
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA biocatalytic approach towards sustainable furanic–aliphatic polyesters†
Yi
Jiang
ab,
Albert J. J.
Woortman
a,
Gert O. R.
Alberda van Ekenstein
a and
Katja
Loos
*ab
aDepartment of Polymer Chemistry, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: k.u.loos@rug.nl
bDutch Polymer Institute (DPI), P.O. Box 902, 5600 AX Eindhoven, The Netherlands
First published on 20th May 2015
Abstract
An eco-friendly approach towards furanic–aliphatic polyesters as sustainable alternatives to aromatic–aliphatic polyesters is presented. In this approach, biobased dimethyl 2,5-furandicarboxylate (DMFDCA) is polymerized with various (potentially) renewable aliphatic diols via Candida antarctica Lipase B (CALB)-catalyzed polymerization using a two-stage method in diphenyl ether. A series of furanic–aliphatic polyesters and oligoesters is successfully produced via enzymatic polymerization. Some products reach very high 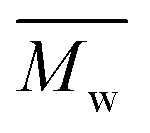 (weight average molecular weight) values of around 100
(weight average molecular weight) values of around 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. Studies on the effect of the diol structure on the enzymatic polymerization indicate that CALB prefers long-chain alkane-α,ω-aliphatic linear diols containing more than 3 carbons. We also found that the molecular weights of the obtained furanic–aliphatic polyesters increase steadily with the increase of reaction temperature from 80 to 140 °C. MALDI-ToF MS analysis reveals that five polyester species may be present in the final products. They were terminated with the ester/–OH, ester/ester, –OH/–OH, no end groups (cyclic), and ester/aldehyde groups, respectively. Furthermore, the structure–property relationships were studied by comparing the crystalline/thermal properties of a series of relevant furanic–aliphatic polyesters.
000 g mol−1. Studies on the effect of the diol structure on the enzymatic polymerization indicate that CALB prefers long-chain alkane-α,ω-aliphatic linear diols containing more than 3 carbons. We also found that the molecular weights of the obtained furanic–aliphatic polyesters increase steadily with the increase of reaction temperature from 80 to 140 °C. MALDI-ToF MS analysis reveals that five polyester species may be present in the final products. They were terminated with the ester/–OH, ester/ester, –OH/–OH, no end groups (cyclic), and ester/aldehyde groups, respectively. Furthermore, the structure–property relationships were studied by comparing the crystalline/thermal properties of a series of relevant furanic–aliphatic polyesters.
Introduction
As concerns on energy shortage and greenhouse gas emissions increase in recent years, the research on sustainable materials based on yearly biomass feedstocks has become a hot appealing topic both in academic and industry fields.1–6 Currently, much attention in this research is paid to 2,5-furandicarboxylic acid (FDCA) and FDCA-based polymers.2,4,5,7 FDCA is a biobased monomer derived from 5-(hydroxymethyl)furfural (HMF) that is produced from various sources of carbohydrates.7,8 It is an interesting rigid compound with the furan ring, resembling the benzene ring in petroleum-based terephthalic acid (TPA). Nowadays, technology pathways to biobased TPA are still under development with big challenges in the academic field,9 but FDCA is readily produced from renewable resources. Therefore, FDCA and its derivatives are advocated to replace TPA or other aromatic monomers in polymer synthesis in the near future.1,4,5 The rigid furan rings render FDCA-based materials with similar and even better thermal and mechanical properties than those of their TPA-based counterparts.10–14Among FDCA-based polymers, furanic–aliphatic polyesters derived from FDCA and aliphatic diols are of great interest. They are promising sustainable analogues of aromatic–aliphatic polyesters that are widely used as commodity thermoplastic engineering polymers such as poly(ethylene terephthalate) (PET), poly(propylene terephthalate) (PPT), poly(butylene terephthalate) (PBT), etc. The ground-breaking work in this field was done by Moore and Kelly in the late 1970s.15,16 They synthesized poly(hexamethylene furanoate) (PHF) by melt polymerization of dimethyl 2,5-furandicarboxylate (DMFDCA) and 1,6-hexanediol (1,6-HDO). Later, in 2009, Gandini et al. prepared high molecular weight poly(ethylene furanoate) (PEF) by polytransesterification of ethylene glycol with a FDCA-based diester diol.17 Subsequently, various furanic–aliphatic polyesters have been prepared,10,11,14,18–21e.g., PEF, poly(propylene furanoate) (PPF), poly(butylene furanoate) (PBF), poly(2,3-butylene furanoate) (P23BF), PHF, 1,4:3,6-dianhydrohexitol-based FDCA polyesters, etc. Novel high molecular weight co-polyesters containing FDCA units are also produced.22,23 Moreover, the chemical, thermal, crystalline, and mechanical properties of these FDCA-based polyesters are earnestly investigated.10–12,20,24
However, elevated temperatures above 200 °C and conventional metal catalysts are commonly applied in the aforementioned synthesis approach. This will accelerate the consumption of the finite fossil feedstocks along with increasing the generation of hazardous waste and pollution emissions. Moreover, undesirable side reactions like decomposition and discoloration may occur at such high temperatures.11 Besides, the metal residues from the catalysts may be harmful to the environment. Therefore, it is necessary to develop an eco-friendly pathway for the production of furanic–aliphatic polyesters.
Enzymatic polymerization is an emerging alternative approach towards polymeric materials, which can compete with conventional techniques.25–31 It involves mild reaction conditions and renewable non-toxic enzyme catalysts, providing a great opportunity to achieve future sustainability in the polymer industry.32 Currently, many kinds of polymers have already been synthesized via enzymatic polymerization; polyesters are the most intensively studied.33–37 In recent years, enzymatic synthesis of sustainable polyesters from biobased monomers has attracted much attention.5,32 Plenty of biobased polyesters have been readily prepared via enzymatic polymerization, e.g., succinate-based aliphatic polyesters,38–40 1,4:3,6-dianhydrohexitol-based polyesters,41,42 vegetal oil-based polyesters,43 sugar derived diol-based polyesters,44,45etc. Recently we successfully synthesized a series of renewable 2,5-bis(hydroxymethyl)furan-based polyesters by two-stage enzymatic polycondensation in diphenyl ether.46 Boeriu et al. prepared biobased furanic–aliphatic oligoesters via one-stage enzymatic polymerization in anhydrous reaction medium (70 wt% toluene and 30 wt% tert-butanol).47 They found that the maximum degree of polymerization ( ) of these oligoesters was only 6. However, as far as we know, the enzyme-catalyzed synthesis of high molecular weight furanic–aliphatic polyesters has not been studied yet.
) of these oligoesters was only 6. However, as far as we know, the enzyme-catalyzed synthesis of high molecular weight furanic–aliphatic polyesters has not been studied yet.
In this study, we present a green pathway to sustainable furanic–aliphatic polyesters. They are enzymatically polymerized from biobased dimethyl 2,5-furandicarboxylate (DMFDCA) and (potentially) renewable aliphatic diols via a two-stage method in diphenyl ether. DMFDCA is preferred to FDCA since DMFDCA possesses better solubility in the reaction media under mild conditions and lower melting temperature. Novozym 435, which is an immobilized form of Candida antarctica Lipase B (CALB) on acrylic resin, is applied as the biocatalyst since CALB has broad substrate specificity and stable performance.28 It is the most favorable enzyme catalyst in biocatalytic polyester synthesis. Moreover, the immobilized CALB functions even at elevated temperatures up to 150 °C and tolerates some other extreme conditions.48–51
Our research starts with investigating the effect of the diol structure on the enzymatic polymerization of furanic–aliphatic polyesters. Then we optimize the enzymatic polymerization conditions to achieve higher molecular weights. We characterize the molecular weights, chemical structures, microstructures and end groups, crystallinity properties, and thermal behavior of the obtained furanic–aliphatic polyesters by SEC, NMR, ATR-FTIR, MALDI-ToF MS, WAXD, TGA and DSC. In addition, we discuss the effect of chemical structures on the crystallinity and thermal properties of the obtained furanic–aliphatic polyesters.
Experimental
Chemicals
The following chemicals were purchased from Sigma-Aldrich: 1,3-propanediol (1,3-PDO, 98%), 1,4-butanediol (1,4-BDO, 99%), 1,6-hexanediol (1,6-HDO, 99%), 1,8-octanediol (1,8-ODO, 98%), 1,10-decanediol (1,10-DDO, 98%), 2,3-butanediol (2,3-BDO, 98%), diethylene glycol (DEG, 99+%), isosorbide (98%), D-sorbitol (98+%), glycerol (99+%), chloroform (HPLC grade), diphenyl ether (99%), and Novozym 435 (an immobilized form of Candida antarctica lipase B (CALB) on acrylic resin, 5000+ U g−1). Dimethyl 2,5-furandicarboxylate (DMFDCA, 97%) was ordered from Fluorochem. The solvent 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, 99+%) was purchased from TCI Europe. CALB was pre-dried overnight in the presence of phosphorus pentoxide (P2O5) at room temperature under high vacuum. Diphenyl ether was dried with calcium hydride (CaH2), vacuum distilled, and stored in 4 Å molecular sieves under a nitrogen environment. The rest of the chemicals were used as received.General procedures for CALB-catalyzed polycondensation of DMFDCA and aliphatic diols in diphenyl ether via a two-stage method
DMFDCA (5.4304 mmol), an aliphatic diol (5.4304 mmol), and diphenyl ether (6 g) were added into a 25 mL round-bottom flask with pre-dried CALB (0.4 g). The reaction was magnetically stirred in an oil bath. A two-stage method was applied to the enzymatic polymerization. In this method, the reactants were reacted at 80 °C for 2 h under an atmospheric nitrogen environment at the first stage. Then at the second stage, polycondensation was performed at 80 °C under reduced pressure of 2 mmHg for the other 72 h.After the polymerization, chloroform (20 mL) was added into the reaction flask to dissolve the final products. CALB was filtered off by normal filtration and then washed three times with chloroform (15 mL). All solutions were then combined, concentrated, and added dropwise into excess of methanol (or hexane). The methanol (or hexane) solution with the precipitated products was firstly stirred at room temperature for several hours and then stored overnight at −20 °C. After that the precipitated products were collected by vacuum filtration and washed three times with methanol (or hexane). Finally, the products were dried in a vacuum oven at room temperature for 2–3 days before analysis.
General procedures for CALB-catalyzed polycondensation of DMFDCA and aliphatic linear diols in diphenyl ether via a temperature-varied two-stage method
DMFDCA (5.4304 mmol), an aliphatic linear diol (5.4304 mmol), and diphenyl ether (6 g) were added into a 25 mL round-bottom flask with pre-dried CALB (0.4 g). A temperature-varied two-stage method was applied to the enzymatic polymerization. In this method, the first stage was performed at 80 °C for 2 h under a nitrogen atmosphere. Then at the second stage, the pressure was reduced to 2 mmHg while maintaining the reaction temperature at 80 °C for the first 24 h, whereafter the reaction temperature was increased to 95 °C for another 24 h. Finally, the reaction temperature was regulated at 95, 120 or 140 °C for the last 24 h.After the polymerization, furanic–aliphatic polyesters were obtained via the same purification and drying procedures as described above.
Furthermore, CALB was recycled from the enzymatic polymerization according to the following procedures. The used CALB was collected in a beaker after filtration. Then chloroform (20 mL) was added and the solution with CALB was magnetically stirred for 30 minutes. After that CALB was filtered off, washed with chloroform (10 mL) twice and collected in a beaker again. Then methanol (20 mL) was added and the solution with CALB was magnetically stirred for 30 minutes. After that CALB was filtered off, washed with methanol (10 mL) twice and collected in a beaker again. Then deionized water (20 mL) was added and the solution with CALB was magnetically stirred for 30 minutes. After that CALB was filtered off, washed with deionized water (10 mL) twice and collected in a beaker. Finally, the recycled CALB was stored in a refrigerator at 0–8 °C before use.
Control reactions
DMFDCA and alkane-α,ω-aliphatic linear diols (1,4-BDO, 1,6-HDO, 1,8-ODO, or 1,10-DDO) were reacted without CALB via the same two-stage method as described before. No polyester was obtained after precipitation in methanol.In addition, DMFDCA and 1,10-DDO were reacted without CALB according to the temperature-varied two-stage method. The reaction temperature was set to 140 °C at the last 24 h. No polyester was obtained after precipitation in methanol.
Moreover, enzymatic ring-opening polymerization of ε-caprolactone (CL) was performed to verify the enzymatic catalytic reactivity of the recycled CALB. At first, CL, 4 Å molecular sieves, anhydrous toluene and pre-dried CALB (fresh or used) were mixed and sealed in a 25 mL round-bottom flask. Then the reactants were stirred at 70 °C for 24 h under the nitrogen environment. After the polymerization, chloroform (20 mL) was added into the reaction flask to dissolve the final products. CALB was filtered off by normal filtration and washed with chloroform (15 mL) three times. All obtained solutions were then combined, concentrated, and added dropwise into excess of hexane. The hexane solution with the precipitated polycaprolactone (PCL) was stirred at room temperature for several hours and then stored at −20 °C overnight. Then the PCL was isolated by centrifugation and washed with hexane three times. Finally, it was dried in a vacuum oven at 40 °C for 2–3 days before analysis.
Furthermore, we testified the enzymatic catalytic reactivity of the used CALB that had the same thermal treatment as those applied in the temperature-varied two-stage polymerization at 120/140 °C. Firstly, pre-dried CALB (fresh) and diphenyl ether were added into a 25 mL round-bottom flask filled with nitrogen. Then the reaction flask was sealed and heated up at: (1) 80 °C for the first 26 h; (2) 95 °C for another 24 h; and (3) 120 or 140 °C for the final 24 h. After cooling down to room temperature, CL and 4 Å molecular sieves were added into the flask immediately. Then the ring-opening polymerization of CL was performed at 70 °C for 24 h. After the reaction, PCL was obtained via the same purification and drying procedures as described above.
Instrumental methods
1H- and 13C-NMR spectra were recorded on a Varian VXR spectrometer (400 MHz for 1H-NMR and 100 MHz for 13C-NMR analysis), using CDCl3-d1 or DMSO-d6 as the solvent. The chemical shifts reported were referenced to the resonances of tetramethylsilane (TMS) or the solvent. The number average molecular weight (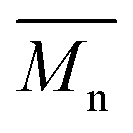 ) was calculated from the 1H-NMR spectra using the equation:
) was calculated from the 1H-NMR spectra using the equation:  , where IF is the integral intensity of the singlet assigned to the furanic protons (
, where IF is the integral intensity of the singlet assigned to the furanic protons (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–, around 7.2 ppm), ID is the integral intensity of the broad resonances ascribed to the methylene protons from the diol units (–CO–O–CH2–, around 4.4 ppm), IE–F is the integral intensity of the singlet assigned to the methoxyl end groups (–O–CH3, around 3.9 ppm), IE–D is the integral intensity of the broad peaks belonging to the methylene protons of the hydroxymethyl end groups (–CH2–OH, around 3.7 ppm), MF is the molecular mass of the FDCA units, and MD is the molecular mass of the diol units. Here we assumed that all the tested polyesters were terminated with the methoxyl and hydroxymethyl groups.
CH–, around 7.2 ppm), ID is the integral intensity of the broad resonances ascribed to the methylene protons from the diol units (–CO–O–CH2–, around 4.4 ppm), IE–F is the integral intensity of the singlet assigned to the methoxyl end groups (–O–CH3, around 3.9 ppm), IE–D is the integral intensity of the broad peaks belonging to the methylene protons of the hydroxymethyl end groups (–CH2–OH, around 3.7 ppm), MF is the molecular mass of the FDCA units, and MD is the molecular mass of the diol units. Here we assumed that all the tested polyesters were terminated with the methoxyl and hydroxymethyl groups.
Attenuated Total Reflection-Fourier Transform Infrared (ATR-FTIR) spectra were recorded on a Bruker IFS88 FT-IR spectrometer. 128 scans were performed for each sample.
The molecular weights (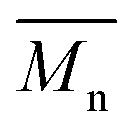 and
and  ) and dispersity (Đ,
) and dispersity (Đ,  ) were measured at 30 °C by using a Viscotek SEC (Size Exclusion Chromatograph) equipped with three detectors (LS detector: Viscotek Ralls detector; VS detector: Viscotek Viscometer Model H502; RI detector: Shodex RI-71 Refractive Index detector), using a guard column (PLgel 5 μm Guard, 50 mm) and two columns (PLgel 5 μm MIXED-C, 300 mm, Agilent Technologies). Chloroform of HPLC grade was used as the eluent, with a flow rate of 1.0 mL min−1. The molecular weight calculations were performed based on the universal calibration method using the universal calibration curve generated by narrow polydispersity polystyrene standards (Agilent and Polymer Laboratories) with the
) were measured at 30 °C by using a Viscotek SEC (Size Exclusion Chromatograph) equipped with three detectors (LS detector: Viscotek Ralls detector; VS detector: Viscotek Viscometer Model H502; RI detector: Shodex RI-71 Refractive Index detector), using a guard column (PLgel 5 μm Guard, 50 mm) and two columns (PLgel 5 μm MIXED-C, 300 mm, Agilent Technologies). Chloroform of HPLC grade was used as the eluent, with a flow rate of 1.0 mL min−1. The molecular weight calculations were performed based on the universal calibration method using the universal calibration curve generated by narrow polydispersity polystyrene standards (Agilent and Polymer Laboratories) with the 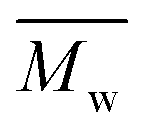 values ranging from 645 to 3001000 g mol−1.
values ranging from 645 to 3001000 g mol−1.
Matrix-Assisted Laser Desorption/Ionization-Time of Flight Mass Spectrometry (MALDI-ToF MS) measurements were performed on a Biosystems Voyager-DE PRO spectrometer. trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) was used as the matrix. The polyester sample (0.5–1.0 mg mL−1) was dissolved in HFIP with DCTB (18.18 mg mL−1) and potassium trifluoroacetate (0.46 mg mL−1). Then the mixture was subsequently hand-spotted on a stainless steel plate and left to dry. Spectra were recorded in the positive and linear mode. The masses of polyesters (MP) correspond to MP = MEG + (n × MRU) + MK+, where MEG is the mass of the end groups, n is the number of the repeating unit, MRU is the mass of the repeating unit, and MK+ is the mass of the potassium cation.
The thermal transitions were characterized by using a TA-Instruments Q1000 Differential Scanning Calorimeter (DSC). The heating and cooling rates were 10 °C min−1. The glass transition temperature (Tg) was determined by Temperature Modulated Differential Scanning Calorimetry (TMDSC). For the TMDSC test, the heating rate was 2 °C min−1, with the temperature modulated at ±0.50 °C for every 60 seconds. The TMDSC test started immediately after the second heating of the normal DSC measurement.
Thermal Gravimetric Analysis (TGA) was performed on a Perkin Elmer ThermoGravimetric Analyzer TGA7. Samples were measured at 10 °C min−1 under a nitrogen environment.
Wide-Angle X-ray Diffraction (WAXD) measurements were performed at room temperature using a Bruker D8 Advance diffractometer (Cu Kα radiation, λ = 0.1542 nm) in the angular range of 5–50° (2θ). The degree of crystallinity (χc) was calculated from the WAXD spectra according to the method established in our previous reports.40,46
NMR analysis of the obtained furanic–aliphatic polyesters
Fig. 1 depicts the 1H- and 13C-NMR spectra of the obtained furanic–aliphatic polyesters. The detailed NMR assignments are as follows:, furan), 4.38 (4H, m, –CO–O–CH2–, from 1,4-BDO), 1.90 (4H, m, –CO–O–CH2–CH2–, from 1,4-BDO), 3.91 (s, –O–CH3, end group from DMFDCA), 3.70 (t, –CH2–OH, end group from 1,4-BDO); 13C-NMR (100 MHz, CDCl3-d1, ppm): 157.79 (–CO), 146.72 (C(C)–O–, furan), 118.45 (C–, furan), 64.76 (–CO–O–CH2–), 25.31 (–CO–O–CH2–CH2–), 62.19 (–CH2–OH, end group), 52.36 (–O–CH3, end group of DMFDCA), 29.01 (–CH2–CH2–OH, end group).
, furan), 4.32 (4H, m, –CO–O–CH2–, from 1,6-HDO), 1.77 (4H, m, –CO–O–CH2–CH2–, from 1,6-HDO), 1.47 (4H, m, –CO–O–CH2–CH2–CH2–, from 1,6-HDO), 3.91 (s, –O–CH3, end group from DMFDCA), 3.64 (t, –CH2–OH, end group from 1,6-HDO); 13C-NMR (100 MHz, CDCl3-d1, ppm): 157.95 (–CO), 146.50 (C(C)–O–, furan), 118.26 (C–, furan), 65.38 (–CO–O–CH2–), 28.45 (–CO–O–CH2–CH2–), 25.50 (–CO–O–CH2–CH2–CH2–), 62.71 (–CH2–OH, end group), 52.34 (–O–CH3, end group of DMFDCA), 32.52 (–CH2–CH2–OH, end group).
, furan), 4.30 (4H, m, –CO–O–CH2–, from 1,8-ODO), 1.74 (4H, m, –CO–O–CH2–CH2–, from 1,8-ODO), 1.36 (8H, m, –CH2–, from 1,8-ODO), 3.90 (s, –O–CH3, end group from DMFDCA), 3.62 (t, –CH2–OH, end group from 1,8-ODO); 13C-NMR (100 MHz, CDCl3-d1, ppm): 158.11 (–CO), 146.74 (C(C)–O–, furan), 118.06 (C–, furan), 65.31 (–CO–O–CH2–), 29.04 (–CH2–), 28.53 (–CH2–), 25.43 (–CH2–), 62.93 (–CH2–OH, end group), 52.33 (–O–CH3, end group of DMFDCA), 32.70 (–CH2–CH2–OH, end group).
, furan), 4.28 (4H, m, –CO–O–CH2–, from 1,10-DDO), 1.73 (4H, m, –CO–O–CH2–CH2–, from 1,10-DDO), 1.38 (4H, m, –CH2–, from 1,10-DDO), 1.29 (8H, m, –CH2–, from 1,10-DDO), 3.91 (s, –O–CH3, end group from DMFDCA), 3.63 (t, –CH2–OH, end group from 1,10-DDO); 13C-NMR (100 MHz, CDCl3-d1, ppm): 158.12 (–CO), 146.91 (C(C)–O–, furan), 118.16 (C–, furan), 65.63 (–CO–O–CH2–), 29.36 (–CH2–), 29.15 (–CH2–), 28.56 (–CH2–), 25.78 (–CH2–), 62.99 (–CH2–OH, end group), 32.75 (–CH2–CH2–OH, end group).
, furan), 4.47 (4H, t, –CO–O–CH2–, from DEG), 3.83 (4H, t, –CH2–O–CH2–, from DEG), 3.91 (s, –OCH3, end group from DMFDCA), 3.74 (m, –CO–O–CH2–CH2–OH, end group from DEG), 3.64 (m, –O–CH2–CH2–OH, end group from DEG), 2.43 (m, –OH, end group from DEG); 13C-NMR (100 MHz, CDCl3-d1, ppm): 157.79 (–CO), 146.57 (C(C)–O–, furan), 118.68 (C–, furan), 68.82 (–CH2–O–CH2–), 64.27 (–CO–O–CH2–), 72.47 (–O–CH2–CH2–OH, end group), 61.67 (–O–CH2–CH2–OH, end group), 52.39 (–O–CH3, end group from DMFDCA).
, furan), 5.31 (2H, m, –CO–O–CH(CH3)–, from 2,3-BDO), 1.39 (6H, m, –CH(CH3)–, from 2,3-BDO), 4.63 (–CO–O–CH(CH3)–, end group from 2,3-BDO), 4.03 (–CO–O–CH(CH3)–CH(CH3)–OH, end group from 2,3-BDO), 3.90 (s, –O–CH3, end group from DMFDCA), 2.99 (–OH); 13C-NMR (100 MHz, CDCl3-d1, ppm): 158.39 and 157.14 (–CO), 146.74 (C(C)–O–, furan), 118.62 (C–, furan), 72.66 (–CH–), 15.21 (–CH3), 52.33 (–O–CH3, end group of DMFDCA), 16.17 (–CH3, end group of 2,3-BDO).
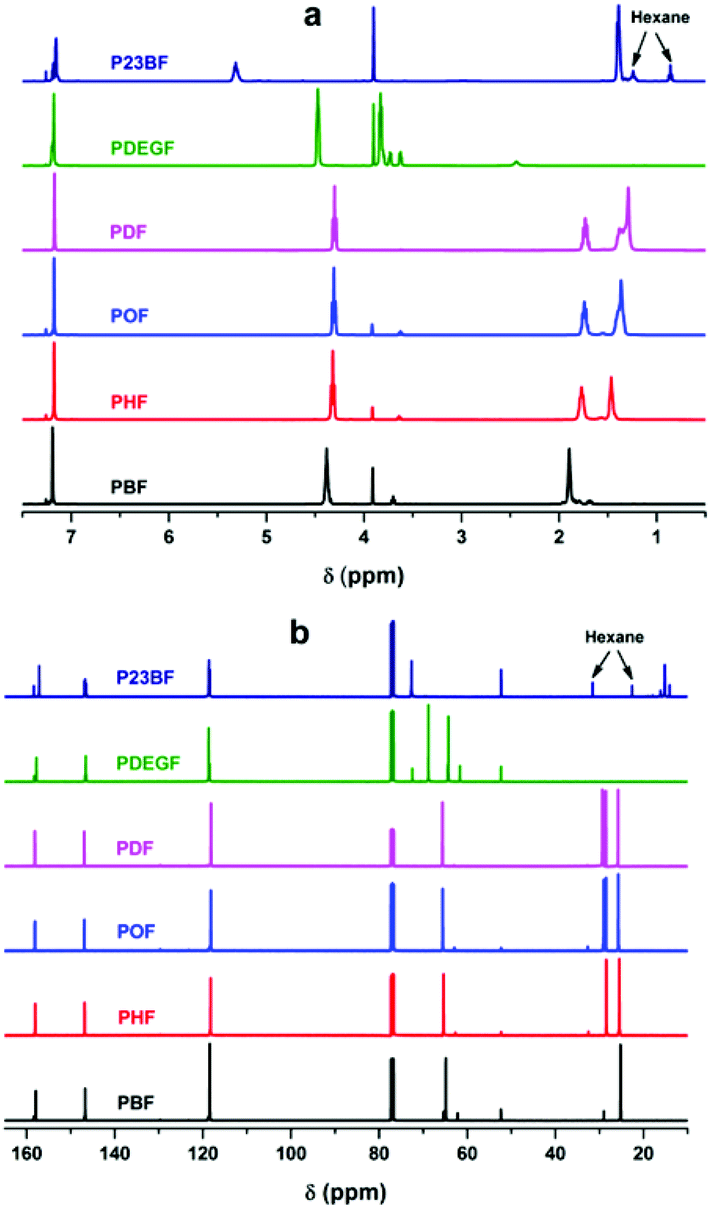 | ||
| Fig. 1 (a) 1H-NMR and (b) 13C-NMR spectra of the obtained furanic–aliphatic polyesters. | ||
ATR-FTIR analysis of the obtained furanic–aliphatic polyesters
The ATR-FTIR spectra of furanic–aliphatic polyesters are illustrated in Fig. 2. The characteristic absorption bands of the obtained furanic–aliphatic polyesters are assigned as follows.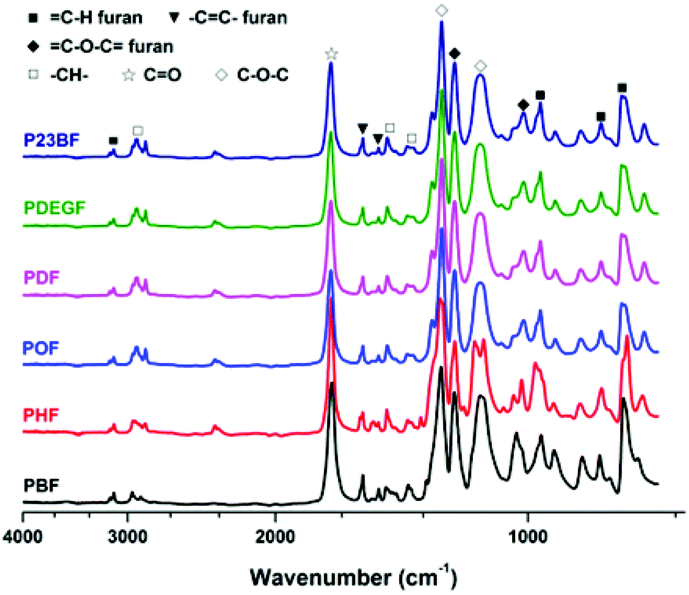 | ||
| Fig. 2 ATR-FTIR spectra of the obtained furanic–aliphatic polyesters. | ||
C–H stretching vibrations of the furan ring), 2780–3021 (C–H stretching vibrations), 1718 (CO stretching vibrations), 1574, 1506 (CC ring stretching vibrations of the furan rings), 1501, 1469, 1328–1414 (–CH– deformation and wagging vibrations), 1268 and 1140 (asymmetric and symmetric stretching vibrations of the ester C–O–C groups), 1010 and 1223 (C–O–C = ring vibrations of the furan ring), 967, 819, 768 (C–H out-of-plane deformation vibrations of the furan ring).
Results and discussion
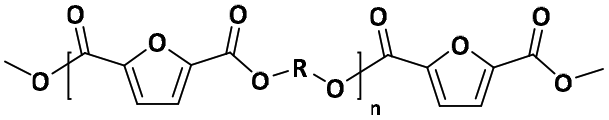
The two-stage enzymatic polycondensation of DMFDCA with aliphatic diols yielded sustainable furanic–aliphatic polyesters/oligoesters with different chemical structures, as illustrated in Scheme 1. The aliphatic diols used were: α,ω-aliphatic linear diols with two primary hydroxyl groups (1,3-PDO, 1,4-BDO, DEG, 1,6-HDO, 1,8-ODO and 1,10-DDO); diols with two secondary hydroxyl groups (2,3-BDO and isosorbide); and polyols with more than two hydroxyl groups (glycerol and D-sorbitol). The number of carbon and oxygen atoms between the two hydroxyl groups in the tested α,ω-aliphatic linear diols is 3, 4, 5, 6, 8 and 10, respectively. This number defines the diol chain length (n) in this study.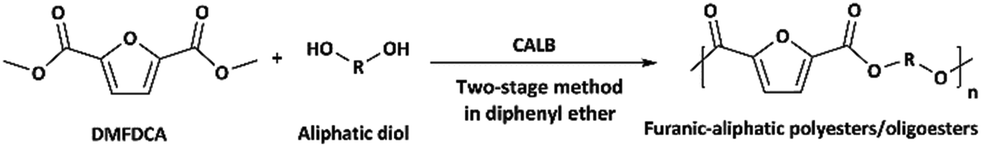 | ||
| Scheme 1 CALB-catalyzed synthesis of furanic–aliphatic polyesters/oligoesters from biobased 2,5-furandicarboxylate (DMFDCA) and aliphatic diols via a two-stage method. | ||
CALB-catalyzed synthesis of furanic–aliphatic polyesters from DMFDCA and aliphatic diols via the two-stage method
The two-stage enzymatic polymerization of DMFDCA with longer-chain alkane-α,ω-aliphatic linear diols resulted in higher molecular weight furanic–aliphatic polyesters, indicating that CALB prefers alkane-α,ω-aliphatic linear diols having a longer chain length. This result agreed well with the previous studies reported by Gross et al.,52 Linko et al.,53 and Morrow,54 in which they also found that enzymatic polymerization involving alkane-α,ω-aliphatic linear diols with longer alkylene chains gave higher values of or
or  (average degree of polymerization). As shown in Table 1, only the oligomer (
(average degree of polymerization). As shown in Table 1, only the oligomer (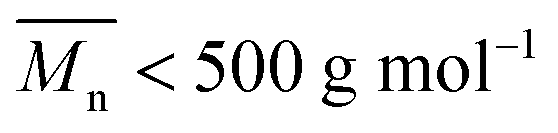 ) was obtained from the enzymatic polymerization of DMFDCA with 1,3-PDO with the shortest chain length (n = 3), even if the reaction time was extended to 194 h. Changing to 1,4-BDO/1,6-HDO/1,8-ODO with longer chain lengths (n = 4, 6, 8, respectively) resulted in a steady increase in molecular weights. The
) was obtained from the enzymatic polymerization of DMFDCA with 1,3-PDO with the shortest chain length (n = 3), even if the reaction time was extended to 194 h. Changing to 1,4-BDO/1,6-HDO/1,8-ODO with longer chain lengths (n = 4, 6, 8, respectively) resulted in a steady increase in molecular weights. The 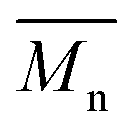 and
and 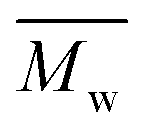 values increased from 1200 and 1700 g mol−1 to 3300 and 4800 g mol−1, respectively, when the chain length of the alkane-α,ω-aliphatic linear diol increased from 4 to 8. Upon further increasing the chain length up to 10 (1,10-DDO), the
values increased from 1200 and 1700 g mol−1 to 3300 and 4800 g mol−1, respectively, when the chain length of the alkane-α,ω-aliphatic linear diol increased from 4 to 8. Upon further increasing the chain length up to 10 (1,10-DDO), the 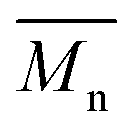 and
and  values increased significantly to 23700 and 48700 g mol−1, respectively. The possible reasons for this phenomenon are discussed below. On the one hand, alkane-α,ω-aliphatic linear diols with longer alkylene chains possess higher enzymatic reactivity; therefore polyesters with higher
values increased significantly to 23700 and 48700 g mol−1, respectively. The possible reasons for this phenomenon are discussed below. On the one hand, alkane-α,ω-aliphatic linear diols with longer alkylene chains possess higher enzymatic reactivity; therefore polyesters with higher  can be produced.52 On the other hand, the solubility of the obtained furanic–aliphatic polyesters showed the following trend in the reaction media: PDF > POF ≈ PHF > PBF. Furthermore, the Tm of the obtained PBF/POF/PHF (around 145 °C) was much higher than that of PDF (Tm around 111 °C) and the enzymatic polymerization temperature (80 °C). As a result, for the enzymatic polymerization of DMFDCA with 1,4-BDO/1,6-HDO/1,8-ODO, low molecular weight products already precipitated from the reaction media after 26 h reaction because of their high Tm and low solubility. In this case, the accessibility of CALB to the polymers was hindered, which led to low polymerization efficiency. In contrast, the enzymatic polymerization of DMFDCA with 1,10-DDO remained consistently in the homogeneous state, where CALB functioned continuously to produce high molecular weight products. Therefore, the molecular weights of furanic–aliphatic polyesters from enzymatic polymerization of DMFDCA with alkane-α,ω-aliphatic linear diols increased steadily with the diol chain length because of the reasons mentioned above.
can be produced.52 On the other hand, the solubility of the obtained furanic–aliphatic polyesters showed the following trend in the reaction media: PDF > POF ≈ PHF > PBF. Furthermore, the Tm of the obtained PBF/POF/PHF (around 145 °C) was much higher than that of PDF (Tm around 111 °C) and the enzymatic polymerization temperature (80 °C). As a result, for the enzymatic polymerization of DMFDCA with 1,4-BDO/1,6-HDO/1,8-ODO, low molecular weight products already precipitated from the reaction media after 26 h reaction because of their high Tm and low solubility. In this case, the accessibility of CALB to the polymers was hindered, which led to low polymerization efficiency. In contrast, the enzymatic polymerization of DMFDCA with 1,10-DDO remained consistently in the homogeneous state, where CALB functioned continuously to produce high molecular weight products. Therefore, the molecular weights of furanic–aliphatic polyesters from enzymatic polymerization of DMFDCA with alkane-α,ω-aliphatic linear diols increased steadily with the diol chain length because of the reasons mentioned above.
| NMRb | SEC | |||||
|---|---|---|---|---|---|---|
| Diola | Chemical structure |
|
|
|
Đ
|
Yield (%) |
a 1,3-PDO = 1,3-propanediol, 1,4-BDO = 1,4-butanediol, 1,6-HDO = 1,6-hexanediol, 1,8-ODO = 1,8-octanediol, 1,10-DDO = 1,10-decanediol, DEG = diethylene glycol, 2,3-BDO = 2,3-butanediol.
b The number average molecular weight (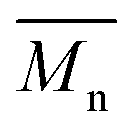 ) was determined from the 1H-NMR spectra.
c The polymerization was performed for 74 h and 194 h, respectively.
d The polymerization was performed for 74 h.
e The number average molecular weight ( ) was determined from the 1H-NMR spectra.
c The polymerization was performed for 74 h and 194 h, respectively.
d The polymerization was performed for 74 h.
e The number average molecular weight ( ), weight average molecular weight ( ), weight average molecular weight (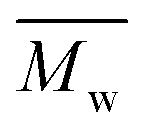 ), and dispersity (Đ, ), and dispersity (Đ,  ) were measured by SEC in DMSO.
f The molecular weights and dispersity were determined by SEC in CHCl3.
g Isolation yield. The product was purified with hexane.
h Isolation yield. The product was purified with methanol. ) were measured by SEC in DMSO.
f The molecular weights and dispersity were determined by SEC in CHCl3.
g Isolation yield. The product was purified with hexane.
h Isolation yield. The product was purified with methanol.
|
||||||
| 1,3-PDOc |
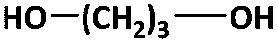
|
200 | <500e | — | — | 62g |
| 1,4-BDOd |
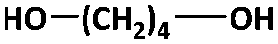
|
1100 | 1200f | 1700 | 1.42 | 77h |
| 1,6-HDOd |
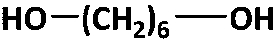
|
2200 | 2500f | 3800 | 1.52 | 93h |
| 1,8-ODOd |

|
2400 | 3300f | 4800 | 1.45 | 88h |
| 1,10-DDOd |
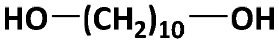
|
10100 |
23700f | 48700 |
2.05 | 94h |
| DEGd |

|
900 | 1000f | 1100 | 1.10 | 53h |
| 2,3-BDOc |
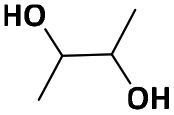
|
500 | 700f | 800 | 1.14 | 97g |
| Isosorbidec |

|
— | <500f | — | — | 58g |
| Glycerolc |
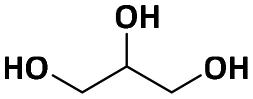
|
— | <500f | — | — | 30g |
| D-sorbitolc |
|
— | <500e | — | — | 44g |



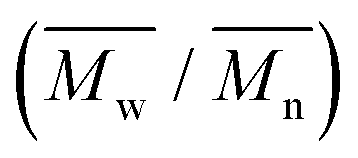
However, Boeriu et al.47 studied the CALB-catalyzed polymerization of DMFDCA with alkane-α,ω-aliphatic linear diols differing in the chain length from 2 to 12. Furanic–aliphatic oligoesters with a  of 6 were obtained after 24 h reaction. They also found that the production showed the following trend with respect to the diol chain length: 2 < 12 < 10 < 3 < 8 < 4 < 6. This was not in agreement with our results. It should be noted that they applied different enzymatic polymerization conditions: (1) different solvent (70 wt% toluene and 30 wt% tert-butanol), (2) shorter reaction time (24 h), and (3) one-stage method. We suspected that this could be the reason. Firstly, the reactivity of alkane-α,ω-aliphatic linear diols to CALB could be varied in diphenyl ether and the mixture of toluene and tert-butanol; and the solubility of the tested diols and the final products could be different. Secondly, the reaction time we applied was 3 times longer. Finally, the polycondensation efficiency is much higher by using the two-stage method under high vacuum.
of 6 were obtained after 24 h reaction. They also found that the production showed the following trend with respect to the diol chain length: 2 < 12 < 10 < 3 < 8 < 4 < 6. This was not in agreement with our results. It should be noted that they applied different enzymatic polymerization conditions: (1) different solvent (70 wt% toluene and 30 wt% tert-butanol), (2) shorter reaction time (24 h), and (3) one-stage method. We suspected that this could be the reason. Firstly, the reactivity of alkane-α,ω-aliphatic linear diols to CALB could be varied in diphenyl ether and the mixture of toluene and tert-butanol; and the solubility of the tested diols and the final products could be different. Secondly, the reaction time we applied was 3 times longer. Finally, the polycondensation efficiency is much higher by using the two-stage method under high vacuum.
Moreover, the enzymatic polymerization of DMFDCA with 2,3-BDO/isosorbide resulted in low molecular weight products since the secondary hydroxyl group is not favored by CALB. Regarding isosorbide, this may be also due to the fact that the hydroxyl groups of isosorbide can be self-condensed with CALB to form ethers as reported by Catalani et al.41
Besides, the enzymatic polymerization of DMFDCA with the tested polyols gave oligomers due to the low solubility of glycerol and D-sorbitol in the reaction media. This obstacle might be overcome by performing the enzymatic polycondensation in bulk. As reported by Gross et al.,55 high molecular weight polyol-based aliphatic polyesters were successfully prepared by CALB-catalyzed enzymatic polycondensation without the solvent. However, more experiments should be performed to study the enzymatic melt polymerization of DMFDCA with polyols since the monomers might be immiscible at mild temperatures.
CALB-catalyzed synthesis of furanic–aliphatic polyesters from DMFDCA and α,ω-aliphatic linear diols via the temperature-varied two-stage method
As discussed above, the major obstacle for enzymatic synthesis of furanic–aliphatic polyesters at mild temperatures is the phase separation caused by the high Tm and the low solubility of the final products. This problem could be circumvented by performing the enzymatic polymerization at higher reaction temperatures. Generally speaking, the enzyme catalysts possess the highest catalytic reactivity at temperatures below 100 °C. However, the immobilized CALB can also function at elevated temperatures up to 150 °C as reported by Zelisko et al.,50 Landfester et al.,49 and Iborra et al.48 Inspired by this, we performed enzymatic polymerizations at temperatures ranging from 80 to 140 °C via a modified two-stage method with varied temperatures. In this method, the polymerization conditions were maintained in the same way as for the normal two-stage method at the first stage. Then at the second stage, the reaction pressure was regulated at 2 mmHg, also in the same way as for the normal two-stage method. But the reaction temperature was gradually increased as follows: (1) 80 °C for the first 24 h; (2) 95 °C for another 24 h; (3) 95, 120, and 140 °C for the final 24 h. In the following discussion of the temperature-varied two-stage enzymatic polymerization, the reaction temperature we mentioned is the temperature during the last 24 h reaction.Moreover, polyesterification is generally boosted at elevated temperatures since the reactivity of the reactants and the reaction speed normally increase with reaction temperature.
The enzymatic polymerization at higher reaction temperatures gave higher molecular weight products (Fig. 3, 4; and S1–S4 and Table S1 in the ESI†). As the representative SEC traces depicted in Fig. 3, furanic–aliphatic polyester produced at higher reaction temperature had a lower retention volume, e.g., the retention volume of the obtained PBF decreased from 15.0–19.2 mL to 13.5–19.2 mL when the reaction temperature was increased from 80 °C to 140 °C. The corresponding 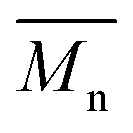 and
and  values increased from 1200 and 1700 g mol−1 to 1600 and 5500 g mol−1, respectively. The increase of molecular weights with reaction temperature can be explained by three reasons. The first reason is that phase separation was delayed at higher reaction temperatures. The second reason is that the elimination of alcohol byproducts and the residual water from enzymatic polymerization became easier at higher temperatures, especially when the reaction media were extremely viscous because of the high molecular weight polyesters produced. Therefore, the efficiency of enzymatic polycondensation was improved. The third reason is that the mobility of the amorphous phase of furanic–aliphatic polyesters was enhanced at higher temperatures, which facilitated the CALB-catalyzed solid-state polymerization.
values increased from 1200 and 1700 g mol−1 to 1600 and 5500 g mol−1, respectively. The increase of molecular weights with reaction temperature can be explained by three reasons. The first reason is that phase separation was delayed at higher reaction temperatures. The second reason is that the elimination of alcohol byproducts and the residual water from enzymatic polymerization became easier at higher temperatures, especially when the reaction media were extremely viscous because of the high molecular weight polyesters produced. Therefore, the efficiency of enzymatic polycondensation was improved. The third reason is that the mobility of the amorphous phase of furanic–aliphatic polyesters was enhanced at higher temperatures, which facilitated the CALB-catalyzed solid-state polymerization.
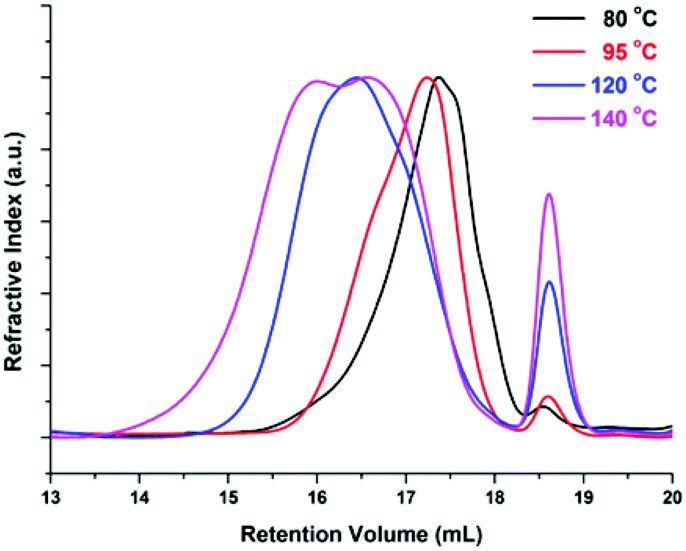 | ||
| Fig. 3 Representative SEC retention curves of poly(butylene furanoate) (PBF) from the temperature-varied two-stage enzymatic polymerization. The temperature indicated here is the reaction temperature during the last 24 h reaction. | ||
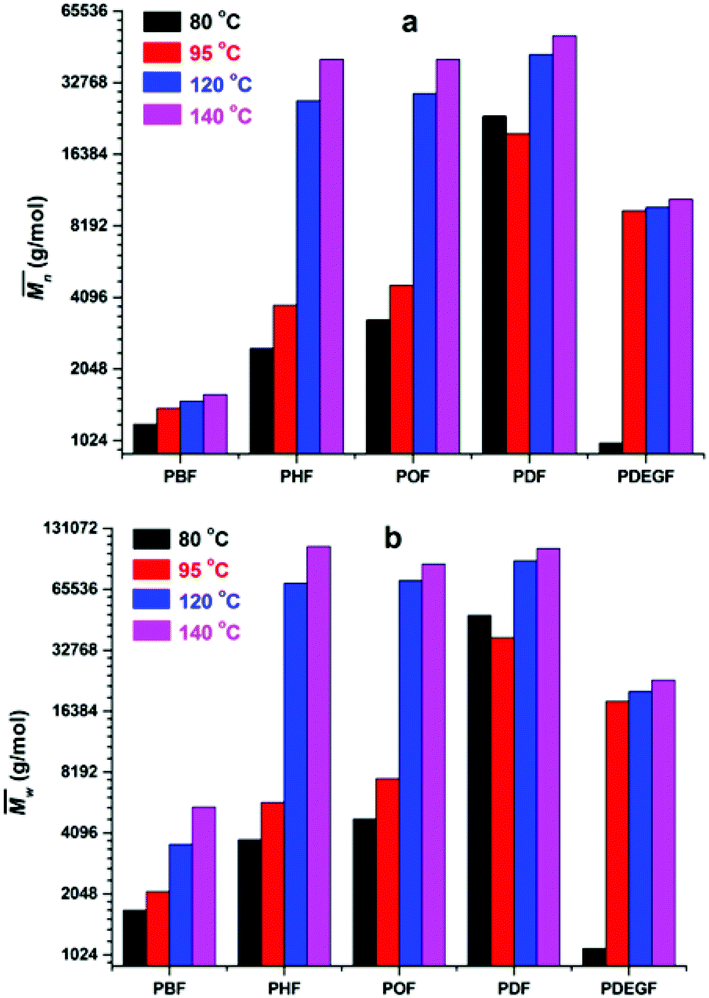 | ||
Fig. 4 The number average molecular weight ( ) and weight average molecular weight ( ) and weight average molecular weight ( ) of the obtained furanic–aliphatic polyesters from the temperature-varied two-stage enzymatic polymerization. The temperature indicated here is the reaction temperature during the last 24 h reaction. ) of the obtained furanic–aliphatic polyesters from the temperature-varied two-stage enzymatic polymerization. The temperature indicated here is the reaction temperature during the last 24 h reaction. | ||
We noticed that the molecular weights of PBF/PDF did not increase too much when the reaction temperature was increased from 80 to 140 °C, but those of PHF/POF increased significantly at the same time. We observed that phase separation still occurred during the enzymatic polymerization of DMFDCA with 1,4-BDO, although it was delayed a bit at higher reaction temperatures. As the DSC results shown later, the Tm of the final product PBF increased significantly from 145 to 168 °C when its  value increased from 1700 g mol−1 to 5500 g mol−1. Therefore, the chain growth of PBF was extremely limited at the tested temperatures. This is the reason why the molecular weights of the obtained PBF increased with reaction temperature, but cannot reach higher values. Meanwhile, the enzymatic polymerization of DMFDCA with 1,10-DDO remained always monophasic at the tested temperatures as discussed before. Hence, the tested reaction temperature had no significant influences on the molecular weights of the obtained PDF. However, phase separation was circumvented at higher reaction temperatures during the enzymatic polymerization of DMFDCA with 1,6-HDO/1,8-ODO. Thus, the enzymatic polymerization yielded significantly higher molecular weight PHF/POF at higher temperatures above 80 °C.
value increased from 1700 g mol−1 to 5500 g mol−1. Therefore, the chain growth of PBF was extremely limited at the tested temperatures. This is the reason why the molecular weights of the obtained PBF increased with reaction temperature, but cannot reach higher values. Meanwhile, the enzymatic polymerization of DMFDCA with 1,10-DDO remained always monophasic at the tested temperatures as discussed before. Hence, the tested reaction temperature had no significant influences on the molecular weights of the obtained PDF. However, phase separation was circumvented at higher reaction temperatures during the enzymatic polymerization of DMFDCA with 1,6-HDO/1,8-ODO. Thus, the enzymatic polymerization yielded significantly higher molecular weight PHF/POF at higher temperatures above 80 °C.
We also found that some oligomers, oligo(butylene furanoate) (OBF), were clearly present in the tested PBF. The proportion of OBF in the final products increased with reaction temperature from 80 to 140 °C. As shown in Fig. 3, two peaks appeared in the SEC traces of the tested PBF (Fig. 3). The dominant peak was ranging from 13.2 mL to 18.3 mL, which was ascribed to PBF with relatively higher  values of more than 1000 g mol−1. The other peak with a rather low integral intensity was ranging from 18.3 to 19.2 mL. This was attributed to the small amount of OBF with a
values of more than 1000 g mol−1. The other peak with a rather low integral intensity was ranging from 18.3 to 19.2 mL. This was attributed to the small amount of OBF with a  value of around 500 g mol−1. As the cumulative weight fractions calculated from SEC in Table S2 (see the ESI†), the proportion of OBF in the final products increased from 2.3% to 10.0% when the reaction temperature was increased from 80 °C to 140 °C. This phenomenon is attributed to three reasons: (1) the high crystallization ability of OBF; (2) the lower catalytic reactivity of CALB at elevated temperatures above 90 °C;56 and (3) the lower conversion of short OBF chains to long PBF chains at higher reaction temperatures. As the WAXD results shown later, the degree of crystallinity of the tested PBF decreased with the increase of molecular weight. According to this, we can expect that OBF can reach a higher degree of crystallinity of more than 56%. During enzymatic polymerization, OBF crystallized and isolated from the reaction media at the first stage. Therefore, the enzymatic polymerization was dominated by the solid-state polymerization mechanism at the tested temperatures; the chain growth was limited to the amorphous phase. Meanwhile, the catalytic reactivity of CALB decreased because of the occurrence of protein denaturation and deactivation at elevated temperatures above 90 °C. As a result, the polymerization efficiency decreased at higher reaction temperature, although OBF melted easily at the same time. Moreover, the conversion of short OBF chains to long PBF chains became lower at higher reaction temperatures due to the excessive evaporation of the diphenyl ether solvent under vacuum.
value of around 500 g mol−1. As the cumulative weight fractions calculated from SEC in Table S2 (see the ESI†), the proportion of OBF in the final products increased from 2.3% to 10.0% when the reaction temperature was increased from 80 °C to 140 °C. This phenomenon is attributed to three reasons: (1) the high crystallization ability of OBF; (2) the lower catalytic reactivity of CALB at elevated temperatures above 90 °C;56 and (3) the lower conversion of short OBF chains to long PBF chains at higher reaction temperatures. As the WAXD results shown later, the degree of crystallinity of the tested PBF decreased with the increase of molecular weight. According to this, we can expect that OBF can reach a higher degree of crystallinity of more than 56%. During enzymatic polymerization, OBF crystallized and isolated from the reaction media at the first stage. Therefore, the enzymatic polymerization was dominated by the solid-state polymerization mechanism at the tested temperatures; the chain growth was limited to the amorphous phase. Meanwhile, the catalytic reactivity of CALB decreased because of the occurrence of protein denaturation and deactivation at elevated temperatures above 90 °C. As a result, the polymerization efficiency decreased at higher reaction temperature, although OBF melted easily at the same time. Moreover, the conversion of short OBF chains to long PBF chains became lower at higher reaction temperatures due to the excessive evaporation of the diphenyl ether solvent under vacuum.
One may argue that dehydration of diol and β-scission of polyesters could occur at elevated temperatures, which also lead to low molecular weights. We thought that the tested temperatures were not high enough to induce such side-reactions. Also as suggested by the MALDI-ToF MS analysis shown later, no acid and alkene end groups were observed in the obtained PBF. However, there is no clear proof that the dehydration of diol did not occur during enzymatic polymerization. More study should be performed to investigate the enzymatic polymerization at elevated temperatures.
We found that DEG behaved differently in the enzymatic polymerization compared to the tested alkane-α,ω-aliphatic linear diols (1,4-BDO, 1,6-HDO, 1,8-ODO, and 1,10-DDO). As shown in Table 1, the two-stage enzymatic polymerization at 80 °C yielded PDEGF with a 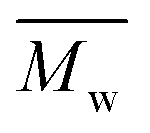 value of 1100 g mol−1, lower than that of PBF (
value of 1100 g mol−1, lower than that of PBF (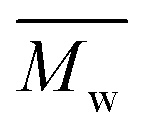 = 1700 g mol−1) synthesized under the same reaction conditions. This was unexpected since DEG has a longer chain length (n = 5) than 1,4-BDO (n = 4). We suspected that the esterification/transesterification reactivity of the hydroxyl groups of DEG is much lower at such a temperature because the electropositivity of the carbon chain is reduced by the oxygen atom within the middle of DEG. However, PDEGF with significantly higher molecular weights than PBF was obtained at temperatures above 80 °C. This could be attributed to two reasons. Firstly, the esterification/transesterification reactivity of the hydroxyl groups of DEG is significantly enhanced at higher temperatures. Secondly, the enzymatic polymerization with PDEGF was monophasic since PDEGF has better solubility in diphenyl ether and low Tm or even no Tm (see DSC results below). As a result, the enzymatic polymerization of DMFDCA with DEG gave high molecular weight products at temperatures above 80 °C.
= 1700 g mol−1) synthesized under the same reaction conditions. This was unexpected since DEG has a longer chain length (n = 5) than 1,4-BDO (n = 4). We suspected that the esterification/transesterification reactivity of the hydroxyl groups of DEG is much lower at such a temperature because the electropositivity of the carbon chain is reduced by the oxygen atom within the middle of DEG. However, PDEGF with significantly higher molecular weights than PBF was obtained at temperatures above 80 °C. This could be attributed to two reasons. Firstly, the esterification/transesterification reactivity of the hydroxyl groups of DEG is significantly enhanced at higher temperatures. Secondly, the enzymatic polymerization with PDEGF was monophasic since PDEGF has better solubility in diphenyl ether and low Tm or even no Tm (see DSC results below). As a result, the enzymatic polymerization of DMFDCA with DEG gave high molecular weight products at temperatures above 80 °C.
Meanwhile, changing the reaction temperature from 95 to 140 °C had no significant influence on the molecular weights of PDEGF (Fig. 4, S4, and Table S4†). The  and
and 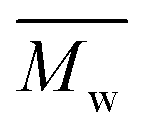 values slightly increased from 9500 and 18300 g mol−1 to 10600 and 23300 g mol−1, respectively. This is because the esterification/transesterification reactivity of DEG increased with reaction temperature, but the catalytic reactivity of CALB decreased at the same time.
values slightly increased from 9500 and 18300 g mol−1 to 10600 and 23300 g mol−1, respectively. This is because the esterification/transesterification reactivity of DEG increased with reaction temperature, but the catalytic reactivity of CALB decreased at the same time.
Furthermore, the polymerization time had a significant influence on the enzymatic polymerization of PDEGF. As shown in Fig. 5 and S5,† the 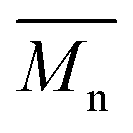 and
and  values increased significantly from 9500 and 18300 g mol−1 to 31400 and 69400 g mol−1, respectively, by extending the reaction time from 48 h to 172 h while maintaining the reaction temperature at 95 °C. This is reasonable because the immobilized CALB is very stable at mild temperatures below 100 °C. At such temperatures it can function well for a very long time without losing too much catalytic reactivity.
values increased significantly from 9500 and 18300 g mol−1 to 31400 and 69400 g mol−1, respectively, by extending the reaction time from 48 h to 172 h while maintaining the reaction temperature at 95 °C. This is reasonable because the immobilized CALB is very stable at mild temperatures below 100 °C. At such temperatures it can function well for a very long time without losing too much catalytic reactivity.
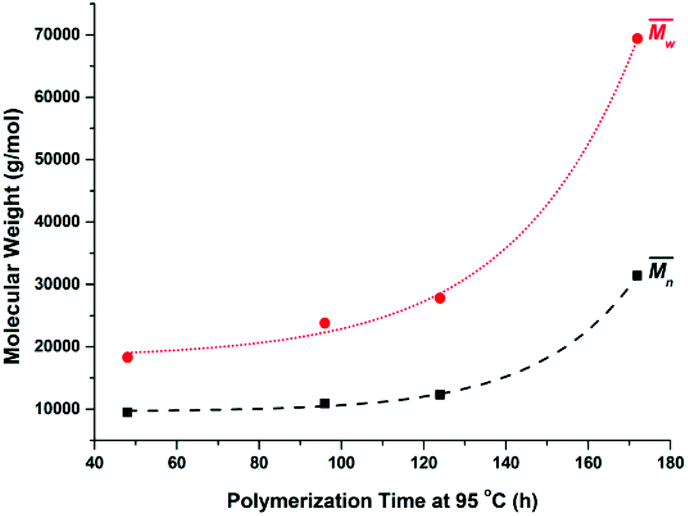 | ||
Fig. 5 The number average molecular weight ( ) and weight average molecular weight ( ) and weight average molecular weight ( ) of poly(diethylene glycol furanoate) (PDEGF) from the temperature-varied two-stage enzymatic polymerization at 95 °C. The reaction time started to count after keeping the reaction at 80 °C for 26 h. ) of poly(diethylene glycol furanoate) (PDEGF) from the temperature-varied two-stage enzymatic polymerization at 95 °C. The reaction time started to count after keeping the reaction at 80 °C for 26 h. | ||
One disadvantage was identified for the two-stage solution enzymatic polymerization at the elevated reaction temperatures (120/140 °C): the reaction media boiled and the solvent diphenyl ether evaporated excessively under such conditions. This could lead to higher dispersity of the polymers.
Control reactions suggested that the catalytic reactivity of the used CALB still retained but decreased (Table S3†). The used CALB tested included: (1) the recycled CALB from the enzymatic polymerization at 120/140 °C; and (2) the CALB treated under the same conditions as those applied in the temperature-varied two-stage enzymatic polymerization at 120/140 °C. It should be noted that the decrease of catalytic reactivity of CALB at elevated temperatures is due to the unfolding and/or inactivating of enzymes. In recent years, many approaches have been developed to address this problem,57–60 such as genetic engineering, chemical modifications, improving immobilization methods, employing nanomaterials as enzyme supporters, etc. Our group also did a lot of studies on immobilization of CALB to improve its stability and catalytic reactivity.61–68 However, the denaturation and deactivation of CALB seem to be inevitable at temperature above 100 °C, although the thermal stability of CALB at mild temperatures can be improved by many methods like using magnetic nanoparticles,60 double immobilization,69 hierarchical interfacial assembly,70 and co-immobilization with ionic liquids.57,71 Therefore, developing immobilized CALB working fully at elevated temperatures are still challenging and demanding. However, elevated temperatures is not necessarily required if enzymatic polymerization is homogeneous at mild temperatures using good solvents for monomers and polymers, e.g. ionic liquids.57 Ionic liquids are considered as eco-friendly solvents since they are non-volatile. Nevertheless, it is also challenging to design suitable ionic liquids for enzymatic polymerization.
Besides, all the tested polymerizations were indeed catalyzed by using enzyme CALB. As confirmed by the blank reactions, no polyester was obtained without CALB by using the same two-stage method and temperature-varied two-stage method.
MALDI-ToF MS analysis of the obtained furanic–aliphatic polyesters: microstructures and end groups
The microstructures and end groups of the synthesized furanic–aliphatic polyesters were characterized by MALDI-ToF MS. The representative MS spectra with peak interpretations are illustrated in Fig. 6 and S6–S15 (see ESI†). Five polyester species with different end groups were identified (Tables 2, and S4 in the ESI†). Four of them were terminated with ester/–OH, ester/ester, –OH/–OH, and ester/aldehyde groups, respectively. The rest was cyclic without end groups. Initially we postulated that the aldehyde end group might be originated from the impurity methyl 5-formyl-2-furoate consisting in the starting material DMFDCA. However, no extra resonance was observed in the 1H-NMR spectrum of DMFDCA (see Fig. S16†). In addition, the mass peaks attributed to the polyester species with ester/aldehyde groups showed rather low intensities, and in most of the tested samples they were undetected. Therefore, we suspected that there might be only a tiny amount of impurity of methyl 5-formyl-2-furoate in DMFDCA, which cannot be detected by NMR.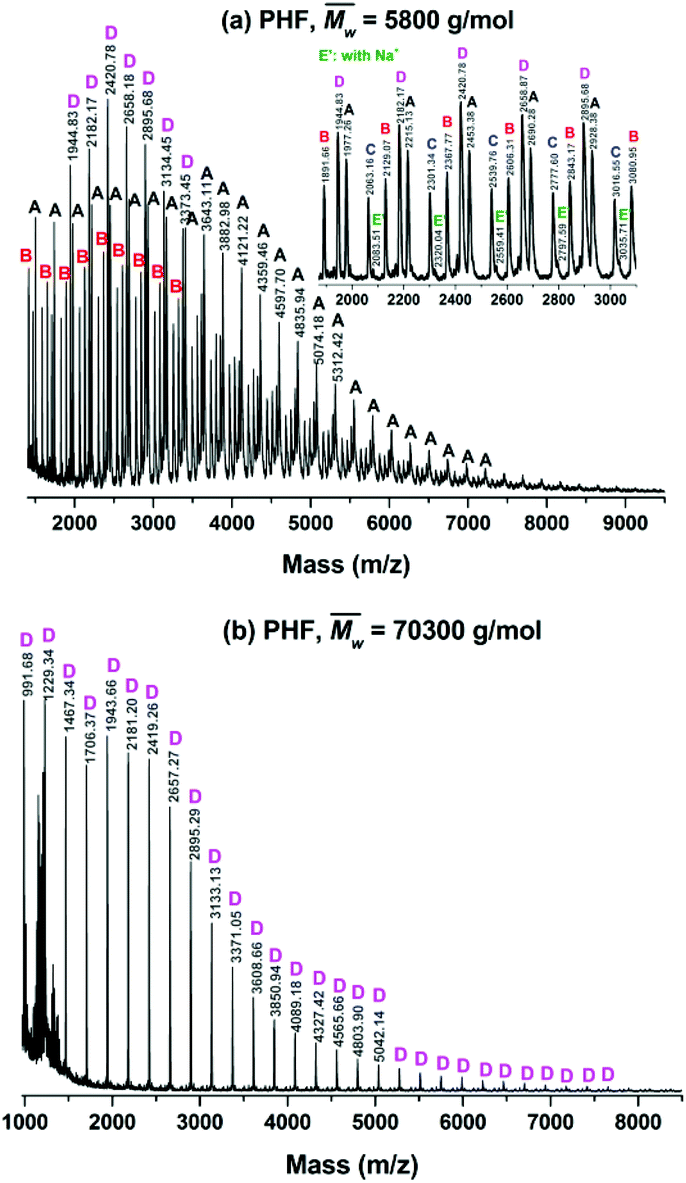 | ||
| Fig. 6 The representative MALDI-ToF MS spectra of poly(hexamethylene furanoate) (PHF) with peak interpretations. A, B, C, D, and E represent the five polyester species ionized by K+. They are terminated with the ester/–OH, ester/ester, –OH/–OH, no end groups (cyclic), and ester/aldehyde groups, respectively. E′ represents the polyesters having the ester/aldehyde end groups that are ionized by Na+. | ||
| Polyester species | End groups | Remaining mass (amu) | |
|---|---|---|---|
| A |

|
Ester/–OH | 32.04 |
| B |
|
Ester/ester | 184.15 |
| C |
|
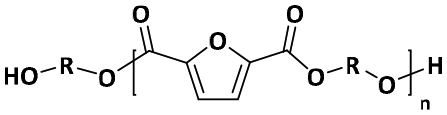
–OH/–OH | R = (CH2)4: 90.12 |
| R = CH(CH3)–CH(CH3): 90.12 | |||
| R = (CH2)6: 118.18 | |||
| R = (CH2)8: 146.23 | |||
| R = (CH2)10: 174.28 | |||
| R = (CH2)2–O–(CH2)2: 106.12 | |||
| D |
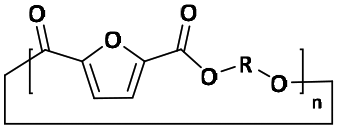
|
Cyclic | 0 |
| E |
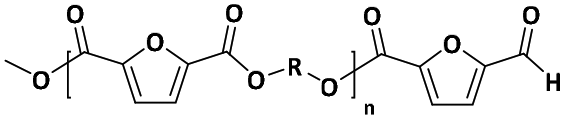
|
Ester/aldehyde | 154.12 |
The MALDI-ToF MS pattern of all the tested low molecular weight furanic–aliphatic polyesters (normally 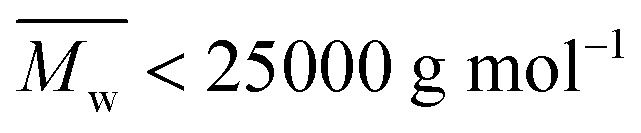 ) was similar, as the representative mass spectrum shown in Fig. 6a. All the identified microstructures were normally present, which might differ in the abundance (see Table S4, ESI†). We observed that furanic–aliphatic polyesters with the ester/–OH and ester/ester end groups were generally the most dominant species if we assume that all polyesters with different end groups and molecular weights behave the same in MALDI. However, this conclusion is not necessarily true since MALDI is not a quantitative technique. The ionization of polymer chains highly depends on their end groups, and polymer chains with low molecular weights are normally overestimated compared to those with higher molecular weights.46,72
) was similar, as the representative mass spectrum shown in Fig. 6a. All the identified microstructures were normally present, which might differ in the abundance (see Table S4, ESI†). We observed that furanic–aliphatic polyesters with the ester/–OH and ester/ester end groups were generally the most dominant species if we assume that all polyesters with different end groups and molecular weights behave the same in MALDI. However, this conclusion is not necessarily true since MALDI is not a quantitative technique. The ionization of polymer chains highly depends on their end groups, and polymer chains with low molecular weights are normally overestimated compared to those with higher molecular weights.46,72
However, the MALDI-ToF MS pattern of the tested high molecular weight furanic–aliphatic polyesters (normally 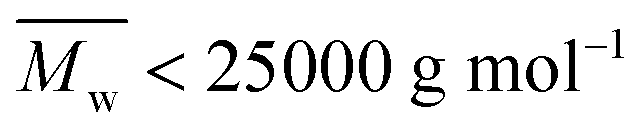 ) was different from those of their low molecular weight counterparts. As the representative mass spectrum depicted in Fig. 6b, cyclic polyesters became the most dominant species. The other four species were rarely observed. However, this was not in agreement with the NMR results in that the methoxyl and hydroxymethyl end groups were clearly observed. We thought this is because the cyclic polyesters had much lower molecular weights. Therefore, the ionized cyclic polyester chains flied easily and they were well detected by MALDI. In contrast, the flight of the other ionized polyester species was extremely limited because of their significantly higher molecular weights. This is the reason why they were absent in the mass spectra.
) was different from those of their low molecular weight counterparts. As the representative mass spectrum depicted in Fig. 6b, cyclic polyesters became the most dominant species. The other four species were rarely observed. However, this was not in agreement with the NMR results in that the methoxyl and hydroxymethyl end groups were clearly observed. We thought this is because the cyclic polyesters had much lower molecular weights. Therefore, the ionized cyclic polyester chains flied easily and they were well detected by MALDI. In contrast, the flight of the other ionized polyester species was extremely limited because of their significantly higher molecular weights. This is the reason why they were absent in the mass spectra.
Crystallinity properties of the obtained furanic–aliphatic polyesters
The tested PBF is a semicrystalline material. It crystallized into a triclinic α-form crystal (Table 3), similar to PBT. This was in good agreement with the previous results reported in the literature.11,20 We also noticed that several extra peaks appeared with low intensities. One possible explanation is that these peaks belong to other crystal structures which were formed by the OBF. As shown in Fig. S17 (see ESI†), the intensity of these peaks increased with , indicating that there was more OBF in proportion to PBF synthesized at higher reaction temperatures. This agreed well with the SEC results discussed above.
, indicating that there was more OBF in proportion to PBF synthesized at higher reaction temperatures. This agreed well with the SEC results discussed above.
| Polymer | Crystal type | 2θ (°)a | d-Spacing (Å)b | Lattice plane |
|---|---|---|---|---|
| a w = weak, s = strong, m = medium. b d-spacing: spacing between the planes in the atomic lattice. | ||||
| PBF | Triclinic α-form | 10.66 (w) | 8.29 | α(001) |
| 17.80 (s) | 4.98 | α(010) | ||
| 22.47 (m) | 3.95 | α(−110) | ||
| 25.03 (s) | 3.55 | α(100) | ||
| PHF | Triclinic β-form | 13.46–13.66 (w) | 6.48–6.57 | β(002) |
| 16.91–16.96 (s) | 5.22–5.24 | β(0–11) | ||
| 24.59–24.74 (s) | 3.60–3.62 | β(100) | ||
| POF | Triclinic β-form | 12.93–13.02 (w) | 6.79–6.84 | β(002) |
| 16.62–16.76 (s) | 5.29–5.33 | β(0–11) | ||
| 24.05–24.10 (s) | 3.69–3.70 | β(100) | ||
| Triclinic β-form | 11.65–11.69 (w) | 7.56–7.59 | β(002) | |
| 16.81–16.86 (s) | 5.25–5.27 | β(0–11) | ||
| 23.80–23. 84 (s) | 3.73–3.74 | β(100) | ||
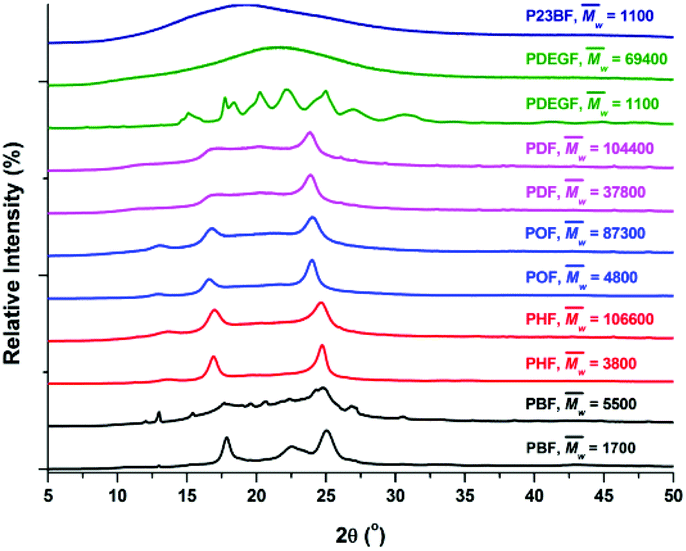
The tested PHF/POF/PDF showed a similar WAXD pattern (Fig. 7, Table 3, and S18–S20†) that was different from PBF. We suspected that the tested PHF/POF/PDF might crystallize into a triclinic β-form crystal, similar to those of the aromatic–aliphatic polyesters with long methylene chains in the diol units (n = 6, 8, 10, 20).73,74 Unfortunately, the unit cells of PHF/POF/PDF have not been reported yet, which require detailed studies in the future. Moreover, we noticed that the diffraction peaks shifted to lower values when the chain length of the diol units in PHF/POF/PDF increased, and the corresponding spacing between the planes in the atomic lattice (d) increased at the same time. This is reasonable because more space is required to form the same crystal lattices if the alkylene chain of the diol units is longer.
 | ||
| Fig. 7 WAXD spectra of the obtained furanic–aliphatic polyesters. | ||
The tested low molecular weight PDEGF (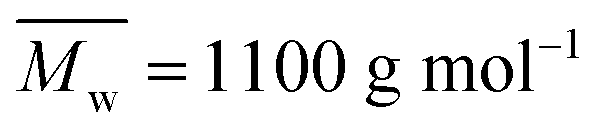 ) is a semicrystalline material. It had eight diffraction peaks, similar to the WAXD pattern of poly(diethylene glycol terephthalate).75 However, all the tested high molecular weight PDEGF are amorphous materials at room temperature. They showed a broad amorphous halo in the WAXD spectra (Fig. S21†). This could be due to the fact that the crystallization rate is very slow for such a high molecular weight PDEGF. In this case, the crystallization was not visible at the tested time scale. The slow crystallization rate of PDEGF could be explained by two reasons. The first reason is that the repeating unit (diethylene glycol furanoate) of PDEGF is more structurally irregular among those in the tested furanic–aliphatic polyesters. This is due to the odd chain length of the DEG units. The second reason is that the two skeletal CH2–CH2 bonds in the DEG unit prefer the gauche conformation to the trans. But only the trans conformations may be present in the crystal as reported in the literature.75 However, the change from the gauche conformation to the trans may be slow in the solid state. Moreover, it should be noted that the tested PDEGF has a similar crystallization behavior to the aromatic–aliphatic polyesters containing the same odd chain length in the diol units. The crystallization rates of these polyester were also quite slow.75,76
) is a semicrystalline material. It had eight diffraction peaks, similar to the WAXD pattern of poly(diethylene glycol terephthalate).75 However, all the tested high molecular weight PDEGF are amorphous materials at room temperature. They showed a broad amorphous halo in the WAXD spectra (Fig. S21†). This could be due to the fact that the crystallization rate is very slow for such a high molecular weight PDEGF. In this case, the crystallization was not visible at the tested time scale. The slow crystallization rate of PDEGF could be explained by two reasons. The first reason is that the repeating unit (diethylene glycol furanoate) of PDEGF is more structurally irregular among those in the tested furanic–aliphatic polyesters. This is due to the odd chain length of the DEG units. The second reason is that the two skeletal CH2–CH2 bonds in the DEG unit prefer the gauche conformation to the trans. But only the trans conformations may be present in the crystal as reported in the literature.75 However, the change from the gauche conformation to the trans may be slow in the solid state. Moreover, it should be noted that the tested PDEGF has a similar crystallization behavior to the aromatic–aliphatic polyesters containing the same odd chain length in the diol units. The crystallization rates of these polyester were also quite slow.75,76
Moreover, the tested P23BF is an amorphous material, showing a broad diffraction halo in its WAXD spectrum (Fig. 7). This can be explained by the strong steric-hindrance effect caused by the appending methyl groups in the polymer main chain.
The degree of crystallinity (χc) and the enthalpy of fusion (ΔHm) of the obtained furanic–aliphatic polyesters were calculated from the WAXD and DSC spectra, respectively (see Table S5 in the ESI†). The χc and ΔHm values in general decreased significantly with increasing  value from around 2000 g mol−1 to around 40000 g mol−1 (see Fig. S22–25†). This is because the mobility of polymer chains reduces fast with
value from around 2000 g mol−1 to around 40000 g mol−1 (see Fig. S22–25†). This is because the mobility of polymer chains reduces fast with  , and the entanglement of polymer chains enhances rapidly at the same time. However, the χc and ΔHm reached constant values when the
, and the entanglement of polymer chains enhances rapidly at the same time. However, the χc and ΔHm reached constant values when the  value further increased. This is due to the fact that the mobility and entanglement of the polymer chains are almost the same when the
value further increased. This is due to the fact that the mobility and entanglement of the polymer chains are almost the same when the  value reaches a critical value.
value reaches a critical value.
The χc values of the obtained furanic–aliphatic polyesters are plotted as a function of the chain length of the alkane-α,ω-aliphatic linear diol units in Fig. 8. For the tested furanic–aliphatic polyesters with high 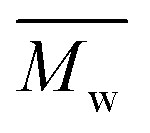 values of around 100000 g mol−1, the χc value decreased from 37% to 21% when the diol chain length increased from 6 to 10. This is due to the fact that the chain regularity decreases with the increase of chain length in the diol units. In addition, we expect that the χc value of PBF with the similar high
values of around 100000 g mol−1, the χc value decreased from 37% to 21% when the diol chain length increased from 6 to 10. This is due to the fact that the chain regularity decreases with the increase of chain length in the diol units. In addition, we expect that the χc value of PBF with the similar high  could be much higher than those of PHF/POF/PDF, although we did not obtain a high molecular weight PBF. This is because the chain regularity of the repeating unit (butylene furanoate) of PBF is the highest among those in the tested furanic–aliphatic polyesters. In conclusion, the χc value of furanic–aliphatic polyesters with similar high molecular weights decreased with increasing chain length of the alkane-α,ω-aliphatic linear diol units. In other words, the crystallization ability of high molecular weight furanic–aliphatic polyesters decreases with increasing chain length of the alkane-α,ω-aliphatic linear diol units.
could be much higher than those of PHF/POF/PDF, although we did not obtain a high molecular weight PBF. This is because the chain regularity of the repeating unit (butylene furanoate) of PBF is the highest among those in the tested furanic–aliphatic polyesters. In conclusion, the χc value of furanic–aliphatic polyesters with similar high molecular weights decreased with increasing chain length of the alkane-α,ω-aliphatic linear diol units. In other words, the crystallization ability of high molecular weight furanic–aliphatic polyesters decreases with increasing chain length of the alkane-α,ω-aliphatic linear diol units.
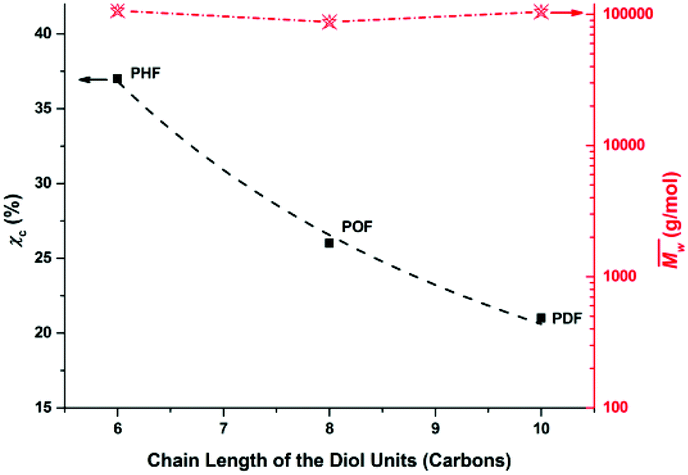 | ||
Fig. 8 The degree of crystallinity (χc) of furanic–aliphatic polyesters as a function of the chain length of the alkane-α,ω-aliphatic linear diol units. The tested furanic–aliphatic polyesters have similar  (weight average molecular weight) values of around 100000 g mol−1. (weight average molecular weight) values of around 100000 g mol−1. | ||
Furthermore, we calculated the ΔH0m of the furanic–aliphatic polyesters with alkane-α,ω-aliphatic linear diol units by linear fitting of ΔHm as a function of χc and then extrapolating to 100% crystallinity (see Fig. S26–S29†). We found that the ΔH0m value increased with increasing chain length of the diol units (Fig. S30†). Moreover, the ΔH0m of PBF was 123 J g−1 according to our calculation. This is in good agreement with the previous value reported in the literature, which was 129 J g−1.13
Thermal stability of the obtained furanic–aliphatic polyesters
The thermal stability of the obtained furanic–aliphatic polyesters was characterized by TGA. As shown in Fig. S31 and Table S5,† the tested P23BF and PBF were less thermal stable than the other tested furanic–aliphatic polyesters. They displayed an extra decomposition step before 350 °C. This can be ascribed to the decomposition of the oligomers consisting in the tested PBF/P23BF due to their low molecular weights ( ).
).
The thermal stability of the tested PHF, POF and PDF with high  values was similar. As shown in Table S5,† the decomposition temperature at 5% weight loss (Td-5%), decomposition temperature at 10% weight loss (Td-10%), and temperature at a maximum rate of decomposition (Td-max) of these furanic–aliphatic polyesters were ranging from 361 to 369 °C, 370 to 378 °C, and 397 to 402 °C, respectively.
values was similar. As shown in Table S5,† the decomposition temperature at 5% weight loss (Td-5%), decomposition temperature at 10% weight loss (Td-10%), and temperature at a maximum rate of decomposition (Td-max) of these furanic–aliphatic polyesters were ranging from 361 to 369 °C, 370 to 378 °C, and 397 to 402 °C, respectively.
To sum up, all the tested high molecular weight furanic–aliphatic polyesters are thermally stable up to 360 °C, indicating that they possess a very broad processing window.
Thermal transitions of the obtained furanic–aliphatic polyesters
The thermal transitions of the obtained furanic–aliphatic polyesters were measured by DSC. The results are summarized in Table S5 (ESI†). As shown in Fig. S32,† no melting peaks appeared in the DSC heating curves of the tested P23BF and high molecular weight PDEGF. However, the tested low molecular weight PDEGF ( ) showed two small melting peaks in the first heating scan (see Fig. S33†). This suggested that the short PDEGF chains were able to crystalize, which can be attributed to their high chain flexibility. However, this low molecular weight PDEGF showed no crystallization peak (Tc) in the cooling step and no melting peak in the second heating scan. This indicated that the crystallization rate of these PDEGF chains was quite slow at the tested time scale. Moreover, the PDEGF chains were still able to crystallize after long-time annealing at temperatures above the Tg (see Fig. S34†).
) showed two small melting peaks in the first heating scan (see Fig. S33†). This suggested that the short PDEGF chains were able to crystalize, which can be attributed to their high chain flexibility. However, this low molecular weight PDEGF showed no crystallization peak (Tc) in the cooling step and no melting peak in the second heating scan. This indicated that the crystallization rate of these PDEGF chains was quite slow at the tested time scale. Moreover, the PDEGF chains were still able to crystallize after long-time annealing at temperatures above the Tg (see Fig. S34†).
The Tm of the tested furanic–aliphatic polyesters with similar high molecular weights decreased with increasing chain length of the alkane-α,ω-aliphatic linear diol units (See Fig. 9). This agreed well with the previous study reported by Noordover et al.72 They investigated the thermal properties of the isoidide dicarboxylic acid-based aliphatic polyesters that consist of the rigid isoidide units and alkane-α,ω-aliphatic linear diol units. These polyesters are similar to the tested furanic–aliphatic polyesters with respect to the rigid-soft structure pattern in the repeating units. It was found that the Tm of the isoidide dicarboxylic acid-based aliphatic polyesters showed a continuous decrease with increasing chain length of the alkane-α,ω-aliphatic linear diol units from 2 to 10. They thought that this is caused by the “polyethylene effect” since the thermal properties of polyesters approach those of polyethylene with increasing number of methylene units incorporated into the polyester main chain.
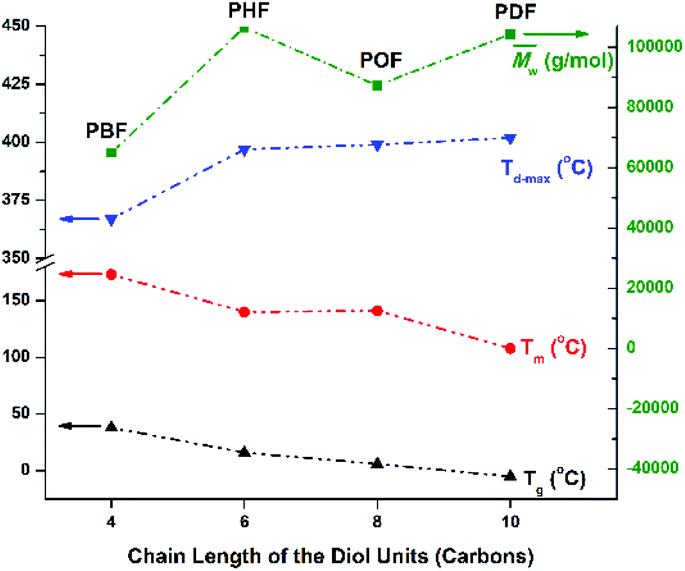 | ||
| Fig. 9 Structure–property relationships of the obtained furani–aliphatic polyesters containing alkane-α,ω-aliphatic linear diol units. The properties of the high molecular weight PBF are obtained from the literature14. | ||
The Tg of the high molecular weight furanic–aliphatic polyesters showed a continuous decrease with increasing chain length of the alkane-α,ω-aliphatic linear diol units (Fig. 9 and S35†). Again this agreed well with the previous results reported in the literature.72 It was found that the Tg of polyadipates, polygalactarates, and isoidide dicarboxylic acid-based aliphatic polyesters steadily decreased with increasing chain length of the aliphatic diol units. This can be explained by the similar “polyethylene effect” as discussed above.
Conclusions
In the present study, we prove that enzymatic polymerization is a robust approach for the synthesis of biobased furanic–aliphatic polyesters. P23BF, PBF, PDEGF, and PHF/POF/PDF are successfully prepared via CALB-catalyzed polymerization, with the value of 1100, 5500, 69400, and around 100000 g mol−1, respectively. These furanic–aliphatic polyesters are promising sustainable alternatives to the petroleum-based aromatic–aliphatic polyesters.
value of 1100, 5500, 69400, and around 100000 g mol−1, respectively. These furanic–aliphatic polyesters are promising sustainable alternatives to the petroleum-based aromatic–aliphatic polyesters.
Higher molecular weight furanic–aliphatic polyesters are produced by enzymatic polymerization of DMFDCA with longer-chain alkane-α,ω-aliphatic linear diols. Only oligomers are obtained from enzymatic polymerization of DMFDCA with 2,3-BDO/isosorbide/glycerol/D-sorbitol. These results reveal that CALB prefers longer-chain alkane-α,ω-aliphatic linear diols.
Moreover, enzymatic polymerization is significantly boosted at elevated temperatures up to 140 °C. We found that the molecular weights of furanic–aliphatic polyesters increase steadily with increasing reaction temperature from 80 to 140 °C.
In addition, five polyester species with different end groups are identified by MALDI-TOF MS. They were terminated with the ester/–OH, ester/ester, –OH/–OH, no end groups (cyclic), and ester/aldehyde groups, respectively. These five polyester species are present in the mass spectra of the low molecular weight products (normally  ). However, cyclic polyesters are the most dominated microstructures in the MS spectra of high molecular weight products (normally
). However, cyclic polyesters are the most dominated microstructures in the MS spectra of high molecular weight products (normally  ).
).
Furthermore, the crystalline and thermal properties of high molecular weight furanic–aliphatic polyesters with alkane-α,ω-aliphatic linear diol units are significantly influenced by the chain length of the diol units. The χc, Tm, and Tg of furanic–aliphatic polyesters decrease steadily with increasing chain length of the alkane-α,ω-aliphatic linear diol units, assuming they have similar high molecular weight values.
Acknowledgements
This research forms part of the research programme of the Dutch Polymer Institute (DPI), project #727c polymers go even greener. Great thanks to Binbing Liu for the photographs in the TOC file.Notes and references
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- A. Gandini, Green Chem., 2011, 13, 1061–1083 RSC.
- M. M. Reddy, S. Vivekanandhan, M. Misra, S. K. Bhatia and A. K. Mohanty, Prog. Polym. Sci., 2013, 38, 1653–1689 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. J. Coelho, C. S. R. Freire and A. J. D. Silvestre, Polym. Chem., 2014, 5, 3119–3141 RSC.
- S. Laurichesse and L. Averous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS PubMed.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- A. Corma Canos, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef PubMed.
- D. I. Collias, A. M. Harris, V. Nagpal, I. W. Cottrell and M. W. Schultheis, Ind. Biotechnol., 2014, 10, 91–105 CrossRef CAS.
- R. J. I. Knoop, W. Vogelzang, J. van Haveren and D. S. van Es, J. Polym. Sci., Polym. Chem., 2013, 51, 4191–4199 CrossRef CAS PubMed.
- J. Zhu, J. Cai, W. Xie, P.-H. Chen, M. Gazzano, M. Scandola and R. A. Gross, Macromolecules, 2013, 46, 796–804 CrossRef CAS.
- S. K. Burgess, J. E. Leisen, B. E. Kraftschik, C. R. Mubarak, R. M. Kriegel and W. J. Koros, Macromolecules, 2014, 47, 1383–1391 CrossRef CAS.
- G. Z. Papageorgiou, V. Tsanaktsis, D. G. Papageorgiou, S. Exarhopoulos, M. Papageorgiou and D. N. Bikiaris, Polymer, 2014, 55, 3846–3858 CrossRef CAS PubMed.
- S. Thiyagarajan, W. Vogelzang, R. J. I. Knoop, A. E. Frissen, J. van Haveren and D. S. van Es, Green Chem., 2014, 16, 1957–1966 RSC.
- J. A. Moore and J. E. Kelly, Abstr. Pap., Am. Chem. Soc., 1974, 85–85 Search PubMed.
- J. A. Moore and J. E. Kelly, Macromolecules, 1978, 11, 568–573 CrossRef CAS.
- A. Gandini, A. J. D. Silvestre, C. P. Neto, A. F. Sousa and M. Gomes, J. Polym. Sci., Polym. Chem., 2009, 47, 295–298 CrossRef CAS PubMed.
- M. Gomes, A. Gandini, A. J. D. Silvestre and B. Reis, J. Polym. Sci., Polym. Chem., 2011, 49, 3759–3768 CrossRef CAS PubMed.
- M. Jiang, Q. Liu, Q. Zhang, C. Ye and G. Zhou, J. Polym. Sci., Polym. Chem., 2012, 50, 1026–1036 CrossRef CAS PubMed.
- J. Ma, X. Yu, J. Xu and Y. Pang, Polymer, 2012, 53, 4145–4151 CrossRef CAS PubMed.
- E. Gubbels, L. Jasinska-Walc and C. E. Koning, J. Polym. Sci,. Polym. Chem., 2013, 51, 890–898 CrossRef CAS PubMed.
- L. Wu, R. Mincheva, Y. Xu, J. M. Raquez and P. Dubois, Biomacromolecules, 2012, 13, 2973–2981 CrossRef CAS PubMed.
- A. F. Sousa, M. Matos, C. S. R. Freire, A. J. D. Silvestre and J. F. J. Coelho, Polymer, 2013, 54, 513–519 CrossRef CAS PubMed.
- G. Z. Papageorgiou, V. Tsanaktsis and D. N. Bikiaris, Phys. Chem. Chem. Phys., 2014, 16, 7946–7958 RSC.
- R. A. Gross, A. Kumar and B. Kalra, Chem. Rev., 2001, 101, 2097–2124 CrossRef CAS PubMed.
- S. Kobayashi, H. Uyama and S. Kimura, Chem. Rev., 2001, 101, 3793–3818 CrossRef CAS PubMed.
- S. Kobayashi and A. Makino, Chem. Rev., 2009, 109, 5288–5353 CrossRef CAS PubMed.
- R. A. Gross, M. Ganesh and W. Lu, Trends Biotechnol., 2010, 28, 435–443 CrossRef CAS PubMed.
- J. i. Kadokawa and S. Kobayashi, Curr. Opin. Chem. Biol., 2010, 14, 145–153 CrossRef CAS PubMed.
- Y. Yang, J. X. Zhang, D. Wu, Z. Xing, Y. L. Zhou, W. Shi and Q. S. Li, Biotechnol. Adv., 2014, 32, 642–651 CrossRef CAS PubMed.
- J. X. Zhang, H. Shi, D. Wu, Z. Xing, A. J. Zhang, Y. Yang and Q. S. Li, Process Biochem., 2014, 49, 797–806 CrossRef CAS PubMed.
- K. Loos, in Biocatalysis in Polymer Chemistry, ed. K. Loos, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, pp. i–xxix Search PubMed.
- N. Miletić, K. Loos and R. A. Gross, in Biocatalysis in Polymer Chemistry, ed. K. Loos, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, pp. 83–129 Search PubMed.
- E. Stavila, G. O. R. Alberda van Ekenstein and K. Loos, Biomacromolecules, 2013, 14, 1600–1606 CrossRef CAS PubMed.
- E. Stavila, R. Z. Arsyi, D. M. Petrovic and K. Loos, Eur. Polym. J., 2013, 49, 834–842 CrossRef CAS PubMed.
- E. Stavila and K. Loos, Tetrahedron Lett., 2013, 54, 370–372 CrossRef CAS PubMed.
- E. Stavila, G. O. R. Alberda van Ekenstein, A. J. J. Woortman and K. Loos, Biomacromolecules, 2014, 15, 234–241 CrossRef CAS PubMed.
- H. Azim, A. Dekhterman, Z. Jiang and R. A. Gross, Biomacromolecules, 2006, 7, 3093–3097 CrossRef CAS PubMed.
- Y. Jiang, A. J. J. Woortman, G. O. R. Alberda van Ekenstein and K. Loos, Biomolecules, 2013, 3, 461–480 CrossRef CAS PubMed.
- Y. Jiang, G. O. R. Alberda van Ekenstein, A. J. J. Woortman and K. Loos, Macromol. Chem. Phys., 2014, 215, 2185–2197 CrossRef CAS PubMed.
- D. Juais, A. F. Naves, C. Li, R. A. Gross and L. H. Catalani, Macromolecules, 2010, 43, 10315–10319 CrossRef CAS.
- A. F. Naves, H. T. C. Fernandes, A. P. S. Immich and L. H. Catalani, J. Polym. Sci., Polym. Chem., 2013, 51, 3881–3891 CrossRef CAS PubMed.
- T. Tsujimoto, H. Uyama and S. Kobayashi, Biomacromolecules, 2001, 2, 29–31 CrossRef CAS PubMed.
- A. Kumar, A. S. Kulshrestha, W. Gao and R. A. Gross, Macromolecules, 2003, 36, 8219–8221 CrossRef CAS.
- C. Japu, A. Martinez de Ilarduya, A. Alla, Y. Jiang, K. Loos and S. Munoz-Guerra, Biomacromolecules, 2015, 16, 868–879 CrossRef CAS PubMed.
- Y. Jiang, A. J. Woortman, G. O. R. Alberda van Ekenstein, D. M. Petrovic and K. Loos, Biomacromolecules, 2014, 15, 2482–2493 CAS.
- Á. Cruz-Izquierdo, L. A. M. van den Broek, J. L. Serra, M. J. Llama and C. G. Boeriu, Pure Appl. Chem., 20158759–69 Search PubMed.
- P. Lozano, T. De Diego, D. Carrie, M. Vaultier and J. L. Iborra, Biotechnol. Prog., 2003, 19, 380–382 CrossRef CAS PubMed.
- L. Ragupathy, U. Ziener, R. Dyllick-Brenzinger, B. von Vacano and K. Landfester, J. Mol. Catal. B: Enzym., 2012, 76, 94–105 CrossRef CAS PubMed.
- M. B. Frampton and P. M. Zelisko, Chem. Commun., 2013, 49, 9269–9271 RSC.
- S. Park and R. J. Kazlauskas, Curr. Opin. Biotechnol., 2003, 14, 432–437 CrossRef CAS.
- A. Mahapatro, B. Kalra, A. Kumar and R. A. Gross, Biomacromolecules, 2003, 4, 544–551 CrossRef CAS PubMed.
- Y.-Y. Linko, Z.-L. Wang and J. Seppälä, Enzyme Microb. Technol., 1995, 17, 506–511 CrossRef CAS.
- C. J. Morrow, MRS Bull., 1992, 17, 43–47 CAS.
- R. Gross, A. Kumar and B. Kalra, United States Pat, US20040019178A1, 2004 Search PubMed.
- A. Kumar and R. A. Gross, Biomacromolecules, 2000, 1, 133–138 CrossRef CAS.
- V. Stepankova, S. Bidmanova, T. Koudelakova, Z. Prokop, R. Chaloupkova and J. Damborsky, ACS Catal., 2013, 3, 2823–2836 CrossRef CAS.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- R. K. Singh, M. K. Tiwari, R. Singh and J. K. Lee, Int. J. Mol. Sci., 2013, 14, 1232–1277 CrossRef CAS PubMed.
- K. Min and Y. J. Yoo, Biotechnol. Bioprocess Eng., 2014, 19, 553–567 CrossRef CAS.
- N. Miletić and K. Loos, Aust. J. Chem., 2009, 62, 799–805 CrossRef.
- N. Miletic, R. Rohandi, Z. Vukovic, A. Nastasovic and K. Loos, React. Funct. Polym., 2009, 69, 68–75 CrossRef CAS PubMed.
- N. Miletić, Z. Vuković, A. Nastasović and K. Loos, J. Mol. Catal. B: Enzym., 2009, 56, 196–201 CrossRef PubMed.
- N. Miletic, V. Abetz, K. Ebert and K. Loos, Macromol. Rapid Commun., 2010, 31, 71–74 CrossRef CAS PubMed.
- N. Miletic, Fahriansyah, L. T. T. Nguyen and K. Loos, Biocatal. Biotransform., 2010, 28, 357–369 CrossRef CAS.
- T. Dai, N. Miletić, K. Loos, M. Elbahri and V. Abetz, Macromol. Chem. Phys., 2011, 212, 319–327 CAS.
- N. Miletic, Z. Vukovic, A. Nastasovic and K. Loos, Macromol. Biosci., 2011, 11, 1537–1543 CAS.
- N. Miletic, A. Nastasovic and K. Loos, Bioresour. Technol., 2012, 115, 126–135 CrossRef CAS PubMed.
- C. Jun, B. W. Jeon, J. C. Joo, Q. A. T. Le, S. A. Gu, S. Byun, D. H. Cho, D. Kim, B. I. Sang and Y. H. Kim, Process Biochem., 2013, 48, 1181–1187 CrossRef CAS PubMed.
- J. N. Talbert, L. S. Wang, B. Duncan, Y. Jeong, S. M. Andler, V. M. Rotello and J. M. Goddard, Biomacromolecules, 2014, 15, 3915–3922 CrossRef CAS PubMed.
- M. L. Verma, C. J. Barrow and M. Puri, Appl. Microbiol. Biotechnol., 2013, 97, 23–39 CrossRef CAS PubMed.
- J. Wu, P. Eduard, S. Thiyagarajan, L. Jasinska-Walc, A. Rozanski, C. F. Guerra, B. A. J. Noordover, J. van Haveren, D. S. van Es and C. E. Koning, Macromolecules, 2012, 45, 5069–5080 CrossRef CAS.
- M. Tasaki, H. Yamamoto, T. Yoshioka, M. Hanesaka, T. H. Ninh, K. Tashiro, H. J. Jeon, K. B. Choi, H. S. Jeong, H. H. Song and M. H. Ree, Polymer, 2014, 55, 1228–1248 CrossRef CAS PubMed.
- I. H. Hall and B. A. Ibrahim, Polymer, 1982, 23, 805–816 CrossRef CAS.
- J. Guzmán and J. G. Fatou, Eur. Polym. J., 1978, 14, 943–949 CrossRef.
- M. Gilbert and F. J. Hybart, Polymer, 1972, 13, 327–332 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The SEC traces, molecular weights, MALDI-ToF MS analysis, WAXD spectra, and physical properties of furanic-aliphatic polyesters; the cumulative weight fractions of poly(butylene furanoate); the molecular weights of polycaprolactone from control reactions; plots of the degree of crystallinity and the enthalpy as a function of the weight average molecular weight; plots of linear fit of enthalpy of fusion as a function of the degree of crystallinity; the DSC, TMDSC and TGA curves of furani-aliphatic polyesters. See DOI: 10.1039/c5py00629e |
| This journal is © The Royal Society of Chemistry 2015 |