
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hypercrosslinked materials: preparation, characterisation and applications
N.
Fontanals
*a,
R. M.
Marcé
a,
F.
Borrull
a and
P. A. G.
Cormack
b
aDepartament de Química Analítica i Química Orgànica, Universitat Rovira i Virgili, Campus Sescelades Marcel·lí Domingo, s/n, 43007 Tarragona, Spain. E-mail: nuria.fontanals@urv.cat; Fax: (+34) 977 55 84 46; Tel: (+34) 977 55 86 29
bWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Thomas Graham Building, 295 Cathedral Street, Glasgow, G1 1XL, Scotland, UK
First published on 23rd July 2015
Abstract
This review article provides an overview of hypercrosslinking technology. In particular, it covers the preparation and characterisation of hypercrosslinked materials and their applications. The synthesis section examines the different monomers, precursor polymers and reagents used to prepare hypercrosslinked materials, but also the different synthetic approaches disclosed in the literature. The various chemical modification reactions relevant to this area are also reviewed. Several examples of applications for hypercrosslinked materials are described; these applications are grouped into thematic areas such as chromatography, gas storage and the trapping of organic contaminants.
 N. Fontanals | Dr Núria Fontanals received her PhD at Universitat Rovira i Virgili (URV) in 2005 in the Chromatography and Environmental Applications research group under the supervision of Prof. Francesc Borrull and Prof. Rosa M. Marcé, following a postdoctoral period with Prof. D. C. Sherrington and Prof. Peter A. G. Cormack in the University of Strathclyde in the Polymer Research Group. In 2007, Fontanals reincorporated in the same research group at URV, where in collaboration with the Strathclyde research group, leads a research line devoted to the development of novel materials to be applied in different analytical methods in order to enhance the sensitivity and selectivity in the determination of challenging compounds in complex samples. |
 R. M. Marcé | Dr Rosa M. Marcé is Full Professor in Analytical Chemistry at the Universitat Rovira i Virgili since 2010. Her main interest is the development of new chromatographic methods to determine contaminants in environmental samples. This interest includes the development of new materials for the extraction techniques and, since 1999 she has collaborated with Prof. D.C. Sherrington and Prof. P.A.G. Cormack in the development and application of new materials including molecular imprinted polymers, and polar and mixed-mode hypercrosslinked polymers. These materials have been used in different sorptive extraction techniques and combined with high-tech liquid chromatography-mass spectrometry techniques, to solve environmental analytical problems. She has supervised several PhD theses in collaboration with Strathclyde University's group. |
 F. Borrull | Dr Francesc Borrull is Full Professor in Analytical Chemistry at the Universitat Rovira i Virgili (URV) since 2003 and he is the head of the research group of Chromatography and Environmental Applications of the URV since its formation in 1992. His research interests are focussed on the separation techniques, including gas and liquid chromatography and capillary electrophoresis, mainly applied to the environmental analysis. He has also significantly contributed to the development and application of new materials for sample treatment techniques to improve analytical methods, and the fruitful collaboration with Prof. D.C. Sherrington and Prof. P.A.G. Cormack has involved the supervision of several PhD theses in collaboration with their group. |
 P. A. G. Cormack | Peter Cormack is Professor of Polymer Chemistry in WestCHEM, Department of Pure and Applied Chemistry, at the University of Strathclyde in Glasgow. He graduated from the University of Strathclyde with BSc (Hons) (1992) and PhD (1996) degrees, completing his PhD degree under the supervision of Professor David C. Sherrington and Dr Barry D. Moore. He spent postdoctoral research periods at the Universities of Cambridge and Lund, before returning to the University of Strathclyde as ICI Lecturer in Polymer Chemistry (1998). His research interests concern the synthesis of functional organic polymers. He works closely with the URV scientists on polymer applications. |
1. Introduction
Macroporous poly(styrene-co-divinylbenzene)s (PS-DVB) with high specific surface areas (SSAs) can be prepared by suspension polymerisation, provided that higher levels of crosslinker (DVB) are used (typically >50 vol%) together with an appropriate amount of a suitable solvent (porogen). However, when very high levels of crosslinker are used then a significant number of vinyl groups remain unreacted and are present in the final materials as pendent vinyl groups; this is because steric congestion and phase separations are an impediment to their complete consumption through crosslinking. Thus, the efficiency of the crosslinking process diminishes significantly as polymerisations ensue, and the products of polymerisation have lower effective levels of crosslinking than one would anticipate based upon the monomer feed.1 Davankov introduced a distinctly new method for the production of porous polystyrenes, a method which comprised the extensive post-crosslinking of linear PS chains by means of a Friedel–Crafts (F–C) reaction. This process installed numerous structural bridges between neighbouring aromatic rings whilst the PS was in a highly swollen state, and yields resins which have a hypercrosslinked (HXL) structure, high micropore content and very high SSAs (up to ∼2000 m2 g−1). Another very characteristic, and unusual, feature is that the HXL products display a high propensity for solvent uptake of both polar and non-polar solvents.2 Their porous characteristics – high SSA in combination with a high micropore content – make HXL resins much more retentive than conventional macroporous polymers when applied in sorption processes. The Davankov hypercrosslinking procedure just described, which relies upon external electrophiles, was developed further by Jerábek3 and Sherrington4 in studies which involved the production of HXL resins via internal electrophiles. More specifically, a reactive comonomer, vinylbenzyl chloride (VBC), was exploited, wherein pendent chloromethyl residues present within a swellable precursor polymer served as a source of internal electrophiles in hypercrosslinking reactions. This strategy allows for better control over the physical format of the hypercrosslinked products.Since their original discovery, HXL materials have evolved, and more sophisticated properties have been installed to enable the materials to fulfil a yet broader range of applications, and this includes the storage of gases,5 the retention of organic compounds present in water6 in sorption extraction techniques, and as novel stationary phases in chromatography.7–9
A number of research groups have worked in the HXL field. David Sherrington contributed considerably to the field through both the development of novel synthetic approaches and through his involvement in the applications of HXL materials. In this review we outline the field as a whole, but draw particular attention to the innovations and developments which have come out of the Sherrington research group over the past years.
2. Synthetic approaches
Originally, HXL networks were obtained via the post-crosslinking of polystyrenic chains/structures in an expanded/swollen state by using external crosslinkers. These external crosslinkers bridge between adjacent aromatic residues, setting in place short, rigid (hyper)crosslinks, and these are very often installed through F–C reactions. Fig. 1A shows the different steps involved during a post-crosslinking reaction of PS. The structure of a typical HXL polymer is a rigid and expanded three-dimensional network. The main structural element of this network is a spatially non-planar cycle formed by crosslinking bridges, and very short chain segments confined between the branching points. By using an external crosslinker together with a F–C catalyst almost all of the aromatic rings can be crosslinked/bridged and ultra-high SSAs achieved. The resulting hypercrosslinked network displays a high degree of crosslinking, low packing density due to the number of rigid spacers holding the polymer chains apart, and an extremely high SSA (up to ∼2000 m2 g−1) arising from a big population of very small pores (mean pore diameter = 2–4 nm).7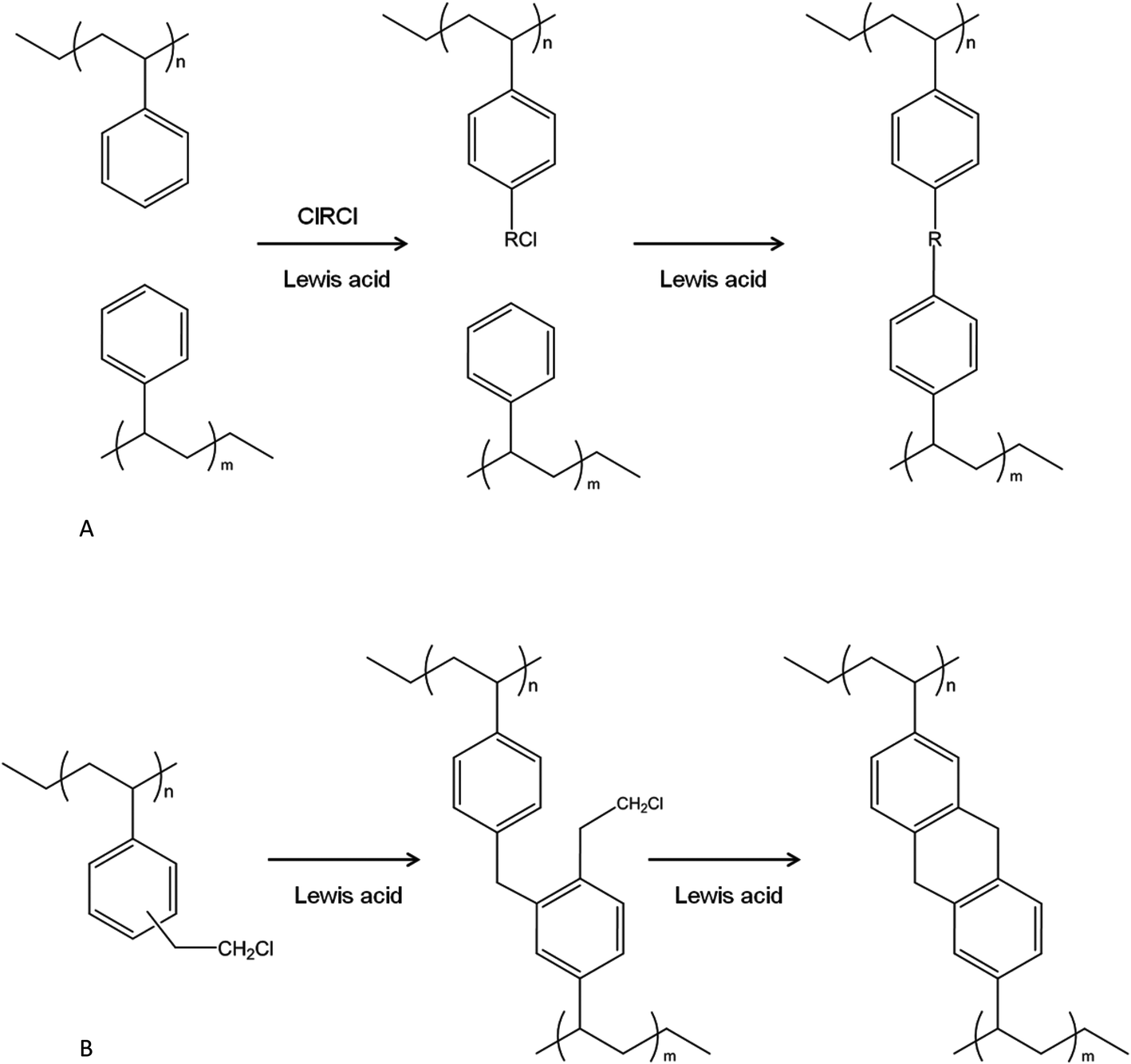 | ||
| Fig. 1 HXL reaction: (A) of PS via external crosslinker; (B) of VBC-DVB via internal electrophile. | ||
The typical morphological properties of HXL materials are as outlined above; however, since the original synthetic procedure was disclosed by Davankov, several variables have been considered and tested. This section will review the different variables and reagents used during the preparation of HXL materials. In addition, the changes in polymer properties which can arise from these modifications to the original procedure will be discussed; these modifications include systematic variations in the reagents used, the electrophiles, the catalysts and the solvents. These parameters, and how they can influence the synthesis and the final properties of HXL materials, will be discussed in the following sections. Table 1 provides illustrative examples of HXL materials which have been synthesised using distinct synthetic approaches. This table also identifies the intended application for each HXL material.
| Precursor | Crosslinker | Solvent | Catalyst | Properties | Application | Ref. | |
|---|---|---|---|---|---|---|---|
| SSA (m2 g−1) | Gas uptake | ||||||
| PS: polystyrene; PS-DVB: polystyrene-divinylbenzene; VBC-DVB: vinylbenzyl chloride-divinylbenzene; DCX: dichloroxylene; CME: chloromethyl methyl ether; BCMBP: 4,4′-bis(chloromethyl)-1,1′-biphenyl; FDA: formaldehyde dimethyl acetal; BCMA: bis-chloromethylantracene; DCE: dichloroethylene; DCB: dichlorobutene; DCH: dichlorohexane; DMF: dimethylformamide; DMSO: dimethyl sulfoxide. | |||||||
| PS linear | DCX | DCE | SnCl4 | 600–1000 | Synthesis | 33 | |
| PS linear | CME | DCE | SnCl4 | 500–1000 | Synthesis | 40 | |
| PS-DVB | BCMBP | ||||||
| PS-DVB | CCl4 | CCl4 | AlCl3 | 400–700 | LC column packing | 43 | |
| PS-DVB | BCMBP | DCE | FeCl3 | Up to 900 | Monolithic column | 48 | |
| DCX | |||||||
| FDA | |||||||
| Fe3O4/pDVB | DCE | DCE | FeCl3 | ∼1300 | Retention contaminants | 37 | |
| VBC-DVB | (VBC) | CCl4 | AlCl3 | ∼1000 | Synthesis | 12 | |
| VBC-DVB | (VBC) | DCE | SnCl4 | ∼1000 | Synthesis | 3 | |
| VBC-DVB | (VBC) | DCE | FeCl3 | 1000–2000 | Synthesis | 4 | |
| AlCl3 | |||||||
| SnCl4 | |||||||
| VBC-DVB | (VBC) | DCE | FeCl3 | 1000–1800 | Synthesis & retention compounds | 11, 15 and 16 | |
| VBC-DVB | (VBC) | DCE | FeCl3 | 1400 | 3 wt% H2 77 K/15bar | H2 storage | 13 |
| VBC-DVB | (VBC) | DCE | FeCl3 | 1300–1900 | 1.5 wt% H2 77 K/1bar | H2 storage | 67 |
| PS-VBC-DVB | (VBC) | DCE | FeCl3 | No data | Monolithic column | 14 | |
| DCB | AlCl3 | ||||||
| DCH | SnCl4 | ||||||
| VBC-triphenylbenzene | BCMBP | DCE | FeCl3 | ∼1200 | 6 wt% CO2 278 K/1 atm | Gas storage | 68 |
| Carbazole bromopheny-lmethanol | 1,10-Phenantroline | DMF | CuI | ∼1000 | 2.39 wt% H2 77 K/1 bar | Gas storage | 22 |
| 16.7 wt% CO2 273 K/1 atm | |||||||
| Carbazoles | FDA | DMF | FeCl3 | 1000–1800 | 1.94 wt% H2 77 K/1 bar | Gas storage | 21 |
| 0.21 wt % CO2 273 K/1 atm | |||||||
| DCX | DCE | FeCl3 | 600–1900 | 0.9–17.7 wt% H2 77 K/1 bar 0.016 wt% CO2 273 K/1 atm | Gas storage | 17 | |
| BCMBP | |||||||
| BCMA | |||||||
| Fluorene and derivatives | BCMBP | DCE | FeCl3 | 1800 | 0.099 wt% CH4 298 K/35 bar | Gas storage | 24 |
| Polyanilines | CH2I2 | DMSO | ∼500 | 2.2 wt% H2 77 K/3.0 MPa | Gas storage | 19 | |
| CH3CHI2 | |||||||
| CH3CH2CHI2 | |||||||
| Polypyrroles | CH2I2 | DMSO | ∼20–700 | 0.6–1.6 wt% H2 77 K/0.4 MPa | Gas storage | 20 | |
| CHI3 | DMF | ||||||
| BI3 | |||||||
| 1,4-Phenylendiamine, 1,3,5-tris-(4′-aminophenylbenzene) | BX3 | DCE | ∼500–1400 | 1.3 wt% H2 77 K/0.4 MPa | Gas storage | 23 | |
| Tetraphenylethylene/1,1,2,2-tetraphenylethylene-1,2-diol | FDA | DCE | FeCl3 | 600–1900 | 0.17 wt% CO2 273 K/1 bar | Gas storage | 29 |
2.1 Polymer precursors
The first polymer precursor used was linear PS, dissolved in solution in a swollen/expanded state arising from the rational selection and use of a good solvent.2 During the course of the crosslinking reaction, the initial solution of polymer in solvent is transformed into a gel and eventually in a solid, monolithic block, which, if so desired, is subsequently ground to produce HXL particles which are irregular in shape and size. Given an objective to produce spherical particles from this protocol rather than irregular particles, the PS precursor can be a lightly crosslinked, gel-type bead, with DVB (0.3–2%) as crosslinker. Such gel-type beads will also swell in the presence of good solvents although they will not normally dissolve. The low DVB content allows the beads to swell, whilst their spherical shape is maintained as the hypercrosslinking reaction proceeds. In a different approach, PS-DVB macroporous polymers (containing up to 30% of DVB) were also hypercrosslinked.2,10 This makes it possible to install crosslinking points additional to those set in place already by the crosslinking agent (DVB). In addition, such HXL polymers will be expected to exhibit bimodal pore size distributions, since micropores will be derived from the hypercrosslinking process and macropores from the synthetic protocol used to prepare the precursor polymer.4 In any case, the degree of crosslinking achievable from a macroporous precursor is lower due to steric impediments.Poly(vinylbenzyl chloride-co-DVB) (VBC-DVB) has been shown to be another excellent precursor polymer for the synthesis of HXL materials, where the chloromethyl (–CH2Cl) substituent is the source of the internal electrophile used to install bridges between adjacent aromatic rings.3,11Fig. 1B shows the procedure used to obtain HXL materials from VBC-DVB precursors. As can be seen in Fig. 1B, when VBC-DVB is employed as precursor the crosslinking process is intramolecular, and this leads to extremely efficient reactions as reported by Ahn et al.4 They found that a gel-type, VBC-DVB resin prepared by suspension polymerisation could be almost fully hypercrosslinked within 15 minutes only, as evidenced by a considerable decrease in the chlorine content, from ∼19 wt% to ∼2 wt%, and a concomitant increase in the SSA from ∼0 m2 g−1 to ∼1200 m2 g−1. They suggested that the last step in this hypercrosslinking process (Fig. 1B) is extremely favourable since the aromatic ring to be substituted is already doubly alkylated and thus electron rich. More importantly, formation of the final product is further enhanced as the ensuing cyclisation reaction results in the formation of a highly favoured six-membered ring. Over the past years, most of the HXL resins prepared were synthesised using VBC-DVB polymers as precursors.3,11–15
Sherrington's group16 tested different isomers of VBC in the production of swellable polymer precursors and reported substantial differences between the isomers when suspension polymerisation was used. In this regard, when polymers were prepared from the pure para-isomer of VBC (pVBC) or a 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 mixture of para- and meta-isomers (mixVBC), differences in the chlorine content of the polymers were found (∼8.5 wt% Cl for pVBC and ∼22.8 wt% Cl for mixVBC). The low chlorine content of the former was ascribed to preferential hydrolysis of the para-isomer of VBC under the synthesis conditions used.
:30 mixture of para- and meta-isomers (mixVBC), differences in the chlorine content of the polymers were found (∼8.5 wt% Cl for pVBC and ∼22.8 wt% Cl for mixVBC). The low chlorine content of the former was ascribed to preferential hydrolysis of the para-isomer of VBC under the synthesis conditions used.
The use of different isomers of dichloroxylene (DCX) (i.e., oDCX, mDCX and pDCX) as self-condensing precursors for the preparation of high SSA materials for H2 storage was also explored.17 The incorporation of o-DCX was consistently detrimental to the development of SSA in these materials, both when used individually or in mixtures with the p- and m-isomers. This finding is associated with the lower degree of condensation (i.e., crosslinking) achieved with o-DCX compared to the other two DCX isomers. In general, both m-DCX and p-DCX gave rise to materials with quite similar SSAs under comparable reaction conditions.
Polyaniline18,19 and polypyrrole20 precursors were evaluated by the Fréchet and Svec groups. They selected these precursors since a preliminary study showed that polyaniline was a potential H2 storage material; however, the SSAs reported prior to their reports were not higher than 100 m2 g−1. In their hypercrosslinking approach, polyaniline was crosslinked using either diiodomethane or paraformaldehyde to form methylene-bridged networks; they obtained HXL porous polyanilines with SSAs up to 632 m2 g−1.19 Later, polyanilines were crosslinked via Ullman and Buchwald coupling reactions, reactions which yielded networks linked by aromatic rings.18 Finally, they prepared different HXL resins by targeting the highest SSA achievable for each methodology (632 m2 g−1 for the diiodomethane route, 480 m2 g−1 for the formaldehyde route, and 316 m2 g−1 for the Ullman and Buchwald strategy). After the synthesis, the most promising HXL materials were tested for H2 adsorption at elevated pressures. They found that the HXL polyaniline material prepared using the diodomethane-based procedure displayed an adsorption capacity of 2.2 wt% H2 at 77 K/3.0 MPa. Another interesting finding was that protonation of HXL polyaniline severely diminished its ability to physisorb H2. The explanation given to rationalise this observation was that aromatic rings which contain electron-donating functionalities adsorb H2 more readily than those bearing electron-withdrawing groups. Thus, while the aniline functionality is strongly electron-donating, protonated anilines are electron-withdrawing and thus decrease the H2 sorption capacity.19
Cognate strategies were used for the hypercrosslinking of polypyrroles20 using diiodomethane, triiodomethane, and triiodoborane, to produce porous structures with CH2, CH and B crosslinks, respectively. The highest SSA reported was only 732 m2 g−1. In spite of this, the authors found these strategies to be valuable, since precursors that contain aryl amines provided shorter crosslinkes and the presence of electron-donating groups (amine) in the aromatic rings enhance the H2-polymer interactions (i.e., increases their ability to physisorb hydrogen).19 Recently, polycarbazole has been proposed as a precursor resin for hypercrosslinking reactions through FeCl3-promoted carbazole-based oxidative coupling polymerisations. The resulting polycarbozole-based HXL resins provide very interesting values of surface area (up to 1800 m2 g−1) as well as H2 (1.94 wt% at 77 K/1.13 bar) and CO2 (0.21 wt% at 77 K/1.13 bar) uptake, and the resins were synthesised using a protocol that the authors claimed to be cost-effective, high yielding, and which involved the use of an inexpensive catalyst and a single monomer only.21,22 Reich and co-workers23 prepared different borozine-linked polymers through the thermolysis of aryl-amine-boron trihalides by employing 1,4-phenylenediamine or 1,3,5-tri-(4′-aminophenyl)benzene in dichloroethane (DCE) as solvent with BCl3 or BBr3. The resulting polymers were highly porous (SSA up to 1400 m2 g−1), but offer moderate low-pressure H2 storage capacity (0.7 to 1.3 wt% at 77 K/1 bar).
Rigid, non-functional aromatic precursors such as fluorene and derivatives (9,9′-spirobi(fluorine), dibenzofuran and dibenzothiophene were also used in the generation of porous copolymer networks obtained via co-condensation with 4,4′-bis(chloromethyl)biphenyl (BCMBP) under F–C catalytic conditions.24 The monomers were incorporated successfully into the polymers, as shown by FT-IR spectroscopy and 13C CP-MAS NMR spectroscopy. The polymers showed high CH4 (up to 0.099 wt% at 298 K/35 bar) and H2 (0.016 wt% at 77 K/1 bar) uptake, as well as a highly porous structures (up to 1800 m2 g−1). Aromatic precursors, including benzene,25,26 polythiophene, polypyrrole and polyfuran,27 aniline26 and hydroxylated and bishydroxymethyl monomers28 were also employed using a similar strategy to deliver HXL materials. All these materials were evaluated for their gas storage potential.
Tetraphenylethylene-based HXL networks were synthesised successfully via F–C alkylation using a dimethyl acetal crosslinker. Moreover, the material properties, in terms of SSA and CO2 uptake, outperformed the corresponding polymer based on 1,1,2,2-tetraphenylethane-1,2-diol (modified with two hydroxyl moieties).29 1,7-Dicarbadodecaborane was reacted further with two chloromethyl functional groups (DCE or BCMBP); the resultant precursor was subsequently self-condensed under F–C conditions. The resulting HXL materials displayed SSAs around 1000 m2 g−1, and their maximum H2 up-take at 77 K/1 bar ranged from 0.021 to 0.028 wt%, depending upon the specific precursor.30
Other strategies and precursors have been developed in order to fine tune the final properties of HXL resins according to their final intended applications. For instance, benzyl chloride-terminated double-four-ring cubic siloxane cages were evaluated as suitable precursors since the HXL networks gave extremely high SSAs (ca. 2500 m2 g−1). The realisation of such high SSA values was attributed to the simultaneous polymerisation of the organic functional groups and the destruction of the siloxane cages during synthesis.31 In addition, this porous siloxane–organic hybrid material provided H2 uptake (∼1.2 wt% at 77 K/1 bar) similar to other materials designed for storage applications (i.e., metal–organic frameworks and porous organic polymers). Wu et al.32 developed a new class of nanostructured porous network materials, which are based on the use of well-defined hairy nanoparticles as network precursors. Hairy nanoparticles (acted as core–shell network building blocks) were prepared by grafting PS chains from the surface of silica nanoparticles using surface-initiated atom transfer radical polymerisation, followed by intra-/interparticle carbonyl crosslinking, and eventually carbonisation. Thus, the range of precursors which is available and appropriate for use in HXL reactions seems to be ever-expanding.
2.2 Precursor polymerisation
Davankov applied the hypercrosslinking methodology to linear or slightly crosslinked PS precursors. This process yields a monolithic block of polymer as the product. If so desired, this monolithic block can be converted, though grinding procedures, into HXL particles which are irregular in size and shape.2,33 However, other groups have exploited precursor materials (mainly styrenic-based) which are in beaded form and derived from, for example, suspension polymerisation.4,12,16 In these examples, if a porous resin is used as precursor, then the product ought to contain not only the original pores (macropores) but also those generated during the hypercrosslinking process (micropores) and thus a bimodal pore size distribution may be anticipated.4HXL resin particles prepared via suspension polymerisation are usually polydisperse (mean diameter of ca. 10–500 μm), which may limit their practical applications in certain fields. Therefore, polymerisation methods other than suspension polymerisation have been used to prepare HXL materials with more uniform particle size distributions. Fig. 2 shows microscopy images of HXL materials where the precursors were prepared via polymerisations other than suspension polymerisation (Fig. 2A).4 In this way, Sherrington's group pioneered the application of emulsion polymerisation which delivered spherical particles of ∼420 nm in diameter which had a near monodisperse particle size distribution (Fig. 2B).11 Non-aqueous dispersion (NAD) polymerisation (spherical particles with diameters in the range 4–10 μm) (Fig. 2C)15 and precipitation polymerisation (PP) (monodisperse spherical particles with ∼4 μm) (Fig. 2D)15 have been employed in an analogous fashion. In each case, relatively uniform particles with high SSAs and a range of diameters governed by the polymerisation approach employed were obtained.
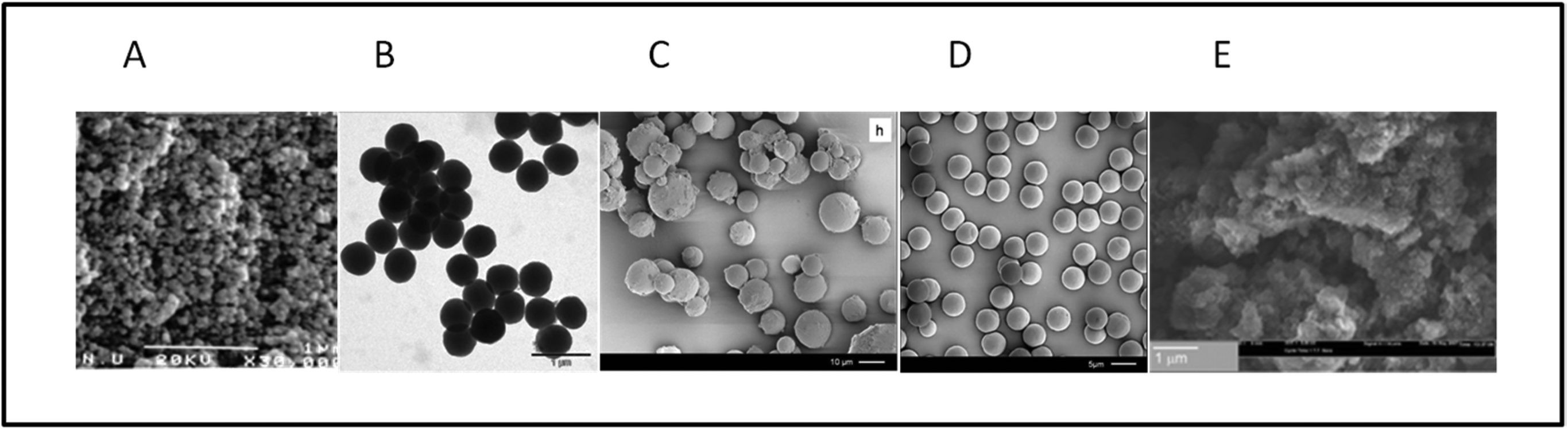 | ||
| Fig. 2 SEM images of different HXL materials, for which the precursors were delivered using different polymerisations: (A) suspension polymerisation; (B) emulsion polymerisation; (C) non-aqueous dispersion polymerisation; (D) precipitation polymerisation; (E) polymerisation in situ in a capillary column. Reprinted from: (A) Macromolecules, 2006, 39, 627–632; copyright (2006) American Chemical Society. (B) Macromolecules, 2006, 39, 5381–5384; copyright (2006) American Chemical Society. (C) and (D) Adv. Mater., 2008, 20, 1298–1302; copyright (2008) Wiley-VCH; (E) J. Mater. Chem., 2007, 17, 4989–4997; copyright (2007) Royal Society of Chemistry. | ||
The authors have had particular success with HXL materials of low mean particle diameter in the micron range, especially when HXL materials derived from PP precursors (known as HXLPP) were used as SPE packing materials for the on-line enrichment of polar compounds from aqueous samples. These sorbents showed greater efficiency and capacity than the commercially available Lichrolut EN HXL material, which has a considerably larger particle size and broader particle size distribution (40–120 μm).34 Since the morphological properties (hydrophobic and SSA ∼1000 m2 g−1) of the resins that were compared were similar, the only significant difference being the particle size (∼4 μm for HXLPP sorbents and 40–120 μm for Lichrolut EN), it could be concluded that the particle size and shape characteristics of the HXLPP materials led to more efficient packing in the sorbent bed, preventing voids, and eventually leading to a more efficient and reproducible extraction procedure, as it was earlier predicted by the Vam Deemter equation.35
Apart from differences in the particle shape and size, we have also examined how the properties of HXL resins varied as a function of the VBC content depending on the type of polymerisation adopted. In this regard, HXL resins derived from different ratios of VBC to DVB in the monomer feed were prepared. For instance, for polymer precursors in particulate form generated from both non-aqueous dispersion (NAD) polymerisation and precipitation polymerisation (PP), it was found that the VBC content had to be 50 wt% or higher in order to generate HXL materials with SSAs in excess of 1000 m2 g−1.15 In contrast, precursor polymers generated from suspension polymerisation with 20% of VBC in the monomer feed gave porous, HXL products with SSAs over 1000 m2 g−1. Nevertheless, the highest SSAs were obtained from gel-type polyVBC precursors crosslinked with 2% DVB under suspension polymerisation conditions; SSAs of up to 2090 m2 g−1 were reported.4 Therefore, the method of polymerisation also influences in the final morphological properties.
Magnetic HXL microspheres have been developed as well, and have been demonstrated to be highly retentive materials for the extraction of target compounds. Gao et al.36 used the PP approach to obtain magnetic HXL particles based on methacrylic acid (MMA)-VBC-DVB; these particles had a core–shell architecture, with Fe3O4/SiO2 at the core. These magnetic microspheres (mean particle size of ∼800 nm) were applied successfully as a sorptive material to retain illegal drugs present in urine. Zhang et al.37 synthesised uniform magnetic polyDVB particles (∼8 μm) with SSAs up to 1300 m2 g−1via membrane emulsification-suspension polymerisation and post-crosslinking reaction. These magnetic HXL materials were applied successfully to the extraction of a group of organic contaminants.
The Fréchet and Svec groups prepared HXL materials in form of monolith8,19 (Fig. 2E), and later went a step further with the in situ hypercrosslinking of monoliths in capillary columns.38 Their first attempt to obtain a monolithic HXL column was successful in that the polymer in the HXL column exhibited a SSA more than one order of magnitude higher than the value measured for the precursor column. Moreover, since it was a mesoporous material, the column afforded good separation for the selected analytes (alkylbenzenes). Fig. 3 compares the isocratic separation of this group of alkylbenzenes using the monolithic column before and after the hypercrosslinking reaction.
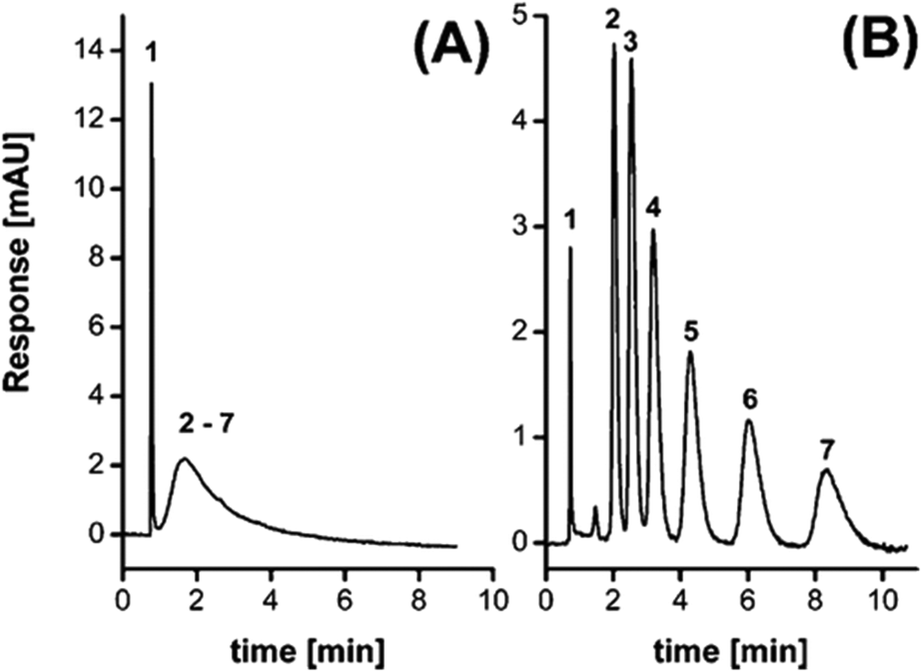 | ||
| Fig. 3 Separation of uracil and different alkylbenzenes using precursor PS-VBC-DVB monolithic column (A) and its HXL counterpart (B). Peaks: uracil (1), benzene (2), toluene (3), ethylbenzene (4), propylbenzene (5), butylbenzene (6), and amylbenzene (7). Reprinted from Anal. Chem., 2010, 82, 1621–1623. Copyright (2010) American Chemical Society. | ||
The morphological properties provided in each polymerisation approach are similar, and this suggests that the hypercrosslinking methodology is generic and can be initiated from precursors derived from distinct polymerisation strategies.
2.3 External crosslinkers
External crosslinkers are exploited whenever precursor polymers are not set-up for internal crosslinking reactions. Initially, the most commonly used external crosslinker was chloromethyl ether (CME),39–42 however, due to its carcinogenic properties it was progressively replaced by other reagents, such as: carbon tetrachloride (CCl4),32,43,44 monochlorodimethyl ether (MCDE),42,45 dichloroxylene (DCX),17,33 4,4′-bis(chloromethyl)biphenyl (BCMBP)10,17,24 or dichloroethylene (DCE)3,13,33 (this reagent can also act as solvent in the synthesis).Varying the external crosslinker can be exploited to modify the nature of the crosslinked bridge in a HXL polymer, both structurally (variations in the bridge length and rigidity impact on the porous morphology) and chemically (installation of a moiety from the external crosslinker). Therefore, in the selection of an external crosslinker one should consider these aspects. Another parameter to bear in mind is the reaction kinetics. For instance, CCl4 reacts more slowly and gives a lower overall conversion than CME.12
The theoretical amount of external crosslinker required for a 100% degree of crosslinking is 0.5 moles of reagent per mole of repeat units (phenyl rings or structure able to crosslink), as reported by Davankov's group45 who studied the influence of the external crosslinker on the crosslinking degree and the SSA. They showed that under nominally identical conditions, the greater the quantity of external crosslinker used the higher the SSA.
Di- and triiodoalkanes (diiodomethane,19,20 diiodoethane19 and diiodopropane19 and triiodomethane20) were also employed to crosslink precursors with aromatic rings that contain electron-donating groups (e.g., polyanilines, polypyrroles, etc.). The authors chose the more reactive diiodoalkanes in comparison to dichloro- or dibromoalkanes to compensate for the limited reactivity of the precursors. All these materials were applied as H2 storage materials; significant progress was reported, although further optimisation needs to be done to compare these materials with PS-based HXL materials.
Four linear diaminoalkanes (1,2-diaminoethane, 1,4-diaminobutane, 1,6-diaminohexane and 1,8-diaminooctane) were tested as external crosslinkers in the hypercrosslinking of precursors based on PS-VBC-DVB which were in form of monolithic stationary phases.46 Apart from the type of crosslinker, other parameters affecting the reaction were optimised. A column with optimal properties was prepared by polymerisation for 2 h at 65 °C followed by hypercrosslinking reaction in the presence of 3% of 1,8-diaminooctane for 2 h at 95 °C.
Li et al.25 proposed a new strategy which consists of the “knitting” of rigid building blocks with an external crosslinker. In so doing, they also selected a more environmentally friendly external crosslinker, formaldehyde dimethyl acetal (FDA), which yields methanol as a by-product of the reaction rather than the typical chloromethyl functionalities which release HCl. Typically, in this one-step approach, the aromatic monomer (including benzene, phenol or chlorobenzene), the crosslinker (FDA) and the catalyst (FeCl3) were dissolved in DCE, and heated at 45 °C for 5 h and then at 80 °C for 19 h to complete the condensation. By adjusting the type of monomer and its molar ratio with respect to FDA, differences in the SSA and H2 and CO2 uptake were observed. The largest values achieved for SSA (∼1400 m2 g−1) and H2 (1.45 wt% at 77.3 K/1.13 bar) and CO2 (13.5 wt% at 273.15 K/1 bar) uptake corresponded to a molar ratio of FDA to benzene of 3. Similar approaches using FDA as external crosslinker were later demonstrated with different monomers such as aromatic heterocyclics,27 hydroxymethylated aromatic molecules,28 aniline and benzene,26 and tetrahedral monomers,29,47 among others.
Recently, Maya et al.48 hypercrosslinked different PS-DVB monoliths using a F–C reaction with FeCl3 involving three external crosslinkers: BCMBP, DCX and also FDA. Apart from the type of external crosslinker used, they also optimised different reaction variables (temperature, time, amount and ratio of reagents). With all three external crosslinkers, the resulting monolith provided an increase in the SSA with respect to the precursor (PS-DVB), which gave benefits in terms of the column efficiency. The polymer with the highest SSA (∼900 m2 g−1) was obtained using a precursor monolith hypercrosslinked with BCMBP. The performance of the new monolithic column was tested by reversed phase liquid chromatography separation of a mixture comprised of acetone and six alkylbenzenes.
2.4 Solvents
As has been outlined already, in a typical hypercrosslinking reaction a linear polymer or a lightly crosslinked polymer is normally dissolved or swollen in a thermodynamically good solvent, since this expands the polymer chains. Rapid crosslinking leaves the polymer chains locked in an expanded form so that when the solvent is removed from the system the collapse of the expanded chains is prevented, the solvent space becomes pore volume and a network of interconnected pores is left behind.49Hildebrand and Hansen parameters provide useful information for the rational selection of solvent, termed a thermodynamically good solvent. DCE is one of the most widely used solvents for hypercrosslinking since it is a good solvent for PS1 and DCE is also compatible with the F–C chemistry, although some other solvents, such as nitrobenzene2,10 and cyclohexane,10 have been employed. Sherrington4 examined the effect of using different solvents (including chlorobenzene, hexane, DCE and DCE/hexane) when the swellable polymer precursor was a macroporous 80%VBC – 20%DVB copolymer. The results showed that DCE yields the highest SSA (∼1200 m2 g−1), but very interestingly hexane and chlorobenzene (which are bad solvents for PS and would thus not be expected to solvate the polymer resin) also gave reasonable SSA values of ∼600 m2 g−1. The resulting HXL materials were characterised by their very high microporosity and very high dry state surface area. The polymers also displayed interesting solvent uptake properties, despite the high degree of crosslinking. Moreover, the resins swelled to similar degrees in both good and bad solvents for PS, in keeping with the argument that all solvents should return each dried network to its lowest energy state formed under the conditions of hypercrosslinking.4 A good example of the solvent uptake behaviour of HXL materials is depicted in Fig. 4, where the solvent uptake of different solvents (including water, methanol, toluene and THF) was monitored for polymers produced during the time course of hypercrosslinking of a VBC-2%DVB gel-type resin. At time zero (no hypercrosslinks present) the solvent uptake for THF was high (THF is a good solvent for PS-based polymers) and extremely low for water (a bad solvent for PS). Nevertheless, as the reaction proceeded from 0.25 h to 18 h all solvents were sorbed at the same level. Different swelling studies4,10,15 have been carried out to test the swelling properties of HXL resins, and the findings from these studies draw similar conclusions: all solvents, including polar protic species such as water and methanol, despite being incompatible with PS-based polymers, are sorbed at almost the same level as toluene and THF (the latter are thermodynamically good solvents for PS).
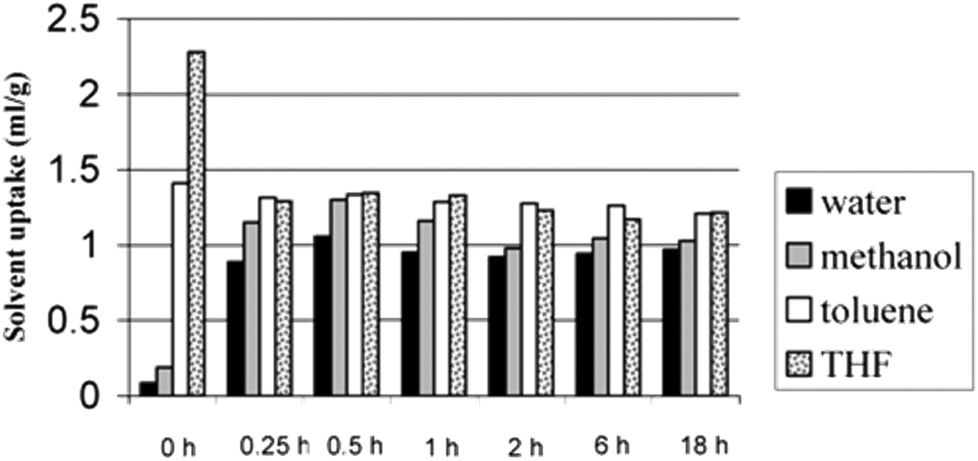 | ||
| Fig. 4 Solvent uptake data of resins isolated at various times during the hypercrosslinking of gel-type VBC-DVB (98:2) precursor resin. Reprinted from Macromolecules, 2006, 39, 627–632. Copyright (2006) American Chemical Society. | ||
Another interesting example was reported by Urban et al.14 wherein different dichloro-based solvents with increasing alkyl chain lengths (which included DCE, 1,4-dichlorobutane (DCB) and 1,6-dichlorohexane (DCH)) were evaluated for the preparation of HXL monoliths in columns. They found that the column permeability decreased when increasing the length of the alkyl chain of the swelling solvent. Therefore, a conclusion which could be drawn is that the solvent may also lead to slightly different properties in the final material.
2.5 Catalysts
The Friedel-Crafts catalyst used in a typical HXL reaction can be either a Lewis acid or a protonic acid such as hydrofluoric acid. In principle, all Lewis acids can potentially catalyse such reactions. Different studies have reported the use of catalysts such as FeCl3,11,13,15,43 aluminium chloride (AlCl3),2,4,12,14 tin chloride (SnCl4),2,3,33,40 zinc chloride (ZnCl2)42 and boron halides (BCl3 or BBr3).23 Nevertheless, FeCl3 is still one of the most widely used. Its use is supported in a study4 where VBC-DVB precursors were hypercrosslinked using a range of catalysts and it was found that FeCl3 was consistently more active than both AlCl3 and SnCl4, giving products with higher SSAs in all cases. The reason for the differences between catalysts is not fully understood, but it was proposed that poor solubility (AlCl3) and steric bulk (SnCl4) may limit the HXL reactions, therefore FeCl3 may offer the best compromise in terms of solubility and molecular size. In Fig. 1B, it can be appreciated how steric bulk may affect the reactions as the first crosslink formed can potentially prevent larger Lewis acids from accessing the second chloromethyl moiety to form the second crosslink. Since the crosslinking process used is intramolecular in nature, the reaction has been shown to be very efficient. It is possible that this step was the limiting step when the relatively large SnCl4 was used as catalyst. FeCl3 is smaller and seemingly not limited in terms of this reaction; this observation, coupled with the enhanced reactivity towards second bridge formation, could well be the cause of the remarkably short reaction times required for HXL reactions involving FeCl3.The optimum amount of Lewis acid required to afford high SSA materials was addressed in a study17 where gel-type VBC-DVB copolymers were hypercrosslinked in the presence of DCX. It was shown that when the ratio of monomers related to the reaction feed was 3.75% (w/v), the optimum FeCl3 ratio was in the range of 0.5–2 mol/mol of monomers. Catalyst ratios below this range led to decreased yields and lower SSAs, whereas much higher catalyst concentrations were detrimental to the SSA of the resulting polymers. If this reaction is catalytic then the FeCl3 concentration should be irrelevant and all chlorine will be lost. However, this observation (which agrees with the earlier report by Sherrington4) would suggest that the action of the Lewis acid within the reaction is at least partially catalytic.
Urban et al.14 studied the effect of the type of catalyst in relation to the final properties of HXL monolithic polymers. In outline, they prepared a series of HXL monoliths based on PS-VBC-DVB using a range of different catalysts (i.e., AlCl3, FeCl3 and SnCl4), and concluded that the column permeability was similar when FeCl3 and SnCl4 were used as catalysts, but decreased significantly when AlCl3 (the most reactive F–C catalyst) was used.
3. Chemical modifications of HXL materials
The HXL materials developed originally were PS-based thus they did not possess any special functional groups besides aromatic residues, and their applications as sorbents rely on simple adsorption via hydrophobic and π–π interactions. Nevertheless, HXL materials bearing special functional groups can be prepared, and this can lead to wider applications for these materials. Functionalised HXL materials can be produced by using post-polymerisation chemical modification reactions, either before or after the hypercrosslinking process, else by direct copolymerisation of functional monomers to produce functionalised polymer precursors. Table 2 lists examples of HXL materials bearing a range of distinct chemical moieties, together with the applications explored for each type material.| HXL material | Functional group | Stage of the modification | Properties | Application | Ref. | |
|---|---|---|---|---|---|---|
| SSA (m2 g−1) | Moiety content | |||||
| PS: polystyrene; PS-DVB: polystyrene-divinylbenzene; VBC-DVB: vinylbenzyl chloride-divinylbenzene; EGDMA: ethylene glycol dimethacrylate; MMA: methyl methacrylic acid; HEMA: hydroxyethyl methacrylatea Ion-exchange capacity.b Already in Table 1. | ||||||
| VBC-DVB (pVBC and mix-VBC) | Hydrolysis under polymer. cond. | During the preparation of the precursor by suspension polymerisation | ∼900 | 3.96 wt% O | Polar sorbent in SPE | 16 |
| ∼1800 | 1.5 wt% O | |||||
| VBC-DVB | Hydrolysis under polymer. cond. | During the preparation of the precursor by suspension polymerisation | 1200 | 2.9 wt% O | Polar sorbent in batch | 51 |
| PS-DVB further chloromethylation | Hydrolysis under polymer. cond. | During the preparation of the precursor by suspension polymerisation | 900 | 5.95 wt% O | Polar sorbent in batch | 42 |
| Commercial PS-DVB | Dimethylamine | Post-modification | 1375 | 1.5 mmol g−1 N−1 | Polar sorbent in batch | 56 |
| VBC-DVB | Dimethylamine | Post-modification | 820 | 1.5 mmol g−1 N−1 | Polar sorbent in batch | 52 |
| PS-DVB | Phthalic anhydride | Post-modification | 1100 | No data | Polar sorbent in batch | 59 |
| PS-DVB | Naphthol | Post-modification | 600 | 2.16 wt% phenolic hydroxyl | Polar sorbent in batch | 62 |
| PS-VBC-DVB | Diaminoalkanes, and taurine | Post-modification | No data | No data | Monolithic column | 46 |
| PS-VBC-DVB-4-acetoxystyrene | Monomer in the polymerisation | Up to 600 | No data | Monolithic column | 66 | |
| VBC-DVB | Piperidine ethylenediamine | Post-modification | ∼1000 | 0.90a mmol N g−1 | WAX sorbent in SPE | 54 |
| 0.75 mmol N g−1 | ||||||
| VBC-DVB | dimethylbutylamine | Pre-modification | 1200–1500 | 0.2–0.4a mmol N g−1 | SAX sorbent in SPE | 53 |
| VBC-DVB | Lauroyl sulfate | Post-modification | 1070–1370 | 1.7–2.8a mmol g−1 | SCX sorbent in SPE | 60 |
| PS-VBC-EGDMA | Sulfuric acid | Post-modification | 330–1020 | 0.6–2.3a mmol g−1 | SCX sorbent in SPE | 55 |
| MMA-VBC-DVB | Monomer in the polymerisation | 1125 | 0.72a mmol g−1 | WCX sorbent in SPE | 65 | |
| HEMA-VBC-DVB | Monomer in the polymerisation | 670–925 | 0.9–1.89 mmol O g−1 | Polar sorbent in SPE | 64 | |
| Polycarbazolesb | Monomer in the polymerisation | ∼1000 | n.d. | Gas storage | 21 and 22 | |
| Polyanilinesb | Monomer in the polymerisation | ∼500 | n.d. | Gas storage | 19 | |
| Polypyrrolesb | Monomer in the polymerisation | ∼20–700 | n.d. | Gas storage | 20 | |
| 1,4-Phenylendiamine,1,3,5-tris-(4′-amino-phenylbenzene)b | Monomer in the polymerisation | ∼500–1400 | n.d. | Gas storage | 23 | |
VBC-derived HXL materials were reported to contain some oxygen due to the partial hydrolysis of chloromethyl groups during the polymer synthesis and/or hypercrosslinking conditions. Sherrington16 reported that the hydrolysis of benzyl chloride moieties would be expected to proceed via an SN1 mechanism, and thus involve the generation of stabilised carbocation intermediates followed by nucleophilic attack of water under the typical conditions of the suspension polymerisation used to prepare VBC-DVB precursors. The degree of hydrolysis of VBC-residues in these HXL materials depended upon the VBC isomer which was employed in the precursor polymer synthesis. Thus, when pVBC was used as monomer the HXL resin contained 4.0% oxygen and had an SSA of ∼900 m2 g−1, whereas when a mixture of VBC isomers (30% pVBC and 70% mVBC) was used as monomer the corresponding HXL resin contained 1.5% oxygen and had a SSA of 1889 m2 g−1. These differences were explained on the basis that the carbocation derived from pVBC would be expected to be more stabilised than that derived from mVBC; the carbocation from pVBC would therefore form more readily and hence the hydrolysis of pVBC would be expected to proceed more rapidly than that of mVBC.
The hydrolysis of VBC residues in VBC-DVB was exploited on purpose to promote the polar properties of the hypercrosslinked products, to deliver HXL materials with higher affinity towards polar compounds.42,50,51 Our research group took full advantage of this phenomenon; HXLGp (derived from pVBC precursor, 4.0 wt% O and 900 m2 g−1) and HXLGmix (derived from a mixture of VBC isomers, 1.5 wt% O and 1900 m2 g−1) were compared to commercial HXL hydrophobic materials, namely: Amberchrom (900 m2 g−1) and Lichrolut EN (1200 m2 g−1) in on-line SPE for a group of polar compounds. In this study, a pre-column was packed with the synthesised HXL resin (40 mg), and 100 mL of sample (spiked at 2 μg L−1 with a mixture of polar compounds) passed through the precolumn in on-line mode. Fig. 5 shows the recoveries from different volumes of sample spiked with phenol. The higher the SSA, the higher was the recovery. However, it was clear that the optimal resin for phenol recovery was the hydrophilic hypercrosslinked resin HXLGp; this resin had an SSA of 900 m2 g−1 and hydrophilic character.
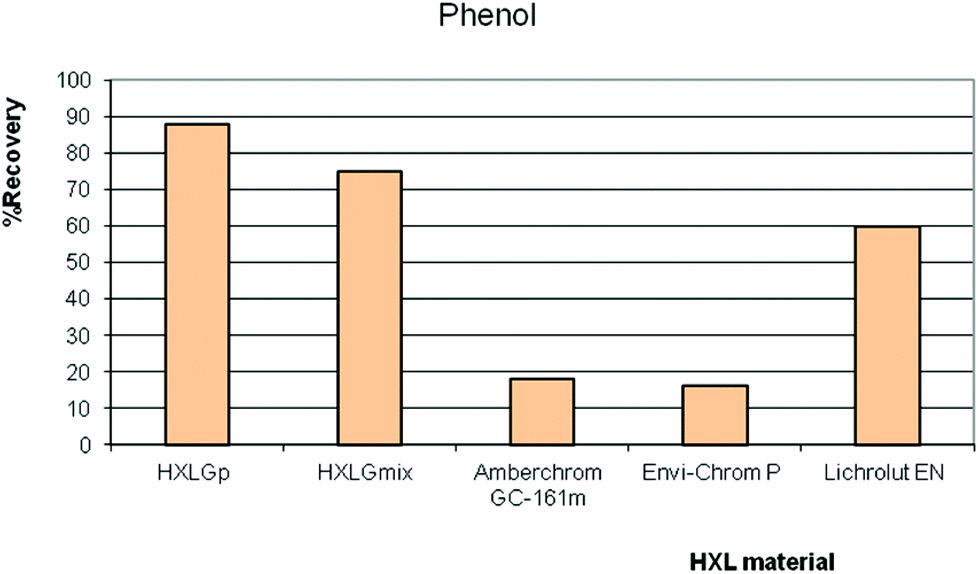 | ||
| Fig. 5 Recovery values when 100 mL of a standard solution spiked with phenol, at a constant amount injected of 0.2 μg, was percolated through a precolumn packed with HXL materials in SPE-LC-UV-Vis. | ||
Li et al.42 reported similar oxygen level contents (6.0 wt%) and SSA (900 m2 g−1) when their precursor polymers were based on chloromethylated PS-DVB. They suggested that the oxygen content could be ascribed to the formation of ethers and ketones; however this suggestion still remains to be verified experimentally.
Regarding post-hypercrosslinking chemical modification reactions, when the HXL precursor is based on VBC-DVB several authors have taken advantage of the incomplete consumption of VBC residues in the hypercrosslinking reaction. A portion of pendent chloromethyl groups survive the hypercrosslinking processes and are thus available for chemical modification.52–55 In this way, Li et al.52,56–58 prepared different HXL materials, either by synthesising the VBC-DVB precursors from first principles, i.e., via copolymerisation of VBC with DVB, or starting from commercially available macroporous resins; these precursors were hypercrosslinked and modified further with dimethylamine to yield HXL materials with amine functionalities which enhanced the sorption properties of the resins toward polar compounds. In a parallel example, the same research group prepared HXL resins bearing carbonyl moieties which displayed enhanced sorption behaviour similar to the amine functional resins.59
We adopted this strategy for the preparation of a family of ion-exchange HXL materials.53–55 Materials were synthesised via post-hypercrosslinking chemical modifications for strong cation exchange (SCX), strong-anion exchange (SAX), and weak-anion exchange (WAX) resins. The ion-exchange materials were prepared from HXL structures, which enhance RP interactions through their high SSA (1000–1500 m2 g−1). Specifically, the HXL materials were prepared from VBC-DVB precursors obtained by PP, which produced low micron size particles that were eminently well-suited for SPE applications. The precursor particles prepared by PP, named as PP, were hypercrosslinked (to give products named HXLPP) by means of F–C reactions which exploited the chemistry of the VBC residues.
With particular regards to the synthesis of the SCX material, the HXLPP resins were post-functionalised with either acetyl sulfate or lauroyl sulfate, with the latter proving to be the most effective reagent for the sulfonation. After optimising different synthetic variables, three HXLPP-SCX resins were prepared with different sulfur contents as governed by the percentage of the sulfonation reagent used in the chemical modification (15%, 20% and 50%). It was found that the HXLPP-SCX sorbent modified with 50% of the sulfonation reagent displayed the highest ion-exchange capacity (IEC) (2.5 meq g−1) and the highest SSA (1370 m2 g−1) even although the possibility of sulfone bridge formation could not be excluded.60 Similar HXL SCX particles were reported by the same authors. In this case, the swellable precursors used were based on polymers with different VBC-ethylene glycol dimethacrylate (EGDMA) ratios, and were synthesised via NAD polymerisation rather than by PP. Later, the HXLNAD particles were sulfonated using sulfuric acid. The resulting HXLNAD materials displayed SSAs from 330 up to 1000 m2 g−1 and IECs from 0.6 to 2.1 mmol g−1. Comparing both types of material (HXLPP-SCX versus HXLNAD-SCX) in terms of retention properties towards ion-exchangeable compounds, both materials provided similar retention behaviour.55
The synthetic procedures used to obtain the HXLPP-SAX resins (modified with 5% and 10% dimethylbutylamine – DMBA) were somewhat different. In this case, DMBA, in an appropriate ratio, was first reacted with the VBC-DVB precursor (obtained via PP) to install the SAX character. The quaternised precursors (PP-SAX) were then hypercrosslinked following the usual procedures and HXLPP-SAX resins obtained. The authors discovered that amination was less efficient after hypercrosslinking due to the bulky nature of the tertiary amine (DMBA) which restricted the access of amine to free chloromethyl groups; therefore, amination was conducted prior to hypercrosslinking.53Fig. 6A outlines the synthetic route used for preparing the HXLPP-SAX sorbents; this is an example of post-polymerisation chemical modification prior to hypercrosslinking.
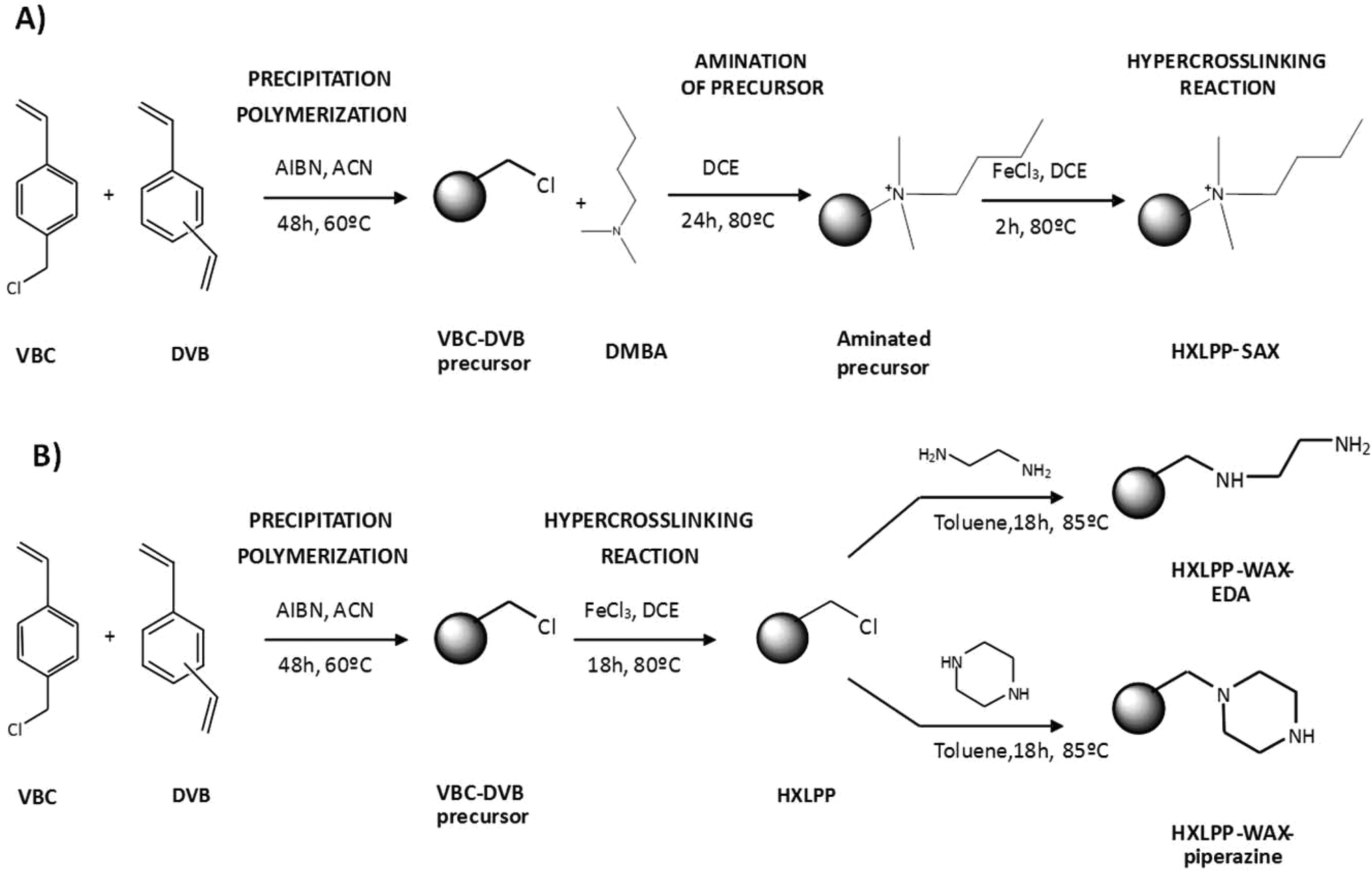 | ||
| Fig. 6 Synthetic approaches to modify different HXL materials with ionic moieties: (A) HXLPP-SAX; (B) HXLPP-WAX-EDA and HXLPP-WAX-piperazine. | ||
In contrast, post-hypercrosslinking chemical modification reactions were used in order to prepare HXLPP-WAX (modified with piperazine, HXLPP-WAX-piperazine, and ethylenediamine, HXLPP-WAX-EDA) (Fig. 6B). In these cases, the amine moieties were introduced via chemical modification of the residual chloromethyl groups in the HXLPP resins For this, the HXLPP resins were wetted with dried toluene and a 5-fold molar excess of the amine (either EDA or piperazine) relative to the chloromethyl group added and reacted for 18 h at 85 °C.54
Recently, an HXL material (900 m2 g−1), synthesised from an F–C alkylation between carbazole and α,α′-dibromo-p-xylene, was subjected to a post-hypercrosslinking synthetic modification via sulfonation to yield an HXL-SO3H material. Upon sulfonation, the material was transformed into a very efficient solid acid catalyst for the production of biodiesels.61
Janků et al.46 hypercrosslinked PS-VBC-DVB monolithic stationary phases by a nucleophilic substitution reaction. Four linear diaminoalkanes (1,2-diaminoethane, 1,4-diaminobutane, 1,6-diaminohexane and 1,8-diaminooctane) were evaluated, as mentioned previously in section 2.3. Furthermore, they also modified the residual chloromethyl groups with 2-aminoethanesulfonic acid (taurine) in order to prepare stationary phases with dual retention mechanisms (hydrophilic and reversed-phase interactions) which were able to separate highly polar compounds such as thiourea. However, as the columns were prepared in situ, the authors did not provide any characterisation information on the HXL materials in terms of IEC or SSA, presumably because they were interested primarily in the properties related to column efficiency. Yuan et al.62 prepared an HXL material from a PS-DVB precursor and used bromoethane as external crosslinker since it is a less toxic reagent than MCDE. Thereafter, they modified the HXL material with 2-naphthol; this installed phenolic moieties, which provided better sorption when contacting to polar compounds.
Other approaches involve the selection of precursor monomers that already contain the polar moiety to enhance the expected properties of the HXL material. As listed in section 2.1, different reagents (aniline,19 pyrrole,20 aminobenzene,63 carbazole22) containing a heteroatom were used successfully to prepare different HXL materials which were then applied primarily as gas storage materials. In these cases, the presence of nitrogen moieties in the network favours the formation of a narrow pore system and a heteroatom-rich pore surface, which leads to high gas adsorption capacity and high selectivity.27
A set of hydrophilic HXL materials were generated from the hypercrosslinking, via F–C alkylation, of a hydrophilic precursor resin based on 2-hydroxyethyl methacrylate (HEMA)-VBC-DVB prepared by PP. The HXL materials had a SSA of ∼900 m2 g−1 and ∼1.5 mmol g−1 O content derived from hydrophilic HEMA residues. This blend of properties meant that they were well-suited for the sorption of polar compounds from aqueous samples, since they provided better recovery values than the commercial materials when used for similar purposes.64
Continuing with the family of ion-exchange HXL materials prepared through PP, for the synthesis of an ion-exchange WCX (HXLPP-WCX) material the carboxylic acid moieties which give rise to WCX character were introduced by copolymerisation of a functional monomer (methacrylic acid – MAA). In this work, the terpolymer MAA-VBC-DVB (10/50/40 w/w% ratio of monomers in the feed) was the precursor polymer in this hypercrosslinking reaction.65 In this way, the carboxylic acid moieties were present from the beginning of the synthetic process and, as their incorporation into the HXLPP-WCX was satisfactory (equivalent to an IEC of 0.72 meq g−1), the authors did not require to evaluate the possibility of introducing the carboxylic group in a post-hypercrosslinking modification step. Moreover, it should be mentioned that the IEC obtained for the HXLPP-WCX (0.72 meq g−1) was similar to that reported for commercially available sorbents such as Oasis WCX (0.75 meq g−1) and Strata-X-CW (0.74 meq g−1). However, the SSA was higher (1125 m2 g−1 for HXLPP-WCX compared to ∼800 m2 g−1 for both Oasis WCX and Strat-X-CW).
Porous polymer monoliths derived from four monomers (PS-VBC-DVB-4-acetoxystyrene) were also hypercrosslinked, which delivered materials containing both chloromethyl and acetoxy groups. The acetoxy groups were then deprotected to convert them into phenolic groups, without affecting the morphology of the material. The ratio of each monomer could be varied to balance the SSA properties with the presence of hydroxyl moieties in the final material. This monolithic column with balanced SSA and hydroxyl moieties delivered useful chromatographic properties since it could establish both reversed-phase and normal phase interactions.66
A series of networks, using benzene and aniline as precursors in the presence of FDA using F–C alkylation, were prepared. Aniline was added to the material since it was reported already that aniline may improve the selectivity for CO2. The SSAs of the materials were measured (Fig. 7). As expected, the SSA decreased with increasing aniline content (the 100% aniline material was essentially non-porous), and the highest SSA (∼1300 m2 g−1) was achieved when 100% benzene was used. However, a significant amount of aniline (up to 40%) could still be incorporated into the polymer without decreasing so markedly the CO2 uptake (Fig. 7). In addition, the difference in the rate at which the SSA decreased and the rate at which the CO2 uptake decreased suggested that these benzene-aniline copolymers had good selectivity for CO2/N2.26 Thus, the modification of the original HXL materials with functional groups may provide different type of final properties, which can be potentially be tuned.
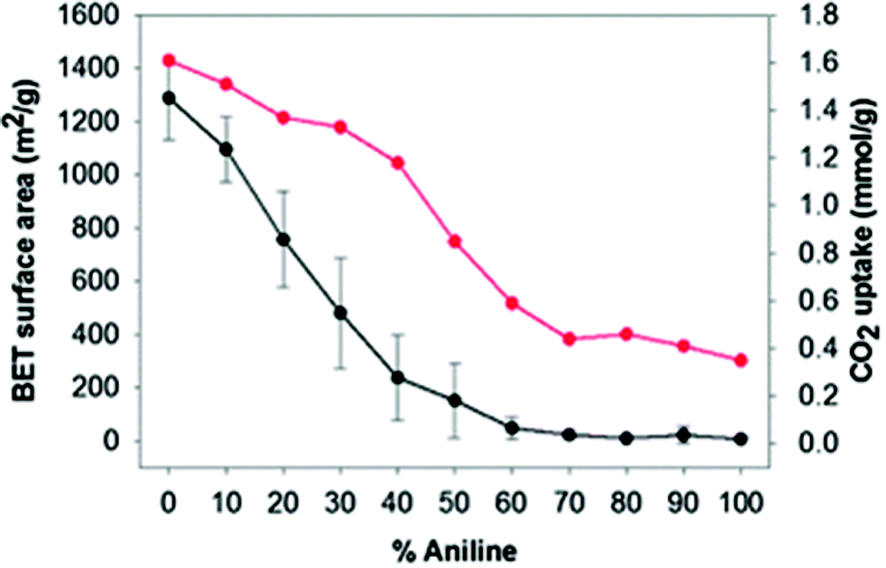 | ||
| Fig. 7 Surface area (black) and CO2 uptakes (red) of the aniline-benzene HXL materials depending on the feed ratio. Error bars calculated from measurements of three nominally identical networks at each % aniline. Reprinted from Polym. Chem., 2012, 3, 2034–2038. Copyright (2012) Royal Society of Chemistry. | ||
4. Conclusions and future perspectives
The HXL material field has experienced a great expansion since the first reports by Davankov. As a proof of this, a number of research groups, including the one led by David Sherrington, have been working in the area with very interesting findings. It is very clear to see that the hypercrosslinking methodology seems to be a generic one, and there is no doubt at all that HXL materials are very useful in terms of applications.Several applications have exploited the high retention properties offered by HXL materials. When functional groups are installed into HXL materials the range of applications can be expanded. For example, installation of polar groups can enhance the hydrophilicity of the materials, a chemical modification which tunes the selectivity of the HXL materials towards the capture of polar compounds.
Further approaches and applications are expected in the coming years, involving a broader range of HXL materials prepared in a range of physical formats with yet more sophisticated properties. One interesting direction for future development will be to enhance the capture selectivity of HXL materials for pollutants/gases. In respect of the selective retention of pollutants, this characteristic will facilitate their use as sorbents in environmental analysis work, however applications in larger-scale purification operations, for example in water treatment works, are an attractive prospect too. In the gas storage field, the capture mechanisms should be addressed in more detail so that the HXL materials are able to work reversibly at practically useful temperatures and not only under cryogenic conditions. We also anticipate that the achievements reported in the capillary monolith field will be expanded to other simple formats, such as thin layers, as suggested by Svec and co-workers.48 Another evolving field of application is the implementation of HXL materials as electrode materials in sensing devices, which is a relatively unexplored field with huge potential. More generally speaking, however, exerting tighter synthetic control over the porous character of the HXL materials (e.g., pore size), expanding the range of chemical functional groups which can be supported, and exploring the potential for the production of HXL materials on a larger scale, are all interesting research challenges just waiting to be tackled in the future.
Acknowledgements
The authors would like to thank the Ministry of Economy and Competitiveness, Spain (Project CTQ2014-52617-P) for the financial support given. We are very proud to be part of this Special Issue dedicated to David Sherrington. More than 15 years ago, Dave brought us the opportunity to collaborate. Since then, we have shared enthusiastically in the field of polymers for analytical applications, and we have enjoyed a scientific trip together which was full of very interesting achievements.References
- D. C. Sherrington, Chem. Commun., 1998, 2275–2286 RSC.
- V. A. Davankov, V. Rogozhin and M. P. Tsyurupa, US Pat, 3729457, 1969 Search PubMed.
- P. Veverka and K. Jerábek, React. Funct. Polym., 1999, 41, 21–25 CrossRef CAS.
- J.-H. Ahn, J.-E. Jang, C.-G. Oh, S.-K. Ihm, J. Cortés and D. C. Sherrington, Macromolecules, 2006, 39, 627–632 CrossRef CAS.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS PubMed.
- N. Fontanals, R. M. Marcé and F. Borrull, J. Chromatogr. A, 2007, 1152, 14–31 CrossRef CAS PubMed.
- V. A. Davankov and M. P. Tsyurupa, in Comprehensive Analytical Chemistry, 2011, vol. 56, pp. 1–648 Search PubMed.
- J. Urban, F. Svec and J. M. J. Fréchet, J. Chromatogr. A, 2010, 1217, 8212–8221 CrossRef CAS PubMed.
- S. Xu, Y. Luo and B. Tan, Macromol. Rapid Commun., 2013, 34, 471–484 CrossRef CAS PubMed.
- V. A. Davankov and M. P. Tsyurupa, React. Polym., 1990, 13, 27–42 CrossRef CAS.
- F. S. Macintyre, D. C. Sherrington and L. Tetley, Macromolecules, 2006, 39, 5381–5384 CrossRef CAS.
- J. Hradil and E. Kralova, Polymer, 1998, 39, 6041–6048 CrossRef CAS.
- J. Y. Lee, C. D. Wood, D. Bradshaw, M. J. Rosseinsky and A. I. Cooper, Chem. Commun., 2006, 2670–2672 RSC.
- J. Urban and V. Škeříková, J. Sep. Sci., 2014, 37, 3082–3089 CrossRef CAS PubMed.
- N. Fontanals, P. Manesiotis, D. C. Sherrington and P. A. G. Cormack, Adv. Mater., 2008, 20, 1298–1302 CrossRef CAS PubMed.
- N. Fontanals, J. Cortés, M. Galià, P. A. G. Cormack, R. M. Marcé, F. Borrull and D. C. Sherrington, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1718–1728 CrossRef CAS PubMed.
- C. D. Wood, B. Tan, A. Trewin, H. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Khimyak, N. L. Campbell, R. Kirk, E. Stockel and A. I. Cooper, Chem. Mater., 2007, 19, 2034–2048 CrossRef CAS.
- H.-L. Wang, J.-L. Chen and Q.-X. Zhang, Adsorpt. Sci. Technol., 2006, 24, 17–28 CrossRef CAS.
- J. Germain, J. M. J. Fréchet and F. Svec, J. Mater. Chem., 2007, 17, 4989–4997 RSC.
- J. Germain, J. M. J. Fréchet and F. Svec, Chem. Commun., 2009, 1526–1528 RSC.
- X. Yang, M. Yu, Y. Zhao, C. Zhang, X. Wang and J.-X. Jiang, RSC Adv., 2014, 4, 61051–61055 RSC.
- L. Pan, Q. Chen, J.-H. Zhu, J.-G. Yu, Y.-J. He and B.-H. Han, Polym. Chem., 2015, 6, 2478–2487 RSC.
- T. E. Reich, K. T. Jackson, S. Li, P. Jena and H. M. El-Kaderi, J. Mater. Chem., 2011, 21, 10629–10632 RSC.
- M. G. Schwab, A. Lennert, J. Pahnke, G. Jonschker, M. Koch, I. Senkovska, M. Rehahn and S. Kaskel, J. Mater. Chem., 2011, 21, 2131–2135 RSC.
- B. Li, R. Gong, W. Wang, X. Huang, W. Zhang, H. Li, C. Hu and B. Tan, Macromolecules, 2011, 44, 2410–2414 CrossRef CAS.
- R. Dawson, T. Ratvijitvech, M. Corker, A. Laybourn, Y. Z. Khimyak, A. I. Cooper and D. J. Adams, Polym. Chem., 2012, 3, 2034–2038 RSC.
- Y. Luo, B. Li, W. Wang, K. Wu and B. Tan, Adv. Mater., 2012, 24, 5703–5707 CrossRef CAS PubMed.
- Y. Luo, S. Zhang, Y. Ma, W. Wang and B. Tan, Polym. Chem., 2013, 4, 1126–1131 RSC.
- S. Yao, X. Yang, M. Yu, Y. Zhang and J.-X. Jiang, J. Mater. Chem. A, 2014, 2, 8054–8059 CAS.
- S. Yuan, D. White, A. Mason and D.-J. Liu, Int. J. Energy Res., 2013, 37, 732–740 CrossRef CAS PubMed.
- W. Chaikittisilp, M. Kubo, T. Moteki, A. Sugawara-Narutaki, A. Shimojima and T. Okubo, J. Am. Chem. Soc., 2011, 133, 13832–13835 CrossRef CAS PubMed.
- D. Wu, C. M. Hui, H. Dong, J. Pietrasik, H. J. Ryu, Z. Li, M. Zhong, H. He, E. K. Kim, M. Jaroniec, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2011, 44, 5846–5849 CrossRef CAS.
- V. A. Davankov, S. V. Rogoshin and M. P. Tsyurupa, J. Polym. Sci., 1974, 47, 95–101 CAS.
- N. Fontanals, R. M. Marcé, P. A. G. Cormack, D. C. Sherrington and F. Borrull, J. Chromatogr. A, 2008, 1191, 118–124 CrossRef CAS PubMed.
- G. Guiochon and F. Gritti, J. Chromatogr. A, 2011, 1218, 1915–1938 CrossRef CAS PubMed.
- Q. Gao, C.-Y. Lin, D. Luo, L.-L. Suo, J.-L. Chen and Y.-Q. Feng, J. Sep. Sci., 2011, 34, 3083–3091 CrossRef CAS PubMed.
- M. Zhang, Q. Zhou, A. Li, C. Shuang, W. Wang and M. Wang, J. Chromatogr. A, 2013, 1316, 44–52 CrossRef CAS PubMed.
- J. Urban, F. Svec and J. M. J. Fréchet, Anal. Chem., 2010, 82, 1621–1623 CrossRef CAS PubMed.
- R. Law, D. C. Sherrington, C. Snape, I. Ando and H. Kurosu, Macromolecules, 1996, 29, 6284–6293 CrossRef CAS.
- R. Joseph, W. T. Ford, S. Zhang, M. P. Tsyurupa, A. V. Pastukhov and V. A. Davankov, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 695–701 CrossRef CAS.
- M. P. Tsyurupa, M. M. Ilyin, A. I. Andreeva and V. A. Davankov, Fresenius’ J. Anal. Chem., 1995, 352, 672–675 CrossRef CAS.
- A. Li, Q. Zhang, G. Zhang, J. Chen, Z. Fei and F. Liu, Chemosphere, 2002, 47, 981–989 CrossRef CAS.
- B. Gawdzik and J. Osypiuk, Chromatographia, 2001, 54, 323–328 CAS.
- V. V. Azanova and J. Hradil, React. Funct. Polym., 1999, 41, 163–175 CrossRef CAS.
- A. V. Pastukhov, M. P. Tsyurupa and V. A. Davankov, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 2324–2333 CrossRef CAS.
- S. Janků, V. Škeříková and J. Urban, J. Chromatogr. A, 2015, 1388, 151–157 CrossRef PubMed.
- R. Dawson, E. Stockel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239–4245 CAS.
- F. Maya and F. Svec, Polymer, 2014, 55, 340–346 CrossRef CAS PubMed.
- J. Germain, J. M. J. Fréchet and F. Svec, Small, 2009, 5, 1098–1111 CrossRef CAS PubMed.
- N. Fontanals, M. Galià, P. A. G. Cormack, R. M. Marcé, D. C. Sherrington and F. Borrull, J. Chromatogr. A, 2005, 1075, 51–56 CrossRef CAS PubMed.
- W. Zhang, C. Hong, B. Pan, Q. Zhang, P. Jiang and K. Jia, J. Hazard. Mater., 2009, 163, 53–57 CrossRef CAS PubMed.
- F. Liu, J. Chen, A. Li, Z. Fei, J. Ge and Q. Zhang, Adsorpt. Sci. Technol., 2004, 22, 13–24 CrossRef CAS.
- D. Bratkowska, A. Davies, N. Fontanals, P. A. G. Cormack, F. Borrull, D. C. Sherrington and R. M. Marcé, J. Sep. Sci., 2012, 35, 2621–2628 CrossRef CAS PubMed.
- N. Fontanals, P. A. G. Cormack and D. C. Sherrington, J. Chromatogr. A, 2008, 1215, 21–29 CrossRef CAS PubMed.
- N. Fontanals, N. Miralles, N. Abdullah, A. Davies, N. Gilart and P. A. G. Cormack, J. Chromatogr. A, 2014, 1343, 55–62 CrossRef CAS PubMed.
- B. C. Pan, Y. Xiong, A. M. Li, J. L. Chen, Q. X. Zhang and X. Y. Jin, React. Funct. Polym., 2002, 53, 63–72 CrossRef CAS.
- A. Li, Q. Zhang, H. Wu, Z. Zhai, F. Liu, Z. Fei, C. Long, Z. Zhu and J. Chen, Adsorpt. Sci. Technol., 2004, 22, 807–820 CrossRef CAS.
- C. Xu, C. Long, A. Li, F. Liu, W. Yang and Q. Zhang, Adsorpt. Sci. Technol., 2006, 24, 65–77 CrossRef CAS.
- F. Zhenghao, C. Jinlong, C. Jianguo, G. Guandao, L. Aimin and Z. Quanxing, Adsorpt. Sci. Technol., 2004, 22, 439–449 CrossRef.
- P. A. G. Cormack, A. Davies and N. Fontanals, React. Funct. Polym., 2012, 72, 939–946 CrossRef CAS PubMed.
- S. Bhunia, B. Banerjee and A. Bhaumik, Chem. Commun., 2015, 51, 5020–5023 RSC.
- X. Yuan, X. Li, E. Zhu, J. Hu, W. Sheng and S. Cao, Carbohydr. Polym., 2008, 74, 468–473 CrossRef CAS PubMed.
- J. Germain, F. Svec and J. M. J. Fréchet, Chem. Mater., 2008, 20, 7069–7076 CrossRef CAS.
- D. Bratkowska, N. Fontanals, P. A. G. Cormack, D. C. Sherrington, F. Borrull and R. M. Marcé, J. Chromatogr. A, 2010, 1217, 3238–3243 CrossRef CAS PubMed.
- D. Bratkowska, R. M. Marcé, P. A. G. Cormack, D. C. Sherrington, F. Borrull and N. Fontanals, J. Chromatogr. A, 2010, 1217, 1575–1582 CrossRef CAS PubMed.
- F. Maya and F. Svec, J. Chromatogr. A, 2013, 1317, 32–38 CrossRef CAS PubMed.
- J. Germain, J. Hradil, J. M. J. Fréchet and F. Svec, Chem. Mater., 2006, 18, 4430–4435 CrossRef CAS.
- R. Vinodh, E. M. Jung, M. Ganesh, M. M. Peng, A. Abidov, M. Palanichamy, W. S. Cha and H. T. Jang, J. Ind. Eng. Chem., 2015, 21, 1231–1238 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |