
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the optoelectronic properties of dual-acceptor based low-bandgap ambipolar polymers by changing the thiophene-bridge length†
Cunbin
An‡
a,
Tomasz
Marszalek‡
a,
Xin
Guo
 *ab,
Sreenivasa Reddy
Puniredd§
a,
Manfred
Wagner
a,
Wojciech
Pisula
ac and
Martin
Baumgarten
*a
*ab,
Sreenivasa Reddy
Puniredd§
a,
Manfred
Wagner
a,
Wojciech
Pisula
ac and
Martin
Baumgarten
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, D-55128, Mainz, Germany
bDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China
cDepartment of Molecular Physics, Faculty of Chemistry, Lodz University of Technology, Zeromskiego 116, 90-924 Lodz, Poland. E-mail: guoxin@dicp.ac.cn; martin.baumgarten@mpip-mainz.mpg.de
First published on 20th July 2015
Abstract
Three very-low-bandgap dual-acceptor based polymers containing diketopyrrolopyrrole (DPP) and a thiadiazoloquinoxaline (TQ) derivative were prepared. Both acceptors in these polymers were separated by one, two and three thiophenes. By only inserting one thiophene between DPP and benzodithiophene condensed TQ, the polymer PDPP-T-TQ shows a very low optical bandgap of 0.60 eV with an electron affinity of −4.23 eV. Optical and electrochemical bandgaps of the polymers were enlarged with increasing the thiophene-bridge length between both acceptors. GIWAXS measurements confirmed that the polymer with three thiophene bridges (PDPP-3T-TQ) showed an ordered arrangement of the crystallites, providing the best ambipolar device performance among these polymers.
Introduction
Conjugated polymers with a very low bandgap (≤1.0 eV) have been drawing substantial attention due to their broad and near-infrared (NIR) absorption, multiple redox states in a small potential window, ambipolar charge carrier transport, and potential use in sensors, batteries, and supercapacitors.1 In a significant breakthrough study, Wudl and his coworkers reported a poly(isothianaphthene) with an energy bandgap of ∼1.0 eV due to a strong quinoidal character in the thiophene ring.2–4 Recently, tailoring donor–acceptor (D–A) interactions in D–A copolymers has proven to be an effective strategy for developing very low bandgap polymers. The combination of strong donors and acceptors with the quinoidal character has produced soluble polymers with optical bandgaps (Eoptg) lower than 0.70 eV, for example, P(DTP-BThBTT)5 and P(CPDT-TQ)6 (Fig. 1).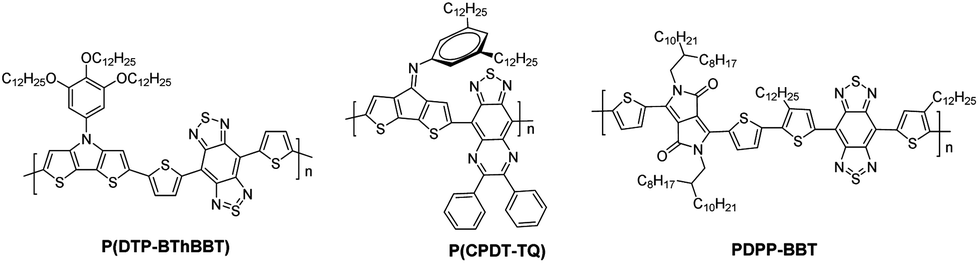 | ||
| Fig. 1 Reported polymers with very low bandgap (≤1.0 eV). | ||
However, many low bandgap polymers containing one strong acceptor carry more holes than electrons, such as those derived from benzobisthiadiazole (BBT)7,8 and diketopyrrolopyrrole (DPP).9,10 Balanced charge carrier transporting polymers are highly desirable for ambipolar OFETs, which allows both p-type and n-type channels to be realized in one device with simple fabrication processes.11 For this purpose, a dual-acceptor design strategy has been proposed to construct D-A1–D-A2 polymers.12–17 For example, PDPP-BBT (Fig. 1) exhibited an Eoptg of around 0.65 eV and ambipolar charge transporting behavior with mobilities of 1.17 cm2 V−1 s−1 for holes and 1.32 cm2 V−1 s−1 for electrons.18 In comparison with the polymers composed of only one strong acceptor, dual-acceptor polymers with very low bandgap and balanced charge carrier transport are rarely studied.
In this paper, we report the combination of two high electron affinity acceptors, DPP and benzodithiophene condensed TQ in one polymer backbone. As previously demonstrated, this condensed TQ was a new strong acceptor and a corresponding D–A copolymer exhibited a very low Eoptg of 0.76 eV and hole-dominant ambipolar behavior.19,20 On the other hand, DPP has been widely studied as a strong acceptor in low bandgap polymers because numerous DPP-based polymers have revealed excellent performance as ambipolar semiconductors.10,21 Therefore, it can be expected that the combination of the two highlighted acceptors could endow the resulting dual-acceptor polymers desired characteristics of both very low bandgap and balanced ambipolarity. Three copolymers (PDPP-T-TQ, PDPP-2T-TQ and PDPP-3T-TQ in Scheme 1) were designed and synthesized, in which the two acceptors are separated in the polymer backbone by oligothiophene bridges with varying lengths in order to elaborately tune the optoelectronic properties and charge carrier transport.
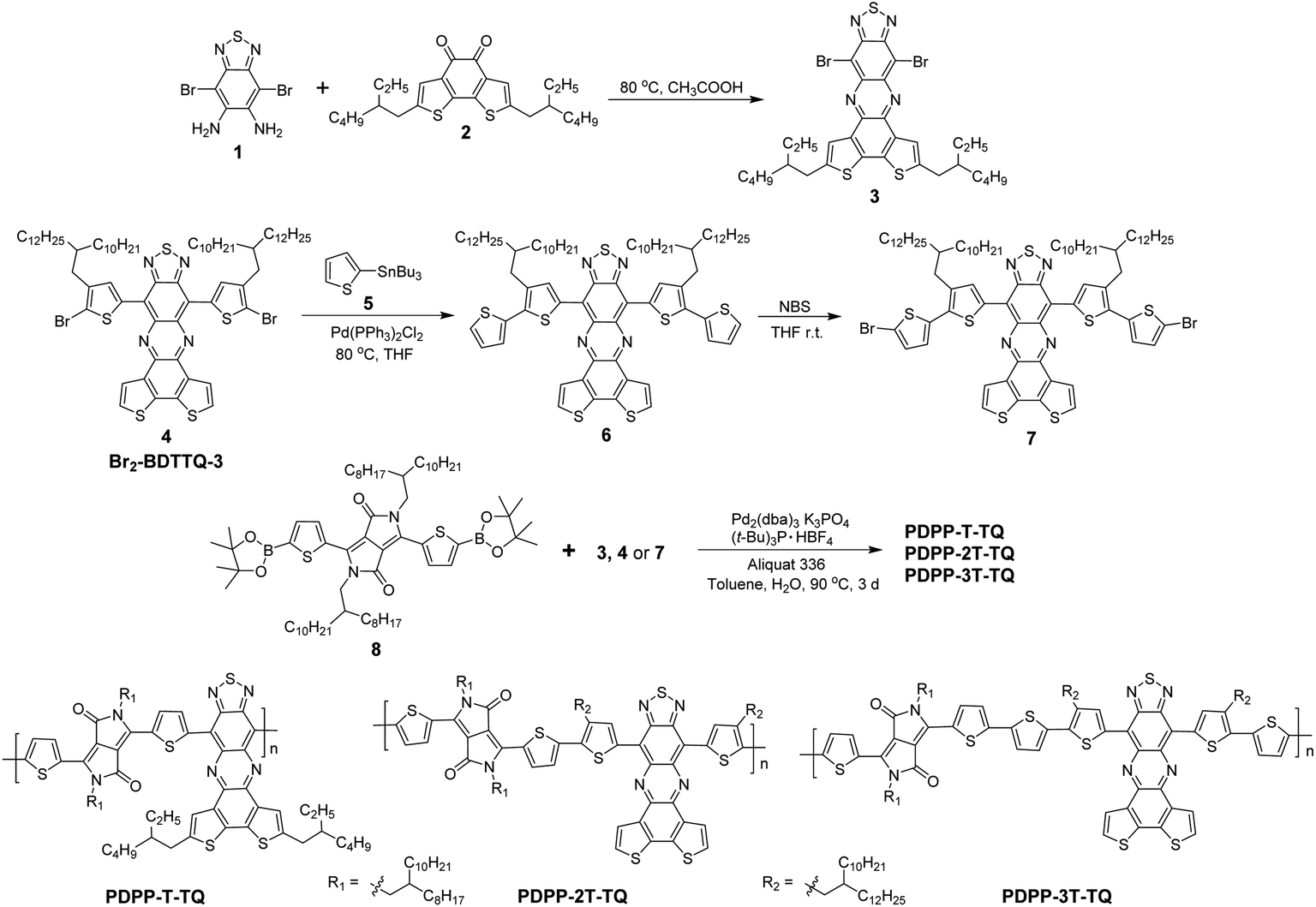 | ||
| Scheme 1 Synthetic routes for the three polymers. | ||
Results and discussion
Synthesis and characterization
The synthesis of the three polymers is outlined in Scheme 1. In order to introduce different oligothiophenes between DPP and condensed TQ, three TQ monomers (3, 4 and 7) were synthesized by flanking thiophenes onto condensed TQ at both sides. Monomer 3 was synthesized from 1 and 2via a condensation reaction. Monomer 7 was prepared from Br2-BDTTQ-3 after two-step reactions, Stille coupling and bromination. The Suzuki coupling reaction was carried out between monomer 3, 4 or 7 and DPP-diboronic ester (8) to give the corresponding PDPP-T-TQ, PDPP-2T-TQ, and PDPP-3T-TQ, respectively. In previous studies,11,22 when using Pd2(dba)3 as a catalyst tri-tert-butylphosphoniumtetrafluoroborate ((t-Bu)3P·HBF4) was usually used as an effective ligand to yield high-molecular-weight polymers in Suzuki coupling based on compound 8. Therefore, the Pd2(dba)3/(t-Bu)3P·HBF4 catalyst system was also chosen here. The number-average molecular weight (Mn) and polydispersity index (PDI) of the three polymers were determined by the gel permeation chromatography (GPC) method using polystyrene as the standard and tetrahydrofuran as the eluent at 30 °C. The data are listed in Table 1. The low Mn of PDPP-T-TQ and PDPP-2T-TQ might arise from the low reaction activity and the steric hindrance in monomers 3 and 4 preventing production of higher Mn during polymerization. A similar low Mn was also reported for PDPP-BBT (Fig. 1).18 Another ligand, tri(o-tolyl)phosphine (P(o-tol)3) was also attempted to prepare PDPP-T-TQ as it was reported that polymers with Mn higher than 10 kg mol−1 could be produced when using the Pd2(dba)3/P(o-tol)3 catalyst system in Suzuki coupling based on DPP-diboronic esters.23–25 The Mn of the resulting polymer (6.3 kg mol−1, PDI: 2.9) is however slightly lower in comparison with that using the ligand of (t-Bu)3P·HBF4. After introducing one more electron-rich thiophene onto monomer 4 at both sides, the steric hindrance between DPP and monomer 7 was reduced so that the PDPP-3T-TQ possessed a higher Mn than the other two polymers. It is thus believed that the sterics around the coupling units play a main role in the limited activity for polymerizations of PDPP-T-TQ and PDPP-2T-TQ. The three polymers exhibited excellent solubility in common organic solvents such as chloroform and tetrahydrofuran at room temperature. Additionally, the decomposition temperature was measured by thermogravimetric analysis (TGA) (Fig. S1†) showing 5% weight loss at 380 °C for PDPP-T-TQ and PDPP-2T-TQ, and 390 °C for PDPP-3T-TQ.| Polymer |
M
w/Mn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (kg mol−1) a (kg mol−1) |
T
db (°C) |
λ
absc (nm) soln. |
λ
absd (nm) film |
E
optgd (eV) |
IPe (eV) | EAe (eV) |
μ
hf (cm2 V−1 s−1) |
μ
ef (cm2 V−1 s−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Determined by GPC in THF using polystyrene standards. b Temperature of decomposition corresponding to 5% weight loss from TGA analysis under N2 with a heating rate of 10 °C min−1. c Dissolved in chloroform (c = 10−5 M). d Drop-cast from chloroform solution (2 mg mL−1). e IP and EA were estimated from the onsets of the first oxidation and reduction peak, while the potentials were determined using ferrocene (Fc) as the standard by the empirical formula IP/EA = −(EonsetOx/Red − E1/2Fc/Fc+ + 4.8) eV, wherein E1/2Fc/Fc+ = 0.40 eV. f Average value of 10 devices. | |||||||||
| PDPP-T-TQ | 26.3/7.4 | 380 | 1434 | 1435 | 0.60 | −5.12 | −4.23 | 2 × 10−5 | 3 × 10−5 |
| PDPP-2T-TQ | 23.1/7.3 | 380 | 1110 | 1115 | 0.75 | −5.06 | −4.13 | 1 × 10−5 | 4 × 10−5 |
| PDPP-3T-TQ | 43.9/13.2 | 390 | 970 | 995 | 0.88 | −5.08 | −4.07 | 5 × 10−4 | 5 × 10−5 |
Optical and electrochemical properties
UV-vis-NIR absorption spectra of the polymers were recorded in chloroform solution (c = 10−5 M) as well as in thin films. The relevant data are summarized in Table 1. In dilute chloroform solution, all polymers exhibit three absorption bands as shown in Fig. 2a. The first intense band between 300 and 500 nm contains double peaks that are typical for many TQ polymers,26–30 suggesting that this band could be contributed by the condensed TQ moiety. The second one ranges from 500 to 800 nm, similar to the maximum absorption (λmax) of some DPP polymers,31,32 indicating its origin from the interaction between DPP and adjacent thiophene units. The last band broadly spans from 800 to 2250 nm, which is attributed to the intramolecular charge transfer (ICT) between donors and acceptors in the polymer main chains.33PDPP-T-TQ exhibits inconspicuous TQ and DPP absorption characters but has a λmax up to 1343 nm, indicating a very strong ICT process within this polymer. Increasing the thiophene bridge to two units between the DPP and condensed TQ leads to a blue shift of 324 nm at λmax for PDPP-2T-TQ. Meanwhile, the absorption profile is much more dominated by the condensed TQ and DPP moieties. After introducing another thiophene between DPP and condensed TQ the λmax of PDPP-3T-TQ further hypsochromically shifts to 970 nm and more remarkable absorption features of condensed TQ and DPP are observed.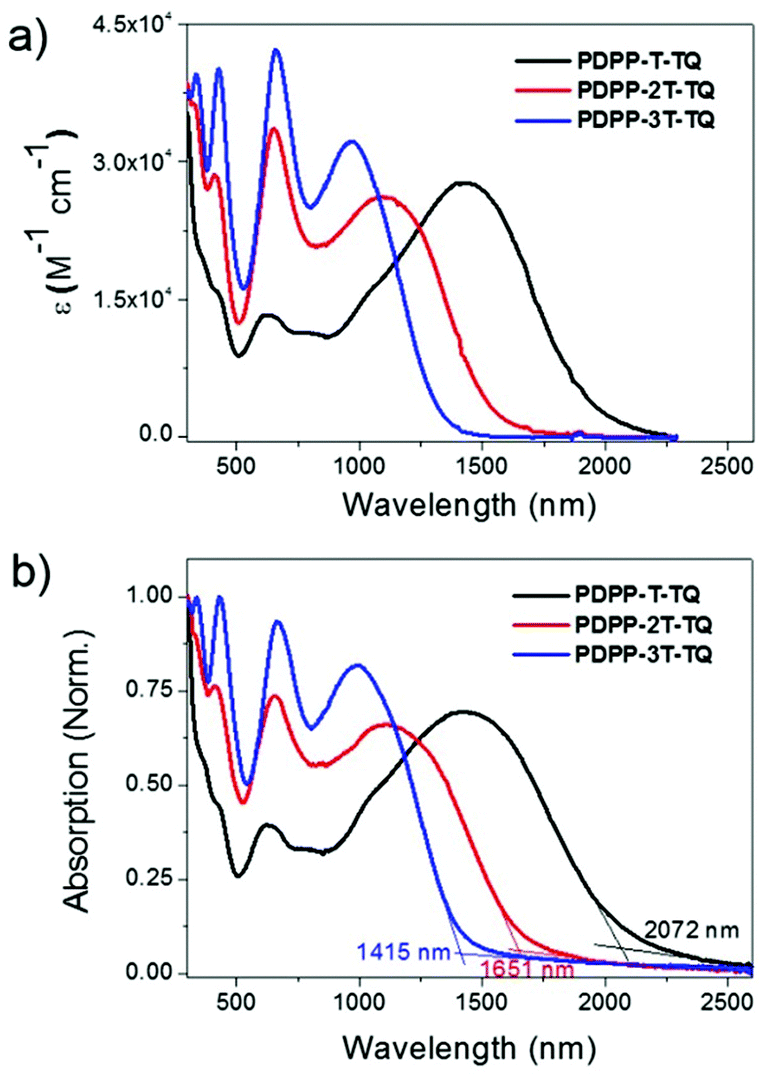 | ||
| Fig. 2 UV-visible-NIR absorption spectra of three polymers in (a) chloroform solutions and (b) films. | ||
Thin films were prepared by drop-casting chloroform solutions of the three polymers onto glass slides. The films display slight red-shifts of 1, 5 and 25 nm at λmax and broadened absorption bands compared with those in solution (Fig. 2b), indicating that the obvious red-shift occurs along with the increased backbone coplanarity of the polymers. The tiny red-shifts of PDPP-T-TQ and PDPP-2T-TQ can be attributed to the disordered morphology of both polymers induced by bulky side chains which retard the efficient packing of polymer chains in the solid state. The optical bandgaps of PDPP-T-TQ, PDPP-2T-TQ and PDPP-3T-TQ were calculated to be 0.60, 0.75 and 0.88 eV, respectively, according to the onset of solid-state absorption spectra. The variation of bandgaps can be explained by the electronic changes in the HOMO and LUMO levels (see the DFT calculations section below for details). The value of PDPP-T-TQ shows the lowest Eoptg among the dual-acceptor polymers reported so far.12,13,34,35 These results demonstrate that changing the distance between DPP and TQ acceptors by changing the oligothiophene bridge length is an effective strategy for tuning the optoelectronic properties of the DPP–TQ dual-acceptor polymers.
The electrochemical properties of three polymers were determined using cyclic voltammetry (CV) from their drop-cast thin films (Fig. 3). The corresponding data are summarized in Table 1. The electron affinities (EAs) and ionization potentials (IPs) of the polymers were calculated from the onset of first reduction and oxidation potentials. The values of EA are −4.23, −4.13 and −4.07 eV for PDPP-T-TQ, PDPP-2T-TQ and PDPP-3T-TQ, respectively, while the corresponding IP values are −5.12, −5.06, and −5.08 eV. Interestingly, the different thiophene numbers can significantly alter the EA values compared to their similar IP values. The PDPP-T-TQ had a lowest EA value which is related to its strongest ICT ability, leading to a narrowest HOMO–LUMO bandgap among these three polymers. The electrochemical bandgaps of three polymers were calculated to be 0.89, 0.93, 1.01 eV for PDPP-T-TQ, PDPP-2T-TQ and PDPP-3T-TQ, respectively. It is the same tendency with their optical gaps. The only difference is that the electronic bandgaps are larger than their optical gap, which is attributed to the exciton binding energy of conjugated polymers.36
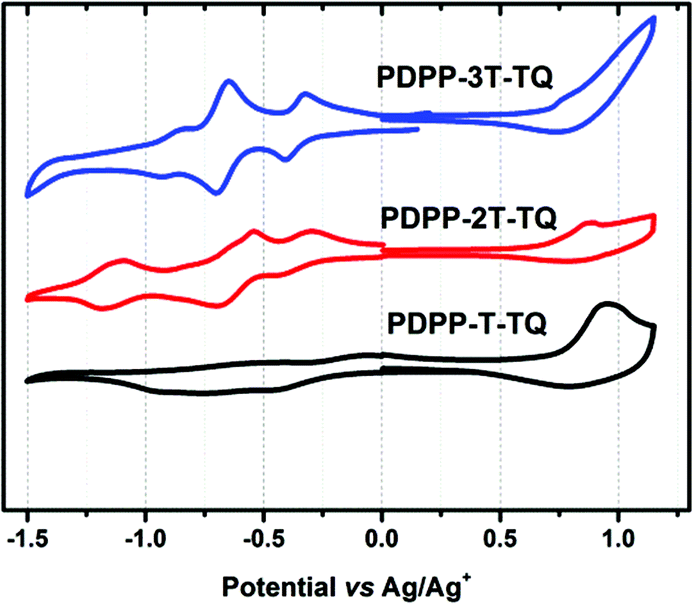 | ||
| Fig. 3 Cyclic voltammograms of PDPP-T-TQ, PDPP-2T-TQ and PDPP-3T-TQ in films. | ||
OFET properties
Bottom-gate, bottom-contact OFETs were fabricated in order to evaluate the charge transport properties of all three polymers. All polymers were deposited by drop-casting 5 mg mL−1 chloroform solution on silicon/silicone dioxide (SiO2) substrates under a nitrogen atmosphere, followed by annealing at 150 °C for 1 h. The 300 nm thick SiO2 dielectric covering the highly doped Si and acting as the gate electrode was functionalized with hexamethyldisilazane (HMDS) to minimize interfacial trapping sites. Representative transfer and output characteristics of PDPP-3T-TQ are shown in Fig. 4. Such curves of the other two polymers are presented in Fig. S2 (ESI†). All polymers show ambipolar device behaviors. In all cases the charge carrier mobilities are calculated from the drain current saturation region. The polymer PDPP-T-TQ exhibits balanced charge transport with hole and electron mobilities of 2 × 10−5 cm2 V−1 s−1 and 3 × 10−5 cm2 V−1 s−1. Additional thiophene units in the bridge between the DPP and TQ acceptor units (PDPP-2T-TQ) in comparison with PDPP-T-TQ do not change the charge carrier mobilities with 4 × 10−5 cm2 V−1 s−1 for electrons and 1 × 10−5 cm2 V−1 s−1 for holes. But a difference in transport behavior is observed for PDPP-3T-TQ, which exhibits a hole mobility of 5 × 10−4 cm2 V−1 s−1 being one order of magnitude higher than that for PDPP-T-TQ and PDPP-2T-TQ. These results implied that the thiophene bridge length between DPP and condensed TQ can transform the DPP–TQ based polymers from well-balanced ambipolar to hole-dominant ambipolar behavior. However, for all polymers a contact resistance is observed in the transfer and output curves.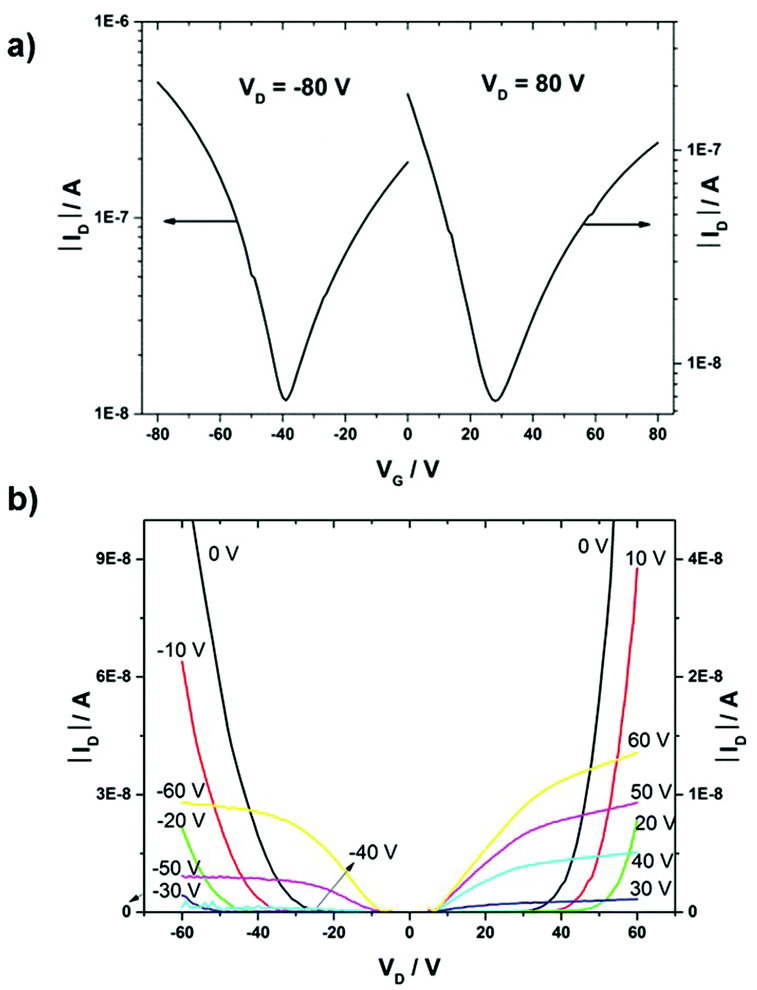 | ||
| Fig. 4 (a) Transfer and (b) output characteristics of PDPP-3T-TQ thin films. | ||
Self-organization in bulk
In order to understand the supramolecular organization of the polymer films, grazing incident wide angle X-ray scattering (GIWAXS) was carried out. GIWAXS gives valuable information about the surface organization of the polymers in thin films.37 For these measurements the same procedure for the film preparation was used as that for the transistor devices. Fig. 5 shows the GIWAXS patterns of the three polymers which reveal slight variations between PDPP-T-TQ and PDPP-2T-TQ. For both polymers an interlayer distance of 1.60 nm and 2.17 nm is determined from the position of the main reflection in a small-angle range. The larger interlayer spacing for PDPP-2T-TQ is directly related to longer side chains. The isotropic distribution of these reflections suggests a lack of long-range order and a random arrangement of the crystallites towards the surface. A characteristic π-stacking reflection is missing indicating a disordered organization of the polymer chains within the layered structures.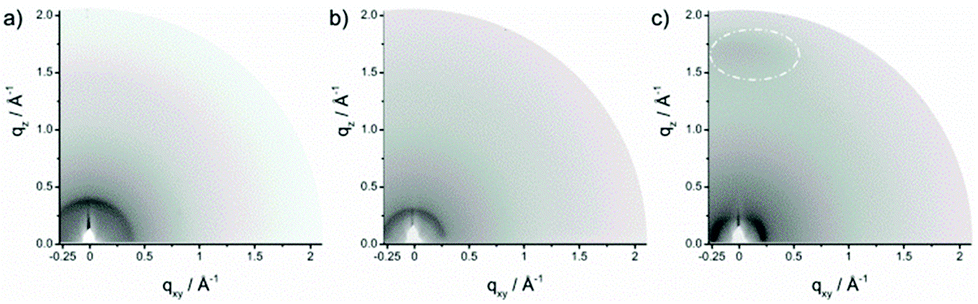 | ||
| Fig. 5 GIWAXS patterns of (a) PDPP-T-TQ, (b) PDPP-2T-TQ and (c) PDPP-3T-TQ thin films. Reflections corresponding to π-stacking are indicated by a dashed circle. | ||
In contrast, the GIWAXS pattern of PDPP-3T-TQ (Fig. 5c) exhibits more distinct reflections. In the small-angle region, the maximum intensity of the reflection is located on the equatorial plane of the pattern, whereby an interlayer distance of 2.65 nm is found. This reflection position in the pattern is indicative of a face-on orientation of the polymer on the surface. This conclusion is verified by the appearance of a π-stacking scattering intensity (Fig. S3†) on the meridional plane at qz = 1.7 Å−1 (0.36 nm). The π-stacking reflection implied a better order arrangement of thin films for PDPP-3T-TQ in comparison with the other two polymers. Increasing the number of thiophene moieties changes the linearity and planarity of the conjugated polymer backbones and reduces the twist between the DPP and TQ groups improving the polymer packing. Additionally, the face-on arrangement allows a 3D transport in the active layer.38 This is the main reason for the higher charge carrier mobility of PDPP-3T-TQ. However, it could not provide an explanation why increasing the thiophene bridge length changes the transistor characteristics from well-balanced ambipolar to hole-dominant ambipolar behavior. Therefore, density functional theory (DFT) calculations were carried out.
Density functional theory calculations
DFT calculations were carried out on a monomeric unit of the polymers, named DPP-T-TQ, DPP-2T-TQ and DPP-3T-TQ, carrying methyl substituents. The distribution of electrons was simulated by density functional theory (DFT) using the Gaussian 03 program39 with the B3LYP functional and 6-31G basis set.40 The electron density distributions of the LUMO and HOMO of the geometry with optimized structures are shown in Fig. 6. All HOMO levels of the three monomeric units were very similar with delocalization along the conjugated backbone. However, the electron density became gradually localized on the TQ segment in the LUMO levels as the thiophene bridge length increased. Such an electron density distribution on the HOMO and LUMO levels forces an unbalanced transport along the conjugated backbone in the case of DPP-3T-TQ. This tendency is quite obvious for the hole transport which increases with increasing the number of thiophene units while the electron transport remains constant. In addition, the results from calculations were well consistent with the observations from CV results, demonstrating that the thiophene bridge length can significantly alter the EA in comparison with IP. It can hence be concluded that the thiophene bridge length not only tunes the organization behavior in thin films but also can strongly influence the ratio of electron and hole carrier mobilities.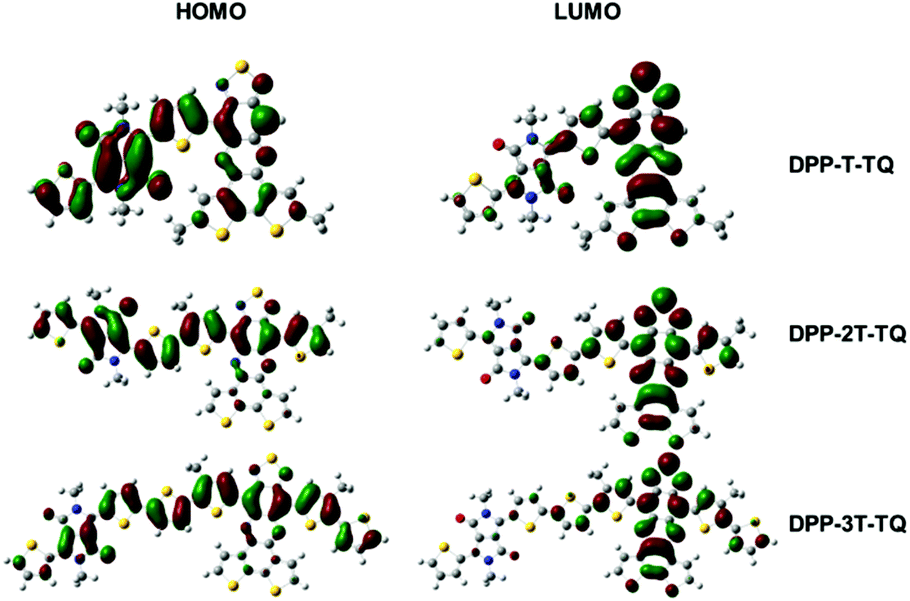 | ||
| Fig. 6 The electron density distribution on one repeat unit of PDPP-T-TQ, PDPP-2T-TQ and PDPP-3T-TQ. | ||
Conclusions
We have successfully synthesized three D-A1–D-A2 dual-acceptor polymers. Both acceptors in these polymers were separated by thiophene bridges with different lengths. The polymer PDPP-T-TQ possessed a very narrow optical bandgap of 0.60 eV. The Eoptg of polymers are significantly increased from 0.60 to 0.88 eV with increasing the thiophene bridge length between both acceptors. The CV measurement indicated high electron affinities of the polymers ranging from −4.07 to −4.23 eV. From polymers PDDP-2T-TQ to PDPP-3T-TQ, although the intramolecular charge transfer abilities of polymers were decreased, the planarity of the polymer was improved as confirmed by GIWAXS. PDPP-3T-TQ exhibited a hole-dominant ambipolar behavior with 5 × 10−4 cm2 V−1 s−1 for holes and 5 × 10−5 cm2 V−1 s−1 for electrons; the hole mobility was one order of magnitude higher than that for PDPP-2T-TQ. The correlations between structures and charge carrier mobilities of DPP–TQ copolymers are beneficial to broaden the understanding of optoelectronic properties of such polymers and to further design new dual-acceptor polymers towards well balanced ambipolarity.Experimental section
General methods
1H NMR and 13C NMR spectra were recorded in deuterated solvents on a Bruker Advance III 500. 1H NMR spectra of the polymers were recorded in C2D2Cl4 solvents on a Bruker DPX 500 at 373 K. High Resolution Mass Spectra (HRMS) were recorded by the Microanalytical Laboratory of Johannes Gutenberg-University, Mainz. Elemental analysis was carried out using a Foss Heraeus Vario EL at the Institute of Organic Chemistry at the Johannes Gutenberg-University, Mainz. UV-Vis-NIR absorption spectra were recorded on a Perkin-Elmer Lambda 9 spectrophotometer at room temperature. Thermogravimetry analysis (TGA) was carried out on a Mettler 500 Thermogravimetry Analyzer. Cyclic voltammetry (CV) was carried out on a computer-controlled GSTAT12 in a three-electrode cell in anhydrous acetonitrile aqueous solution of Bu4NPF6 (0.1 M) with a scan rate of 50 mV s−1 at room temperature under argon. A Pt wire, a silver wire, and a glassy carbon electrode were used as the counter electrode, the reference electrode, and the working electrode, respectively. The molecular weights were determined using a PSS-WinGPC (PSS) (pump: alliance GPC 2000) GPC equipped with a UV or RI detector running in THF at 30 °C using a PLgel MIXED-B column (particle size: 10 mm, dimensions: 0.8 × 30 cm) calibrated against polystyrene standards.OFET device fabrication and measurements
All FETs were fabricated employing the bottom-gate, bottom-contact architecture. The 230 nm thick SiO2 dielectric covering the highly doped Si acting as the gate electrode was functionalized with hexamethyldisilazane (HMDS) to minimize interfacial trapping sites. Polymer thin films were deposited by drop-casting 5 mg mL−1 chloroform solution on FET substrates under a nitrogen atmosphere, followed by annealing at 150 °C for 1 h. The channel lengths and widths are 20 and 1400 mm, respectively. All the electrical measurements (using Keithley 4200 SCS) were performed in a glove box under a nitrogen atmosphere.Grazing incidence wide angle X-ray scattering (GIWAXS)
GIWAXS experiments were performed using a solid anode X-ray tube (Siemens Kristalloflex X-ray source, a copper anode X-ray tube operated at 30 kV and 20 mA), Osmic confocal MaxFlux optics, an X-ray beam with pinhole collimation and a MAR345 image plate detector. The samples were prepared as a thin film with the same procedure as that used in OFET fabrication.Synthetic details
All chemicals and reagents were used as received from commercial sources without further purification unless stated otherwise. Chemical reactions were carried out under ambient atmosphere. The intermediate compounds 4,7-dibromobenzo[c][1,2,5]thiadiazole-5,6-diamine (1),41 2,7-bis(2-ethylhexyl)benzo[2,1-b:3,4-b′]dithiophene-4,5-dione (2),19Br2-BDTTQ-3 (4),20 and 3,6-bis-(5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-thiophen-2-yl))-N,N-bis(octyldodecyl)-1,4-dioxo-pyrrolo[3,4-c]pyrrole (8)22 were prepared according to the published procedures.8,12-Dibromo-2,5-bis(2-ethylhexyl)-[1,2,5]thiadiazolo[3,4-i]dithieno[3,2-a:2′,3′-c]phenazine (3)
A suspension of 1 (0.40 g, 1.23 mmol), 2 (0.55 g, 1.23 mmol) and 35 mL of acetic acid was placed in a 50 mL Schlenk tube. The mixture was heated to 80 °C and stirred overnight. After cooling to room temperature, the product was filtered and washed with methanol, then purified by column chromatography using hexane/dichloromethane (3/1) as the eluent to give 0.77 g of compound 3 (dark green solid, 85%). 1H NMR (250 MHz, CD2Cl2) δ 8.03 (s, 2H), 3.00 (dd, J = 2.5 Hz, J = 5.0 Hz, 4H), 1.76 (m, 2H), 1.41 (br, 16H), 0.95 (br, 12H). 13C NMR (62.5 MHz, CDCl3) δ 151.65, 145.10, 142.45, 137.39, 137.66, 133.30, 123.47, 113.40, 41.74, 34.87, 32.63, 29.09, 25.76, 23.19, 14.35, 11.03. HRMS (ESI+): m/z calcd 731.0547, found 731.0541.8,12-Bis(3-(2-decyltetradecyl)-[2,2′-bithiophen]-5-yl)-[1,2,5]thiadiazolo[3,4-i]dithieno[3,2-a:2′,3′-c]phenazine (6)
Br2-BDTTQ-3 (4) (0.35 g, 0.26 mmol), 2-tributylstanylthiophene 5 (0.30 g, 0.78 mmol) and Pd(PPh3)Cl2 (21 mg, 0.026 mmol) were dissolved in 15 mL of anhydrous THF under argon. The resulting solution was stirred for 16 h at 80 °C. The solvent was removed under reduced pressure to afford a dark-red oil, which was purified by column chromatography to give 0.29 g (dark-green solid, 82%) of compound 6. 1H NMR (250 MHz, CD2Cl2, ppm) δ 8.62 (s, 2H), 7.79 (d, J = 5.0 Hz, 2H), 7.33 (t, J = 5.0 Hz, J = 2.5 Hz, 2H), 7.08 (m, 6H), 2.67 (d, J = 7.50 Hz, 4H), 1.80 (m, 2H), 1.38–1.23 (m, 80H), 0.89–0.84 (m, 12H). 13C NMR (62.5 MHz, CD2Cl2, ppm) δ 150.67, 138.82, 137.89, 137.85, 137.77, 137.72, 137.04, 134.68, 134.45, 134.35, 127.92, 126.88, 125.29, 125.13, 124.40, 119.14, 38.77, 34.43, 33.77, 32.40, 32.37, 30.76, 30.32, 30.28, 30.25, 30.20, 30.15, 29.87, 29.83, 26.82, 23.13, 14.33. HRMS (ESI+): m/z calc. 1351.6854, found 1351.6850.8,12-Bis(5′-bromo-3-(2-decyltetradecyl)-[2,2′-bithiophen]-5-yl)-[1,2,5]thiadiazolo[3,4-i]dithieno[3,2-a:2′,3′-c]phenazine (7)
Compound 6 (0.25 g, 0.18 mmol) was dissolved in 15 mL of THF at room temperature. NBS (72.4 mg, 0.41 mmol) was carefully added into the solution in small batches in the dark. The mixture was stirred for 5 h. After removing the solvent under reduced pressure, the residue was purified by column chromatography to give monomer 7 as a dark-green solid (0.24 g, 86%). 1H NMR (250 MHz, CD2Cl2) 1H NMR (250 MHz, THF-d8, ppm) δ 8.66 (s, 2H), 7.93 (d, J = 5.0 Hz, 2H), 7.38 (d, J = 5.0 Hz, 2H), 7.11 (d, J = 2.5 Hz, 2H), 6.92 (d, J = 5.0 Hz, 2H), 2.67 (d, J = 7.50 Hz, 4H), 1.81 (br, 2H), 1.39–1.22 (m, 80H), 0.88–0.83 (m, 12H). 13C NMR (62.5 MHz, THF-d8, ppm) δ 150.97, 139.75, 139.48, 138.51, 138.13, 138.10, 137.79, 137.27, 135.12, 134.94, 131.38, 127.24, 126.00, 125.44, 119.47, 112.10, 39.21, 34.84, 34.22, 32.76, 32.73, 31.11, 30.67, 30.62, 30.57, 30.51, 30.24, 30.19, 27.26 (overlap by THF-d8) 23.41, 14.33. HRMS (ESI+): m/z calcd 1507.5063, found 1507.5059.Synthesis of PDPP-T-TQ
To a solution of compounds 3 (0.05 mmol) and 8 (0.05 mmol), tri-tert butylphosphoniumtetrafluoroborate ((t-Bu)3P·HBF4, 0.0067 mmol), tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3, 0.00335 mmol) and Aliquat 336 (2 drops) in 6 mL of toluene was added a solution of potassium phosphate (0.074 g, 0.35 mmol) in 0.46 mL of degassed water. The mixture was vigorously stirred at 90 °C for 3 days. The polymer was end-capped with phenyl units by adding phenyl boronic acid and bromobenzene in sequence. After cooling to room temperature, the reaction mixture was poured into vigorously stirred methanol (100 mL). The polymer was filtered and subjected to Soxhlet extraction with methanol, acetone and hexane. The hexane fraction was collected and added 30 mL of sodium diethyldithiocarbamate aqueous solution (1 g per 100 ml) to remove the residual palladium catalyst, and the mixture was heated to 60 °C with vigorous stirring for 2 h. The mixture was separated and the organic phase was washed with water 3 times. The hexane solution was concentrated and precipitated in methanol. The resulting solid was collected by filtration and dried in a vacuum to afford a black solid (55 mg, 77%). Molecular weight by GPC (30 °C): Mn = 7.4 kDa, PDI = 3.54. UV-Vis: λmax (solution in chloroform): 1434 nm, 616 nm, 415 nm and 350 nm; λmax (thin film): 1435 nm, 624 nm and 424 nm. The 1H NMR spectra are shown in Fig. S9.† Elemental analysis: calcd for C86H122N6O2S5: C 72.12, H 8.59, N 5.87, S 11.19; found: C 72.00, H 8.82, N 5.92, S 10.92.Synthesis of PDPP-2T-TQ and PDPP-3T-TQ
These two polymers were prepared using monomers 4 and 7 instead of monomer 3 by a similar procedure and work-up to PDPP-T-TQ.PDPP-2T-TQ (black solid, 75 mg, 74%). Molecular weight by GPC (30 °C): Mn = 7.3 kDa, PDI = 3.18. UV-Vis: λmax (solution in chloroform): 1110 nm, 655 nm, 418 nm and 328 nm; λmax (thin film): 1115 nm, 656 nm, 418 nm and 332 nm. The 1H NMR spectra are shown in Fig. S10.† Elemental analysis: calcd for C126H190N6O2S7: C 73.99, H 9.36, N 4.11, S 10.97; found: C 73.60, H 9.65, N 3.99, S 10.92.
PDPP-3T-TQ (black solid, 88 mg, 80%). Molecular weight by GPC (30 °C): Mn = 13.2 kDa, PDI = 3.31. UV-Vis: λmax (solution in chloroform): 971 nm, 661 nm, 430 nm and 334 nm; λmax (thin film): 995 nm, 664 nm, 418 nm and 339 nm. The 1H NMR spectra are shown in Fig. S11.† Elemental analysis: calcd for C134H194N6O2S9: C 73.84, H 8.85, N 3.80, S 13.06; found: C 72.55, H 9.15, N 3.73, S 12.88.
Acknowledgements
This work was supported by SFB-TR49. C.A. gratefully acknowledges the China Scholarship Council (CSC) for offering a 3 year scholarship.Notes and references
- T. A. Skotheim and J. R. Reynolds, Handbook of conducting polymers, 3rd edn; Conjugated Polymers: Theory, Synthesis, Properties, and Characterization, CRC Press LLC, Boca Raton, FL, 2007 Search PubMed.
- F. Wudl, M. Kobayashi and A. J. Heeger, J. Org. Chem., 1984, 49, 3382–3384 CrossRef CAS.
- J. L. Brédas, A. J. Heeger and F. Wudl, J. Chem. Phys., 1986, 85, 4673–4678 CrossRef PubMed.
- I. Hoogmartens, P. Adriaensens, D. Vanderzande, J. Gelan, C. Quattrocchi, R. Lazzaroni and J. L. Bredas, Macromolecules, 1992, 25, 7347–7356 CrossRef CAS.
- T. T. Steckler, X. Zhang, J. Hwang, R. Honeyager, S. Ohira, X.-H. Zhang, A. Grant, S. Ellinger, S. A. Odom, D. Sweat, D. B. Tanner, A. G. Rinzler, S. Barlow, J.-L. Brédas, B. Kippelen, S. R. Marder and J. R. Reynolds, J. Am. Chem. Soc., 2009, 131, 2824–2826 CrossRef CAS PubMed.
- M. E. Foster, B. A. Zhang, D. Murtagh, Y. Liu, M. Y. Sfeir, B. M. Wong and J. D. Azoulay, Macromol. Rapid Commun., 2014, 35, 1516–1521 CrossRef CAS PubMed.
- J. D. Yuen, R. Kumar, D. Zakhidov, J. Seifter, B. Lim, A. J. Heeger and F. Wudl, Adv. Mater., 2011, 23, 3780–3785 CAS.
- J. Fan, J. D. Yuen, M. F. Wang, J. Seifter, J. H. Seo, A. R. Mohebbi, D. Zakhidov, A. Heeger and F. Wudl, Adv. Mater., 2012, 24, 2186–2190 CrossRef CAS PubMed.
- L. Bürgi, M. Turbiez, R. Pfeiffer, F. Bienewald, H.-J. Kirner and C. Winnewisser, Adv. Mater., 2008, 20, 2217–2224 CrossRef PubMed.
- Z. Y. Chen, M. J. Lee, R. S. Ashraf, Y. Gu, S. Albert-Seifried, M. M. Nielsen, B. Schroeder, T. D. Anthopoulos, M. Heeney, I. McCulloch and H. Sirringhaus, Adv. Mater., 2012, 24, 647–652 CrossRef CAS PubMed.
- X. Guo, S. R. Puniredd, B. He, T. Marszalek, M. Baumgarten, W. Pisula and K. Müllen, Chem. Mater., 2014, 26, 3595–3598 CrossRef CAS PubMed.
- P. Sonar, S. P. Singh, Y. Li, M. S. Soh and A. Dodabalapur, Adv. Mater., 2010, 22, 5409–5413 CrossRef CAS PubMed.
- S. Cho, J. Lee, M. Tong, J. H. Seo and C. Yang, Adv. Funct. Mater., 2011, 21, 1910–1916 CrossRef CAS PubMed.
- R. S. Ashraf, A. J. Kronemeijer, D. I. James, H. Sirringhaus and I. McCulloch, Chem. Commun., 2012, 48, 3939–3941 RSC.
- P. Sonar, T. R. B. Foong, S. P. Singh, Y. N. Li and A. Dodabalapur, Chem. Commun., 2012, 48, 8383–8385 RSC.
- A. J. Kronemeijer, E. Gili, M. Shahid, J. Rivnay, A. Salleo, M. Heeney and H. Sirringhaus, Adv. Mater., 2012, 24, 1558–1565 CrossRef CAS PubMed.
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS PubMed.
- J. D. Yuen, J. Fan, J. Seifter, B. Lim, R. Hufschmid, A. J. Heeger and F. Wudl, J. Am. Chem. Soc., 2011, 133, 20799–20807 CrossRef CAS PubMed.
- C. An, S. R. Puniredd, X. Guo, T. Stelzig, Y. Zhao, W. Pisula and M. Baumgarten, Macromolecules, 2014, 47, 979–986 CrossRef CAS.
- C. An, M. Li, T. Marszalek, D. Li, R. Berger, W. Pisula and M. Baumgarten, Chem. Mater., 2014, 26, 5923–5929 CrossRef CAS.
- J. Lee, A. R. Han, H. Yu, T. J. Shin, C. Yang and J. H. Oh, J. Am. Chem. Soc., 2013, 135, 9540–9547 CrossRef CAS PubMed.
- X. Guo, S. R. Puniredd, M. Baumgarten, W. Pisula and K. Müllen, Adv. Mater., 2013, 25, 5467–5472 CrossRef CAS PubMed.
- C. Kanimozhi, N. Yaacobi-Gross, K. W. Chou, A. Amassian, T. D. Anthopoulos and S. Patil, J. Am. Chem. Soc., 2012, 134, 16532–16535 CrossRef CAS PubMed.
- S. Chen, B. Sun, W. Hong, Z. Yan, H. Aziz, Y. Meng, J. Hollinger, D. S. Seferos and Y. Li, J. Mater. Chem. C, 2014, 2, 1683–1690 RSC.
- J. Yao, Z. Cai, Z. Liu, C. Yu, H. Luo, Y. Yang, S. Yang, G. Zhang and D. Zhang, Macromolecules, 2015, 48, 2039–2047 CrossRef CAS.
- M. Li, C. An, T. Marszalek, X. Guo, Y.-Z. Long, H. Yin, C. Gu, M. Baumgarten, W. Pisula and K. Müllen, Chem. Mater., 2015, 27, 2218–2223 CrossRef CAS.
- C. An, M. Li, T. Marszalek, X. Guo, W. Pisula and M. Baumgarten, J. Mater. Chem. C, 2015, 3, 3876–3881 RSC.
- T. L. Dexter Tam, T. Salim, H. Li, F. Zhou, S. G. Mhaisalkar, H. Su, Y. M. Lam and A. C. Grimsdale, J. Mater. Chem., 2012, 22, 18528–18534 RSC.
- M. L. Keshtov, D. V. Marochkin, V. S. Kochurov, A. R. Khokhlov, E. N. Koukaras and G. D. Sharma, Polym. Chem., 2013, 4, 4033–4044 RSC.
- T. T. Steckler, P. Henriksson, S. Mollinger, A. Lundin, A. Salleo and M. R. Andersson, J. Am. Chem. Soc., 2014, 136, 1190–1193 CrossRef CAS PubMed.
- W. Li, W. S. C. Roelofs, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2012, 134, 13787–13795 CrossRef CAS PubMed.
- S. K. Son, B. S. Kim, C.-Y. Lee, J. S. Lee, J. H. Cho, M. J. Ko, D.-K. Lee, H. Kim, D. H. Choi and K. Kim, Sol. Energy Mater. Sol. Cells, 2012, 104, 185–192 CrossRef CAS PubMed.
- Y. Lee and W. H. Jo, J. Phys. Chem. C, 2012, 116, 8379–8386 CAS.
- G. Zhang, J. Guo, J. Zhang, P. Li, J. Ma, X. Wang, H. Lu and L. Qiu, Polym. Chem., 2015, 6, 418–425 RSC.
- H.-J. Song, D.-H. Kim, E.-J. Lee, S.-W. Heo, J.-Y. Lee and D.-K. Moon, Macromolecules, 2012, 45, 7815–7822 CrossRef CAS.
- N. S. Sariciftci, Primary Photoexcitations in Conjugated Polymers: Molecular Excitons vs. Semiconductor Band Model, World Scientific, Singapore, 1997 Search PubMed.
- J. Rivnay, S. C. B. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488–5519 CrossRef CAS PubMed.
- J. Mei, D. H. Kim, A. L. Ayzner, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2011, 133, 20130–20133 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 03, Revision B.04, Gaussian, Inc., Pittsburgh PA, 2003 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- H. Li, F. Zhou, T. L. D. Tam, Y. M. Lam, S. G. Mhaisalkar, H. Su and A. C. Grimsdale, J. Mater. Chem. C, 2013, 1, 1745–1752 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental part, CV, TGA, DFT calculations, and NMR and HRMS spectra. See DOI: 10.1039/c5py00802f |
| ‡ These authors contributed equally. |
| § Present address: Institute of Materials Research and Engineering, A*STAR (Agency for Science, Technology and Research), 3 Research Link, Singapore 117602. |
| This journal is © The Royal Society of Chemistry 2015 |