
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of polystyrene-grafted cellulose acetate copolymers via nitroxide-mediated polymerization†
Guillaume
Moreira
a,
Elisabetta
Fedeli
a,
Fabio
Ziarelli
b,
Donatella
Capitani
c,
Luisa
Mannina
cd,
Laurence
Charles
a,
Stéphane
Viel
 a,
Didier
Gigmes
a and
Catherine
Lefay
*a
a,
Didier
Gigmes
a and
Catherine
Lefay
*a
aAix Marseille Université, CNRS, Institut de Chimie Radicalaire UMR 7273, campus Saint Jérôme, Avenue Escadrille Normandie-Niemen, Case 542, 13397 Marseille Cedex 20, France. E-mail: catherine.lefay@univ-amu.fr
bAix Marseille Université, Centrale Marseille, CNRS, Fédération des Sciences Chimiques FR 1739, F-13397 Marseille, France
cLaboratorio NMR “Annalaura Segre”, Istituto di Metodologie Chimiche, Centro Nazionale delle Ricerche, I-00015 Monterotondo, Roma, Italy
dDipartimento di Chimica e Tecnologie del Farmaco, Sapienza Università di Roma, Piazzale Aldo Moro 5, I-00185 Rome, Italy
First published on 16th June 2015
Abstract
Cellulose acetate-g-polystyrene (CA-g-PS) grafted copolymers have been synthesized by nitroxide-mediated polymerization (NMP) under homogeneous conditions by using the 1,2-intermolecular radical addition (1,2-IRA) methodology to introduce the SG1-based alkoxyamine BlocBuilder MA (BB) on the polysaccharide backbone. A preliminary study conducted on model sugar-based alkoxyamines proved that whatever the position of the alkoxyamine on the sugar unit, the number of sugar units and the presence of OH groups, one could expect a controlled/living polymerization process. This was confirmed by the graft polymerization of styrene from the BB-modified cellulose acetate backbone with two different grafting ratios (3 and 20%) and different grafted chain lengths (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 80000 g mol−1 targeted Mn). The grafted copolymers were carefully analyzed, in particular, by DOSY NMR to confirm their grafted structure.
000 and 80000 g mol−1 targeted Mn). The grafted copolymers were carefully analyzed, in particular, by DOSY NMR to confirm their grafted structure.
Introduction
Cellulose is the most abundant raw material on earth.1 Despite all its well-established and interesting properties such as biocompatibility and biodegradability, cellulose suffers from inherent drawbacks. To circumvent its poor solubility in usual organic solvents, one method consists in chemically modifying cellulose into cellulose derivatives. Cellulose acetate, for instance, offers the advantage of being soluble in THF and DMF.To improve the mechanical properties (like thermoplasticity and dimensional stability) of cellulose and cellulose derivatives, different strategies of cellulose grafting have been studied: the “grafting from” method based on the growth of synthetic polymer chains from the polysaccharide backbone and the “grafting to” method, which consists of coupling pre-synthesized and functionalized synthetic polymer chains onto the polysaccharide backbone.2,3 The first method is currently the most employed one because it allows higher grafting densities to be reached due to a lower steric hindrance as compared to the “grafting to” strategy. In addition, different methods of graft (co)polymerization of synthetic monomers have been reported, which can be divided into three groups: (i) free radical polymerization, (ii) living ionic and ring-opening polymerization and (iii) controlled/living radical polymerization (CLRP).
The recent advent of CLRP techniques such as nitroxide-mediated polymerization (NMP),4 atom-transfer radical polymerization (ATRP)5,6 and reversible-addition fragmentation chain transfer (RAFT),7 which are compatible with a large range of (functional) monomers, has opened new prospects in the polysaccharide-based material area. By controlling the grafting density, the length and the composition of the grafted chains, new hybrid materials with finely tuned properties are now available that enlarge the range of sustainable materials. In particular, for cellulose (and polysaccharides in general), ATRP2 has been the most employed CLRP technique. To our knowledge, only Daly et al.8 used NMP in a grafting approach to modify a cellulose derivative, hydroxyisopropyl cellulose, by using the 2,2,6,6-tetramethyl-1-piperidinyloxy radical (TEMPO) as a control agent and Barton ester intermediates to initiate the polymer chains from the backbone. Nevertheless, NMP is nowadays maybe one of the most convenient CLRP techniques since it is a monocomponent initiating system that does not require any catalyst. Among the nitroxides developed for NMP, the acyclic β-phosphorylated nitroxide N-(2-methylpropyl)-N-(1-diethylphosphono-2,2-dimethylpropyl)-N-oxyl named SG1 by Tordo et al.9,10 is now recognized as the most potent one since it can be used to control the polymerization of styrenics, acrylates, acrylic acid and methacrylates (in the presence of a small amount of acrylonitrile or styrene).11–13 We have recently reviewed14 that the 1,2-intermolecular radical addition (1,2-IRA) of the SG1-based BlocBuilder MA alkoxyamine (BB) developed by Arkema onto activated olefins is an efficient method to introduce the SG1 nitroxide onto polymers. This method allows elaboration of complex architectures such as block or star copolymers as well as chitosan-based grafted copolymers. In particular, chitosan-g-poly(methyl methacrylate-co-acrylonitrile) and chitosan-g-poly(styrene sulfonate) graft copolymers were synthesized by our group under heterogeneous conditions.15 More recently, Cunningham et al.36 reported the synthesis of chitosan-g-poly(butyl acrylate) copolymers under homogeneous conditions. They also reported the synthesis of chitosan-g-polystyrene copolymers but these precipitated during the polymerization. In conclusion, cellulose derivatives and, in particular, cellulose acetate have never been modified by polystyrene chains synthesized by a SG1-based NMP grafting from method under homogeneous conditions.
In regard to the literature, the main aim of this work was first to prove that the 1,2-IRA is a powerful functionalization method to elaborate under homogeneous conditions cellulose acetate-g-polystyrene grafted copolymers by NMP (Scheme 1).
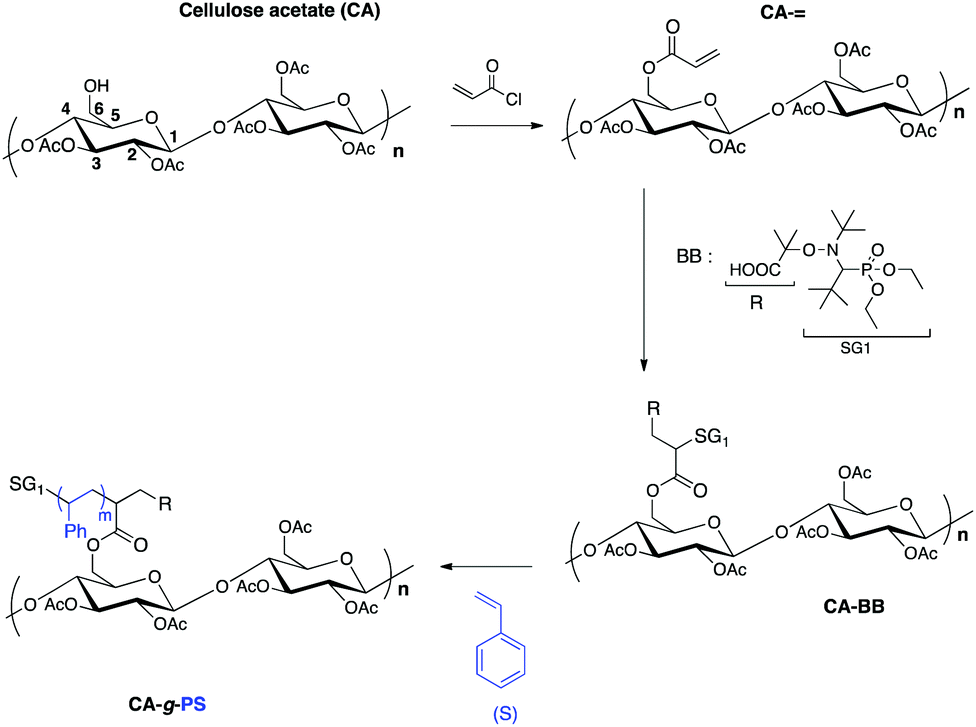 | ||
| Scheme 1 Synthesis of cellulose acetate-g-polystyrene grafted copolymers by SG1-based nitroxide-mediated polymerization via 1,2-intermolecular radical addition of BlocBuilder MA. The grafting has been arbitrarily represented on the C6 but other grafting positions are possible (on C2 or C3). | ||
Another challenge here was to reach, via this grafting methodology, reasonable Mn values (>10000 g mol−1) at a reasonable conversion (>20%). The last goal of this work was to give a convincing proof of the grafted character of the cellulose acetate-g-PS copolymers by using the DOSY NMR technique.
After a preliminary study on sugar and BB-based alkoxyamines to determine the influence of several parameters on the activation energy (Ea(C-ON)) of the homolytic dissociation of the alkoxyamines, we showed that the 1,2-IRA was an efficient method to finely control the grafting density of the alkoxyamine moieties onto the cellulose acetate backbone. The subsequent NMP of styrene enabled the synthesis of well-defined cellulose acetate-g-polystyrene copolymers with different grafted synthetic chain lengths and grafting ratios. DOSY NMR experiments (recorded under high resolution magic angle spinning conditions) clearly proved the grafted structure of the copolymers, whereas the grafting ratio was determined by solid-state NMR. Finally, the SEC analysis of the PS chains after hydrolysis of the polysaccharide backbone proved the controlled/living character of the polymerization.
Experimental part
Materials
Triethylamine (Et3N, Aldrich, 99+%), cellulose acetate (Aldrich, degree of substitution (DS) of 2.45, average Mn 30000 g mol−1), acryloyl chloride (Aldrich, 97+%), D-glucose (Aldrich, 99.5%), D-cellobiose (Alfa Aesar, 98+%), lithium chloride (LiCl, Aldrich, 99+%), lithium bromide (LiBr, Aldrich, 99+%), sodium perchlorate (NaClO4, Aldrich, ACS reagent ≥98%), tetrabutylammoniumhexafluorophosphate (TBAPF6, Fluka, 98+%), (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO, Aldrich, 98%), styrene (Aldrich, 99+%), N,N-dimethylformamide (DMF, SDS, analytical grade), N,N-dimethylacetamide (DMA, Aldrich, 99.5+%), sodium chloride (NaCl, Carlo Erba), ethyl acetate (EtOAc, Carlo Erba), pentane (SDS, technical grade), methanol (MeOH, SDS, technical grade), dichloromethane (CH2Cl2, Carlo Erba), anhydrous ethanol (EtOH, Carlo Erba), glacial acetic acid (Carlo Erba), chloroform (CHCl3, Carlo Erba), diethyl ether (Et2O, Aldrich), pyridine (Alfa Aesar), triethyl phosphite (P(O)(OEt)3, Aldrich, 99+%), tert-butylbenzene (tBuPh, Aldrich, 99%), trityl chloride (Aldrich, ≥97%), acetic anhydride (Aldrich, ≥99%), hydrogen bromide (Aldrich, 33 wt% in acetic acid), di-tert-butyl dicarbonate (Aldrich, ≥99%), acetyl bromide (Aldrich, 99%), silver carbonate (Alfa Aesar, 99.5%), 1,4-dioxane (Aldrich, ≥99%), acetone (Aldrich), perchloric acid (Aldrich), potassium hydroxide (VWR), sodium carbonate (Aldrich), sodium hydrogen carbonate (VWR), sodium hydroxide (Carlo Erba), and magnesium sulfate (Aldrich) were used as received. The SG1 nitroxide and BlocBuilder MA alkoxyamine were kindly supplied by Arkema. Tetrahydrofuran (THF, SDS, Purex for analysis) was distilled over sodium before use.
Analytical techniques
Solid-state NMR was performed on a Bruker Avance 400 MHz spectrometer (magnetic field 9.4 T) operating at 13C and 31P resonance frequency of 101.6 MHz and 162.1 MHz, respectively. All experiments were performed with a commercial Bruker double-bearing probe with zirconium dioxide rotors of 4 mm outer diameter.For the 13C CPMAS experiment, the CP technique was applied during Magic Angle Spinning (MAS) of the rotor at 9 kHz. To obtain the best quantitative results, the contact time was optimized at 3 ms and a ramped 1H-pulse starting at 100% power and decreasing until 50% was used during the polarization transfer. To improve the resolution, a dipolar decoupling GT8 pulse sequence was applied during the acquisition time. A good signal-to-noise ratio in the 13C CPMAS experiment was obtained with 8 K scans using a recycle delay (2.5 s) that was set to 5 times the 1H spin-lattice relaxation time.
Quantitative 31P NMR spectra were obtained with a Single Pulse Experiment (SPE) using a recycle delay (40 s) that was equal to five times the longest 31P spin-lattice (T1) relaxation time. To optimize the quantification, about 20 mg of the sample were placed in the active central volume of 4 mm zirconium rotors by using two PTFE inserts, and spun at a magic angle spinning rate of 10 kHz. The number of scans was set between 128 and 400, depending on the alkoxyamine concentration. The 13C chemical shifts were referenced to tetramethylsilane and calibrated with the glycine carbonyl signal (176.5 ppm); the 31P chemical shifts were referenced to 85% aqueous phosphoric acid (0 ppm).
HRMAS and HRMAS DOSY experiments were performed at 300 K using a BRUKER AVANCE-I liquid-state spectrometer from Bruker BioSpin GmbH (Rheinstetten, Germany), and operating at 600.1 MHz for the 1H Larmor frequency. A commercial BRUKER 4 mm double-resonance 13C/1H High Resolution MAS (HRMAS) probe head was used, which was equipped with a 2H lock channel and a magnetic field gradient coil (aligned along the magic angle spinning axis). All HRMAS experiments were conducted at a spinning rate of 4 kHz. For HRMAS experiments, 4 mm (external diameter) zirconia rotors equipped with Teflon inserts were used. This ensured that the active volume of the NMR coil of the HRMAS probe head (20 μL) was the same as the volume of the sample that was effectively analyzed. The rotors were capped with a Kel-F drive tip. The presence in the HRMAS probe head of a gradient coil allowed the use of a gradient pulse for coherence selection and solvent signal suppression. The proton 90° RF pulse was 8 μs for the HRMAS probe head. Diffusion NMR experiment (DOSY) spectra were recorded using the bipolar gradient longitudinal eddy current delay pulse sequence. The gradient coil of the HRMAS probe head was calibrated to be 6 G cm−1 A−1.16 The diffusion time and the gradient pulse durations were optimized to achieve a decay in the signal intensity at the highest gradient strength of at least 95%. Typically, 16 gradient values were used from 2% to 95% of the maximum gradient strength.
The size-exclusion chromatography (SEC) analyses were performed by using an EcoSEC system from TOSOH equipped with a differential refractometer detector. THF was used as an eluent with 0.25 vol% toluene as a flow marker at a flow rate of 0.3 mL min−1 after filtration on Alltech PTFE membranes with a porosity of 0.2 μm. The column oven was kept at 40 °C, and the injection volume was 20 μL. One ResiPore pre-column (50 mm, 4.6 mm) and two ResiPore columns (250 mm, 4.9 mm) from Polymer Laboratories were used in series. The system was calibrated by using polystyrene standards in the range 100–400000 g mol−1, purchased from Agilent.
Differential scanning calorimetry (DSC) was carried out on a TA DSC Q20 using a heat/cool/heat cycle from 50 to 250 °C. After the cooling cycle, samples were kept at 50 °C for 1 min before being heated to 250 °C. The heating and cooling rates were respectively 10 °C min−1 and 5 °C min−1. Only the second heating cycle was used to compare the different polymer samples.
Methods
000 g mol−1 and 80000 g mol−1 (at 100% conv.). The macroinitiator, the free SG1 and the styrene monomer were dissolved in DMF. The solution was poured into glass prescored ampoules and was degassed by three dry/freeze cycles. Each ampoule was sealed under vacuum and introduced into an oil bath at 120 °C. The time of the reaction was varied from 30 minutes to 24 h in order to study the polymerization kinetics. The reaction was stopped by cooling and breaking the ampoule. The solution was poured into cold methanol and the precipitated CA-g-PS copolymer was collected by filtration and dried under vacuum for several hours. The monomer conversion was measured by 1H NMR and the PS molecular weight was determined by SEC after hydrolysis of the cellulose acetate backbone. The typical NMR characterization of the grafted CA-g-PS copolymer is given in the ESI.†
Results and discussion
Influence of the alkoxyamine structure on its homolytic bond dissociation activation energy (Ea)
Since it was impossible to know whether each hydroxyl function (OH group on carbon 2, 3 or 6, Scheme 1) was acetylated in the commercially available cellulose acetate polysaccharide, it was consequently impossible to determine which residual hydroxyl function would be modified into an acrylate function and then modified by the 1,2-IRA of BB. The homolytic dissociation rate constant (kd) of alkoxyamines is governed by the structure of the alkyl moiety, with a combination of polar, steric, and stabilization effects.18,19 This means that depending on how the alkoxyamine is linked to the polysaccharide backbone, it could be possible to obtain different kd values, i.e., different activation energy values. Thus, before performing the 1,2-IRA of BB units onto acrylate-functionalized cellulose acetate chains, we investigated the influence of several parameters on the homolytic bond dissociation energy Ea(C-ON) of glucose-based model alkoxyamines, namely (i) the position of the SG1 in the sugar-based alkoxyamine (i.e. acrylation of a primary or secondary hydroxyl function), (ii) the influence of the presence of an unprotected hydroxyl function that can influence the polarity of the alkyl moiety and (iii) the influence of the number of sugar units (glucose or cellobiose-based alkoxyamines).It has been shown that, for a given monomer and nitroxide, the dissociation rate constant of the initiating alkoxyamine determines the degree of control over the polymerization.20,21 If all the alkoxyamine units (on a given multialkoxyamine compound) exhibit distinct Ea values (hence distinct bond dissociation rates), it will be impossible to obtain a controlled polymerization process because all the polymer chains will not be initiated at the same time.
To study the influence of the alkoxyamine structure onto the Ea(C-ON) value, four acetylated glucose-based alkoxyamines (Fig. 1) have been prepared from D-glucose. The synthesis and NMR characterization of the alkoxyamines 1, 2, 3 and 4 are described in the ESI.† The ESR analysis22 of the first two alkoxyamines gave the same Ea(C-ON) (123–124 kJ mol−1) proving that the alkoxyamine position on the sugar unit (i.e., mimicking the coupling of the acryloyl chloride onto primary and secondary alcohols respectively) had no influence on the kinetics of the homolytic bond dissociation. The number of sugar units in the alkoxyamine (alkoxyamines 2 and 3) as well as the protection or not of the hydroxyl functions by acetylation (alkoxyamines 3 and 4) did not affect the Ea(C-ON) value either (123–124 kJ mol−1).
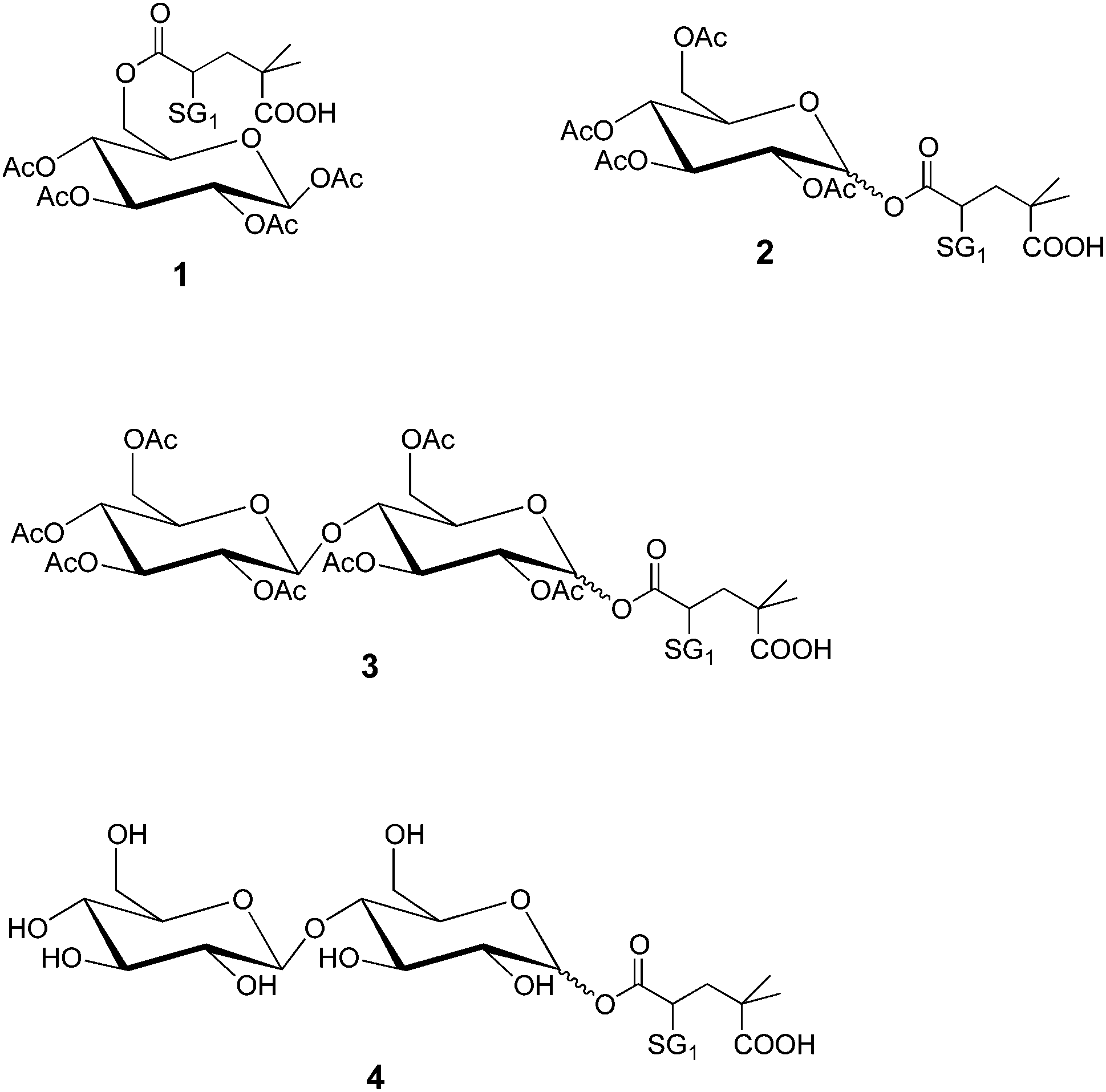 | ||
| Fig. 1 BB and glucose-based alkoxyamine. | ||
It is here worth noting that these Ea(C-ON) values are significantly higher than those corresponding to the free BlocBuilder MA (112 kJ mol−1)4 and are in good agreement with those measured in the case of the 1,2-IRA of the BB onto n-butyl acrylate (125 kJ mol−1).
In conclusion, even though it is not possible to control the position of the SG1-based alkoxyamine onto the sugar units or along the polysaccharide chain, and the number of free hydroxyl functions on the cellulose acetate-based macromolecular multialkoxyamine, this should not be detrimental to the success of the further NMP process and the elaboration of well-defined grafted copolymers.
Whatever the position, the bond dissociation energy Ea(C-ON) of all the grafted alkoxyamines is close to 123–124 kJ mol−1. This point is of prime importance to achieve a controlled/living nitroxide-mediated polymerization process. This conclusion should be valid whatever the polysaccharide used, provided the grafting of the alkoxyamine occurred via 1,2-IRA onto acrylate functionality.
Synthesis of the macromolecular multiinitiator cellulose acetate-BB (CA-BB)
Two macromolecular multialkoxyamines CA-BB with two different SG1 grafting ratios have been prepared by a two-step procedure: (i) acrylation of the hydroxyl functions to introduce activated olefin onto the polysaccharide backbone (CA-=) and (ii) 1,2-IRA of the BB alkoxyamine onto the so-grafted acrylate functions to give CA-BB (Scheme 1). This method is based on the decomposition of the starting BB alkoxyamine to generate alkyl radicals that will then add onto an activated double bond (here an acrylate function) that is then trapped irreversibly by the in situ generated SG1 nitroxide. The final secondary alkoxyamine is therefore more stable than the starting tertiary one at the temperature used to perform the 1,2-IRA as shown in Scheme 2. In this study, two acrylated ratios have been prepared, namely 3 and 20%, which means that 3 and 20 acrylate functions have been grafted per 100 sugar units respectively.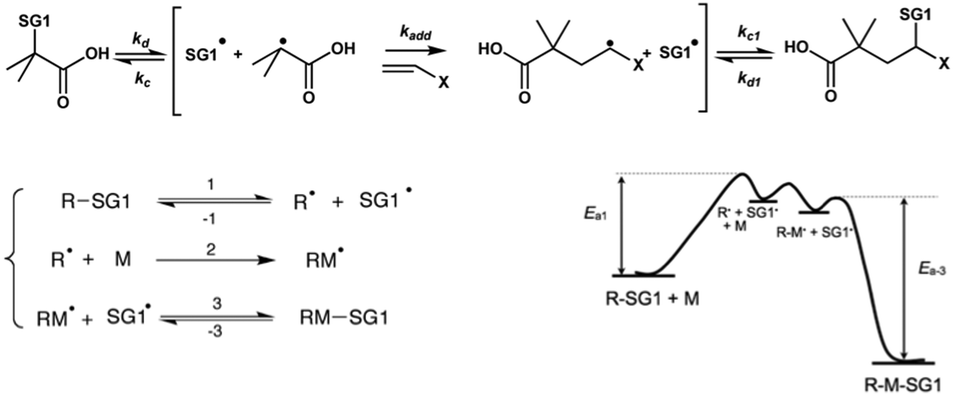 | ||
| Scheme 2 Mechanism of the 1,2-intermolecular radical addition. | ||
Since the cellulose acetate we used in this work has a degree of substitution (DS) of 2.45, this means that the sugar unit is at 55% diacetylated and at 45% triacetylated. The highest activated olefin functionalization degree that could be obtained is then 55%. We decide not to target such a high grafting ratio to avoid problems of steric hindrance when polymerizing. The acrylate grafting ratio has been measured by 13C solid state NMR by integrating the two vinylic carbons (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C at 130 ppm) versus the anomeric carbon C1 of the polysaccharide at 100 ppm (Fig. 2a and c). The peaks at 70–80, 60 and 20 ppm correspond respectively to the carbons C2–5, C6 and CH3 of the initial cellulose acetate. The precise peak assignment of acrylated cellulose acetate (CA-=) and BlocBuilder-modified cellulose acetate (CA-BB) is given in the ESI.†
C at 130 ppm) versus the anomeric carbon C1 of the polysaccharide at 100 ppm (Fig. 2a and c). The peaks at 70–80, 60 and 20 ppm correspond respectively to the carbons C2–5, C6 and CH3 of the initial cellulose acetate. The precise peak assignment of acrylated cellulose acetate (CA-=) and BlocBuilder-modified cellulose acetate (CA-BB) is given in the ESI.†
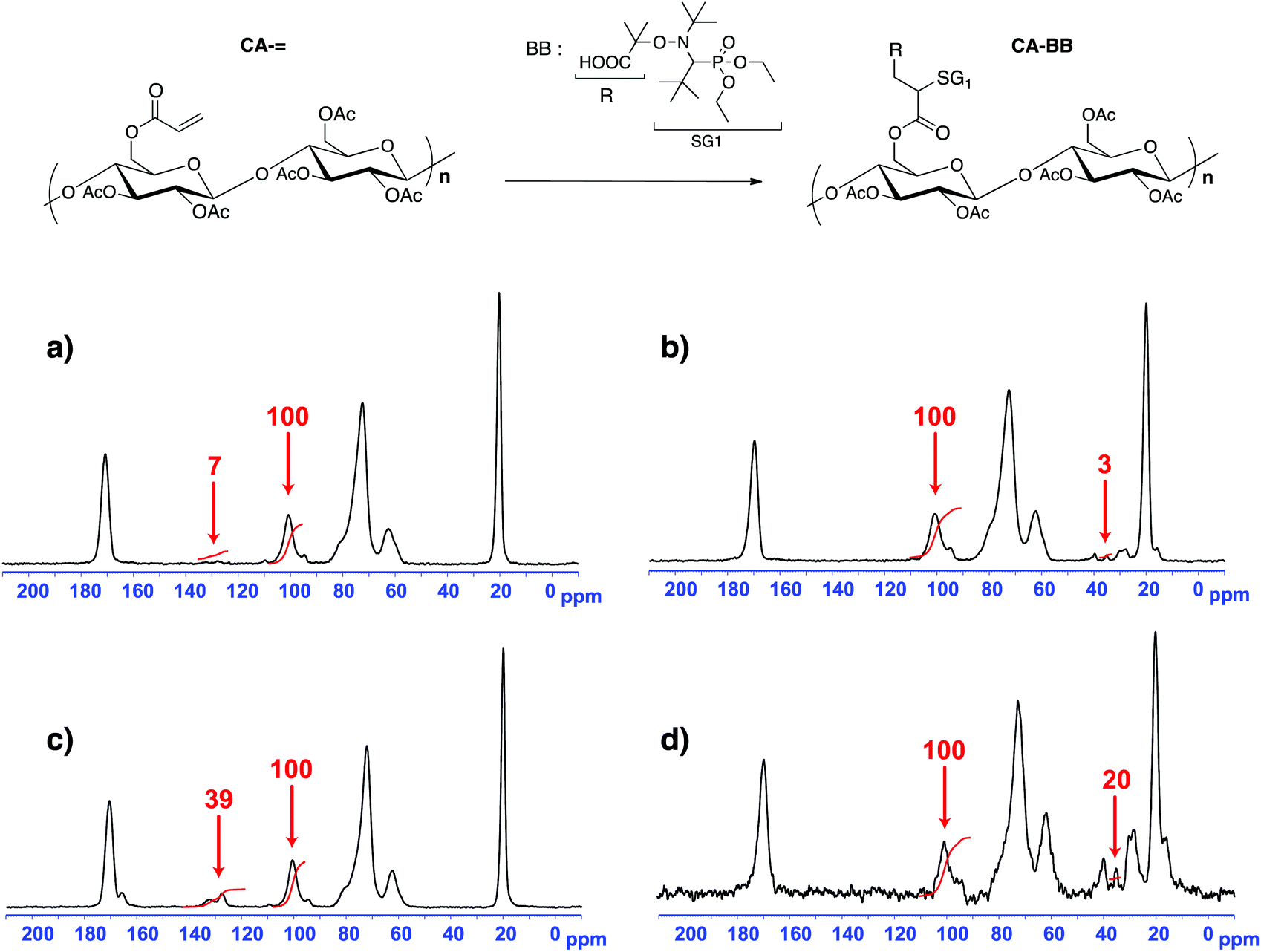 | ||
| Fig. 2 13C solid state NMR of cellulose acetate with a grafting ratio of (a) 3% of acrylate functions; (b) 3% of SG1 moieties; (c) 20% of acrylate functions and (d) 20% of SG1 moieties. The red numbers correspond to the integration values. The integration value of the anomeric carbon C1 of the polysaccharide at 100 ppm was set at 100. | ||
The acrylated cellulose acetate with a grafting ratio of 3% has been prepared with an excess of acryloyl chloride (7.4 mmol) and Et3N (9.3 mmol) compared to the theoretical value for such a grafting ratio, namely 1.1 mmol for 10 g of CA (DS of 2.45). With a DS of 2.45, the molar mass of one sugar unit of the initial cellulose acetate was evaluated to be 265 g mol−1. Once the experimental conditions have been set up for the 3% acrylated degree, conditions to obtain a 20% grafting ratio were obtained by multiplying the previous acryloyl chloride and Et3N excesses by the corresponding ratio (i.e. 20/3).
The 1,2-IRA of the BB onto the acrylate functions gave the corresponding CA-BB macromolecular multialkoxyamines with a yield close to 100% as measured by 13C solid state NMR by integrating one carbon of the BB versus the anomeric carbon C1 of the cellulose acetate (Fig. 2b and d). This methodology based on the 1,2-IRA is therefore an efficient synthesis method to precisely control the functionalization degree of cellulose acetate chains and the number of BB-based alkoxyamines per polysaccharide chain. Such a control is necessary in order to prepare grafted copolymers with a well-controlled architecture by NMP. It has to be noted that, within experimental precision, there was a total consumption of the vinylic functions, which is required to avoid cross-linking of the polymer chains.
The 1,2-IRA method is thus an efficient method to prepare macromolecular multialkoxyamines with different grafting ratios and after several purification steps, 13C solid NMR is a method of choice to properly characterize the synthesized macroinitiators.
NMP of styrene initiated by the macromolecular multialkoxyamine CA-BB. Synthesis of the cellulose acetate-g-polystyrene grafted copolymers
The synthesis and characterization of cellulose and cellulose derivative grafted copolymers under homogeneous conditions by CLRP techniques are not straightforward. To our knowledge, only Daly et al.8 reported the grafting of PS chains by NMP under homogeneous conditions onto a cellulose derivative, namely hydroxyisopropyl cellulose. The authors used Barton ester intermediates to subsequently introduce the TEMPO nitroxide onto the polysaccharide backbone. Nevertheless, the controlled/living character of the polymerization was not proven because there was no comparison between the experimental and theoretical Mn values. Concerning the RAFT technique, Stenzel et al.23,24 showed that the synthesis of hydroxyisopropyl cellulose-g-PS grafted copolymers was limited by (i) comb–comb termination events in the case of the R-group approach and (ii) the reduced accessibility of the RAFT group with increasing conversion with the Z-group approach. The ATRP technique has been reported as well to graft PS chains onto ethyl cellulose25,26 and cellulose acetate.27,28 However no real proof of the grafted structure of the copolymer was provided. This is actually a very important point because it is typically difficult to ensure that the bromide derivative has indeed been properly grafted and not simply absorbed on the polysaccharide. It is consequently difficult to prove that the PS chains, which are usually analyzed after hydrolysis of the (supposedly) grafted copolymer, were not simply absorbed as well. Another milestone when synthesizing a grafted copolymer by a CLRP technique is to be able to obtain grafted chains with reasonable molar masses (higher than 10000 g mol−1) and with low PDI values (<1.5).
Two methods can be used to reach this goal whatever the CLRP technique: (i) target a high Mn value and stop the polymerization at low conversion (typically around 5–10%), (ii) target lower Mn values and reach higher conversion (>10%). The first method is usually easier to set up but suffers from spoiling a large amount of monomer.
The aim of this part is to prove that the NMP technique via the 1,2-IRA is an efficient method (i) to prepare well-defined grafted copolymers (low PDI values and experimental Mn values in good agreement with theoretical ones) and (ii) to reach reasonable Mn values at reasonable conversion. The grafted structure of the copolymer will be studied in the next part.
Several CA-g-PS grafted copolymers have been synthesized from the two macromolecular multiinitiator CA-BB (SG1 grafting ratios of 3 and 20%) in the presence of 5 mol% of free SG1. The influence on the polymerization controlled character of the grafting ratio and the initiator concentration was studied by targeting Mn 40000 and 80000 g mol−1. Even if the polymerization of styrene mediated by the SG1 nitroxide usually does not require extra free nitroxide,20 the use of 5 mol% of free SG1 decreases the occurrence of self-termination at the beginning of the polymerization. This is needed to limit the coupling between the polysaccharide chains. 31P NMR was used to determine the molar quantity of alkoxyamine in a given weight of CA-BB. This value was used to determine the monomer quantity and to use it to target a polystyrene molecular weight of 40000 or 80000 g mol−1.
One of the main problems when synthesizing grafted copolymers is to limit the formation of side homopolymer chains and then to eliminate them. We tried to limit the presence of free polystyrene chains in the grafted copolymer by carefully purifying the CA-BB to eliminate eventually adsorbed BB. In addition, after styrene polymerization, the grafted copolymer was carefully purified by Soxhlet extraction with cyclohexane (whatever the experimental conditions, a limited amount of free PS, i.e. <5 wt%, was recovered).
The efficiency of the Soxhlet extraction step in eliminating free PS chains from the grafted copolymer sample is demonstrated by the 13C CPMAS NMR data shown in Fig. 3. This sample was obtained after NMP of styrene (targeted Mn = 80000 g mol−1) initiated by BB at 120 °C for 2 h in DMF in the presence of CA. At the end of the polymerization, the polymers were precipitated in cold methanol. The 13C CPMAS NMR analysis of the residue obtained after 24 h of Soxhlet extraction did not show the presence of any polystyrene chain.
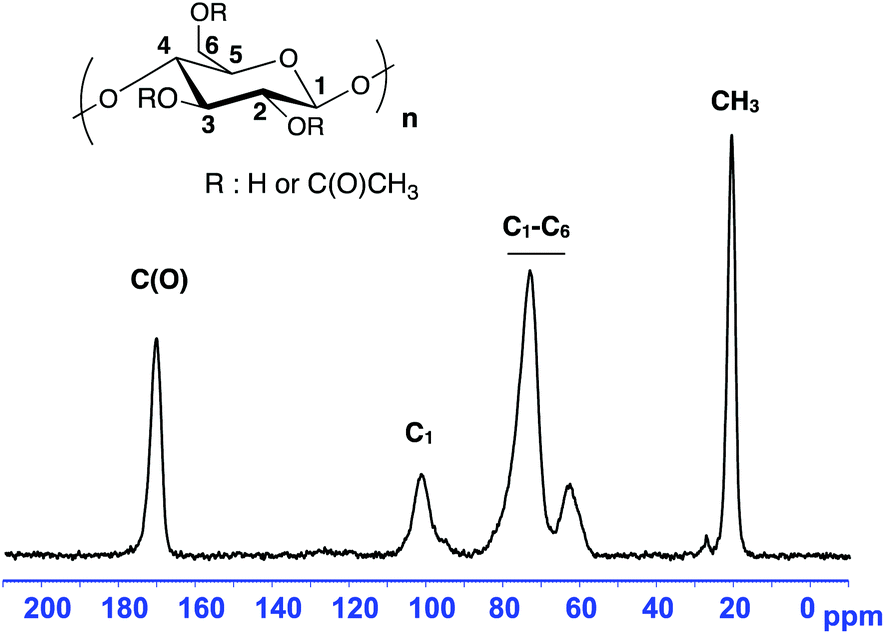 | ||
| Fig. 3 13C CPMAS NMR of the residue (CA) after Soxhlet extraction with cyclohexane. | ||
After the CA-g-PS grafted copolymer purification step, the polysaccharide backbone was hydrolyzed and the released PS chains were analyzed by SEC. The 13C CPMAS of the residue proved the efficiency of the hydrolysis because only PS peaks were observed (see Fig. S23 in the ESI†).
Whatever the grafting ratio (3 or 20%, Fig. 4) or the initial macromolecular initiator concentration (2.46 or 1.86 mmol L−1 in the case of CA-BB3%, Fig. 5), the evolution of the experimental Mn is linear with the conversion and is in good agreement with the theoretical values, and the PDI values remain low (<1.5) up to 30% of conversion when targeting 40000 or 80000 g mol−1 at 100% conv. The polymerization can thus be qualified as controlled up to 30% conv. and living as shown by the shift of the molecular weight distribution towards high values. At higher conversions (>40%), the PDI values increase (>1.6) and the molecular weight distribution broadens (broad distribution at 16 h, Fig. 6) even if the experimental Mn values are still in relatively good agreement with the theoretical ones. The loss of the control and living character could be explained by transfer and/or termination reactions that become predominant after 40% conv.
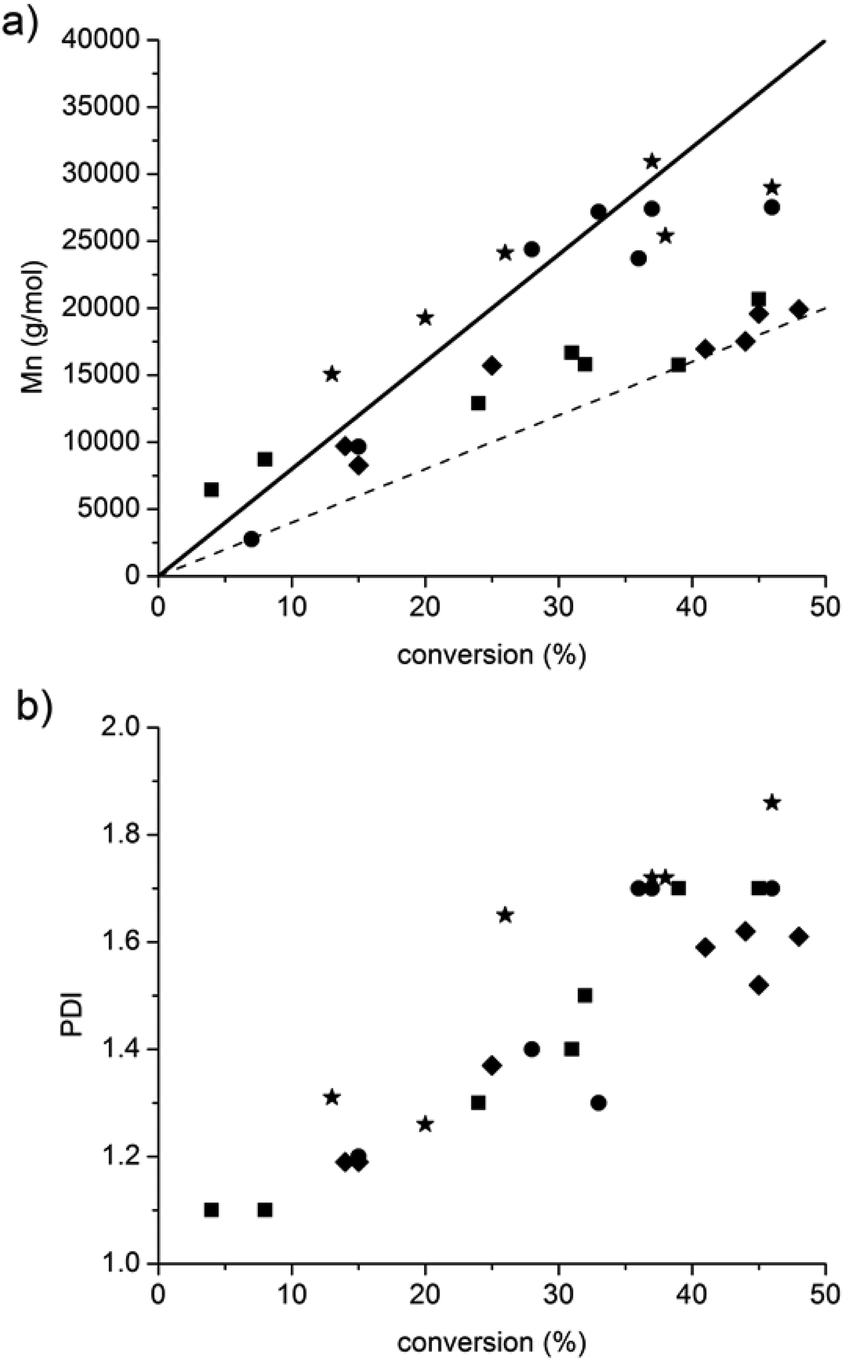 | ||
| Fig. 4 Influence of the grafting ratio (3% and 20%) on the evolution of the Mn and PDI values versus conversion (CA-BB at 2.46 mmol L−1). Molecular weight targeted Mn at 100% conv. in the case of ■: grafting ratio of 3% and targeted Mn at 100% conv. = 40000 g mol−1, ◆: grafting ratio of 20% and targeted Mn at 100% conv. = 40000 g mol−1, ●: grafting ratio of 3% and targeted Mn at 100% conv. = 80000 g mol−1 and ★: grafting ratio of 20% and targeted Mn at 100% conv. = 80000 g mol−1. Dashed and full lines are the theoretical lines when targeting 40000 and 80000 g mol−1 respectively. | ||
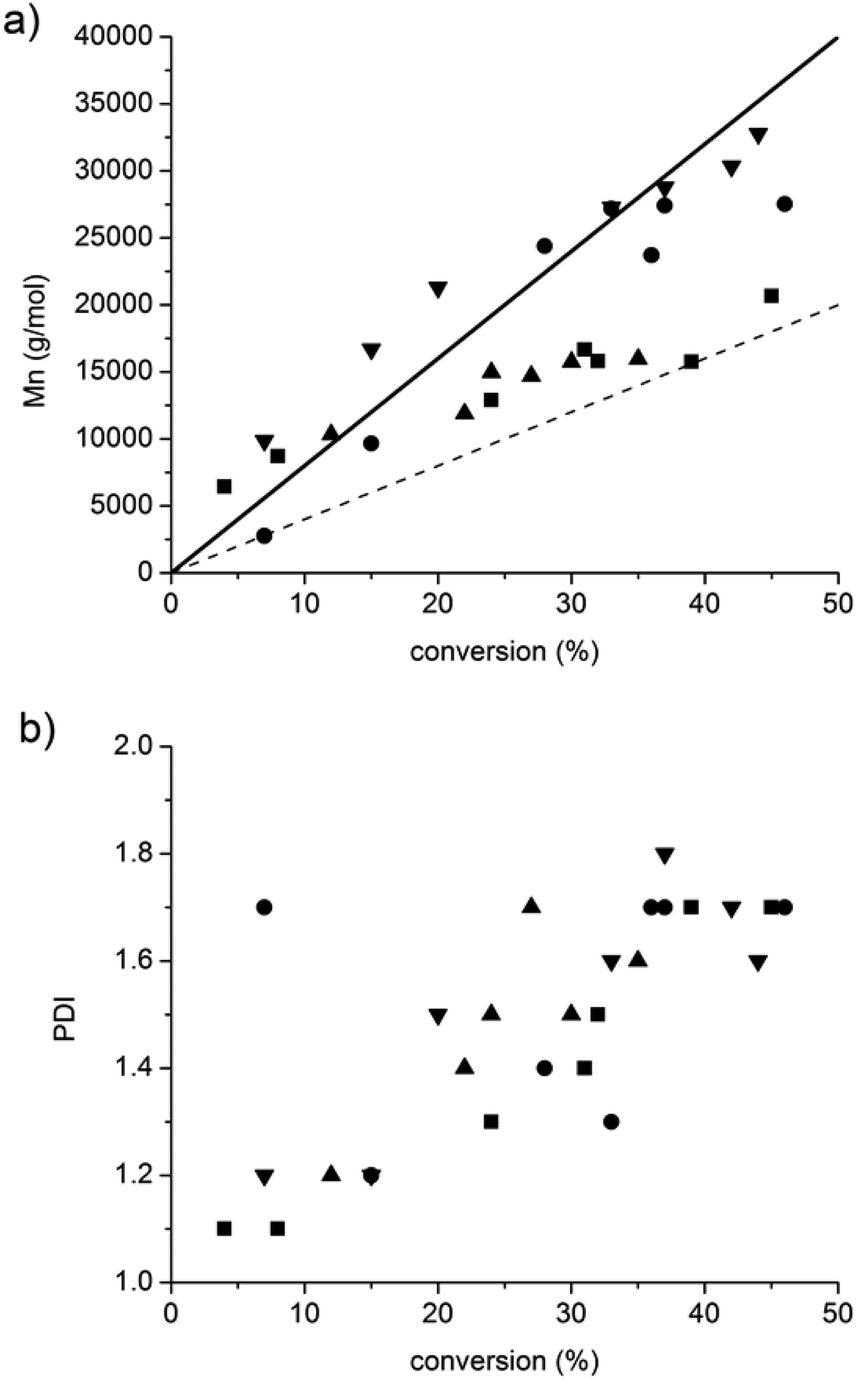 | ||
| Fig. 5 Influence of macromolecular concentration (CA-BB3% at 1.86 or 2.46 mmol L−1) on the evolution of the Mn and PDI values versus conversion. CA-BB3% initial concentration (mmol L−1) and molecular weight targeted at 100% conv. in the case of ■: 2.46 and 40000 g mol−1, ●: 2.46 and 80000 g mol−1, ▲: 1.86 and 40000 g mol−1 and ▼: 1.86 and 80000 g mol−1. Dashed and full lines are the theoretical lines when targeting 40000 and 80000 g mol−1, respectively. | ||
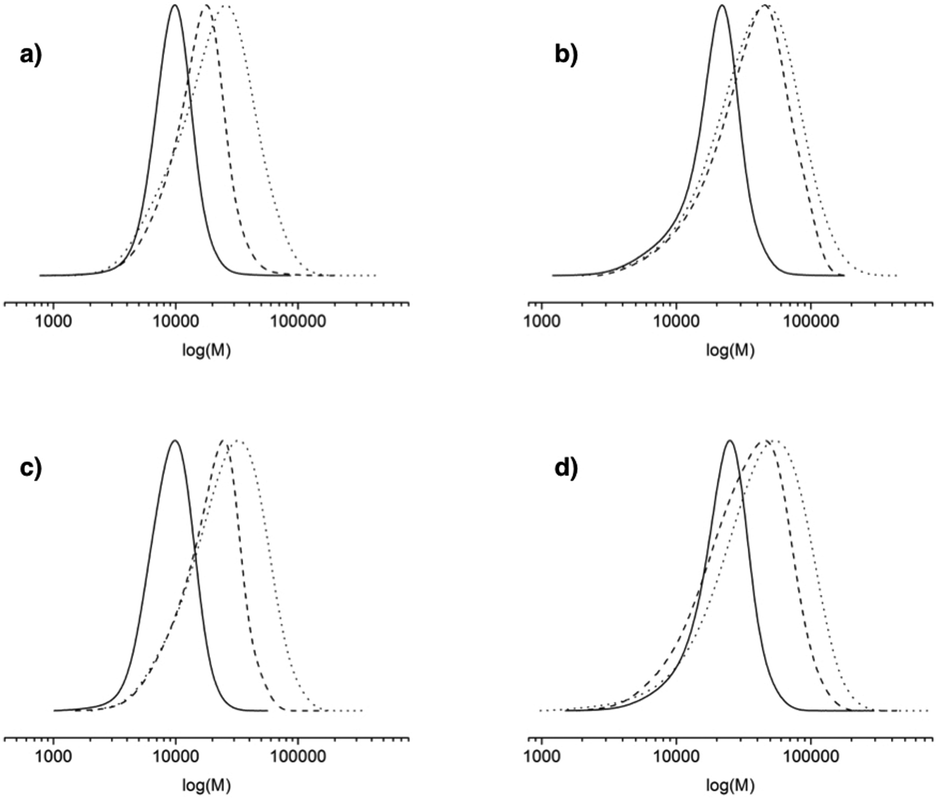 | ||
| Fig. 6 Evolution of the molecular weight distribution versus log(M) for various grafting ratios and targeted Mn at 100% conv.: (a) grafting ratio of 3% and 40000 g mol−1, (b) grafting ratio of 3% and 80000 g mol−1, (c) grafting ratio of 20% and 40000 g mol−1, and (d) grafting ratio of 20% and 80000 g mol−1. Full line: 1 h, dash line: 3 h and dotted line: 16 h. | ||
Fig. 4 and 5 also show that whatever the grafting ratio or the initial macromolecular initiator concentration, it is possible to reach reasonable Mn values (30000 g mol−1) with reasonable conversion (40% when targeting 80000 g mol−1). This point was of high importance to demonstrate the efficiency of the grafting methodology.
Characterization of the grafted copolymer
After the purification step by Soxhlet extraction, the grafted copolymers were analysed by 13C CPMAS solid state NMR. This technique has the advantage of eliminating any issue related to (partial) solubility of the copolymer in a given solvent and hence gives a complete analytical description of the sample. Whatever the grafting ratio and the grafted chain lengths, the 13C CPMAS data proved the presence of both CA and PS in the samples. A typical 13C CPMAS spectrum is given in Fig. 7.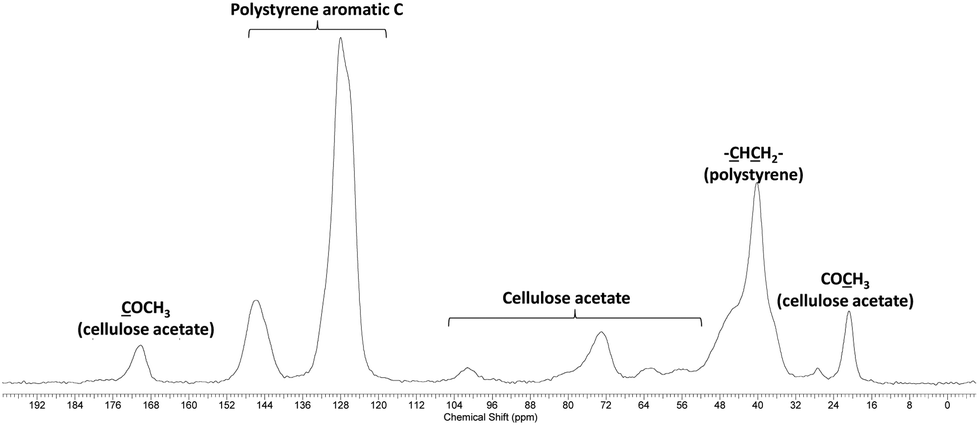 | ||
| Fig. 7 13C CPMAS NMR of the graft copolymer CA-g-PS. | ||
Two model grafted copolymers, namely CA-g-PS (20%, 13000) and CA-g-PS (3%, 21000), were analysed by DSC as well (Fig. 8). The percentage represents the grafting ratio and the second value (13000 or 21000) the Mn of the grafted PS chains. The glass transition temperature (Tg) of PS at approximately 100 °C is clearly observed in the two graft copolymers. Concerning the CA part, the Tg around 190 °C is observed in CA-g-PS3%,21000 but not in CA-g-PS20%,13000 probably because of a too high proportion of PS in the copolymer. The fusion temperature (Tf) of CA around 225 °C is not observed as expected in CA-g-PS20%,13000 and not in CA-g-PS3%,21000 either probably because of a trend toward the amorphous state due to the presence of PS as observed in the case of a blend of CA with PS.29,30
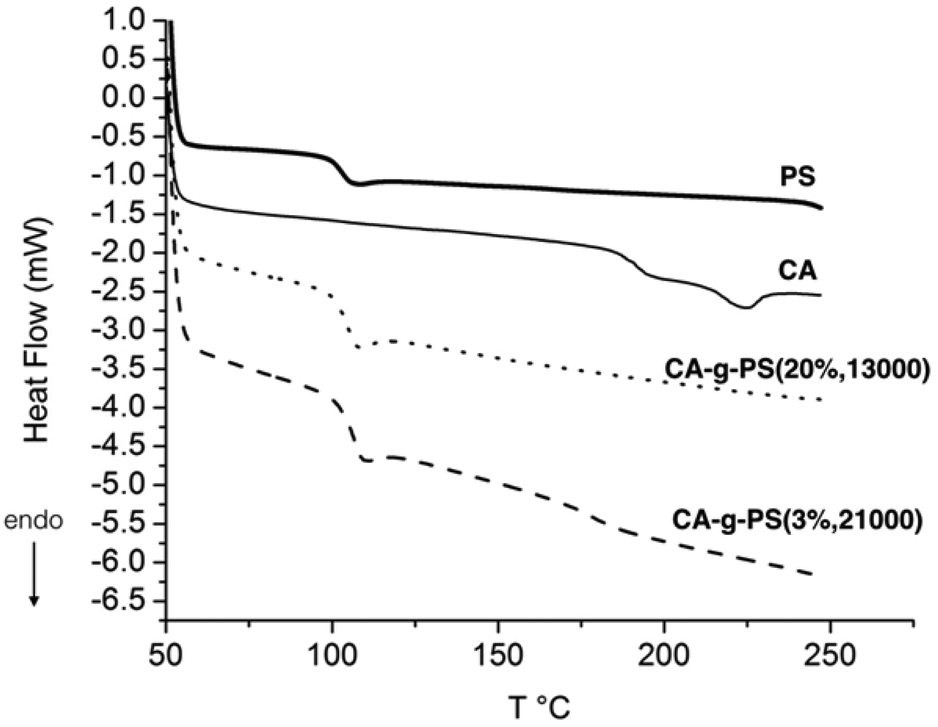 | ||
| Fig. 8 DSC thermograms in the second scan for cellulose acetate (thin line), PS (15000 g mol−1, thick line), CA-g-PS20%,13000 (dotted line, grafting ratio 20%, Mn(PS) = 13000 g mol−1) and CA-g-PS3%,21000 (dashed line, grafting ratio 3%, Mn(PS) = 21000 g mol−1). | ||
One of the challenges when synthesizing graft copolymers is to ascertain the grafted character of the material. While DSC and CPMAS experiments clearly proved the presence of both PS and CA units in the copolymers, which were analysed after Soxhlet extraction (vide supra), they could only provide indirect indications of the grafted structure of the copolymer.
Evidence of the graft structure of the copolymers was provided here by DOSY-NMR. This analytical technique produces two-dimensional correlation maps showing chemical shifts and diffusion coefficients respectively on the horizontal and vertical axes. The NMR signals due to compounds of different molecular sizes can thus be clearly distinguished. DOSY-NMR has already been successfully used to prove the formation of block copolymers31–33 and graft copolymers.34,35
Fig. 9 shows the DOSY maps recorded at each step of the synthesis of CA-g-PS3% grafted copolymers. The DOSY NMR spectra of grafted copolymers with a higher grafting ratio are reported in the ESI.† In the case of the grafted copolymer (Fig. 9c), the correlation spots in the DOSY map of the 1H NMR resonances due to the PS and CA units are all aligned on the same horizontal trace (within experimental errors), which proves that CA and PS chains have the same apparent translational mobility (and hence are linked together). This situation was not observed in the case of a mixture of CA with a PS homopolymer (with a molecular weight similar to that of the grafted PS units in the CA-g-PS copolymer), since CA and PS spots were not aligned (Fig. 9d). Altogether, DOSY NMR provides reliable evidence of the success of the grafted copolymer synthesis (as opposed to simply a mixture of two distinct polymers).
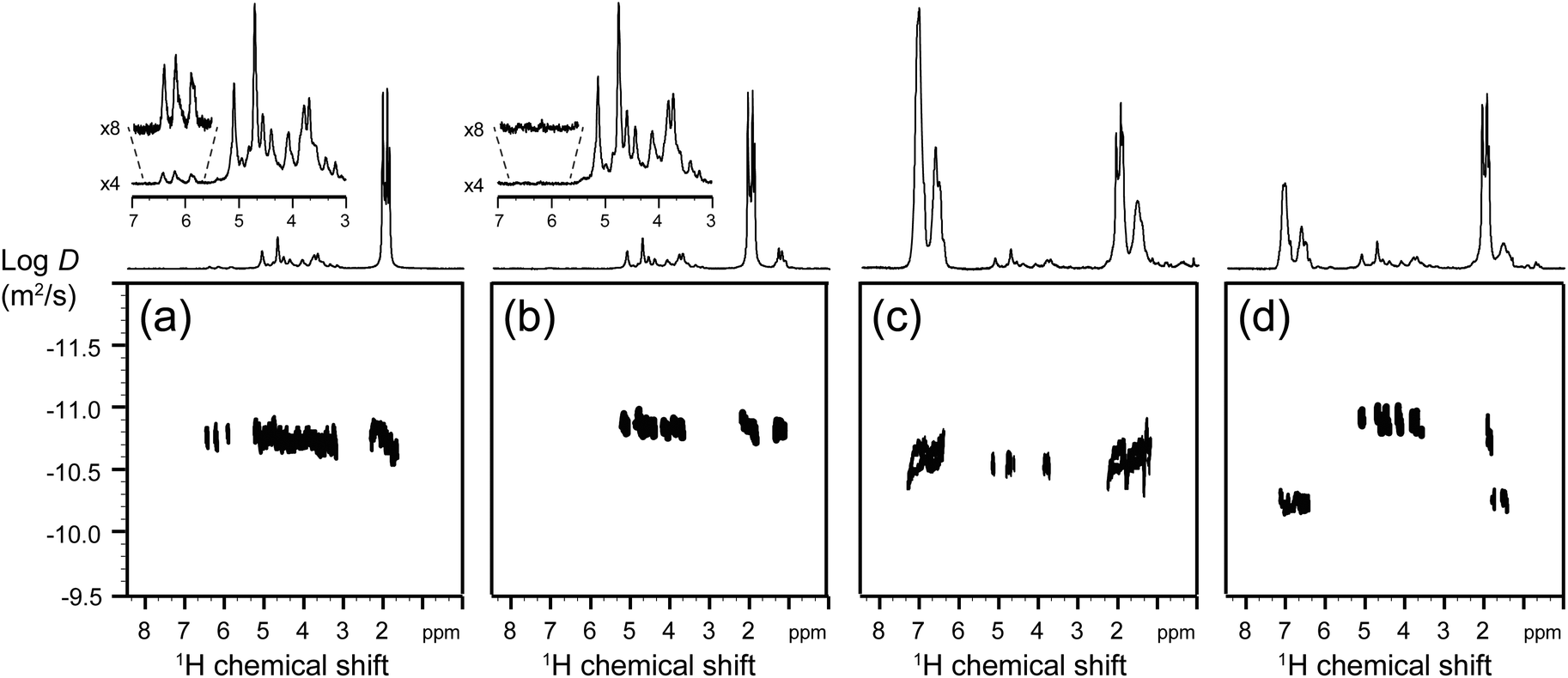 | ||
| Fig. 9 DOSY NMR in the case of 3% grafting ratio of (a) CA-=, (b) CA-BB, (c) CA-g-PS3%, and (d) a mixture of CA and a commercial PS (Mn = 30000 g mol−1). | ||
Conclusions
This work reported that 1,2-IRA is a powerful functionalization method to prepare, under homogeneous conditions, cellulose acetate-g-polystyrene graft copolymers. A preliminary study on different model alkoxyamines showed that whatever the alkoxyamine position on the glucose unit, the number of sugar units and the eventual presence of free hydroxyl groups, all the alkoxyamines have the same homolytic bond dissociation activation energy (Ea = 123–124 kJ mol−1). This was compulsory to aim at synthesizing well-defined graft copolymers by NMP.After the coupling by 1,2-IRA of the SG1-based alkoxyamine called BlocBuilder MA onto acrylate groups previously introduced onto the polysaccharide backbone, the nitroxide-mediated polymerization of styrene allowed well-defined grafted copolymers to be obtained. In addition, whatever the targeted molecular weight (40000 or 80000 g mol−1), it was possible to reach a reasonable molecular weight (typically higher than 15000–25000 g mol−1) with a reasonable styrene conversion (30%) and at the same time with a polydispersity index below 1.5. Up to 30% of conversion, the polymerization of styrene from the cellulose acetate backbone fulfilled the criteria of a controlled/living polymerization (good agreement between experimental and theoretical Mn values; PDI below 1.5; linear increase of the Mn with conversion). While SEC and usual NMR techniques were used to study the controlled/living character of the polymerization, the DOSY NMR technique gave a convincing proof of the grafted structure of the copolymers.
Acknowledgements
ARKEMA is acknowledged for kindly providing BlocBuilder MA alkoxyamine. The authors thank Agence Nationale de la Recherche for financial support, grant number ANR-10-CDII-09. SV acknowledges financial support provided by an invited professorship position from Sapienza Università di Roma.Notes and references
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- M. Tizzotti, A. Charlot, E. Fleury, M. Stenzel and J. Bernard, Macromol. Rapid Commun., 2010, 31, 1751–1772 CrossRef CAS PubMed.
- D. Roy, M. Semsarilar, J. T. Guthrie and S. Perrier, Chem. Soc. Rev., 2009, 38, 2046–2064 RSC.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS PubMed.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- W. H. Daly, T. S. Evenson, S. T. Iacono and R. W. Jones, Macromol. Symp., 2001, 174, 155–163 CrossRef CAS.
- D. Benoit, S. Grimaldi, S. Robin, J. P. Finet, P. Tordo and Y. Gnanou, J. Am. Chem. Soc., 2000, 122, 5929–5939 CrossRef CAS.
- J. P. Finet, F. Le Moigne, Y. Gnanou, P. Nicol, S. Grimaldi, N. Plechot and P. Tordo, Patent PCT WO96/24620, 1996 Search PubMed.
- J. Nicolas, S. Brusseau and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 34–47 CrossRef CAS PubMed.
- B. Charleux, J. Nicolas and O. Guerret, Macromolecules, 2005, 38, 5485–5492 CrossRef CAS.
- J. Nicolas, C. Dire, L. Mueller, J. Belleney, B. Charleux, S. R. A. Marque, D. Bertin, S. Magnet and L. Couvreur, Macromolecules, 2006, 39, 8274–8282 CrossRef CAS.
- D. Gigmes, P. E. Dufils, D. Gle, D. Bertin, C. Lefay and Y. Guillaneuf, Polym. Chem., 2011, 2, 1624–1631 RSC.
- C. Lefay, Y. Guillaneuf, G. Moreira, J. J. Thevarajah, P. Castignolles, F. Ziarelli, E. Bloch, M. Major, L. Charles, M. Gaborieau, D. Bertin and D. Gigmes, Polym. Chem., 2013, 4, 322–328 RSC.
- S. Viel, F. Ziarelli, G. Pages, C. Carrara and S. Caldarelli, J. Magn. Reson., 2008, 190, 113–123 CrossRef CAS PubMed.
- M. Girod, M. Mazarin, T. N. T. Phan, D. Gigmes and L. Charles, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3380–3390 CrossRef CAS PubMed.
- D. Bertin, D. Gigmes, S. R. A. Marque and P. Tordo, Macromolecules, 2005, 38, 2638–2650 CrossRef CAS.
- D. Bertin, D. Gigmes, S. R. A. Marque and P. Tordo, Chem. Soc. Rev., 2011, 40, 2189–2198 RSC.
- F. Chauvin, P. E. Dufils, D. Gigmes, Y. Guillaneuf, S. R. A. Marque, P. Tordo and D. Bertin, Macromolecules, 2006, 39, 5238–5250 CrossRef CAS.
- D. Gigmes, D. Bertin, C. Lefay and Y. Guillaneuf, Macromol. Theory Simul., 2009, 18, 402–419 CrossRef CAS PubMed.
- S. Marque, C. Le Mercier, P. Tordo and H. Fischer, Macromolecules, 2000, 33, 4403–4410 CrossRef CAS.
- M. H. Stenzel, T. P. Davis and A. G. Fane, J. Mater. Chem., 2003, 13, 2090–2097 RSC.
- M. Hernandez-Guerrero, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Eur. Polym. J., 2005, 41, 2264–2277 CrossRef CAS PubMed.
- D. Shen, H. Yu and Y. Huang, Cellulose, 2006, 13, 235–244 CrossRef CAS.
- D. W. Shen, H. Yu and Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4099–4108 CrossRef CAS PubMed.
- P. Vlcek, M. Janata, P. Latalova, J. Dybal, M. Spirkova and L. Toman, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 564–573 CrossRef CAS PubMed.
- P. Vlcek, M. Janata, P. Latalova, J. Kriz, E. Cadova and L. Toman, Polymer, 2006, 47, 2587–2595 CrossRef CAS PubMed.
- C. da Silva Meireles, G. R. Filho, R. M. N. de Assuncao, M. Zeni and K. Mello, J. Appl. Polym. Sci., 2007, 104, 909–914 CrossRef PubMed.
- G. R. Filho, R. C. da Silva, C. D. Meireles, R. M. N. da Assuncao and H. Otaguro, J. Appl. Polym. Sci., 2005, 96, 516–522 CrossRef PubMed.
- C. Lefay, D. Gle, M. Rollet, J. Mazzolini, D. Bertin, S. Viel, C. Schmid, C. Boisson, F. D'Agosto, D. Gigmes and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 803–813 CrossRef CAS PubMed.
- C. Barrere, M. Mazarin, R. Giordanengo, T. N. T. Phan, A. Thevand, S. Viel and L. Charles, Anal. Chem., 2009, 81, 8054–8060 CrossRef CAS PubMed.
- S. Viel, M. Mazarin, R. Giordanengo, T. N. T. Phan, L. Charles, S. Caldarelli and D. Bertin, Anal. Chim. Acta, 2009, 654, 45–48 CrossRef CAS PubMed.
- H. Ohkawa, G. B. W. L. Ligthart, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2007, 40, 1453–1459 CrossRef CAS.
- X. Pan, C. Chen, J. Peng, Y. Yang, Y. Wang, W. Feng, P. Deng and L. Yuan, Chem. Commun., 2012, 48, 9510–9512 RSC.
- O. Garcia-Valdez, R. Champagne-Hartley, E. Saldivar-Guerra, P. Champagne and M. F. Cunningham, Polym. Chem., 2015, 6, 2827–2836 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of alkoxyamines 1, 2, 3 and 4, characterization of acrylated cellulose acetate (CA-=, 13C NMR), characterization of CA-BB (13C and 31P NMR, DOSY NMR). 13C NMR of hydrolysed PS chains, DOSY NMR of CA-g-PS copolymers with a 20% grafting ratio. See DOI: 10.1039/c5py00752f |
| This journal is © The Royal Society of Chemistry 2015 |