
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermoresponsive, well-defined, poly(vinyl alcohol) co-polymers†
Thomas
Congdon
b,
Peter
Shaw
a and
Matthew I.
Gibson
 *b
*b
aSynthomer (UK) Ltd, Central Road, Templefields, Harlow, Essex, CM20 2BH, UK
bDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: m.i.gibson@warwick.ac.uk; Fax: +44(0) 247 652 4112
First published on 1st June 2015
Abstract
Thermoresponsive polymers have attracted huge interest as adaptable biomaterials based on their reversible solubility behaviour which can be exploited for controlled drug delivery or cellular uptake. The most famous and successful of these is poly(ethylene glycol) (PEG), but the thermal transition temperatures that are practically accessible are not physiologically useful. There are some notable examples of synthetic, responsive, polymers that are highly tunable over a physiologically relevant range, but there is still a need for these to be clinically validated in terms of toxicology and immunogenity for in vivo usage, in addition to their widely used in vitro applications. Poly(vinyl alcohol), PVA, is an appealing biocompatible polymer which is already used for a huge range of biomedical applications. Here, PVA is shown to be a highly tunable, thermoresponsive polymer scaffold. RAFT/MADIX polymerization is used to obtain a library of well-defined polymers between 8 and 50 kDa. Selective alkanoylation of the obtained PVA enabled the effect of side-chains, end-groups and molecular weight on the observable transition temperatures to be studied by turbidimetry. It was found that increasingly hydrophobic side chains (acetyl, propanoyl, butanoyl), or increasing their density led to corresponding decreases in cloud point. PVA with just 10 mol% butanoylation was shown to have a thermal transition temperature close to physiological temperatures (37 °C), compared to 70 mol% for acetylation, with temperatures in between accessible by controlling both the relative degree of functionalization, or by altering the chain length. Finally, a secondary response to esterase enzymes was demonstrated as a route to ‘turn off’ the responsive behaviour on demand. This study suggests that PVA-derived polymers may be a useful platform for responsive biomaterials.
Introduction
The development of synthetic polymer materials which can (dynamically) respond to an applied external stimulus has attracted huge interest.1 This is inspired by the adaptable properties of natural biomacromolecules, such as proteins, which can undergo significant structural changes in response to stimuli. Polymer materials have been developed to respond to a plethora of triggers, such as light,2 temperature,3 pH,4 magnetic fields5 or changes in ion concentration.6 These responses usually result in changes in the solubility of the material, but complex responses can be triggered using more topographically advanced materials causing changes in morphology,7 self-assembly8 or disassembly of micellar structures,9 or a change in surface characteristics.10 Of all these stimuli, temperature-responsive materials are by far the most studied, due to their synthetic accessibility. Two types of behaviour are observed, depending of the chemical and physical properties of the thermoresponsive material. Either the polymer can undergo a lower critical solution temperature (LCST) type transition, where above a specific temperature the system will demix, precipitating the polymer as a solid, or an upper critical solution temperature (UCST) type transition, where above a specific temperature the polymer and water phases will mix, and the polymer will dissolve into the solution.This reversible solution behaviour has been widely explored for biomedical applications using polymer therapeutics. For example, conjugation of a responsive polymer to a protein not only endows enzymatic resistance and improved pharmacokinetic profiles, but can also make them ‘smarter’ as the polymer phase transition can modulate the protein function providing an ‘on/off’ switch.11 A particularly attractive target for responsive polymers is cancerous tissue which due to its increased metabolic activity,12,13 is often several degrees warmer than the surrounding tissue providing a specific trigger for the smart nanomaterials. It has also been shown that the LCST transition can be used to not only make the polymer insoluble, but to trigger a hydrophilic to lipophilic transition. This is highly desirable as drug carriers must be hydrophilic to promote long circulation times and stability in the blood stream, but lipophilic to engage with and permeate though cellular membranes. Chilkoti et al. showed that LCST thermoresponsive polymers better penetrate tissues.14 Gibson et al. have studied the interactions between model lipid membranes and POEGMAS.15 Alexander et al. have demonstrated that gold nanoparticles with responsive polymers can enter cells above their LCST.16 In all these applications, a key consideration is the precise tuning of the observable transition temperature, which is typically achieved through control of the polymer molecular weight distribution, or though addition of comonomers. In particular Lutz demonstrated that different molecular weights of POEGMAS can be tuned by copolymerisation, due to the monomer incorporating a chain transfer agent.17 More recently it has been shown that the transition temperature can be tuned by the addition of a secondary external stimuli, enabling thermo-responsive polymers to be employed as scaffolds for ‘isothermal’ transitions.1,18 Pyridyl-disulfide terminated polymers have been employed by Phillips et al. to induce glutathione end-group exchange to trigger the phase transition.19 Other isothermal shifts have been triggered by the binding of bacteria to vancomycin-terminated branched polymers,20,21 or specific metal ions,22 or indeed by simple blending procedures using polymer particles with co-operative transitions.23,24
Despite the huge range of studies described above, responsive polymers have yet to be widely employed for in vivo applications, which is in part because, in many cases, their pharmacokinetic, toxicological and immunological properties are not well understood, precluding many of them from reaching the clinic. Nevertheless some examples exist, such as commercially available pNiPAm sheets used to culture cell sheets, which Canavan et al. concluded to have no inherent toxicity.25
A particularly interesting biocompatible polymer is poly(vinyl alcohol), PVA. PVA is produced on an industrial scale and is most commonly used for is adhesive properties, and as an additive and stabilizer in paint and other emulsions. PVA is also widely employed in the medical and pharmaceutical coatings industry because it has FDA approval for several in vivo applications. Furthermore, PVA is degradable in the environment (by microoganisms),26 and has even been shown to be a potent inhibitor of ice crystal growth,27,28 and can enhance blood cryopreservation.29 In addition to the above, there are a few reports of PVA/PVAc copolymers that show thermo-responsive behaviour. To the authors’ knowledge the first report on the thermoresponsive behaviour of PVA was published by Timasheff et al.30 Miya et al. reported that fractionated, butanoylated PVA displayed both LCST and UCST thermal transitions.31 Tagawa et al. reported that 88 kDa PVA with 20 mol% residual acetate groups displayed LCST type cloud point transitions that showed a strong concentration dependence.32 Later reports by Crowther et al. examined the effect that the molar mass of PVA,33 and the size and sequence of acetate and hydroxyl functionality has on the thermoresponsive behaviour,34 surmising that random regions of hydroxyl/acetate groups would contribute less to the thermodynamic drive towards demixing compared to the blocky regions of either group. Both of these reports used high molecular weight PVA with degrees of acetylation ranging from 6–20 mol%. Ivanova et al. prepared and tested a range of acetal functionalised PVAs that displayed thermoresponsive behaviour.35 More recently Chen et al. reported that the incorporation of small amounts of glycine, alanine and valine along a PVA chain afforded thermoresponsive behaviour, dependent on the amino acid installed.36 More commonly PVA is used or incorporated into materials, usually hydrogels, alongside more well-known thermoresponsive polymers.37 Xiao et al. utilised glutaraldehyde cross-linked PVA as a thermoresponsive hydrogel.38
However, despite its biocompatible nature, PVA has not been widely explored for application as a stimuli-responsive biomaterial and there are no established studies detailing structure–property relationships on its transition temperatures. In part this is due to the previous difficulties in synthesising it in a controlled manner, which has been overcome by advances in controlled radical polymerisation. Therefore, this manuscript describes a detailed study into the synthesis and characterisation of a library of PVAs and alkanoylated PVAs. This library is used to study their thermoresponsive behaviour, and in particular the ability to fine-tune the transition temperature such that it is applicable in a physiological range. Finally, the ability to modulate the transition temperature by application of enzymes is demonstrated as a potential secondary-stimuli response.
Experimental
Materials
Phosphate-buffered saline solutions were prepared using preformulated tablets (Sigma-Aldrich) in 200 mL of MilliQ water (>18 Ω mean resistivity) to give a buffered pH of 7.4. Acetic acid (glacial), 4,4′-azobis(4-cyanovaleric acid), butanoic acid, hydrazine hydrate solution (78–82%), lypholysed porcine liver esterase, propionic acid and vinyl acetate were all purchased from Sigma Aldrich. O-Ethyl-S-1-phenyl carbonodithioate (CTA 1) and ethyl 2-(ethoxycarbonothioylthio)propanoate (CTA 2) were both prepared using previously reported methods (see ESI†).39Analytical methods
1H and 13C NMR spectra were recorded on Bruker DPX-300 and DPX-400 spectrometers using deuterated solvents purchased from Sigma-Aldrich. Chemical shifts are reported relative to residual non-deuterated solvent. Infrared data was recorded on a Bruker Vector 22 GI003097. Thermal transitions were measured using an Agilent Technologies Cary60 UV/Vis spectrometer equipped with a Quantum Northwest TC1 temperature controller, with a heating rate of 2 °C min−1 and absorbance at 650 nm used to measure turbidity and determine cloud points. Recorded data was normalised in each case, except in Fig. 4, where the data was normalised to the maximum absorbance of P80Ac29. Size exclusion chromatography (SEC) was used to examine and differentiate between the molecular weights and dispersity of the synthesized carbohydrates. The THF GPC system comprised of a Varian 390-LC-Multi detector suite fitted with differential refractive index (DRI), light scattering (LS) and ultra-violet (UV) detectors equipped with a guard column (Varian Polymer Laboratories PLGel 5 μm, 50 × 7.5 mm) and two mixed D columns of the same type. The mobile phase was THF with 5% triethylamine (TEA) eluent at a flow of 1.0 mL min−1, and samples were calibrated against Varian Polymer Laboratories Easi0Vials linear poly(styrene) and poly(methylmethacrylate) standards (162–2.4 × 105 g mol−1) using Cirrus v3.3.Polymerisation of vinyl Acetate using CTA 1
As a representative example CTA 1 (0.21 g, 0.99 mmoles), vinyl acetate (4.67 g, 2.64 mmol) and ACVA (4,4′-azobis(4-cyanovaleric acid)) (0.013 g, 0.046 mmoles) were added to a stoppered vial. The solution was thoroughly degassed under a flow of N2 for 20 min and the reaction mixture was then allowed to polymerise at 68 °C for typically 15 h. The yellow solutions were then cooled to room temperature. Poly(vinyl acetate) was then recovered as a yellow sticky solid after precipitation into hexane. The hexane was decanted and the poly(vinyl acetate) was re-dissolved in THF, which was then concentrated in vacuo and thoroughly dried under vacuum at 40 °C for 24 h, forming a white crystalline solid. Representative characterisation data for PVAc80: 1H NMR (400 MHz, CDCl3): δ = 4.61 (–CHO–CH2 br 1H), δ = 1.74 (–CO–CH3 br 3H), δ = 1.53 (–CH2– br 2H), MSECn (THF) = 6900 Da, Mw/Mn = 1.17.Hydrolysis of poly(vinyl acetate) to poly(vinyl alcohol)
As a representative example, poly(vinyl acetate) (1.5 g, Mn 6900 Da, Đ = 1.17) was dissolved in a methanol (20 mL) and hydrazine hydrate solution (15 mL, 80% in water) in a round bottom flask. The reaction mixture was stirred at 30 °C for 2 h. The reaction mixture was then dialysed using distilled water and poly(vinyl alcohol) was recovered as a spongy white solid by freeze drying the dialysis solution. Degree of hydrolysis was determined by 1H NMR. Representative characterisation data for PVA80: 1H NMR (400 MHz, D2O): δ = 4.00 (–CHOH– br 1H), δ = 1.68–1.60 (–CH2– br 2H).Acetylation of poly(vinyl alcohol)
As a representative example, poly(vinyl alcohol) (0.5 g, Mn 29 kDa, Đ = 1.28) was dissolved in water (2.4 mL), acetic acid (7.6 mL) and HCl (0.1 mL, 3 M solution in water) in a vial equipped with a stir bar. The reaction mixture was stirred at 40 °C for 4 days. The reaction mixture was then dialysed and partially acetylated poly(vinyl alcohol) was recovered by freeze drying the dialysis solution. Conversion was determined by 1H NMR integration of the acetate methyl protons (δ = 2.08) and the –CH2– backbone protons (δ = 1.93–1.50) and IR by examining the –OH stretch at 3340 cm−1 and the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch at 1738 cm−1. Representative characterisation data for P350Ac0.7: 1H NMR (400 MHz, D2O): δ = 4.00 (–CHOH– br 1H), δ = 3.82 (–CHO–CH2 br 1H), δ = 2.08 (–CO–CH3 br 3H), δ = 1.93–1.50 (–CH2– br 2H).
O stretch at 1738 cm−1. Representative characterisation data for P350Ac0.7: 1H NMR (400 MHz, D2O): δ = 4.00 (–CHOH– br 1H), δ = 3.82 (–CHO–CH2 br 1H), δ = 2.08 (–CO–CH3 br 3H), δ = 1.93–1.50 (–CH2– br 2H).
Post polymerisation modification of PVA to PVA·PVButanoate stat copolymers
As a representative example, poly(vinyl alcohol) (0.1 g, Mn 29 kDa, Đ = 1.28) was dissolved in water (3.0 mL), butanoic acid (2.0 mL) and HCl (0.1 mL, 3 M solution in water) in a vial equipped with a stir bar. The reaction mixture was stirred at 40 °C for 4 days. The reaction mixture was then dialysed and partially butyanoated poly(vinyl alcohol) was recovered by freeze drying the dialysis solution. Conversion was determined by 1H NMR integration of the butanoyl –CH3 protons (δ = 0.93, t) and the –CH– backbone protons (δ = 4.1–3.4), and IR by examining the –OH stretch at 3340 cm−1 and the –CO stretch at 1738 cm−1. Representative characterisation data for P350Bu0.03: 1H NMR (400 MHz, D2O): δ = 4.00 (–CHO– br m 1H), δ = 2.31 (–CH2–CH3 t 2H), δ = 1.93–1.50 (–CH2– and CH2–CH2–CH3 br m 4H,), δ = 0.93 (–CH2–CH3 t 3H).
Post polymerisation modification of PVA to PVA·PVPropanoate stat polymers
As a representative example, poly(vinyl alcohol) (0.1 g, Mn 11.1 kDa, Đ = 1.21) was dissolved in water (1.5 mL), propanoic acid (1.5 mL) and HCl (0.1 mL, 3 M solution in water) in a vial equipped with a stir bar. The reaction mixture was stirred at 40 °C for 4 days. The reaction mixture was then dialysed and partially propanoated poly(vinyl alcohol) was recovered by freeze drying the dialysis solution. Conversion was determined by 1H NMR integration of the propanoyl –CH2 protons (δ = 2.27, t) and the –CH– backbone proton (δ = 4.1–3.4), and IR by examining the –OH stretch at 3340 cm−1 and the –CO stretch at 1738 cm−1. Representative characterisation data for P250Pr0.10: 1H NMR (400 MHz, D2O): δ = 4.00 (–CHO– br m 1H), δ = 2.27 (–CH2–CH3 t 2H), δ = 1.93–1.50 (–CH2– br 2H).
Enzymatic degradation of copolymers
P350Ac0.7 (30 mg, Mn 29 kDa, Đ 1.39) was dissolved in PBS solution (5 mL) in a stoppered vial equipped with a stir bar, to make up a final solution of 10 mg mL−1 of polymer. Lypholysed porcine liver esterase (5 mg) was added and the mixture was stirred vigorously for 7 days at 37 °C. Aliquots were taken and the turbidimetry curves were recorded using a Cary60 UV/Vis spectrometer.Results
In order to probe the cloud point behaviour of PVA/PVAc a range of copolymers with different molecular weights and degrees of acetylation were required. Previous studies on the responsive behaviour of PVAs have been limited to high molecular weight polymers with large dispersties, derived from free radical polymerization.38 Due to the deactivated nature of vinyl acetate, xanthates (rather than the more commonly used di/tri thio esters) were required to enable RAFT/MADIX polymerization. The MADIX agent O-ethyl-S-1-phenylmethyl carbonodithioate (CTA 1) was chosen as it has been reported to control the radical polymerisation of vinyl acetate.40 Here, vinyl acetate was polymerised in bulk, and then subsequently hydrolysed using hydrazine hydrate, followed by dialysis to afford well defined PVA with no residual acetate groups, Scheme 1.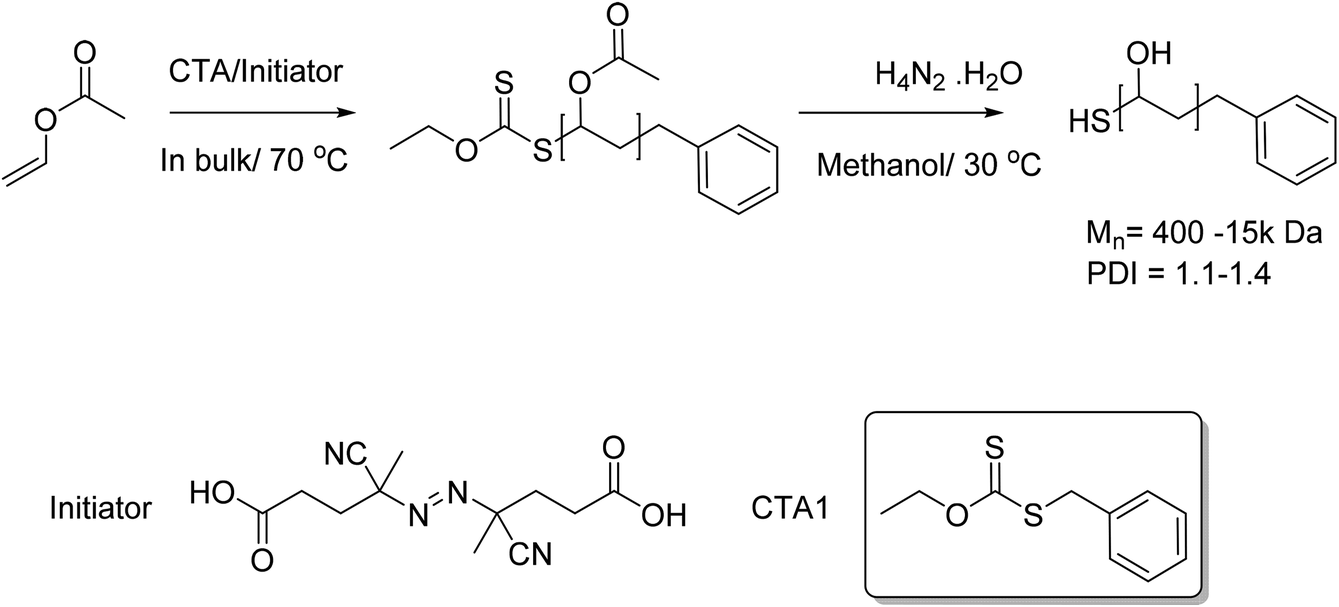 | ||
| Scheme 1 MADIX controlled polymerisation of vinyl acetate and its subsequent conversion to PVA using hydrazine hydrate solution. | ||
The polymers obtained by bulk RAFT/MADIX polymerization of vinyl acetate are shown in Table 1. SEC analysis showed that the polymers prepared were of low dispersity and predictable molecular weights (Fig. 1), and PVAcs with higher molecular weight ranges displayed correspondingly higher dispersity; as expected for a MADIX polymerization of deactivated monomers.
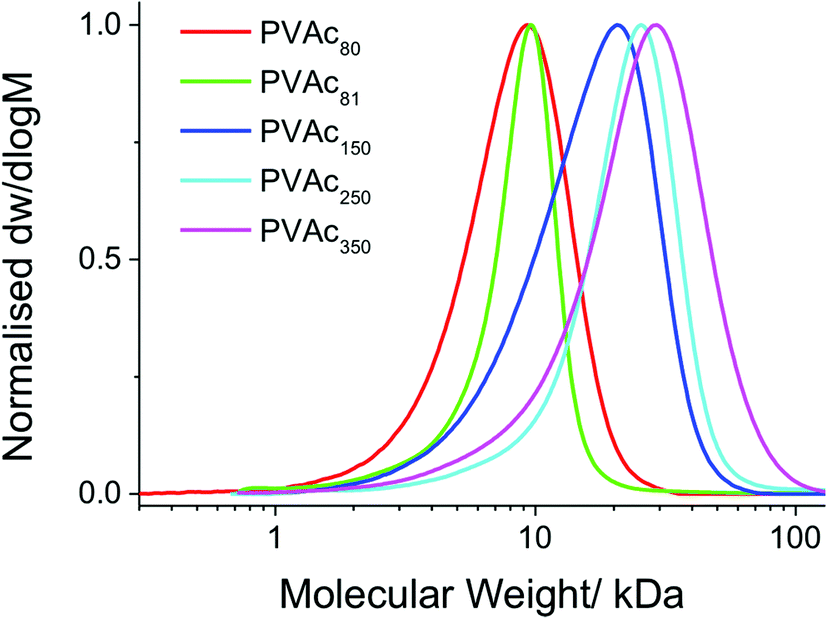 | ||
| Fig. 1 Size exclusion chromatography traces of poly(vinyl acetate) polymers. | ||
| Entry | [M]/[CTA] | Conv.a | M n (theo)b (g mol−1) | M n (SEC)c (g mol−1) | Đ (−) | DPNd | PVAe |
|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR of the reaction mixture. b Theoretical Mn assuming 100% conversion. c Determined by SEC in THF using PMMA polymer standards. d Number average degree of polymerisation, determined from conversion of monomer to polymer by 1H NMR. e Corresponding PVA prepared by complete hydrolysis of the PVAc, determined by 1H NMR. f Prepared using MADIX agent CTA 2 to afford a carboxylic acid functionalised end group, required for determining end group effects. All other entries prepared using CTA1. * Denotes carboxylic acid functional end group on the PVA polymer afforded by using CTA 2. | |||||||
| PVAc80 | 100 | 82.9 | 8600 | 7100 | 1.17 | 83 | PVA80 |
| PVAc81f | 100 | 81.4 | 8600 | 7000 | 1.18 | 81 | PVA80* |
| PVAc150 | 200 | 73.8 | 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 200 |
12700 |
1.45 | 148 | PVA150 |
| PVAc250 | 300 | 80.1 | 25800 |
20700 |
1.21 | 240 | PVA250 |
| PVAc350 | 500 | 68.8 | 43000 |
29600 |
1.39 | 344 | PVA350 |
Typically PVA/PVAc copolymers would be obtained by partial basic (NaOH) deprotection of PVAc, but this has been shown to give rise to ‘blocky’ polymers rather than true random distribution of the acetate groups.41 Therefore, hydrazine hydrate was used as a strong nucleophile to completely remove the acetates, without affecting the chain length. Acetic acid/HCl solutions were then used to re-introduce acetate groups in a statistical and controlled fashion (Fig. S3†), due to the inherent reversibility of the reaction, as we have previously demonstrated (Scheme 2).28 The degree of acetylation was quantified by 1H NMR and acetylation was corroborated by IR spectroscopy (Fig. S2†). This process did not affect the chain length of the polymer (Fig. S6†). Using PVA350 a library of eight sequentially acetylated polymers were obtained with degrees of acetylation ranging from 0–70 mol% (Table 2).
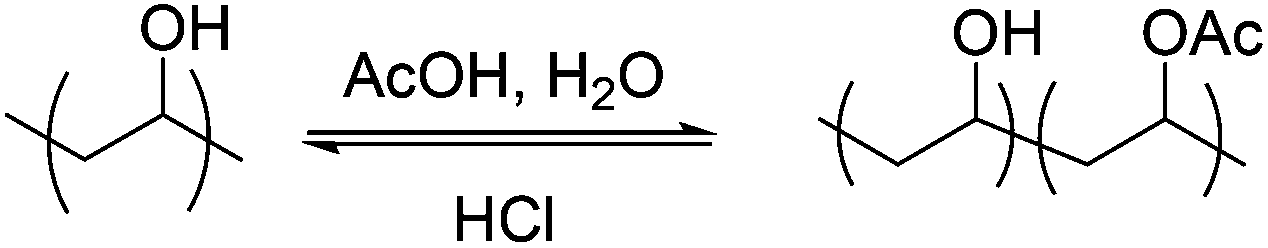 | ||
| Scheme 2 Partial acetylation of PVA using a 75:25 v:v mixture of acetic acid and water with catalytic HCl. | ||
| Entrya | [H2O]:[acetic acid]:[HCl]b |
Acetylationc/mol% | DP |
|---|---|---|---|
|
a For simplicity and ease of cross-referencing, polymers are denoted firstly their size, then by the degree of alkanoylation as a fraction.
b Ratios given as v:v:v; 1 mL to 0.1 g PVA·3 M HCl solution.
c Determined by 1H NMR, by comparing the integrals of the PVA α-H (δ = 4.00 ppm) and PVAc –CH3 (δ = 1.74 ppm) shifts of the purified copolymer. * Denotes carboxylic acid functional end group on the PVA/PVAc polymer afforded by using CTA 2.
|
|||
| P350Ac0.7 | 17:83:10 |
70 | 350 |
| P350Ac0.44 | 32:68:10 |
44 | 350 |
| P350Ac0.27 | 50:50:10 |
27 | 350 |
| P350Ac0.22 | 45:55:10 |
22 | 350 |
| P80Ac0.29 | 25:75:1 |
29 | 80 |
| P150Ac0.27 | 25:75:1 |
27 | 150 |
| P250Ac0.25 | 25:75:1 |
25 | 250 |
| P80Ac0.33* | 19:67:12 |
33 | 80 |
With this library of polymers to hand, the thermoresponsivity of the polymers was evaluated by turbidimetry, using a UV/Vis spectrometer equipped with a temperature control unit. All experiments were conducted at 10 mg mL−1 (to remove concentration associated effects).42 The cloud point was defined as being the point where absorbance (or strictly, scattering) is 50% of the maximum measured intensity at 650 nm. It should be noted that this method gives the cloud point only. The LCST, (Lower Critical Solution Temperature), is the lowest temperature at which a binary system will demix into two separate phases, and is governed by the ratio of the two components in that system. Fig. 2 shows the turbidimetry curves for PVAs with the same degree of polymerization, but with varying degrees of acetylation.
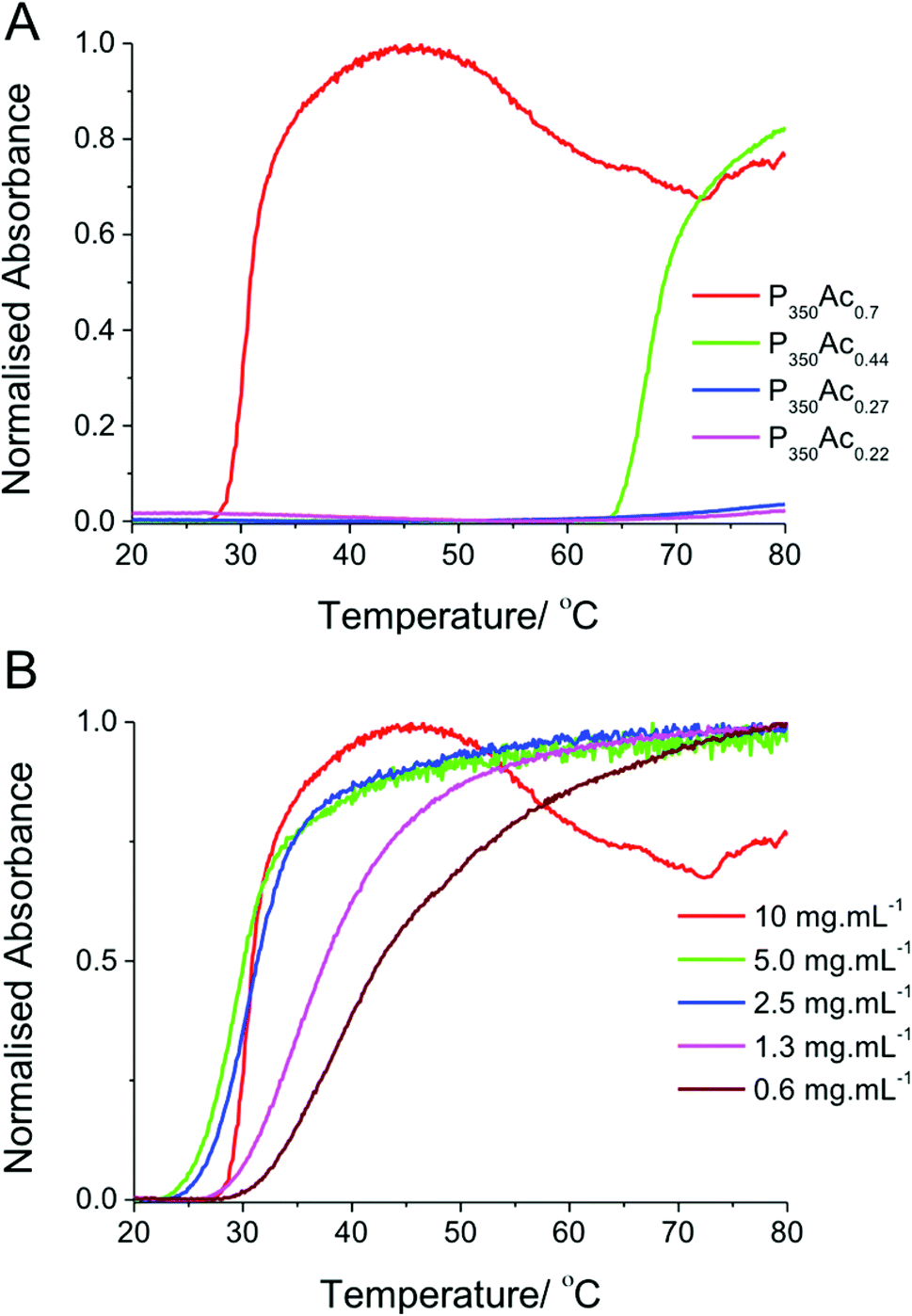 | ||
| Fig. 2 (A) Turbidimetry curves of P350, with different degrees of acetylation. Measurements were carried out using 10 mg ml−1 solutions of each polymer. (B) The concentration dependent behaviour of P350Ac0.7. | ||
Fig. 2 shows that at degrees of acetylation below 40% there is no thermally induced demixing and no cloud point is observed at this temperature range in distilled water. The slight increase in absorbance at 75 °C seen in P350Ac0.27 and P350Ac0.22 is likely due to the solution coming close to boiling point and the formation of bubbles in the solution. P350Ac0.44 shows a clear transition between 63–80 °C, with a cloud point of 69 °C. P350Ac0.7 has a much higher degree of acetylation, and hydrophobicity, hence a comparably lower cloud point transition of 32 °C, which is close to physiologically relevant temperatures. These results compare well to other reported thermoresponsive polymers such as PNIPAM (and others) which have been hypothesised to be useful for biomedical applications. It should again be highlighted that a key advantage of PVA/PVAc is its wide use in many FDA approved biomedical applications which may aid clinical translation of such materials.
P350Ac0.7 with the thermal transition closest to physiological temperatures was further investigated as a function of concentration. As expected, the cloud point transition temperature increased with a corresponding decrease in concentration. However this increase only became apparent below 2.5 mg mL−1. Thermoresponsive activity at these dilute concentrations is vital for any practical in vivo application which these polymers could be applied to.43
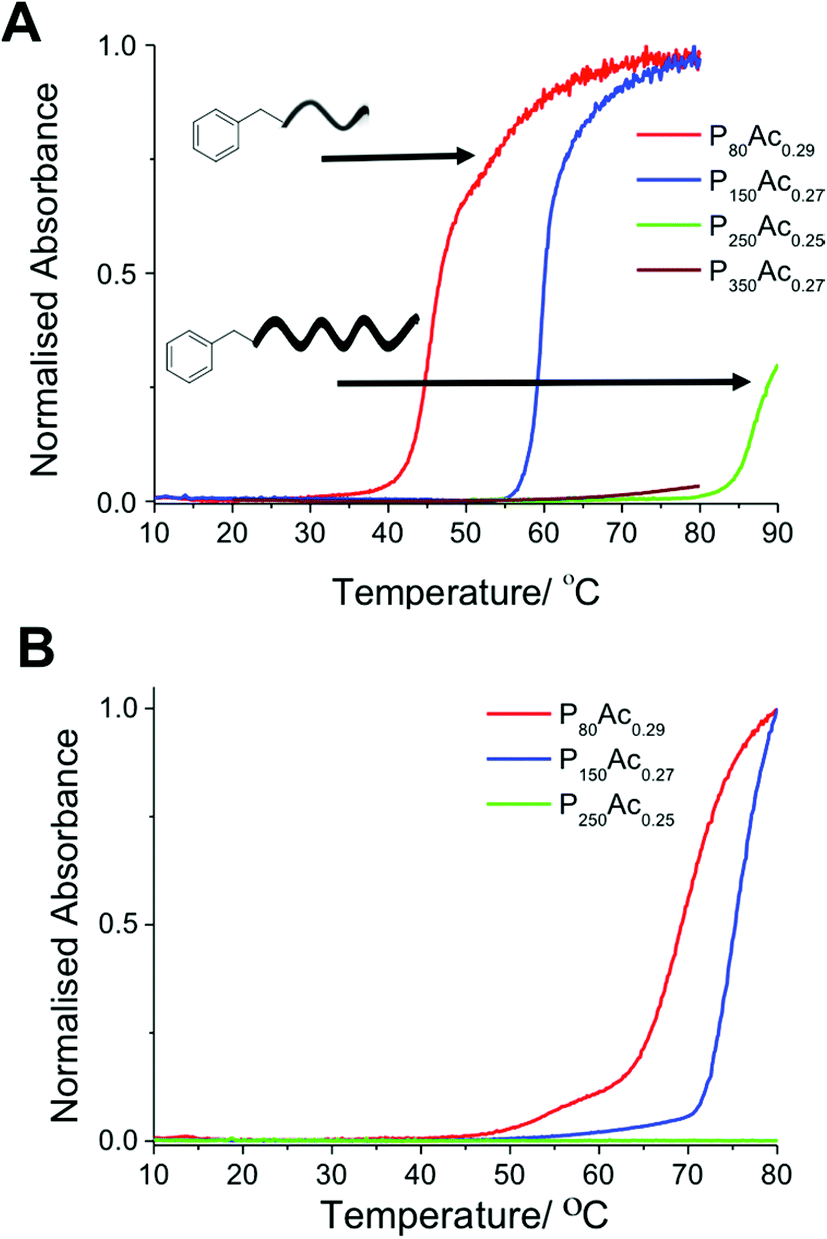
Flory-Huggins solution theory postulates that longer polymers will have lower transition temperatures caused by the increased weight fraction driving the energetics of the system towards demixing. A series of polymers with degrees of polymerisation ranging from 80 to 350 with a fixed degree of acetylation of ∼30% were evaluated. These results (Fig. 3) show that at this low degree of acetylation, and high concentration of 30 mg mL−1 the cloud point actually increased with chain length, the opposite of what Flory-Huggins theory predicts. P80Ac0.29, with a DP of 80, has a cloud point of 47 °C whereas P150Ac0.27, with a DP of 150, has a cloud point of 59 °C. This trend holds at lower concentrations, where the cloud point for each polymer is correspondingly higher but the difference in temperature is roughly the same. Our initial hypothesis was that this was due to the formation of ‘pseudo’ micelles; a rearrangement of hydrophilic and hydrophobic regions in the polymer, a property which makes PVA(Ac) copolymers so useful as stabilizers and emulsifiers in water based paints. DLS and dye inclusion assays, however, ruled this out; in terms of size or absorbance of dye into a hydrophobic region, there was no difference in any of the polymers used in this study. Therefore it was hypothesised that this phenomenon could be attributed to the larger effect the end groups would have on shorter polymers compared to larger polymers. We have previously shown the crucial importance of end-groups on the transitions of pNIPAMs. To test this a different MADIX agent, ethyl 2-(ethoxycarbonothioylthio)propanoate (CTA 2) was used to incorporate a carboxylic acid end group in to a PVA(Ac) copolymer with the same degree of polymerisation and degree of acetylation. This polymer (P80Ac0.33*) showed no detectable cloud point, with turbidity slightly decreasing with temperature (Fig. 4), compared to the benzyl-terminated polymers which showed cloud points in this range. This serves to highlight that when using CRP-derived polymers, the whole polymer structure, not just side-chain composition should be considered when assessing responsivity.
 | ||
| Fig. 3 (A) Turbidimetry curves showing the molecular weight dependent cloud point behaviour of PVA(Ac) statistical copolymers with 30 mol% acetylation, at 30 mg mL−1. (B) Cloud point behaviour of PVA(Ac) statistical copolymers with 30 mol% acetylation, at 10 mg mL−1. | ||
 | ||
| Fig. 4 Turbidimetry curves showing the cloud point behaviour of P80Ac29 and P80Ac0.33* with hydrophobic and hydrophilic end groups at 10 mg mL−1. | ||
The above data showed that chain length and degree of acetylation enabled control over transition temperature, and highlighted the importance of end group selection. To provide a wider ‘toolbox’ of transitions and to potentially reduce the degree of substitution which is required, the effect of incorporating increasingly hydrophobic side chains was studied. Using a similar method as for acetate modification PVA was modified using water/propanoic acid or butanoic acid mixtures, Table 3. The solubility of these polymers was (predictably) much lower than corresponding PVA/PVAc copolymers, meaning lower degree of functionalization were employed. Pleasingly, these copolymers showed sharp cloud point transitions between 25–35 °C for the propanoyl functionalised PVA and 40–50 °C for the butanoyl functionalised PVA (Fig. 5). Interestingly P350Bu0.10, with 10 mol% butanoyl functional groups displayed a cloud point of 50 °C, whereas P250Pr0.10 displayed no observable cloud point at the same concentration, indicating that lower degrees of substitution are needed for the butanoyl functionalised PVAs. This simple procedure offers a practical route to fine-tuning PVA transition temperature and does not require copolymerisation (unlike for example NIPAM, PEGMA, 2-oxazolines)44 which often gives rise to non-statistical monomer distributions.
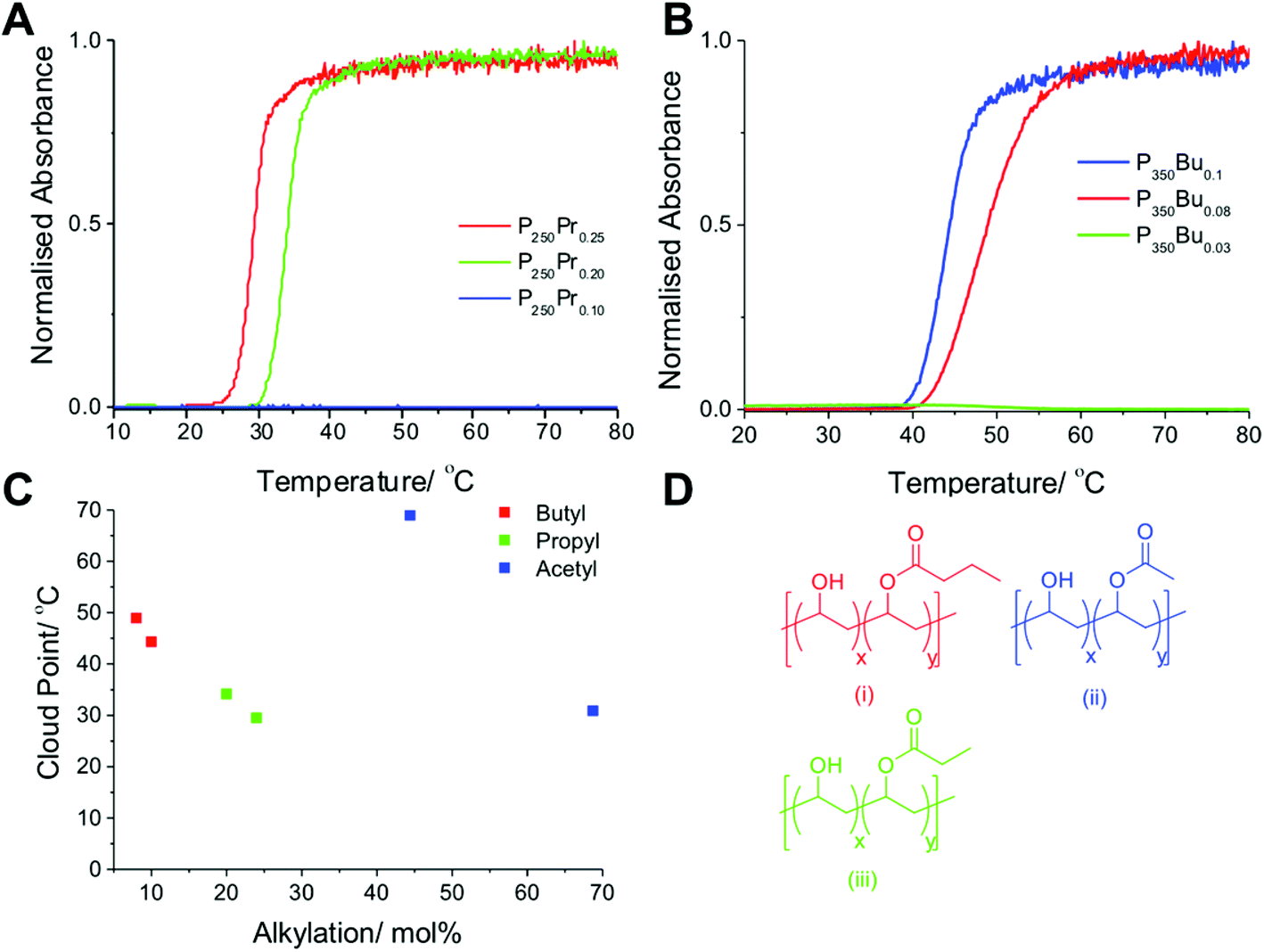 | ||
| Fig. 5 (A) Turbidimetry curves showing the cloud point behaviour of PVA·propanoyl statistical copolymers at 10 mg mL−1. (B) Turbidimetry curves showing the cloud point behaviour of PVA·Bu copolymers at 10 mg mL−1 (C) comparison of cloud points (the temperature at 50% of normalised absorbance) of differently alkanoylated PVA copolymers. (D) Structures of statistical copolymers used in D (i) PVA·Butanoyl (ii) PVA·PVAc (iii) PVA·Propanoyl. | ||
| Entry | [H2O]:[alkanoic acid]:[HCl]a |
Alkanoylation (NMR)b/mol% | DP | Cloud point/°C |
|---|---|---|---|---|
|
a Ratios given as v:v:v; 1 mL to 0.1 g PVA·3 M HCl solution. Alkanoic acid is either propanoic or butanoic acid, depending on entry.
b Determined by 1H NMR, by comparing the integrals of the PVA α-H (δ = 4.00 ppm) and –CH3 shifts of the purified copolymers.
|
||||
| P250Pr0.25 | 15:85:1 |
25 | 250 | 29.51 |
| P250Pr0.20 | 40:60:1 |
20 | 250 | 34.18 |
| P250Pr0.10 | 50:50:3 |
10 | 250 | None |
| P350Bu0.10 | 60:40:2 |
10 | 350 | 48.9 |
| P350Bu0.08 | 70:30:2 |
8 | 350 | 44.3 |
| P350Bu0.03 | 80:20:2 |
3 | 350 | None |
As a final demonstration of the utility of this PVA system as responsive polymers, its susceptibility to enzymatic degradation was studied, to make the polymers ‘doubly’ responsive, and enable the LCST behaviour to be ‘switched off’ under specific biochemical conditions (such as intracellularly). P350Ac0.7 was prepared to a dilution of 10 mg mL−1 in PBS solution to which was added porcine liver esterase and the solutions were stirred vigorously at 37 °C for 6 days, with aliquots taken and analysed every 24 hours (Fig. 6). A linear increase in the observable cloud point was seen, as acetate groups were removed by the esterase and the solubility of the polymer increased at higher temperatures, without any change in the total polymer concentration. Without the enzyme, no significant hydrolysis was observed over the same time period. In summary, we have shown that PVA/PVAc represents a versatile scaffold for assembling stimuli-responsive polymers. We anticipate that these will find application as biocompatible, but importantly, regulatory acceptable biomaterials for applications such as drug delivery and biosensing.
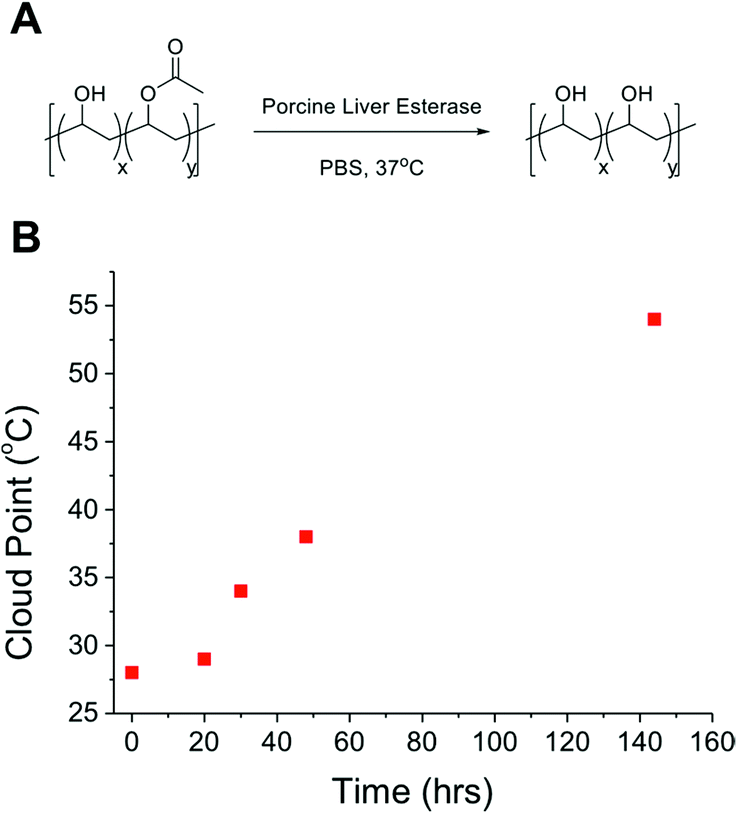 | ||
| Fig. 6 (A) Hydrolysis of acetate functionality of P350Ac0.7 using porcine liver esterase in PBS solution. (B) The cloud point behaviour of P350Ac0.7 after addition of porcine liver esterase, showing increase in cloud point over time as acetate groups are enzymatically hydrolysed. | ||
Conclusions
Here the application of alkoxylated poly(vinyl alcohol) as a highly tunable, and biocompatible, thermoresponsive polymer scaffold is studied in detail. Using RAFT/MADIX polymerization it was possible to obtain polymers with chain lengths between 80 and 350 units and low dispersity. Statistical copolymers were obtained by first removing the acetates from PVAc and then selective incorporation of either acetyl, propanoyl or butanoyl esters. This diverse library was evaluated to show that longer PVAs have an inverse relationship between cloud point (LCST) and chain length. Several formulations were shown to have transitions temperatures in the physiological range, with the more hydrophobic side groups (butanoyl) only requiring 10 mol% functionalization compared to 60 mol% for acetate. The importance of the end groups was also studied, with hydrophobic end-groups shown to dominate the phase transition such that shorter polymers appeared to give lower LCST than longer, which must be considered when investigating such properties. Finally, the ability of an esterase to selectively remove the acetate groups, to enable the LCST to be ‘switched’ off was demonstrated. These results show that PVA is a valid alternative to traditionally used PNIPAMs, POEGMAs or poly(oxazolines) owing to their highly tunable nature and established in vivo biocompatibility.Acknowledgements
Equipment used was supported by the Innovative Uses for Advanced Materials in the Modern World (AM2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). This work was supported by a Research Project Grant from the Leverhulme Trust (RPG-144) and a Research Grant from the Royal Society. Dr Peter Shaw (Synthomer UK) is thanked for helpful discussions.References
- D. J. Phillips and M. I. Gibson, Polym. Chem., 2015, 6, 1033 RSC.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468 RSC.
- R. Liu, M. Fraylich and B. Saunders, Colloid Polym. Sci., 2009, 287, 627 CAS.
- E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173 CrossRef CAS PubMed.
- C. d. l. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276 RSC.
- G. Pasparakis and M. Vamvakaki, Polym. Chem., 2011, 2, 1234 RSC.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042 RSC.
- R. Tong, L. Tang, L. Ma, C. Tu, R. Baumgartner and J. Cheng, Chem. Soc. Rev., 2014, 43, 6982 RSC.
- J. F. Mano, Adv. Eng. Mater., 2008, 10, 515 CrossRef CAS PubMed.
- A. Castellanos, S. J. DuPont, A. J. Heim, G. Matthews, P. G. Stroot, W. Moreno and R. G. Toomey, Langmuir, 2007, 23, 6391 CrossRef CAS PubMed.
- T. Shimoboji, E. Larenas, T. Fowler, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2003, 14, 517 CrossRef CAS PubMed.
- N. Arora, D. Martins, D. Ruggerio, E. Tousimis, A. J. Swistel, M. P. Osborne and R. M. Simmons, Am. J. Surg., 2008, 196, 523 CrossRef PubMed.
- M. Gautherie, Ann. N. Y. Acad. Sci., 1980, 335, 383 CrossRef CAS PubMed.
- D. E. Meyer, B. C. Shin, G. A. Kong, M. W. Dewhirst and A. Chilkoti, J. Controlled Release, 2001, 74, 213 CrossRef CAS.
- Y. Saaka, R. C. Deller, A. Rodger and M. I. Gibson, Macromol. Rapid Commun., 2012, 33, 779 CrossRef CAS PubMed.
- S. Salmaso, P. Caliceti, V. Amendola, M. Meneghetti, J. P. Magnusson, G. Pasparakis and C. Alexander, J. Mater. Chem., 2009, 19, 1608 RSC.
- J.-F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046 CrossRef CAS PubMed.
- M. J. Summers, D. J. Phillips and M. I. Gibson, Chem. Commun., 2013, 49, 4223 RSC.
- D. J. Phillips and M. I. Gibson, Chem. Commun., 2012, 48, 1054 RSC.
- J. Shepherd, P. Sarker, K. Swindells, I. Douglas, S. MacNeil, L. Swanson and S. Rimmer, J. Am. Chem. Soc., 2010, 132, 1736 CrossRef CAS PubMed.
- P. Sarker, J. Shepherd, K. Swindells, I. Douglas, S. MacNeil, L. Swanson and S. Rimmer, Biomacromolecules, 2010, 12, 1 CrossRef PubMed.
- Q. Yan, J. Yuan, Y. Kang, Z. Cai, L. Zhou and Y. Yin, Chem. Commun., 2010, 46, 2781 RSC.
- D. J. Phillips, J. P. Patterson, R. K. O'Reilly and M. I. Gibson, Polym. Chem., 2014, 5, 126 RSC.
- M. Liu, P. Tirino, M. Radivojevic, D. J. Phillips, M. I. Gibson, J.-C. Leroux and M. A. Gauthier, Adv. Funct. Mater., 2013, 23, 2007 CrossRef CAS PubMed.
- M. A. Cooperstein and H. E. Canavan, Langmuir, 2009, 26, 7695 CrossRef PubMed.
- E. Chiellini, A. Corti, S. D'Antone and R. Solaro, Prog. Polym. Sci., 2003, 28, 963 CrossRef CAS.
- C. Budke and T. Koop, ChemPhysChem, 2006, 7 Search PubMed.
- T. Congdon, R. Notman and M. I. Gibson, Biomacromolecules, 2013, 14, 1578 CrossRef CAS PubMed.
- R. C. Deller, M. Vatish, D. A. Mitchell and M. I. Gibson, Nat Commun, 2014, 5, 3244 Search PubMed.
- F. F. Nord, M. Bier and S. N. Timasheff, J. Am. Chem. Soc., 1951, 73, 289 CrossRef CAS.
- T. Shiomi, K. Imai, C. Watanabe and M. Miya, J. Polym. Sci., Polym. Phys. Ed., 1984, 22, 1305 CrossRef CAS PubMed.
- K. Furusawa and T. Tagawa, Colloid Polym. Sci., 1985, 263, 353 CAS.
- D. Eagland and N. J. Crowther, Eur. Polym. J., 1991, 27, 299 CrossRef CAS.
- N. J. Crowther, D. Eagland, F. F. Vercauteren and W. A. B. Donners, Eur. Polym. J., 1993, 29, 1553 CrossRef CAS.
- D. Christova, S. Ivanova and G. Ivanova, Polym. Bull., 2003, 50, 367 CrossRef CAS PubMed.
- R.-C. Wang, H.-J. Liu, J.-G. Tong and Y. Chen, Polym. Chem., 2014, 5, 2417 RSC.
- L. Wei, C. Cai, J. Lin and T. Chen, Biomaterials, 2009, 30, 2606 CrossRef CAS PubMed.
- Q. An, C. Beh and H. Xiao, J. Appl. Polym. Sci., 2014, 131, 39720 Search PubMed.
- J. Skey and R. K. O'Reilly, Chem. Commun., 2008, 4183 RSC.
- M. H. Stenzel, L. Cummins, G. E. Roberts, T. P. Davis, P. Vana and C. Barner-Kowollik, Macromol. Chem. Phys., 2003, 204, 1160 CrossRef CAS PubMed.
- I. Piirma, Polymeric surfactants, M. Dekker, New York, N.Y., 1992 Search PubMed.
- N. S. Ieong, M. Hasan, D. J. Phillips, Y. Saaka, R. K. O'Reilly and M. I. Gibson, Polym. Chem., 2012, 3, 794 RSC.
- K. Bebis, M. W. Jones, D. M. Haddleton and M. I. Gibson, Polym. Chem., 2011, 2, 975 RSC.
- C. Weber, R. Hoogenboom and U. S. Schubert, Prog. Polym. Sci., 2012, 37, 686 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py00775e |
| This journal is © The Royal Society of Chemistry 2015 |