
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign of thiol- and light-sensitive degradable hydrogels using Michael-type addition reactions†
Prathamesh M.
Kharkar
a,
Kristi L.
Kiick
*abc and
April M.
Kloxin
*ad
aDepartment of Materials Science and Engineering, University of Delaware, Newark, DE 19716, USA. E-mail: kiick@udel.edu; akloxin@udel.edu
bDepartment of Biomedical Engineering, University of Delaware, Newark, DE 19716, USA
cDelaware Biotechnology Institute, University of Delaware, Newark, DE 19716, USA
dDepartment of Chemical and Biomolecular Engineering, University of Delaware, Newark, DE 19716, USA
First published on 2nd July 2015
Abstract
Injectable depots that respond to exogenous and endogenous stimuli present an attractive strategy for tunable, patient-specific drug delivery. Here, the design of injectable and multimodal degradable hydrogels that respond to externally applied light and physiological stimuli, specifically aqueous and reducing microenvironments, is reported. Rapid hydrogel formation was achieved using a thiol-maleimide click reaction between multifunctional poly(ethylene glycol) macromers. Hydrogel degradation kinetics in response to externally applied cytocompatible light, reducing conditions, and hydrolysis were characterized, and degradation of the gel was controlled over multiple time scales from seconds to days. Further, tailored release of an encapsulated model cargo, fluorescent nanobeads, was demonstrated.
Synthetic hydrogels have been widely employed for a range of applications from sensors,1 membranes,2,3 and lithography4 to sealants,5 adhesives,6,7 and controlled cell culture and drug delivery devices.8–12 In particular, hydrogels formed by step growth mechanisms have garnered significant attention owing to their homogenous network structure, robust mechanical properties, and ease of property modulation using responsive and orthogonal chemistries.13–15 For example, nearly ideal step-growth hydrogels have been formed by copper-catalyzed azide–alkyne,14,16 ring strained alkyne–azide,17,18 tetrazine trans-cyclooctene,19,20 photoinitiated thiol–ene,15,21 and thiol–maleimide reactions22,23 with stoichiometric amount of reactive functional groups. In biomedical applications, control of mechanical and biochemical properties in time and in situ within these materials is key and can be achieved with various combinations of these chemistries for independent control of hydrogel formation and modification24,25 or by engineering hydrogel degradation.8 Degradation (e.g., ester or enzymatic hydrolysis or other mechanisms) is particularly important for the site-specific delivery of encapsulated therapeutics, including proteins, small molecules, and cells, for administering a desired ‘dose’ with high efficacy while mitigating off-target effects.26,27 Complete degradation of the material also alleviates the need for implant ‘removal’ as the cleavage products are cleared after matrix dissolution.28,29 Further, tunable control over degradation in a noninvasive manner achieved with labile chemistries responsive to endogenous (e.g., hydrolysis, pH, thiol concentration) or exogenous (e.g., light) stimuli, as will be described here, may provide a mechanism for tailoring release profiles for patient-specific treatments.
Control over degradation of hydrogels that are covalently crosslinked often has been achieved by incorporation of degradable groups that can undergo ester hydrolysis or enzymatic degradation.8 Recently, hydrogel degradation using chemistries such as retro Michael-type additions with thiol-exchange,30,31 retro Diels–Alder reactions,32,33 or photocleavable chemistries34,35 has received considerable attention as each provides a responsive synthetic handle for engineering rates of degradation. While these reversible or irreversible cleavage reactions provide control over material degradation and cargo release, a hydrogel system that degrades in response to multiple stimuli would provide a unique tool to create complex cargo release profiles. In recent years, a few groups including ours have developed dually degradable hydrogels for modulating drug release profiles based on degradation kinetics.36,37 While these materials allow microenvironment-responsive release, hydrogel-based drug carriers that degrade in response to multiple triggers, both exogenous (e.g., light) and endogenous (e.g., reducing and aqueous microenvironments), would allow for sustained and complex therapeutic release profiles with spatial and temporal control post fabrication. Several pioneering studies have demonstrated the synthesis of different water-soluble photodegradable macromers with various reactive functionalities, including acrylates, azides, alcohols, amines, halides, and carboxylic acids.38–41 Building upon this, we sought to create injectable, photodegradable and microenvironment-responsive hydrogels that can be crosslinked in situ under mild cytocompatible conditions appropriate for in vitro and in vivo applications.
In this communication, we report a degradable hydrogel, sensitive to multiple stimuli, as an injectable cargo carrier with properties that can be tuned in situ. To achieve this, a novel maleimide end-functionalized, photodegradable macromer containing an o-nitrobenzyl ether (o-NB) group was synthesized and characterized. Thiol–maleimide click reactions were utilized for hydrogel formation, as they are highly efficient, occur under mild conditions, and do not require a catalyst;23,42,43 additionally, selection of the thiol-containing functional group imparts degradability in response to specific stimuli found in cellular microenvironments (e.g., reducing conditions and water). The o-NB photolabile group was selected for its susceptibility to degradation over a wide range of cytocompatible irradiation conditions, including long wavelength UV, visible, and two-photon IR light, for user-controlled degradation.34,44 Taken together, the resulting hydrogel undergoes ester hydrolysis in response to an aqueous microenvironment, retro Michael-type reaction with thiol-exchange in response to a reducing microenvironment, and photocleavage in response to externally applied light. This approach provides spatiotemporal control of the properties of cytocompatible hydrogels formed using a catalyst-free Michael-type reaction. Degradation in response to light or aqueous and reducing microenvironments was characterized by monitoring temporal evolution of hydrogel mechanical properties. For proof-of-concept, we also demonstrate how the mode of degradation mediates the release of a model cargo by the incorporation and responsive release of polymeric nanobeads.
Design and synthesis of building blocks with different degradable functional groups
Several multiarm poly(ethylene glycol) (PEG)-based macromers with different reactive functionalities were created for the rational design of biocompatible, responsive hydrogels with distinct modes of controlled degradation, from preprogrammed and responsive to externally tunable. Specifically, four-arm PEG end functionalized with aryl thiols (PEG-4-arylSH) was reacted with four-arm photodegradable PEG end-functionalized with maleimides (PEG-4-PD-MI) by a Michael-type addition reaction (Fig. 1A and B). PEG, a hydrophilic polymer that is FDA-approved for various applications, is easily modified with appropriate reactive functional groups for tailoring of the hydrogel properties while limiting any non-specific protein-material interaction.37 Crosslinking of the photodegradable and thiol-sensitive PEG macromers produces a hydrogel whose main degradable functional units are o-NB and mercaptophenylacetic acid (MPA)-based thioether succinimide linkages, respectively. Upon the application of cytocompatible doses of light, the photolabile o-NB linkage undergoes irreversible cleavage due to photochemically induced photoisomerization yielding ketone and carboxylic acid–based cleavage products (Fig. 1C).45 Nitrobenzyl-based photolabile groups have been used in several applications as protective groups for uncaging of proteins,46 spatiotemporally controlling hydrogel properties,34,38 and the release of live cells or bioactive proteins.41,47,48 Aryl–thiol based thioether succinimide linkages are susceptible to thiol exchange in the presence of glutathione (GSH), which provides a reducing microenvironment (Fig. 1D). The degradation kinetics of mercatophenyl-based thiols in response to reducing microenvironments have been investigated previously by Kiick and coworkers.30,31,37 Since the concentration of glutathione is elevated in carcinoma tissues compared to surrounding healthy tissues,49,50 the incorporation of aryl-thiol-based linkages that cleave in response to glutathione may provide increased release of therapeutics in carcinoma tissues and thus provide higher therapeutic efficacy. In addition, both macromers (PEG-4-PD-MI and PEG-4-arylSH) contain an ester linkage allowing for hydrolysis of the resulting polymeric network under aqueous conditions ultimately leading to complete degradation of the hydrogel in aqueous environments. Overall, the incorporation of multiple cleavable groups that can degrade in response to endogenous and exogenous stimuli presents an attractive strategy for designing drug delivery systems with complex release profiles that could be tuned for the needs of an individual patient.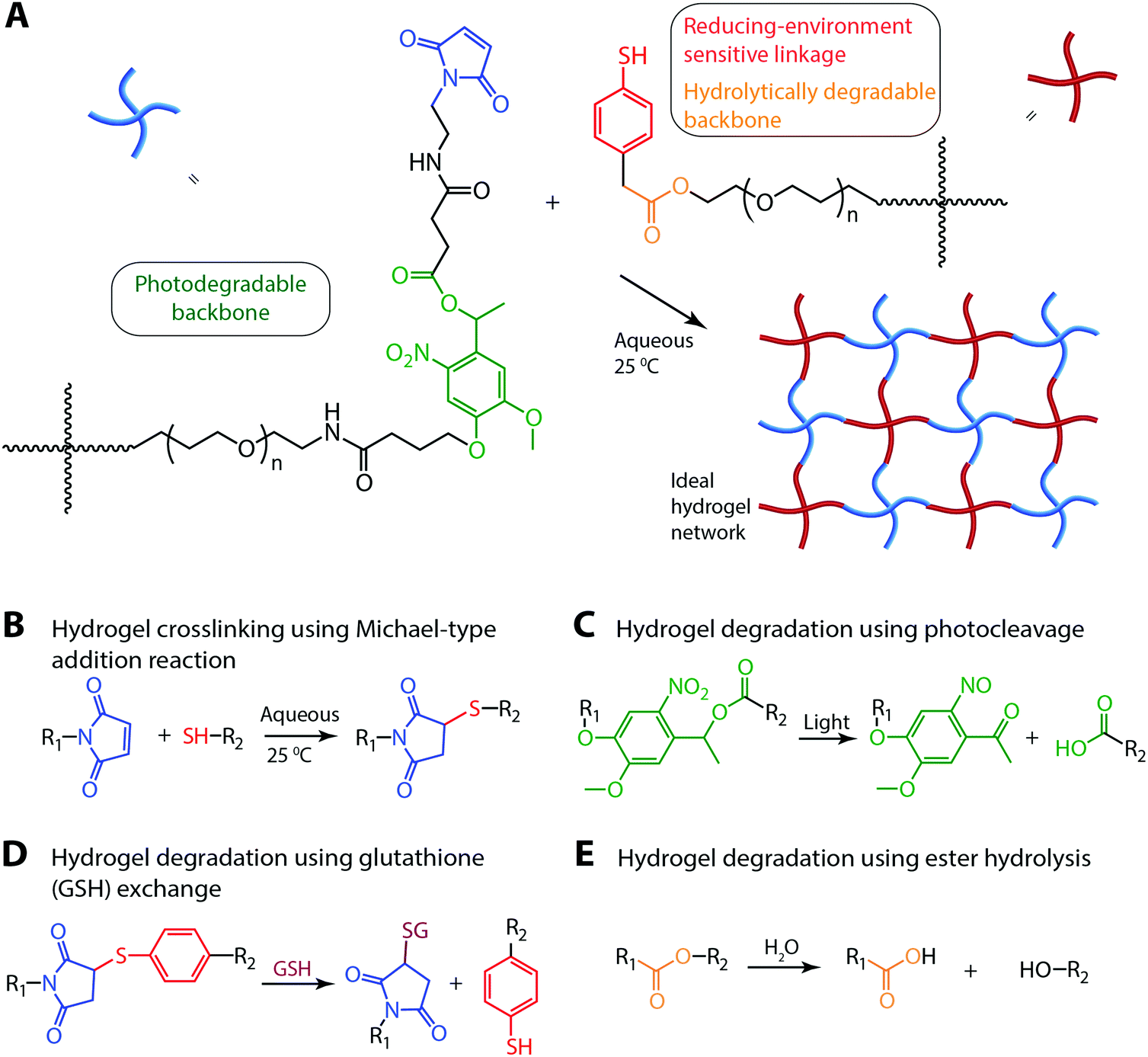 | ||
| Fig. 1 Multimodal degradable hydrogel formation and degradation. (A) Multimodal degradable hydrogels were formed by reacting maleimide and thiol end functionalized four-arm poly(ethylene glycol) macromolecular precursors using (B) a Michael-type addition reaction. (C) An o-nitrobenzyl ether based photodegradable functional group that undergoes irreversible cleavage upon irradiation with UV, visible, or two-photon IR light were incorporated into the backbone of the network, providing externally-triggered, rapid degradation of thick hydrogels by surface erosion. (D) Presence of arylthiol based thioether succinimide linkages allowed responsive, bulk degradation by retro Michael-type reaction, where thiol exchange with exogenous glutathione (GSH) present in a reducing microenvironment leads to irreversible cleavage. (E) Ester linkages were incorporated within both macromolecular precursors for bulk degradation of hydrogels by hydrolysis over longer timescales. | ||
PEG-4-arylSH was synthesized via esterification by reacting mercaptophenylacetic acid with the hydroxyl end groups of four-arm PEG (Scheme S1†). Two primary methods can be used for synthesis of the PEG-4-PD-MI: (i) synthesizing a small photodegradable maleimide monomer for functionalizing the end groups of PEG (or other macromolecules) or (ii) modifying the end groups of PEG by sequential reactions to conjugate the polymer with the photodegradable maleimide group. Our preliminary efforts focused on the former based on prior successful syntheses of an acrylated photodegradable monomer.39 However, conjugation of the photodegradable maleimide monomer to a PEG-bis-amine was challenging and led to limited modification of the amine end groups of the polymer (∼5–10% modification) [data not shown]. Consequently, we pursued the latter and synthesized a small precursor of the photolabile group, coupled it with the amine end groups of PEG, and subsequently modified the PEG-photolabile precursor with maleimide end groups. This unique approach, as elaborated below, was used to create a PEG crosslinker end-functionalized with a photodegradable maleimide (Scheme 1). We first synthesized an intermediate, 4-(4-(1-hydroxyethyl)-2-methoxy-5-nitrophenoxy) butanoic acid (V, Scheme S2†), using a protocol published by Kloxin et al. with one modification.39 Briefly, the nitration step was carried out continuously on an ice bath to minimize any side reactions (i.e., nitration at multiple positions), and the purification of the nitrated product was carried out using flash chromatography (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 ethyl acetate:hexane to 6:4 ethyl acetate:hexane) (see ESI† for detailed protocol). The carboxylic acid groups from intermediate V were activated with 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) in the presence of N,N-diisopropylethylamine (DIPEA) to form an amide linkage with four-arm PEG amine (PEG-4-NH2). This reaction yielded a polymer-photolabile precursor intermediate with hydroxyl end groups (pII) with 99% modification as assessed from integration of the area of the aromatic protons in the 1H NMR spectrum. Multiarm PEG (four-arm) maleimide was employed instead of linear PEG to provide higher functionality per crosslinker and facilitate encapsulation and entrapment of various cargos for delivery applications. The hydroxyl groups from pII were subsequently reacted with succinic anhydride to convert them to carboxylic acids and yield the acid-functionalized photodegradable polymer intermediate (pIII). The reaction yield was ∼79%, and the functionality (∼100%) was quantified by the disappearance of the hydroxyl proton at 5.26 ppm in the 1H NMR spectrum. The carboxylic acid groups were activated using HATU to form an o-acyl(tetramethyl)-active ester that can react with the nucleophilic amine of N-(2-aminoethyl) maleimide (AEM) (or other amine-functionalized reactive groups of interest). This reaction yielded maleimide-functionalized, photodegradable 4-arm PEG (pIV, PEG-4-PD-MI) with a reaction yield of 82% and functionality of 79%, as quantified by monitoring the area of the maleimide ring protons (6.97 ppm) in the 1H NMR spectrum (see ESI†). The lower functionalization with maleimide observed here, as compared to the other end group modification reactions, may be attributed to two side reactions: free amines of AEM may react with maleimides on other AEM molecules, and the maleimide ring may undergo hydrolysis during synthesis and purification. Although the synthesis of a water-insoluble nitrobenzyl moiety with maleimide end group recently has been reported,51 to the best of our knowledge, this is the first reported synthesis of a water-soluble maleimide end-functionalized photodegradable PEG macromer for controlling hydrogel degradation. These methods in principle could be employed for designing cell compatible hydrogels for tissue engineering and regenerative medicine applications.
:7 ethyl acetate:hexane to 6:4 ethyl acetate:hexane) (see ESI† for detailed protocol). The carboxylic acid groups from intermediate V were activated with 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) in the presence of N,N-diisopropylethylamine (DIPEA) to form an amide linkage with four-arm PEG amine (PEG-4-NH2). This reaction yielded a polymer-photolabile precursor intermediate with hydroxyl end groups (pII) with 99% modification as assessed from integration of the area of the aromatic protons in the 1H NMR spectrum. Multiarm PEG (four-arm) maleimide was employed instead of linear PEG to provide higher functionality per crosslinker and facilitate encapsulation and entrapment of various cargos for delivery applications. The hydroxyl groups from pII were subsequently reacted with succinic anhydride to convert them to carboxylic acids and yield the acid-functionalized photodegradable polymer intermediate (pIII). The reaction yield was ∼79%, and the functionality (∼100%) was quantified by the disappearance of the hydroxyl proton at 5.26 ppm in the 1H NMR spectrum. The carboxylic acid groups were activated using HATU to form an o-acyl(tetramethyl)-active ester that can react with the nucleophilic amine of N-(2-aminoethyl) maleimide (AEM) (or other amine-functionalized reactive groups of interest). This reaction yielded maleimide-functionalized, photodegradable 4-arm PEG (pIV, PEG-4-PD-MI) with a reaction yield of 82% and functionality of 79%, as quantified by monitoring the area of the maleimide ring protons (6.97 ppm) in the 1H NMR spectrum (see ESI†). The lower functionalization with maleimide observed here, as compared to the other end group modification reactions, may be attributed to two side reactions: free amines of AEM may react with maleimides on other AEM molecules, and the maleimide ring may undergo hydrolysis during synthesis and purification. Although the synthesis of a water-insoluble nitrobenzyl moiety with maleimide end group recently has been reported,51 to the best of our knowledge, this is the first reported synthesis of a water-soluble maleimide end-functionalized photodegradable PEG macromer for controlling hydrogel degradation. These methods in principle could be employed for designing cell compatible hydrogels for tissue engineering and regenerative medicine applications.
 | ||
| Scheme 1 Synthetic route for preparing a macromolecular crosslinking agent functionalized with photodegradable maleimide. Reagents and conditions are as follows: (a) DIPEA, HATU in DMF under Ar; (b) succinic anhydride, DMAP in DMF under Ar at 50 °C; and (c) N-(2-aminoethyl) maleimide, TFA, DIPEA, HATU in DMF under Ar. | ||
Hydrogel formation and tunable mechanical properties
The mechanical properties of hydrogels formed using the synthesized multifunctional macromers were investigated to demonstrate the utility of Michael-type addition to form hydrogels sensitive to multiple stimuli. In particular, hydrogel gelation kinetics and mechanical properties play a crucial role in the clinical translation of injectable hydrogels for controlled drug delivery applications. Here, dynamic time sweep rheological experiments were conducted to monitor the gelation kinetics and to demonstrate the utility of these functionalized macromolecules to form crosslinked networks on timescales appropriate for injection. Hydrogels were formed in situ (i.e., on the rheometer stage) by mixing precursor solutions of photodegradable PEG-4-PD-MI and a microenvironment-sensitive PEG-4-arylSH at 1:1 ratio of maleimide to thiol. The mixed precursor solution was added to the rheometer stage before any apparent increase in the solution viscosity, and time sweep measurements were acquired under the viscoelastic regime (Fig. S1†). Representative results for the formation of a 5 wt% hydrogel are shown in Fig. 2A. Due to rapid gelation, the crossover point of storage and loss moduli, which is an indirect measurement of the gel point, occurred prior to the first measurement on the rheometer. A consistent increase in storage modulus (G′, from ∼1900 Pa to ∼4000 Pa) without a significant increase in loss modulus (G′′, values ranging from ∼50 Pa to ∼70 Pa) as a function of time was observed, correlating with the crosslinking of the hydrogel (G ∼ ρx). Further, consistent values of storage modulus as a function of frequency highlight the elastic nature of the hydrogels (Fig. S1A†). Properties that are critical for the success of injectable hydrogel-based drug carriers, especially mesh size that dictates diffusion of solutes in and out of hydrogel, are dependent on the mechanical properties and vary with the network crosslink density. The impact of crosslink density on the elastic properties of the hydrogel was investigated by varying the polymer concentration (Fig. 2B). With an increase in the total polymer concentration from 2 to 5 wt%, the storage modulus (G′) increased from 0.2 kPa to 3.7 kPa, which correlates to elastic moduli (E) ranging between ∼0.6 to ∼11 kPa, where G ∼ E/3 based on rubber elasticity theory. This range of elastic moduli matches well with that of various soft tissues, from that of neural to muscular tissues.52,53 The corresponding mesh size, calculated from Flory–Rehner theory,54 varied between 10 nm to 14 nm as a function of polymer concentration, providing a handle to control entrapment, diffusion, and release of cargo molecules (Fig. S2†). The crosslinking time, defined here as the time to reach 90% of the final storage modulus value, ranged from approximately 2 to 10 minutes (Fig. 2C). The equilibrium mass swelling ratio (q) of resulting hydrogels varied from 17 to 32 (Table S1†). The gelation kinetics and elastic properties of these novel hydrogels are consistent with the gelation kinetics of similar Michael-type crosslinked PEG hydrogels.23,55,56 Overall, these results indicate that the gelation time and initial elastic properties of the multimodal degradable hydrogels formed using Michael-type reaction of these modified macromers can be tuned over relevant ranges for the end application.
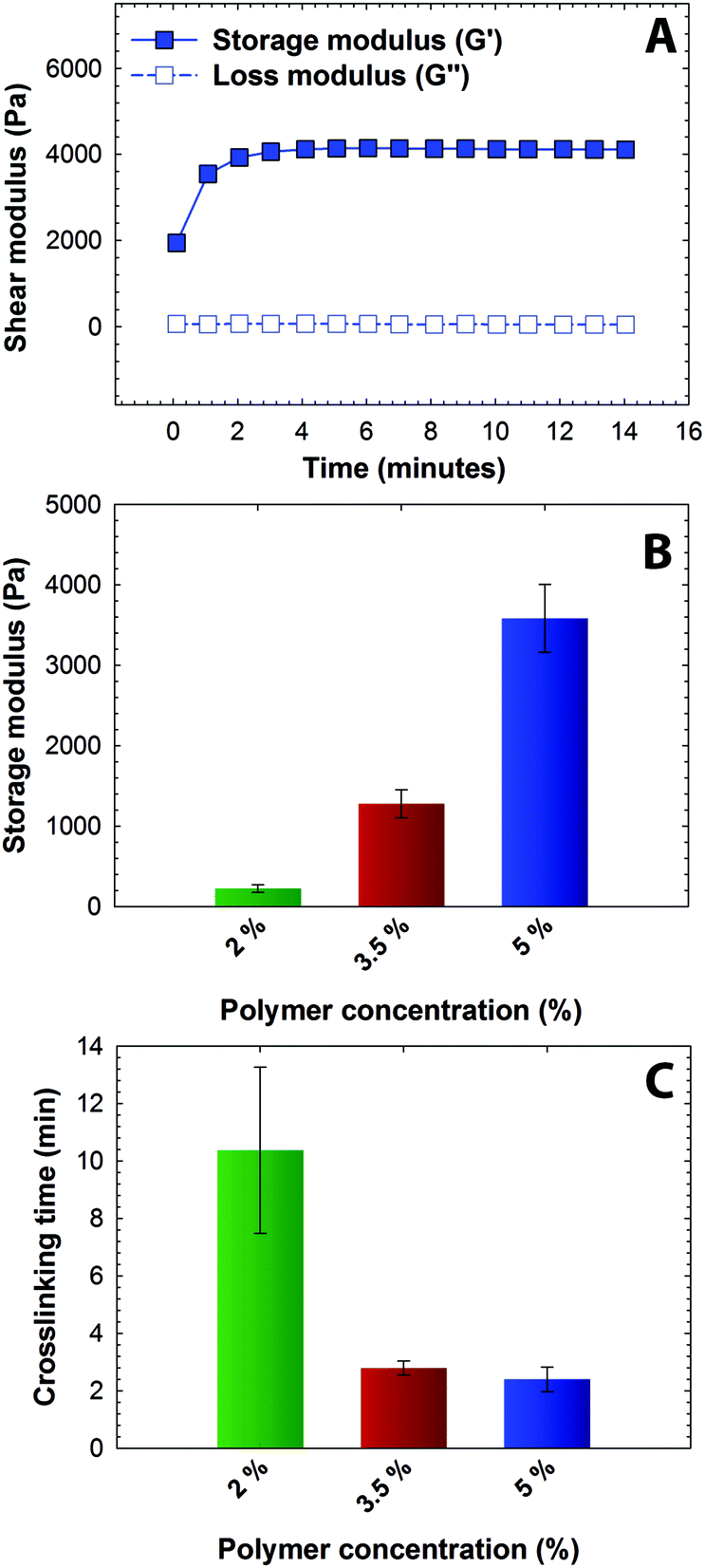 | ||
| Fig. 2 Rheological characterization of hydrogels (A) Hydrogel formation was monitored by dynamic time sweep measurements using an oscillatory rheometer, where all measurements were performed within the linear viscoelastic regime (representative data for 5 wt% hydrogel shown). (B) Polymer concentration was varied to achieve a range of moduli and corresponding mesh sizes (see Fig. S2†), and the impact on crosslinking time was assessed. With increased polymer concentration, the crosslink density increased resulting in increased storage modulus and lowered gelation time. The data shown illustrate the mean (n ≥ 3) with error bars showing the standard error. | ||
Degradation in response to exogenous and endogenous stimuli
We next sought to establish a range of degradation profiles and times that could be achieved with these multifunctional gels in detailed studies of their degradation kinetics in response to light, reducing microenvironments, and aqueous microenvironments. Changes in the elastic properties of the hydrogels were monitored as a function of time upon application of each stimulus, focusing on the 5 wt% composition with a mesh size (∼10 nm) that is appropriate for release of large cargo (e.g., antibodies, nanoparticles, and cells). To study the light-mediated degradation of the multimodal degradable hydrogels, samples formed in situ on a photorheometer were irradiated with cytocompatible light conditions (10 mW cm−2 at 365 nm [long wavelength UV] or 400–500 nm [visible]).34,57,58 The hydrogel shear modulus is directly correlated with the crosslink density per the theory of rubber elasticity (G ∼ ρx),59 and a decrease in the storage modulus thus indicates cleavage of crosslinks and degradation of the hydrogel (eqn S8†). The degradation behavior of the hydrogels was compared with that of a negative control (gels formed using PEG-4-alkylSH and PEG-4-MI that lack the photodegradable group, Fig. S4†), as shown in Fig. 3A. A significant decrease (approximately 15% reduction) in storage modulus for the multimodal degradable hydrogel was observed with the application of each short pulse light (30-second of 10 mW cm−2 at 365 nm), whereas the elastic properties of the control hydrogel remained unchanged. Multimodal degradable hydrogels also degraded with visible irradiation (10 mW cm−2 at 400–500 nm, similar to conditions used clinically) as shown in Fig. 3B inset. These data confirm the triggered degradation of the hydrogels in response to light; the rate of degradation in response to applied light was calculated using continuous degradation data (Fig. 3B) assuming first-order degradation kinetics based on network connectivity and the kinetics of photocleavage (Fig. S5†).60 The rate constant (k) for the initial degradation time period was found to be 3.03 ± 0.13 × 10−1 min−1 (t1/2 = 2.3 min) and compares well with typical rate constants for cleavage of similar o-nitrobenzyl ether based moieties (k ∼ 0.2–0.3 min−1).57,61 In addition, a reduction in storage modulus was observed with low doses of visible light with a rate constant of 2.20 ± 0.03 × 10−2 min−1 (Fig. 3B inset). The order of magnitude difference in degradation rate constant between 365 nm and 400–500 nm can be attributed to the respective differences in absorbance and quantum yield of the o-NB group at these wavelengths (Fig. S3†).39,47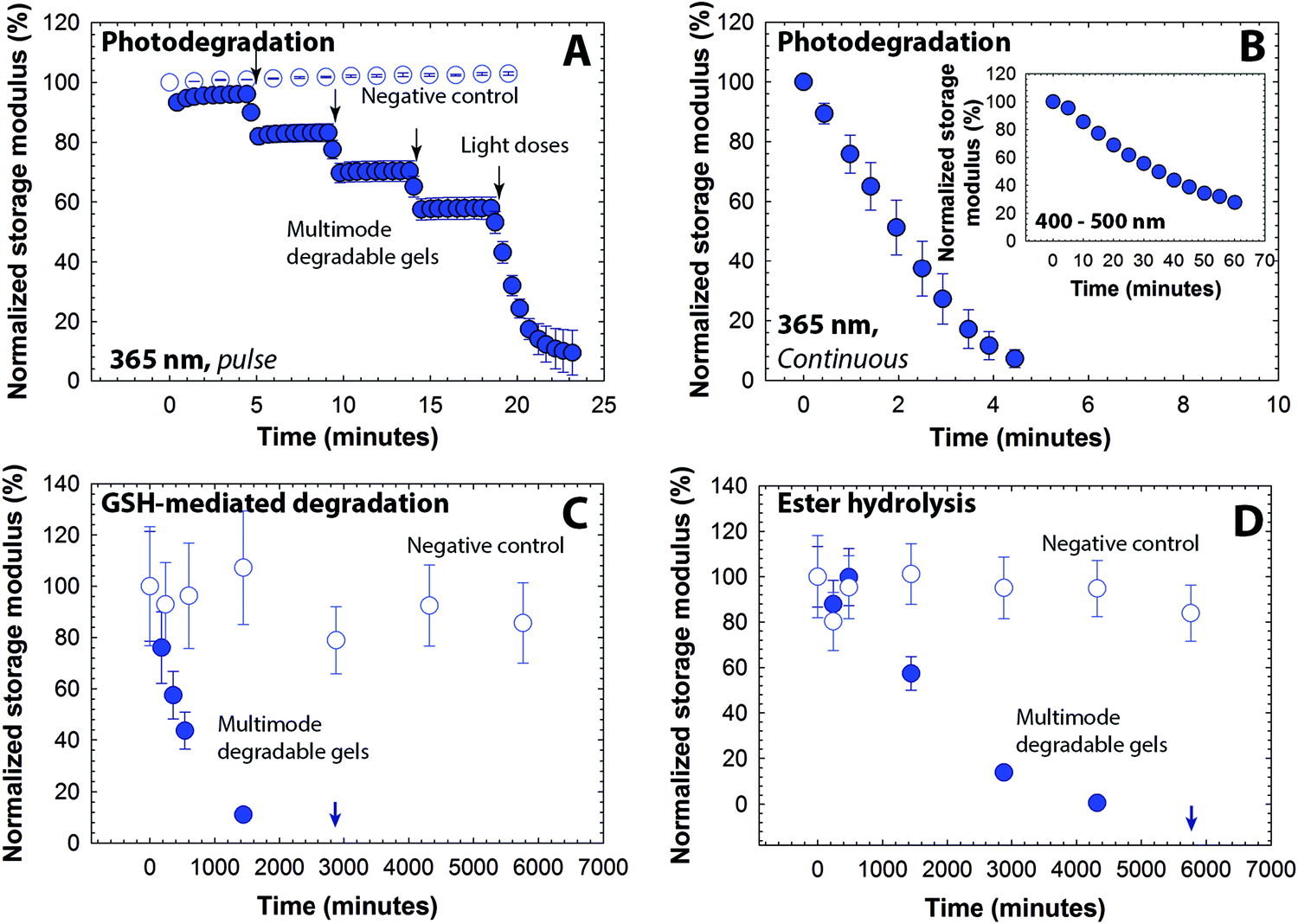 | ||
| Fig. 3 Degradation of multimodal degradable hydrogels. Degradation of hydrogels in response to different stimuli was studied by monitoring the storage modulus as a function of time. (A) Hydrogels exhibited a rapid decrease in the modulus in response to externally applied, low doses of light (10 mW cm−2 at 365 nm, 30 s pulse, closed symbols), and (B) complete degradation is observed after ∼4.5 minutes of continuous exposure. The negative control hydrogels, which lack a photolabile group, show no significant change in modulus over time in response to the same light exposure (open symbols). The hydrogels also were responsive to low intensity visible light (10 mW cm−2 at 400 to 500 nm, continuous irradiation, inset). (C) In a thiol-rich reducing microenvironment like that observed in tumors (ca. 10 mM GSH), a decrease in the modulus of the multimodal degradable hydrogel (closed symbols) was observed, indicating responsive degradation due to reversible click and thiol exchange reactions of aryl thiol based thioether succinimide linkages with GSH under reducing conditions. Complete degradation is observed in approximately 2 days. (D) In an aqueous microenvironment (phosphate buffer), the multimodal degradable hydrogel exhibited slower degradation due to hydrolysis of ester linkages with complete degradation in approximately 4 days (closed symbols). The negative control (no degradable groups), after initial swelling, was relatively stable during the experimental time frame (open symbols). The data shown illustrate the mean (n ≥ 3) with error bars showing the standard error. | ||
Aryl thiol functionalized PEG macromers provide degradability in response to reducing microenvironments within the multimodal degradable hydrogels. GSH is a reducing agent produced at increased levels by highly metabolically active cells, and consequently, is found at elevated concentrations in carcinoma tissues (on the order of 10 mM intracellularly and 10 μm extracellularly).49,50 To study hydrogel degradation in response to a GSH-rich microenvironment, multimodal degradable hydrogels were suspended in buffer with a physiologically-relevant concentration of GSH (10 mM),62 and the elastic properties of the hydrogels were monitored periodically using oscillatory rheometry (Fig. 3C). Nondegradable PEG hydrogels without photolabile or reducing environment sensitive linkages (PEG-4-MI and PEG-4-alkylSH) and similar mechanical properties were used as a negative control (Fig. S4†). As is apparent in Fig. 3C, both control and degradable hydrogels show an initial decrease in storage modulus (approximately 10 to 20% reduction within the first 180 minutes), which can be attributed to initial equilibrium swelling of the hydrogels. Notably, a continuous decrease in modulus for the degradable hydrogels is observed after initial swelling until complete degradation is observed after approximately 2880 minutes (2 days), confirming degradation of these hydrogels in response to the reducing microenvironment. Side reactions, such as maleimide ring hydrolysis that results in a non-degradable crosslink, also can impact the rate and extent of gel degradation; however, our earlier studies indicated that the thiol exchange with the Michael-type adduct occurs on timescales (k = 1.75 × 10−3 min−1) that are orders of magnitude faster than this side reaction (k = 5.5 × 10−5 min−1).30,37 Further, ester hydrolysis provides a third mechanism for degradation, where its rate was expected to be slower than that of the thiol-exchange reaction based on prior work (k = 1.87 × 10−5 min−1).37 Degradation of the multimodal hydrogels in reducing environments without applied light consequently was expected to be dominated by the thiol exchange.30,37 Fits to the data indicated that the observed kinetics for early degradation (first day up to 1440 minutes) were pseudo-first-order with a rate constant of 1.52 ± 0.003 × 10−3 min−1 (t1/2 = 450 min), which is consistent with earlier reported values for similar hydrogels (Fig. S5†).37 Complete hydrogel dissolution (i.e., reverse gelation) was observed after approximately 2 days (at 2880 minutes), which is faster than our earlier studies of dually degradable hydrogels that did not contain a photolabile group (complete dissolution at ∼4 days). We hypothesized that this disparity is due to the elevated rate of hydrolysis of esters present in the PEG-4-PD-MI, since our earlier study indicated that the ester linkage present on the PEG-4-arylSH is relatively stable with a half-life of 14 days. To test this hypothesis, we conducted hydrolytic cleavage studies, characterizing the degradation of multimodal hydrogels over time in an aqueous solution without GSH (Fig. 3D). The rate of ester hydrolysis was found to be 6.84 ± 0.91 × 10−4 min−1 (t1/2 = 1013 min, pseudo-first order kinetics, Fig. S5†), which was an order of magnitude larger than that observed for dually degradable hydrogels without the photolabile group (k = 1.87 × 10−5 min−1).37 In retrospect, these results are not surprising since neighboring functional groups (here, the photodegradable moiety) have been observed to significantly influence the rate of ester hydrolysis.63,64 Additional small-molecule studies will shed light on the impact of neighboring groups on the ester hydrolysis of these macromolecules, thus enabling improved design of biomaterials. Nevertheless, the degradation of the multimodal degradable hydrogels can be controlled over minutes to days, with half-lives ranging three orders of magnitude from ∼2 to ∼1000 minutes (Table 1).
| Degradation mode | Rate constant (min−1) | Half life (t1/2) (min) |
|---|---|---|
| Photodegradation (365 nm) | 3.03 ± 0.13 × 10−1 | 2 |
| Photodegradation (400–500 nm) | 2.20 ± 0.03 × 10−2 | 32 |
| Thiol exchange | 1.52 ± 0.003 × 10−3 | 450 |
| Ester hydrolysis | 6.84 ± 0.91 × 10−4 | 1013 |
Degradation mediated release of model cargo nanobeads
To demonstrate the utility of this multimodal degradable system for tailored release, fluorescently-labeled nanobeads were entrapped within the hydrogel as a model cargo, where similarly sized beads or particles can be laden with various therapeutics of interest.65–67 Since the diameter of these nanobeads (ϕ ≈ 100 nm) is ∼10-fold larger than the estimated mesh size of the hydrogels (ξ ≈ 10 nm), the release of cargo was expected to be driven by hydrogel degradation for the proof of concept studies demonstrating control over the time and dose of released cargo. The release of nanobeads in response to appropriate degradation stimuli was monitored in solution using fluorescence spectroscopy. The fractional release, which is defined as the ratio of cargo released at a particular time point (Mt) to the cargo released at complete degradation (M∞) was plotted as a function of time (Fig. 4). Hydrogels that were incubated in a thiol-rich microenvironment (10 mM GSH) exhibited limited release of nanobeads (≤1%) before complete hydrogel dissolution; release was observed only after reverse gelation after 2 days (2880 minutes). In addition, when incubated in PBS buffer, release was observed only after gel dissolution at 4 days owing to ester hydrolysis (5760 minutes, Fig. S7†). In contrast, hydrogels that were degraded using externally applied light exhibited light-responsive release with approximately 6 to 8% release of the loaded cargo in response to each 10-minute light pulse (10 mW cm−2 at 365 nm), without any observable bulk degradation of the hydrogel as elaborated below. These release profiles are consistent with the degradation mechanisms for the related functional groups, where multimodal hydrogels (height ∼1.8 mm) should degrade in bulk with reducing and aqueous microenvironments and by surface erosion with applied light.57,60,68 For photodegradation studies, the height of the hydrogel decreased as a function of time while no significant changes in the diameter of hydrogel discs were observed, further supporting degradation by surface erosion (Fig. S6†). Surface erosion is expected when using UV and visible light to degrade these o-NB-based hydrogels, owing to the strong absorbance of these wavelengths of light by the photolabile group within these thick hydrogel constructs; for example, at 365 nm, the molar absorptivity of the photolabile group is 3840 L mol−1 cm−1, equating to ∼6% transmittance at thickness of 100 μm and confining degradation to the top of the surface-eroding gel (see ESI†).39,57,60 Interestingly, the release of nanobeads (∼6 to 8%) in response to each applied light pulse was slightly less than expected (∼15% based on the rate of surface erosion in degradation studies, see ESI†). We speculate that this disparity likely arises from increased light attenuation in the presence of nanobeads (∼17% by volume of total gel content) that scatter light and may hinder further the light penetration through the hydrogel. Nanobeads were chosen here for proof of concept where similarly sized particles loaded or decorated with small molecular drugs, proteins, or siRNA can be encapsulated within these hydrogels for localized release for specific applications of interest.69,70 Note, a more continuous release profile upon bulk degradation by thiol exchange or hydrolysis would be expected for biologics (e.g., antibodies and other therapeutic proteins) directly encapsulated within these types of gels; protein release will be driven by diffusion as the mesh size of the hydrogel increases commensurate with degradation, as observed in our earlier work with dually degradable hydrogels.37 Overall, these results support the hypothesis that the release of cargo can be tuned by controlling the degradation rate and, in principle, such a strategy could be employed for spatiotemporal control over release of cargo molecules in biological systems. For example, we envision that this approach could prove useful for the treatment of various skin cancers, where photodynamic therapies are often employed; with these new materials, therapeutics for the treatment of carcinomas could be released in response to the tumor microenvironment and, as needed, adjusted with light on a patient-specific basis.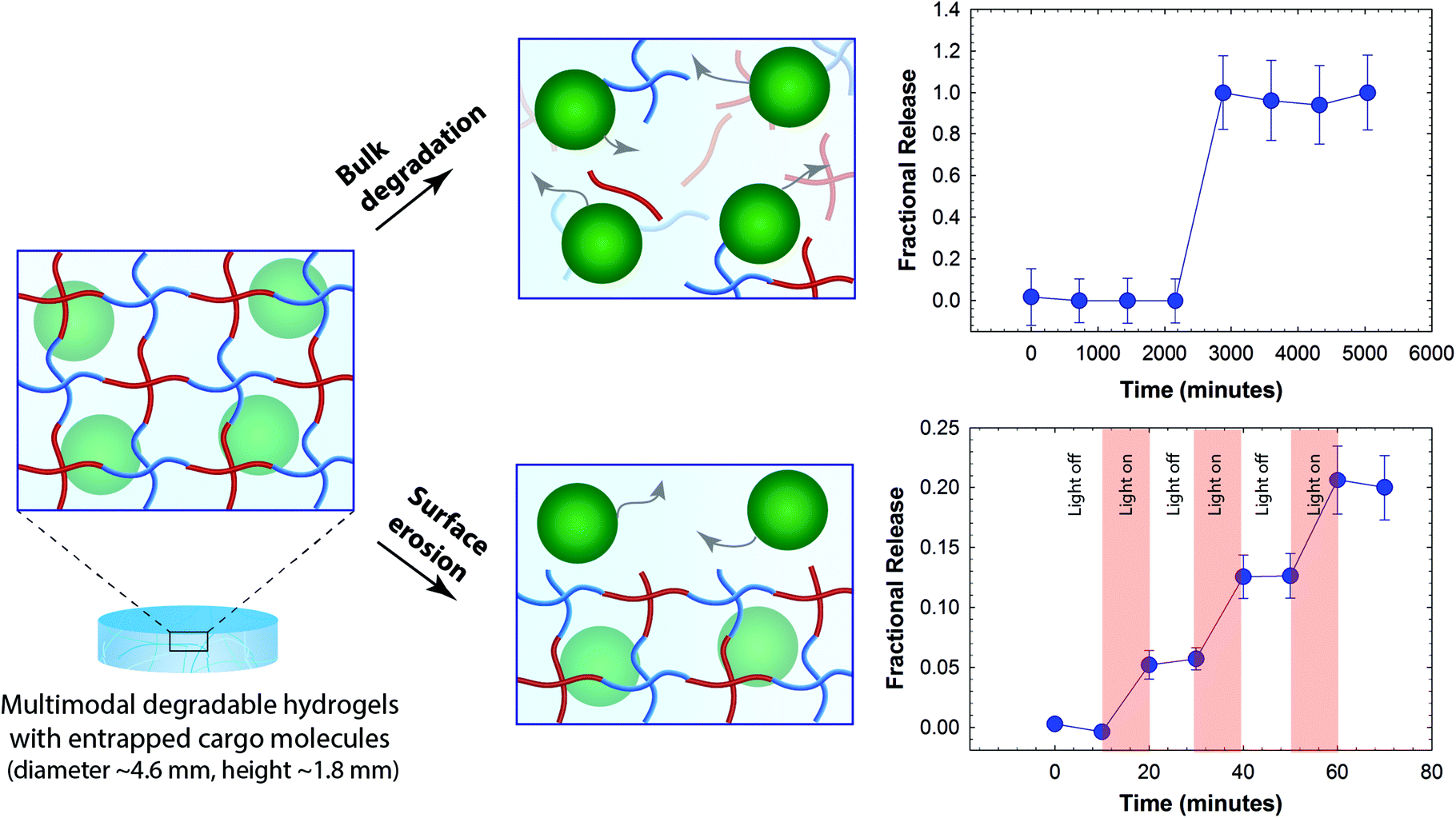 | ||
| Fig. 4 Degradation-mediated release of cargo. Fluorescent nanobeads (diameter ∼100 nm) were encapsulated as a model cargo within the multimodal degradable hydrogel during formation. Since the diameter of cargo molecules is ∼10 times larger than the mesh size, we hypothesized that the release would be controlled by degradation of the hydrogel. Hydrogel samples that were incubated in a reducing microenvironment (10 mM GSH) showed burst release of nanobeads upon hydrogel dissolution (e.g., reverse gelation). When irradiated with pulses of light (10 mW cm−2 at 365 nm periodically for 10 minute intervals), the hydrogel exhibited surface erosion (Fig. S6†), owing to light attenuation within these thick hydrogels (height ∼1.8 mm), and the release of nanobeads correlated with this light-mediated surface erosion of the hydrogels. The data shown illustrate the mean (n ≥ 3) with error bars showing the standard error. | ||
Conclusion
In summary, this work combines three cleavage chemistries to engineer multimodal degradable hydrogels for responsive and triggerable modulation of properties and cargo release. Specifically, a versatile Michael-type addition reaction was employed to synthesize an injectable hydrogel system formed in situ, using PEG macromers functionalized with thiols or a photodegradable maleimide, respectively, where incorporation of an aryl thiol imparted degradability in response to reducing microenvironments. The hydrogels exhibited rapid gelation and consistent mechanical properties between samples, which will be helpful for their development as injectable drug delivery vehicles in vivo. By incorporating a photodegradable o-nitrobenzyl ether group, a thiol-sensitive succinimide thioether linkage, and ester linkages, the hydrogels demonstrated unique controlled degradation via surface erosion or bulk degradation mechanisms, respectively, with degradation rate constants ranging from ∼10−1 min−1 to ∼10−4 min−1. As a proof of concept, the controlled release of nanobeads from the hydrogel was demonstrated in a preprogrammed, stimuli-responsive, or spatiotemporal fashion. In principle, such a strategy could be employed for delivery of multiple therapeutics with precise control over the release and delivery of cargoes, such as drugs, siRNA, drug-loaded nanoparticles, or cells, for creating complex degradation profiles as necessitated by the end application of interest.Acknowledgements
Research reported in this publication was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) (P20GM103541), an IDeA from NIGMS from the NIH (1 P30 GM110758-01), the Burroughs Wellcome Fund (subcontract from University of California, Davis), and the University of Delaware Research Foundation. Additionally, the authors are deeply grateful to Mr Srinivasa Chintala and Prof. Joseph Fox for insightful discussions and for supporting an early version of this study in his laboratory. The authors thank Mr Bassil El-Zaatari, Prof. Christopher Kloxin, and Prof. Wilfred Chen for the training and use of equipment in their respective laboratories. Authors are thankful to Prof. Emanual Maverakis for helpful discussion regarding material design. Additionally, the authors would like to thank Mr Matthew Rehmann for feedback on earlier versions of this manuscript.References
- D. Buenger, F. Topuz and J. Groll, Prog. Polym. Sci., 2012, 37, 1678–1719 CrossRef CAS PubMed.
- I. Tokarev and S. Minko, Adv. Mater., 2010, 22, 3446–3462 CrossRef CAS PubMed.
- Q. Yang, N. Adrus, F. Tomicki and M. Ulbricht, J. Mater. Chem., 2011, 21, 2783–2811 RSC.
- J. K. Chen and C. J. Chang, Materials, 2014, 7, 805–875 CrossRef CAS PubMed.
- E. Cho, J. S. Lee and K. Webb, Acta Biomater., 2012, 8, 2223–2232 CrossRef CAS PubMed.
- P. Chawla, A. R. Srivastava, P. Pandey and V. Chawla, Mini-Rev. Med. Chem., 2014, 14, 154–167 CrossRef CAS.
- C. W. Peak, J. J. Wilker and G. Schmidt, Colloid Polym. Sci., 2013, 291, 2031–2047 CAS.
- P. M. Kharkar, K. L. Kiick and A. M. Kloxin, Chem. Soc. Rev., 2013, 42, 7335–7372 RSC.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef PubMed.
- M. Patenaude, N. M. Smeets and T. Hoare, Macromol. Rapid Commun., 2014, 35, 598–617 CrossRef CAS PubMed.
- M. Guvendiren and J. A. Burdick, Curr. Opin. Biotechnol., 2013, 24, 841–846 CrossRef CAS PubMed.
- S. P. Zustiak and J. B. Leach, Biotechnol. Bioeng., 2011, 108, 197–206 CrossRef CAS PubMed.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury and C. J. Hawker, Chem. Commun., 2006, 2774–2776, 10.1039/B603438a.
- B. J. Adzima, Y. Tao, C. J. Kloxin, C. A. DeForest, K. S. Anseth and C. N. Bowman, Nat. Chem., 2011, 3, 256–259 CrossRef CAS PubMed.
- H. Shih and C. C. Lin, Biomacromolecules, 2012, 13, 2003–2012 CrossRef CAS PubMed.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank and K. Kingsbury, Chem. Commun., 2006, 2774–2776 RSC.
- C. A. DeForest, B. D. Polizzotti and K. S. Anseth, Nat. Mater., 2009, 8, 659–664 CrossRef CAS PubMed.
- J. Zheng, L. A. Smith Callahan, J. Hao, K. Guo, C. Wesdemiotis, R. A. Weiss and M. L. Becker, ACS Macro Lett., 2012, 1, 1071–1073 CrossRef CAS PubMed.
- H. Zhang, K. T. Dicker, X. Xu, X. Jia and J. M. Fox, ACS Macro Lett., 2014, 3, 727–731 CrossRef CAS PubMed.
- D. L. Alge, M. A. Azagarsamy, D. F. Donohue and K. S. Anseth, Biomacromolecules, 2013, 14, 949–953 CrossRef CAS PubMed.
- J. D. McCall and K. S. Anseth, Biomacromolecules, 2012, 13, 2410–2417 CrossRef CAS PubMed.
- Y. K. Liang and K. L. Kiick, Polym. Chem., 2014, 5, 1728–1736 RSC.
- E. A. Phelps, N. O. Enemchukwu, V. F. Fiore, J. C. Sy, N. Murthy, T. A. Sulchek, T. H. Barker and A. J. Garcia, Adv. Mater., 2012, 24, 64–70 CrossRef CAS PubMed.
- W. X. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Drug Delivery Rev., 2014, 24, 2572–2590 CAS.
- C. A. DeForest and D. A. Tirrell, Nat. Mater., 2015, 14, 523–531 CrossRef PubMed.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS PubMed.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2012, 64, 49–60 CrossRef PubMed.
- N. Angelova and D. Hunkeler, Trends Biotechnol., 1999, 17, 409–421 CrossRef CAS.
- S. X. Lu and K. S. Anseth, Macromolecules, 2000, 33, 2509–2515 CrossRef CAS.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS PubMed.
- A. D. Baldwin and K. L. Kiick, Polym. Chem., 2013, 4, 133–143 RSC.
- K. C. Koehler, K. S. Anseth and C. N. Bowman, Biomacromolecules, 2013, 14, 538–547 CrossRef CAS PubMed.
- A. A. Kislukhin, C. J. Higginson, V. P. Hong and M. G. Finn, J. Am. Chem. Soc., 2012, 134, 6491–6497 CrossRef CAS PubMed.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- I. Tomatsu, K. Peng and A. Kros, Adv. Drug Delivery Rev., 2011, 63, 1257–1266 CrossRef CAS PubMed.
- S. Y. Choh, D. Cross and C. Wang, Biomacromolecules, 2011, 12, 1126–1136 CrossRef CAS PubMed.
- P. M. Kharkar, A. M. Kloxin and K. L. Kiick, J. Mater. Chem. B, 2014, 2, 5511–5521 RSC.
- C. A. DeForest and K. S. Anseth, Nat. Chem., 2011, 3, 925–931 CrossRef CAS PubMed.
- A. M. Kloxin, M. W. Tibbitt and K. S. Anseth, Nat. Protoc., 2010, 5, 1867–1887 CrossRef CAS PubMed.
- D. R. Griffin, J. L. Schlosser, S. F. Lam, T. H. Nguyen, H. D. Maynard and A. M. Kasko, Biomacromolecules, 2013, 14, 1199–1207 CrossRef CAS PubMed.
- D. R. Griffin and A. M. Kasko, J. Am. Chem. Soc., 2012, 134, 13103–13107 CrossRef CAS PubMed.
- D. P. Nair, M. Podgorski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2013, 26, 724–744 CrossRef.
- Y. Zhou, W. Nie, J. Zhao and X. Yuan, J. Mater. Sci.: Mater. Med., 2013, 24, 2277–2286 CrossRef CAS PubMed.
- C. P. Holmes, J. Org. Chem., 1997, 62, 2370–2380 CrossRef CAS PubMed.
- C. G. Bochet, J. Chem. Soc., 2002, 125–142, 10.1039/B009522m.
- A. Deiters, ChemBioChem, 2010, 11, 47–53 CrossRef CAS PubMed.
- M. W. Tibbitt, B. W. Han, A. M. Kloxin and K. S. Anseth, J. Biomed. Mater. Res., Part A, 2012, 100, 1647–1654 CrossRef PubMed.
- D. S. Shin, J. You, A. Rahimian, T. Vu, C. Siltanen, A. Ehsanipour, G. Stybayeva, J. Sutcliffe and A. Revzin, Angew. Chem., Int. Ed., 2014, 53, 8221–8224 CrossRef CAS PubMed.
- T. C. Chang, M. J. Chang and S. Hsueh, Gynecol. Obstet. Invest., 1993, 36, 52–55 CrossRef CAS PubMed.
- D. Y. Wong, Y. L. Hsiao, C. K. Poon, P. C. Kwan, S. Y. Chao, S. T. Chou and C. S. Yang, Cancer Lett., 1994, 81, 111–116 CrossRef CAS.
- M. A. Azagarsamy and K. S. Anseth, Angew. Chem., Int. Ed., 2013, 52, 13803–13807 CrossRef CAS PubMed.
- J. Swift, I. L. Ivanovska, A. Buxboim, T. Harada, P. C. Dingal, J. Pinter, J. D. Pajerowski, K. R. Spinler, J. W. Shin, M. Tewari, F. Rehfeldt, D. W. Speicher and D. E. Discher, Science, 2013, 341, 1240104 Search PubMed.
- I. Levental, P. C. Georges and P. A. Janmey, Soft Matter, 2007, 3, 299–306 Search PubMed.
- P. J. Flory, Principles of polymer chemistry, Cornell University Press, 1953 Search PubMed.
- M. Arslan, T. N. Gevrek, A. Sanyal and R. Sanyal, RSC Adv., 2014, 4, 57834–57841 RSC.
- S. Mahadevaiah, K. G. Robinson, P. M. Kharkar, K. L. Kiick and R. E. Akins, Biomaterials, 2015, 62, 24–34 CrossRef CAS PubMed.
- A. M. Kloxin, M. W. Tibbitt, A. M. Kasko, J. A. Fairbairn and K. S. Anseth, Adv. Mater., 2010, 22, 61–66 CrossRef CAS PubMed.
- M. Guvendiren and J. A. Burdick, Nat. Commun., 2012, 3, 792 CrossRef PubMed.
- L. R. G. Treloar, The physics of rubber elasticity, Oxford University Press, 1975 Search PubMed.
- M. W. Tibbitt, A. M. Kloxin, L. Sawicki and K. S. Anseth, Macromolecules, 2013, 46, 2785–2792 CrossRef CAS PubMed.
- D. R. Griffin, J. T. Patterson and A. M. Kasko, Biotechnol. Bioeng., 2010, 107, 1012–1019 CrossRef CAS PubMed.
- G. Wu, Y. Z. Fang, S. Yang, J. R. Lupton and N. D. Turner, J. Nutr., 2004, 134, 489–492 CAS.
- M. L. Maniar, D. S. Kalonia and A. P. Simonelli, J. Pharm. Sci., 1992, 81, 705–709 CrossRef CAS PubMed.
- Y. Shalitin and S. A. Bernhard, J. Am. Chem. Soc., 1964, 86, 2291–2292 CrossRef CAS.
- M. E. Davis, Z. G. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- C. E. Probst, P. Zrazhevskiy, V. Bagalkot and X. Gao, Adv. Drug Delivery Rev., 2013, 65, 703–718 CrossRef CAS PubMed.
- A. Z. Wang, R. Langer and O. C. Farokhzad, Annu. Rev. Med., 2012, 63, 185–198 CrossRef CAS PubMed.
- M. W. Tibbitt, A. M. Kloxin and K. S. Anseth, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1899–1911 CrossRef CAS PubMed.
- S. Dou, X. Z. Yang, M. H. Xiong, C. Y. Sun, Y. D. Yao, Y. H. Zhu and J. Wang, Adv. Healthcare Mater., 2014, 3, 1792–1803 CrossRef CAS PubMed.
- M. H. El-Dakdouki, E. Puré and X. Huang, Nanoscale, 2013, 5, 3895–3903 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experiment details and polymer characterization. See DOI: 10.1039/c5py00750j |
| This journal is © The Royal Society of Chemistry 2015 |