spectrometer in CDCl3, using tetramethylsilane (TMS) as an internal standard. Molecular weights and molar-mass dispersities (ĐM) of the polymers were determined by gel permeation chromatography (GPC) with a Knauer system equipped with 4 PSS-SDV gel columns (particle size = 5 μm) with pore sizes ranging from 102 to 105 Å (PSS, Mainz, Germany) together with a differential refractive index detector (RI-101 from Shodex). THF (HPLC grade) was used as solvent at a flow rate of 1.0 mL min−1. A Mettler thermal analyzer with 851 TG and 821 DSC modules was used for the thermal characterization of copolymers. Thermal stability was determined by recording TGA traces at a heating rate of 10 K min−1 in nitrogen atmosphere (flow rate = 50 ml min−1). DSC scans were recorded in nitrogen atmosphere (flow rate = 80 ml min−1) at a scanning rate of 10 K min−1. A sample mass of 10 ± 1 mg was used in both DSC and TGA analysis.Copolymerization of MPDO and VCP under free radical condition
The mixture of MPDO and VCP with different molar ratios and di-tert-butyl peroxide as initiator were added into a 10 mL Schlenk tube equipped with a magnetic stir bar under argon. After degassing by freeze–vacuum–thaw cycles three times, the tubes were sealed under argon, and then placed in a preheated oil bath at 120 °C for 48 h while stirring. Subsequently, the tubes were opened and the reaction mixture was poured into methanol while stirring to cause polymer precipitation. Finally, the purified polymer was dried under vacuum at 60 °C for 48 h. Copolymerization of MPDO and VCP for reactivity ratios calculation was carried out with various feed ratios at 120 °C and stopped at low conversion. The reactivity ratios were calculated using Kelen–Tüdõs method.51 Details are given in the ESI.†APCN preparation using VCP–MPDO copolymers and OEGMA under free radical condition
VCP–MPDO copolymer, OEGMA and DTBP were dissolved in 0.5 mL dioxane at a molar ratio of carbon–carbon double bonds![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :OEGMA:DTBP = 1:25:1. After degassing with argon for 1 h, the reaction mixture was placed in a preheated oil bath at 140 °C for 24 h without stirring. After the polymerization, the resulting gel was extracted with chloroform overnight to remove the unreacted VCP–MPDO copolymer and linear POEGMA. Finally, the purified APCNs were dried in vacuum at 60 °C for 24 h.
:OEGMA:DTBP = 1:25:1. After degassing with argon for 1 h, the reaction mixture was placed in a preheated oil bath at 140 °C for 24 h without stirring. After the polymerization, the resulting gel was extracted with chloroform overnight to remove the unreacted VCP–MPDO copolymer and linear POEGMA. Finally, the purified APCNs were dried in vacuum at 60 °C for 24 h.
In vitro degradation studies of APCNs
In vitro degradation studies of APCNs were carried out by placing APCNs in pH = 7 PBS buffer solution with the enzyme concentration of 15 U mL−1 (Lipase from Pseudomonas cepacia) at 37 °C. The degradation medium was replaced every 24 h. The remaining mass of APCNs was recorded after freeze drying at different degradation time.Results and discussion
Copolymerization of VCP with MPDO
Various copolymers of VCP with MPDO were prepared by changing the molar ratio of the two monomers in the initial feed (Scheme 1, Table 1).| Entrya | Feed ratio (mol%) | Yield % | M n (g mol−1) |
Đ
M
b |
Copolymer compositionc (mol%) | |||
|---|---|---|---|---|---|---|---|---|
| VCP | MPDO | VCP (ring-opened) | VCP (ring-retained) | MPDO | ||||
| a Initiator: DTBP (1 wt% of monomer); reaction temperature: 120 °C; reaction time: 48 h. b Determined by THF-GPC with RI detector; PS-standards used for calibration (Fig. 3). c Determined by 1H-NMR spectra of resulting VCP–MPDO copolymers (Fig. 1A). | ||||||||
| 1 | 30 | 70 | 33 | 5.4 × 103 | 1.4 | 25 | 20 | 55 |
| 2 | 50 | 50 | 43 | 8.3 × 103 | 1.4 | 31 | 28 | 41 |
| 3 | 70 | 30 | 46 | 1.2 × 104 | 1.8 | 29 | 35 | 36 |
After purification, the resulting copolymers were obtained as white solids. The copolymer structure was analyzed by NMR spectroscopy (Fig. 1). According to the NMR spectra of PVCP (please see ESI, Fig. S1†) and PMPDO50 homopolymer, the characteristic peaks of VCP and MPDO units were confirmed and marked in Fig. 1. In the 13C-NMR spectrum (Fig. 1B), the absence of peaks between δ = 90–110 ppm confirms that all MPDO units were copolymerized as ring-opened structure providing ester units in the polymer backbone (structure 3 in Fig. 1). In the 1H-NMR spectrum (Fig. 1A), the broad peak c at δ = 4.7–5.3 ppm originates from the carbon–carbon double bond –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– of VCP units with ring-opened structure (structure 1 in Fig. 1), the peak a at δ = 0.9–1.1 ppm from the CH3CH2O– group of VCP units with ring-opened and also ring-closed structure (structures 1 and 2 in Fig. 1) and peak b,i between δ = 3.7–4.5 ppm were assigned to the –CH2–CH(Ph)– group of MPDO units and CH3CH2O– group of VCP units in the copolymer. Through comparing the areas of these 3 peaks, the VCP–MPDO copolymer composition was calculated.
CH– of VCP units with ring-opened structure (structure 1 in Fig. 1), the peak a at δ = 0.9–1.1 ppm from the CH3CH2O– group of VCP units with ring-opened and also ring-closed structure (structures 1 and 2 in Fig. 1) and peak b,i between δ = 3.7–4.5 ppm were assigned to the –CH2–CH(Ph)– group of MPDO units and CH3CH2O– group of VCP units in the copolymer. Through comparing the areas of these 3 peaks, the VCP–MPDO copolymer composition was calculated.
 | ||
| Fig. 1 NMR-spectra of VCP–MPDO copolymer (entry 1 in Table 1). A: 1H-NMR spectrum; B: 13C-NMR spectrum. Structure 1: VCP unit with ring-opened structure in copolymer; structure 2: VCP unit with ring-retained structure in copolymer; structure 3: MPDO unit in copolymer. | ||
The 2D HMBC NMR spectrum (Fig. 2) shows correlation of the proton peaks originating from d,e of VCP units (structure 1) at 2.4 ppm with the aromatic carbon signals of MPDO units (structure 3) at 128 ppm, confirming covalent linkage between VCP and MPDO units in the copolymer. The compositions of VCP–MPDO copolymers with different monomer ratio in feed are summarized in Table 1. An increase of VCP in the initial feed led to a higher VCP content in the VCP–MPDO copolymer and slightly increased amount of ring-retained cyclobutane units. The cyclobutane units were formed by the addition of a newly formed radical during propagation via ring-opening of VCP to the double bond of the same unit in the polymer main chain i.e. intramolecular isomerization, a very well-known reaction during ring-opening polymerization of VCP.52 It is a competitive reaction with the reaction of ring-opened propagating VCP radical with a new VCP monomer/MPDO and increases slightly (45% to 55%) with the VCP content in the reaction mixture (Table 1) when all other parameters such as amount and type of the initiator and temperature of polymerization remained the same. The change in the amount of ring-retained structure with feed composition is not very drastic. In our previous work about homopolymerization of VCP using DTBP initiator at 120 °C the ratio of ring-opened and ring-retained structures were about 50:50.52
 | ||
| Fig. 2 HMBC spectrum of VCP–MPDO copolymer (entry 1 in Table 1). Cross-peak A: Coupling between group d,e of VCP unit with ring-opened structure (structure 1) and carbon–carbon double bond c of VCP unit with ring-opened structure (structure 1); cross-peak B: Coupling between group d,e of VCP unit with ring-opened structure (structure 1) and phenyl group of MPDO unit (structure 3). | ||
The number-average molecular weights of the resulting VCP–MPDO copolymers were determined by THF-GPC applying a polystyrene calibration and ranged from 5.4 × 103 to 1.2 × 104 g mol−1 (Table 1). The corresponding GPC-traces show unimodal molecular weight distributions for all copolymers (Fig. 3). An increase of VCP in the initial feed led to a higher molar mass of the resulting copolymer.
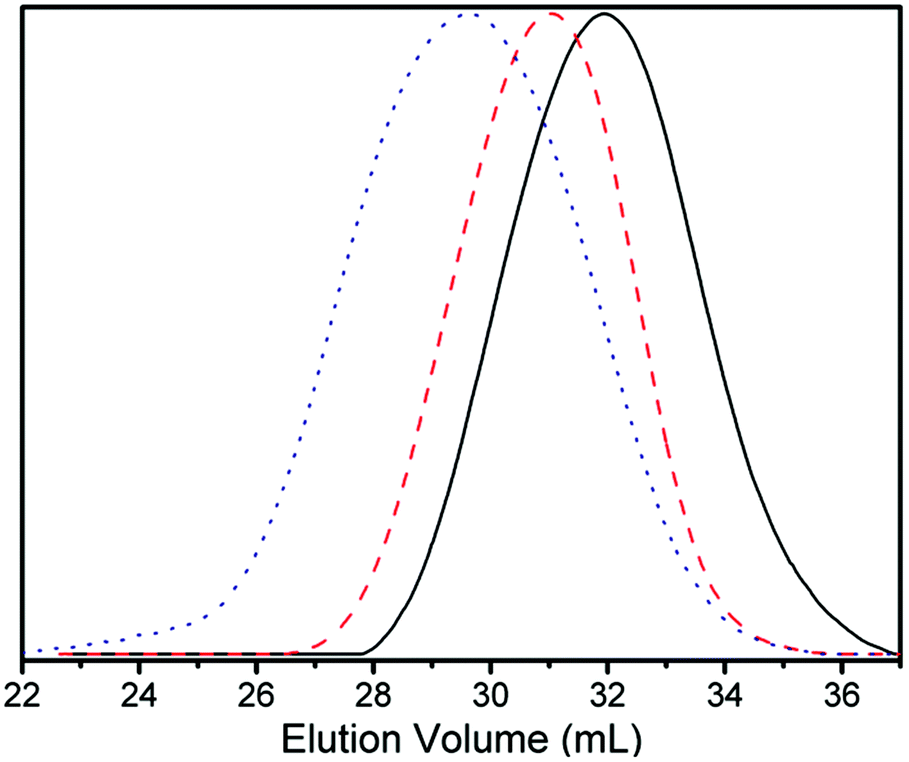 | ||
| Fig. 3 THF-GPC traces of VCP–MPDO copolymers with different comonomer ratios in feed. Reaction temperature: 120 °C, reaction time: 48 h. Composition in feed: black-solid line: VCP:MPDO = 30:70; red-broken line: VCP:MPDO = 50:50; blue-dotted line: VCP:MPDO = 70:30. | ||
The reactivity ratios of the two monomers were calculated using the Kelen–Tüdõs method.51 To that end, a series of VCP–MPDO copolymers with low yields (< 5%) were prepared. The composition of the resulting polymers was determined using 1H-NMR technique as described above. A detailed discussion on the determination of the reactivity ratios can be found in the ESI (Tables S1, S2 and Fig. S3, S4†). From the corresponding Kelen–Tüdõs plot (Fig. S3†) the reactivity ratios of VCP and MPDO were determined to rVCP = 0.23 ± 0.08 and rMPDO = 0.18 ± 0.02. There are no big differences between the reactivity ratios of VCP and MPDO and both values are below 1. Consequently, depending on the monomer unit at the active chain end, i.e., VCP or MPDO, the incorporation of the respective other comonomer, i.e., MPDO or VCP, is slightly preferred (copolymerization diagram in Fig. S4†). This suggests a nearly random distribution of MPDO and VCP units in the copolymer.
Thermal characterization of VCP–MPDO copolymers was carried out using TGA and DSC techniques. The glass transition and decomposition temperatures of the copolymers are listed in Table 2.
| Entrya | Copolymer compositionb (mol %) | T g (°C) | Dec. temp.d (°C) | ||
|---|---|---|---|---|---|
| VCP (ring-opened) | VCP (ring-retained) | MPDO | |||
| a Initiator: DTBP (1 wt% of monomer); reaction temperature: 120 °C; reaction time: 48 h; same as entries 1–3 in Table 1. b Determined by 1H-NMR spectra (Fig. 1A). c Determined by DSC (please see Fig. S6 in ESI). d Determined by TGA (please see Fig. S5 in ESI). | |||||
| 1 | 25 | 20 | 55 | 45 | 402 |
| 2 | 31 | 28 | 41 | 43 | 410 |
| 3 | 29 | 35 | 36 | 49 | 410 |
The copolymers show a high thermal stability (Tmax: temperature at which rate of decomposition was maximum = 400–410 °C) with single step decomposition (please see Fig. S5 in ESI†). Single glass transition was obtained for copolymers with different comonomer ratios (please see Fig. S6 in ESI†), reconfirming the nearly random incorporation of MPDO and VCP in the copolymer chains.
Synthesis of biodegradable amphiphilic conetworks using VCP–MPDO copolymers
Based on the cross-linkable carbon–carbon double bonds and the almost random VCP–MPDO combination on the copolymer backbone (random distribution of ester units), the VCP–MPDO copolymers have the potential to be used for the preparation of biodegradable amphiphilic conetworks (APCNs). As shown in Scheme 2, APCNs were prepared from a reaction mixture of VCP–MPDO copolymer, OEGMA and DTBP in dioxane with a molar ratio of carbon–carbon double bonds:OEGMA:DTBP = 1:25:1 at 140 °C. After 24 h reaction, colorless cross-linked gels with very high gel fractions were obtained. The gel fraction was determined by calculating the ratio of the gel dry mass to polymer precursor mass (Table 3). The FT-IR showed the presence of characteristic peaks of both VPC–MPDO copolymer and POEGMA segments in the APCNs (please see ESI Fig. S2†).
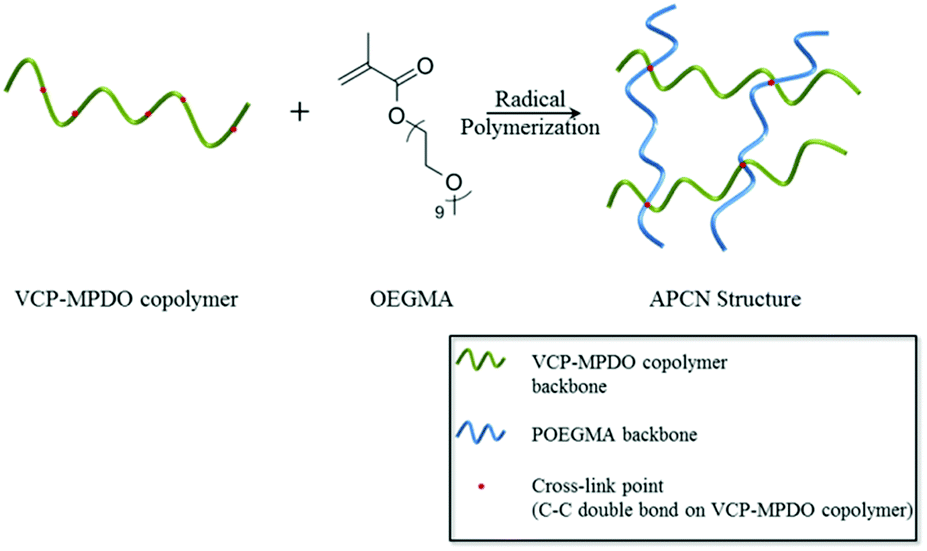 | ||
| Scheme 2 Preparation of biodegradable amphiphilic conetworks. OEGMA = oligo(ethylene glycol) methacrylate, Mn ∼ 500 g mol−1. Di-tert-butyl peroxide was used as initiator and reaction temperature was at 140 °C. | ||
| Sample | Gel fraction (%) | Weight swelling ratio (w/w) after equilibration (%) | |
|---|---|---|---|
| In water | In DMF | ||
|
a The OEGMA:VCP–MPDO (feed molar ratio) was 6.25:1, 7.75:1 and 7.25:1 for gels 1, 2 and 3, respectively.
b APCNs Gel 1: prepared by VCP–MPDO copolymer of entry 1 in Table 1.
c APCNs Gel 2: prepared by VCP–MPDO copolymer of entry 2 in Table 1.
d APCNs Gel 3: prepared by VCP–MPDO copolymer of entry 3 in Table 1.
|
|||
| Gel 1b | 79 | 1130 | 1230 |
| Gel 2c | 91 | 1030 | 1160 |
| Gel 3d | 93 | 1070 | 1150 |
Characterization of biodegradable APCNs
The swelling property is one of the most important properties of APCNs and it was studied by immersing dry gels in different solvents. The swelling ratio was calculated using the following equation and averaged for 3 samples:| Weight swelling ratio = [(weightswollen gel/weightdry gel) − 1]100% (w/w) |
Because of the co-existence of hydrophilic (HI) and hydrophobic (HO) structures in the copolymer network, the APCNs show different swelling properties in various solvents. The swelling properties of APCNs in water and DMF are shown in Fig. 4 and listed in Table 3.
 | ||
| Fig. 4 Weight swelling ratio of APCNs (black: Gel 1, red: Gel 2, blue: Gel 3) in (A) DMF and (B) water. | ||
The gels showed equilibration in water and also DMF in approximately 8 h. DMF is a good solvent for both the hydrophilic and hydrophobic phases in the resulting APCNs and results in an extension of all polymer chains in the networks. However, water is a selective solvent only for the hydrophilic phase and only the poly[oligo(ethylene glycol) methacrylate] (POEGMA) segments extend in water. Due to the lowest content of VCP units with carbon–carbon double bond in the VCP–MPDO copolymer for network preparation, Gel 1 has the lowest concentration of cross-linking points in the network and hence showed the maximum swelling ratio of 1130% in water (Fig. 4B) and 1230% in DMF (Fig. 4A). It was larger than Gel 2 (1030% in water and 1160% in DMF) and Gel 3 (1070% in water and 1150% in DMF). The swelling ratios of all APCNs in water were smaller than in DMF. All APCNs showed a high transparency in DMF (Fig. 5 top) due to the extension of both hydrophobic and hydrophilic parts. Because of the phase separation between the hydrophilic (POEGMA chains) and hydrophobic (VCP–MPDO copolymer) segments, the APCNs appeared opaque in water (Fig. 5 bottom).
 | ||
| Fig. 5 Photographs of APCNs swollen in different solvents. A1: Gel 1 in DMF; A2: Gel 1 in H2O; B1: Gel 2 in DMF; B2: Gel 2 in H2O; C1: Gel 3 in DMF; C2: Gel 3 in H2O. | ||
Due to the influence of polymer crystallinity to the drug permeation and biodegradability of the whole network in the aim of biomedical application,53 the thermal properties of APCNs were studied through DSC technique (Fig. 6). The APCNs showed a low melting temperature around −3 °C due to oligo(ethylene glycol) side-chains. The glass transition temperature of the VCP–MPDO copolymer segments in the conetwork was observed at 45 °C and Tg of POEGMA backbone at 85 °C, respectively. Due to the higher glass transition temperature of the hydrophobic segments in the conetwork, the APCNs could keep their shape at room temperature.
 | ||
| Fig. 6 DSC heating trace of purified APCNs, Gel 3 as an example. | ||
The degradation behavior of APCNs was studied by placing APCNs in pH = 7 PBS buffer solution with the enzyme concentration of 15 U mL−1 at 37 °C (Lipase from Pseudomonas cepacia) (Fig. 7). The degradability of cross-linked APCNs was realized through cleavage of VCP–MPDO copolymer segments, which act as cross-linking chains in the APCNs. The weight loss noted might be due to the release of POEGMA chains or OEGMA fragments or a combination of both after enzymatic degradation of ester units from VCP–MPDO and ester units of POEGMA linking OEGMA to the polymer backbone. The complete degradability of the present APCNs is not possible due to the C–C backbone of POEGMA. Due to the highest content of ester bonds from MPDO-units and the lowest content of carbon–carbon double bonds as cross-linking points from ring-opened VCP-units in the conetwork, Gel 1 exhibited the fastest decomposition rate and was decomposed in 35 d in the presence of Lipase from Pseudomonas cepacia.
 | ||
| Fig. 7 Mass loss of APCNs against enzyme in pH = 7 PBS buffer solution in dependence of degradation time. Black: Gel 1; red: Gel 2; navy: Gel 3. | ||
Conclusions
Cross-linkable unsaturated polyesters were prepared by radical ring-opening copolymerization of VCP and MPDO. The unsaturated and ester units were provided by ring-opening of VCP and cyclic ketene acetal (MPDO), respectively, and were distributed almost randomly along the copolymer backbone. The VCP–MPDO copolymers were used as hydrophobic (HO) precursors for the formation of amphiphilic conetworks (APCNs) via copolymerization with OEGMA, forming the hydrophilic (HI) part. Through the free radical polymerization of OEGMA in the presence of VCP–MPDO copolymer, the APCNs were successfully produced. The concentration of the cross-linking points and hence the swelling property of APCNs in DMF and water depend on the amount of double bonds in the hydrophobic precursor. The APCNs provided unique swelling property with excellent enzymatic degradability. The method provides a simple and versatile route for making functional biodegradable APCNs using radical chemistry in the future.Acknowledgements
Authors would like to thank Deutsche Forschungsgemeinschaft (DFG) for the financial support. The work was carried out in the frame of SFB 840.References
- C. S. Patrickios and T. K. Georgiou, Curr. Opin. Colloid Interface Sci., 2003, 8, 76–85 CrossRef CAS.
- G. Erdodi and J. P. Kennedy, Prog. Polym. Sci., 2006, 31, 1–18 CrossRef CAS PubMed.
- L. Mespouille, J. L. Hedrick and P. Dubois, Soft Matter, 2009, 5, 4878–4892 RSC.
- M. Rikkou-Kalourkoti, E. Loizou, L. Porcar, K. Matyjaszewski and C. S. Patrickios, Polym. Chem., 2012, 3, 105–116 RSC.
- Y. Yuan, A.-K. Zhang, J. Ling, L.-H. Yin, Y. Chen and G.-D. Fu, Soft Matter, 2013, 9, 6309–6318 RSC.
- L. Bromberg, M. Temchenko and T. A. Hatton, Langmuir, 2002, 18, 4944–4952 CrossRef CAS.
- J. Zednik, R. Riva, P. Lussis, C. Jérôme, R. Jérôme and P. Lecomte, Polymer, 2008, 49, 697–702 CrossRef CAS PubMed.
- C. Lin and I. Gitsov, Macromolecules, 2010, 43, 10017–10030 CrossRef CAS.
- J. P. Kennedy, Macromol. Symp., 2001, 175, 127–132 CrossRef CAS.
- I. Gitsov and C. Zhu, J. Am. Chem. Soc., 2003, 125, 11228–11234 CrossRef CAS PubMed.
- I. Gitsov and C. Zhu, Macromolecules, 2002, 35, 8418–8427 CrossRef CAS.
- R. Haigh, N. Fullwood and S. Rimmer, Biomaterials, 2002, 23, 3509–3516 CrossRef CAS.
- S. Rimmer, M. J. German, J. Maughan, Y. Sun, N. Fullwood, J. Ebdon and S. MacNeil, Biomaterials, 2005, 26, 2219–2230 CrossRef CAS PubMed.
- N. Bruns and J. C. Tiller, Nano Lett., 2004, 5, 45–48 CrossRef PubMed.
- G. Savin, N. Bruns, Y. Thomann and J. C. Tiller, Macromolecules, 2005, 38, 7536–7539 CrossRef CAS.
- D. Chen, J. P. Kennedy and A. J. Allen, J. Macromol. Sci., Part A: Pure Appl. Chem., 1988, 25, 389–401 CrossRef PubMed.
- E. Themistou and C. S. Patrickios, Macromolecules, 2004, 37, 6734–6743 CrossRef CAS.
- E. Loizou, A. I. Triftaridou, T. K. Georgiou, M. Vamvakaki and C. S. Patrickios, Biomacromolecules, 2003, 4, 1150–1160 CrossRef CAS PubMed.
- C. N. Walker, K. C. Bryson, R. C. Hayward and G. N. Tew, ACS Nano, 2014, 8, 12376–12385 CrossRef CAS PubMed.
- M. Delerba, J. R. Ebdon and S. Rimmer, Macromol. Rapid Commun., 1997, 18, 723–728 CrossRef CAS PubMed.
- A. S. Sawhney, C. P. Pathak and J. A. Hubbell, Macromolecules, 1993, 26, 581–587 CrossRef CAS.
- I. Barakat, P. Dubois, R. Jérôme, P. Teyssié and E. Goethals, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 2099–2110 CrossRef CAS PubMed.
- I. Barakat, P. Dubois, C. Grandfils and R. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2401–2411 CrossRef CAS.
- T. Chen, T. Cai, Q. Jin and J. Ji, e-Polymers, 2015, 15, 3–13 CrossRef CAS.
- S. Agarwal, Polym. Chem., 2010, 1, 953–964 RSC.
- W. J. Bailey, Z. Ni and S.-R. Wu, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 3021–3030 CrossRef CAS PubMed.
- J. Bailey, J. L. Chou, P. Z. Feng, V. Kuruganti and L. L. Zhou, Acta Polym., 1988, 39, 335–341 CrossRef CAS PubMed.
- W. J. Bailey, Z. Ni and S. R. Wu, Macromolecules, 1982, 15, 711–714 CrossRef CAS.
- L. Ren, C. Speyerer and S. Agarwal, Macromolecules, 2007, 40, 7834–7841 CrossRef CAS.
- H. Wickel, S. Agarwal and A. Greiner, Macromolecules, 2003, 36, 2397–2403 CrossRef CAS.
- J.-F. Lutz, J. Andrieu, S. Üzgün, C. Rudolph and S. Agarwal, Macromolecules, 2007, 40, 8540–8543 CrossRef CAS.
- N. Xiao, H. Liang and J. Lu, Soft Matter, 2011, 7, 10834–10840 RSC.
- G. G. Hedir, C. A. Bell, N. S. Ieong, E. Chapman, I. R. Collins, R. K. O'Reilly and A. P. Dove, Macromolecules, 2014, 47, 2847–2852 CrossRef CAS.
- B. Ochiai and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2827–2834 CrossRef CAS PubMed.
- A. Tardy, V. Delplace, D. Siri, C. Lefay, S. Harrisson, B. de Fatima Albergaria Pereira, L. Charles, D. Gigmes, J. Nicolas and Y. Guillaneuf, Polym. Chem., 2013, 4, 4776–4787 RSC.
- Y. Zhang, M. Zheng, T. Kissel and S. Agarwal, Biomacromolecules, 2011, 13, 313–322 CrossRef PubMed.
- L. Ren and S. Agarwal, Macromol. Chem. Phys., 2007, 208, 245–253 CrossRef CAS PubMed.
- Y. Shi and S. Agarwal, e-Polymers, 2015, 15, 217–226 CrossRef CAS.
- F. Sanda, T. Takata and T. Endo, Macromolecules, 1993, 26, 1818–1824 CrossRef CAS.
- F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 265–276 CrossRef CAS.
- A. de Meijere, V. Bagutski, F. Zeuner, U. K. Fischer, V. Rheinberger and N. Moszner, Eur. J. Org. Chem., 2004, 3669–3678 CrossRef CAS PubMed.
- T. Hirano, A. Tabuchi, M. Seno and T. Sato, Macromolecules, 2001, 34, 4737–4741 CrossRef CAS.
- V. Delplace, S. Harrisson, A. Tardy, D. Gigmes, Y. Guillaneuf and J. Nicolas, Macromol. Rapid Commun., 2014, 35, 484–491 CrossRef CAS PubMed.
- S. Ata, D. Mal and N. K. Singha, RSC Adv., 2013, 3, 14486–14494 RSC.
- D. J. Siegwart, J. K. Oh and K. Matyjaszewski, Prog. Polym. Sci., 2012, 37, 18–37 CrossRef CAS PubMed.
- D. H. Seuyep N, D. Szopinski, G. A. Luinstra and P. Theato, Polym. Chem., 2014, 5, 5823–5828 RSC.
- D. H. Seuyep N, G. A. Luinstra and P. Theato, Polym. Chem., 2013, 4, 2724–2730 RSC.
- E. Behravesh, S. Jo, K. Zygourakis and A. G. Mikos, Biomacromolecules, 2002, 3, 374–381 CrossRef CAS PubMed.
- L. Krishna and M. Jayabalan, J. Mater. Sci.: Mater. Med., 2009, 20, 115–122 CrossRef PubMed.
- W. J. Bailey, S.-R. Wu and Z. Ni, Die Makromolekulare Chemie, 1982, 183, 1913–1920 CrossRef CAS PubMed.
- T. Kelen and F. Tüdõs, J. Macromol. Sci., Part A: Pure Appl. Chem., 1975, 9, 1–27 CrossRef PubMed.
- P. P. Contreras, P. Tyagi and S. Agarwal, Polym. Chem., 2015, 6, 2297–2304 RSC.
- W. Chen, F. Meng, R. Cheng and Z. Zhong, J. Controlled Release, 2010, 142, 40–46 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Homopolymerization and characterization of VCP and reactivity ratio calculations. See DOI: 10.1039/c5py00962f |
| This journal is © The Royal Society of Chemistry 2015 |