
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
RAFT dispersion polymerization: a method to tune the morphology of thymine-containing self-assemblies†
Yan
Kang
,
Anaïs
Pitto-Barry
 ,
Anna
Maitland
and
Rachel K.
O'Reilly
*
,
Anna
Maitland
and
Rachel K.
O'Reilly
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: Rachel.oreilly@warwick.ac.uk
First published on 11th June 2015
Abstract
The synthesis and self-assembly of thymine-containing polymers were performed using RAFT dispersion polymerization. A combination of microscopy and scattering techniques was used to analyze the resultant complex morphologies. The primary observation from this study is that the obtained aggregates induced during the polymerization were well-defined despite the constituent copolymers possessing broad dispersities. Moreover, a variety of parameters, including the choice of polymerization solvent, the degree of polymerization of both blocks and the presence of an adenine-containing mediator, were observed to affect the resultant size and shape of the assembly.
Introduction
Heterogeneous polymerizations, including dispersion, emulsion, suspension, and precipitation polymerizations, are easily performed and widely used in industry.1 Recently the combination of heterogeneous polymerization with reversible-deactivation radical polymerization (RDRP) has become of great interest, given that this approach can combine the advantages of heterogeneous polymerization with the controlled nature of RDRP methods.2,3 RDRP techniques including reversible addition-fragmentation chain-transfer (RAFT) polymerization,3–5 various forms of atom-transfer radical polymerization (ATRP),6,7 nitroxide-mediated polymerization (NMP),8,9 amongst other methods10–12 have been readily applied in this area. A number of these processes offer significant advantages in the design of well-defined nanoobjects through the synthesis of their corresponding block copolymers.13–18 For example, RAFT dispersion polymerization has been widely developed to prepare a solvent-insoluble polymer block from a solvent-soluble monomer in the presence of a solvent-soluble macromolecular chain transfer agent (macro-CTA).15,19–24 In most cases, by simply varying the polymerization conditions, a range of common morphologies (spheres, cylinders, vesicles) and a few novel structures (lumpy rod,25 framboidal morphology,26 concentric vesicle,27 and knot-like structure28) can be obtained. In addition, the resultant block copolymers are generally obtained with good blocking efficiencies and relatively low dispersities.Recently we have demonstrated the synthesis of nucleobase-containing materials using RAFT dispersion polymerization.28 In our previous study, poly(methyl methacrylate) (PMMA) was used as the macro-CTA and only adenine-containing monomers or a mixture of adenine-containing monomers and thymine-containing monomers were used to chain extend the PMMA block. RAFT dispersion polymerizations were investigated in both chloroform and 1,4-dioxane. It was found that using these conditions polymers obtained were generally well-controlled in terms of molecular weight and molecular weight distribution. Moreover, it was also observed that the choice of polymerization solvent played a key role in the final morphologies achieved.
Thymine is one of the natural nucleobases in DNA and binds to adenine via complementary hydrogen bonding interactions to stabilize DNA and enable the formation of a double helix. Moreover, it is also well-known that thymine can form triple hydrogen bonds with diaminopyridine or its derivatives.23,24 Based on these properties, thymine is commonly used for controlling polymer tacticity,29 templating polymerizations,30–32 facilitating molecular self-assembly and aggregation33–36 and protecting active moieties.37 However, there are still few reports on the systematic study of the self-assembly of thymine-containing polymers of various lengths.33,38 Considering the advantages of RAFT dispersion polymerizations, we have applied this approach to readily prepare a variety of thymine-containing nanostructures. However, compared to our previous work on RAFT dispersion polymerization to prepare nucleobase-containing nanostructures,28 contrasting results in terms of morphology and molecular weight distributions of the resultant polymers have been observed. Indeed, in the pure thymine system the produced morphologies are more varied and better defined despite the fact that the resultant polymers are bimodal in most cases. This contradicts current thinking that narrow dispersity block copolymers are a prerequisite for the formation of well-defined assemblies.39 However, this study shows that well-defined nanoobjects can still be attained although the constituent block copolymers possess broad dispersities (ĐM often greater than 1.5). Indeed, the study of the solution behavior of polydisperse block amphiphiles is of growing scientific interest.40–48 It is known that block copolymers with broad dispersities affect the interfacial curvature, which allows structures with non-constant interfacial mean curvatures, but that are thermodynamically stabilized, to form (e.g., prolate-spheroid micelles,40 stabilized vesicles,43 and more complex structures42,49). In order to further extend this concept we have performed RAFT dispersion polymerizations of a thymine-containing monomer, which under varying conditions can undergo hydrogen-bonding to allow for more complex polymerization procedures. To explore this fully a range of polymerization conditions were explored and their effect on the resultant morphologies and polymers investigated. Such functional polymers, capable of intra- and inter-molecular H-bonding interactions are an interesting emerging class of new materials which enable access to novel morphologies and can provide an insight into the self-assembly process of such complex functional polymers.
Experimental
Materials
Methyl methacrylate (MMA) was bought from Aldrich and passed through a column of neutral alumina to remove the inhibitor. 2,2-Azo-bis(isobutyronitrile) (AIBN) was purchased from Molekula and recrystallized twice from methanol. The synthesis of 2-cyano-2-propyl dodecyl trithiocarbonate (CPDT) was based on a previous report.50 Thymine-functionalized monomer, 2-(2-(thymine-1-yl)acetoxyl) ethyl methacrylate (TMA), was synthesized according to our published method.51 Adenine-containing mediator, 9-hexyladenine, was synthesized according to a previous report.52 1,4-Dioxane, chloroform (CHCl3), and other solvents were used as received from Fisher Scientific. Deuterated solvents were bought from Apollo Scientific.Homopolymerization of TMA
The typical procedure for the RAFT polymerization of TMA was as follows: TMA, CPDT (1 eq.) and AIBN (0.1 eq.) were dissolved in 1,4-dioxane. The mixture was degassed via 4 freeze–pump–thaw cycles, filled with nitrogen and then immersed into an oil bath at 60 °C. The polymerization was quenched in a liquid nitrogen bath and exposing to air. The mixture was concentrated and precipitated into cold methanol. The polymer was washed with methanol several times and dried in a vacuum oven. The polymer was characterized by 1H NMR spectroscopy and SEC (DMF eluent, PMMA standards).Polymerization of methyl methacrylate (MMA)
The typical procedure of RAFT polymerizations of MMA was as follows: MMA, CPDT (1 eq.) and AIBN (0.1 eq.) were dissolved in toluene. The mixture was degassed via 4 freeze–pump–thaw cycles, filled with nitrogen and then immersed into an oil bath at 60 °C. The polymerization was quenched by putting into a liquid nitrogen bath and exposing to air. The mixture was precipitated into cold methanol and filtered. The polymer was dissolved in THF and precipitated again. This precipitation procedure was repeated 3 times in total to remove unreacted monomer. The light yellow polymers were dried in a vacuum oven and characterized by 1H NMR spectroscopy and SEC (DMF eluent, PMMA standards).Synthesis of block copolymers using PMMA as macro-CTA
The typical procedure was as follows: PMMA (1 eq.), TMA (X), adenine mediator (X) and AIBN (0.1 eq.) were dissolved in chloroform, 1,4-dioxane or mixtures of the two solvents. The mixtures were thoroughly degassed via 4 freeze–pump–thaw cycles, filled with nitrogen and then immersed into an oil bath at 60 °C. The polymerizations were quenched by exposing to air and cooling down. The mixture was precipitated in methanol and washed with methanol several times. The polymers were dried in a vacuum oven and characterized by 1H NMR spectroscopy and SEC (DMF eluent, PMMA standard). The DP of the block was varied by adding different amounts of TMA.Characterization
1H NMR spectra were recorded on a Bruker DPX-300 or DPX-400 spectrometer with DMSO-d6 or deuterated chloroform (CDCl3) as the solvent. The chemical shifts of protons were reported relative to tetramethylsilane at δ = 0 ppm or solvent residues (CHCl31H: 7.26 ppm; DMSO 1H: 2.50 ppm).Size exclusion chromatography (SEC) was performed in HPLC grade DMF containing 5 mM NH4BF4 at 50 °C, with a flow rate of 1.0 mL per minute, on a set of two PLgel 5 μm Mixed-D columns, plus one guard column. SEC data was analyzed with Cirrus GPC software calibrated using polymethyl methacrylate (PMMA) standards. The SEC was equipped with both refractive index (RI) and UV detectors.
Transmission electron microscopy (TEM) was performed on a JEOL 2000FX electron microscope at an acceleration voltage of 200 kV. All TEM samples were prepared on graphene oxide (GO)-coated carbon grids (Quantifoil R2/2) which allows high contrast images to be acquired without staining.53 Generally, a drop of sample (20 μL) was deposited on a grid which was placed on a piece of filter paper and then left to air dry.
Hydrodynamic diameters (Dh) and size distributions of the self-assemblies were determined by dynamic light scattering (DLS). The DLS instrumentation consisted of a Malvern Zetasizer NanoS instrument operating at 25 °C with a 4 mW He–Ne 633 nm laser module. Measurements were made at a detection angle of 173° (back scattering), and Malvern DTS 6.20 software was used to analyze the data. The viscosity of the mixtures of CHCl3 and 1,4-dioxane is estimated by a simplified equation.54
ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) η = X1lnη1 + X2lnη1 η = X1lnη1 + X2lnη1 |
Static light scattering (SLS) measurements were performed using an ALV CGS3 (λ = 632 nm) at 20 °C. The data was collected from 12° up to 30° with an interval of 2° or 30° up to 150° with an interval of 10° and calibrated with filtered toluene and filtered CHCl3 as backgrounds. The refractive index increment of the polymer in CHCl3 was measured to be 0.053 mL g−1.
Small-angle neutron scattering (SANS) experiments were recorded at the ISIS neutron beam facility, on the sans2d instrument at the Rutherford Appleton Laboratory, Oxford. Samples were measured at 20 mg mL−1 in CDCl3, which provides high contrast in scattering length for the polymer. Small-angle X-ray scattering (SAXS) experiments were carried out on the SAXS-WAXS beamline at the Australian Synchrotron facility at a photon energy of 15 keV. The samples were prepared in 1,4-dioxane and were run using 1.5 mm diameter quartz capillaries. The measurements were collected at 25 °C with a sample to detector distance of 7.160 m to give a q range of 0.0015 to 0.08 Å−1, where q is the scattering vector and is related to the scattering angle (2θ) and the photon wavelength (λ) by the following equation:

All patterns were normalized to fixed transmitted flux using a quantitative beam stop detector. The scattering from the solvent was measured in the same location as sample collection and was subtracted for each measurement. The two-dimensional SAXS images were converted into one-dimensional SAXS profiles (I(q) versus q) by circular averaging, where I(q) is the scattering intensity. ScatterBrain and NCNR Data Analysis IGOR Pro software were used to plot and analyze SAXS and SANS data.55 The scattering length density of the solvents and monomers were calculated using the “Scattering Length Density Calculator” provided by the NIST Center for Neutron Research.56
Results and discussion
The homopolymerizations of TMA in chloroform and DMF were reported in our previous study.51 Although TMA could be polymerized in chloroform, due to the heterogeneous nature of the polymerization it was not well-controlled.51 However, poly(TMA) obtained using DMF as the polymerization solvent was well-defined in terms of molecular weight and molecular weight distribution.51 In this study, the homopolymerization of TMA in 1,4-dioxane was also performed. The solubility of PTMA in 1,4-dioxane was improved compared to that in chloroform, although some precipitation of the polymer still occurred as the polymerization proceeded, however, the molecular weight distribution of the resultant polymer was narrower (1.14 versus 1.86) and the resultant molecular weight was more controlled compared to polymerization in chloroform (polymer 1, Fig. S1 and Table S1†). These observations suggest that it is possible to perform RAFT dispersion polymerizations of TMA using 1,4-dioxane as the solvent. Based on these observations 1,4-dioxane was used a good solvent for the dispersion polymerization with a PMMA macro-CTA.The synthetic route for the PMMA macro-CTA synthesis (2 and 3) is shown in Scheme 1. CPDT was used as the CTA as it is suitable for the polymerization of methacrylates and the polymerizations were stopped at relatively low conversion <60% to ensure good end group fidelity. The DP of the PMMA macro-CTA was determined by 1H NMR spectroscopy by comparing the integration of PMMA backbone signals with those of the CPDT end group. The resulting DPs of PMMA were ca. 70 (polymer 2) and 220 (polymer 3). SEC (DMF as eluent, PMMA standards) was used to further determine the molecular weight and the molecular weight distribution. In addition to the observed narrow molecular weight distributions, the molecular weights from SEC are consistent with the results from 1H NMR spectroscopy. Furthermore, the RI SEC trace and UV SEC trace (λ = 309 nm from trithiocarbonate end group) overlap well, suggesting good end group fidelity (Fig. S2†).
 | ||
| Scheme 1 Synthetic route for the macro-CTAs (PMMA, 2 and 3) and the thymine-containing diblock copolymers using RAFT dispersion polymerization (4–9). | ||
RAFT dispersion polymerizations of TMA were performed at 60 °C using 2 or 3 as the macro-CTA and AIBN as the initiator. The conversions of the polymerizations were determined by 1H NMR spectroscopy and the molecular weight and molecular weight distributions were analysed by SEC (DMF as eluent, PMMA as standards). The morphologies induced during the polymerizations were characterized by both light scattering and microscopy analysis. To explore the polymerization parameters more fully and understand the dispersion polymerization procedure a range of conditions (including solvent quality, nucleobase block length, addition of a mediator) were utilized to prepare the block copolymers and nanostructures.
Effect of solvent
As observed in our previous study, solvent plays a key role in the RAFT dispersion polymerization of nucleobase-containing monomers,28 where different morphologies are observed using different solvents. In this study, chloroform, 1,4-dioxane, and mixtures of the two solvents at various ratios were utilized as the polymerization solvent to study the effect on the RAFT dispersion polymerization of TMA. The initial ratio of macro-CTA:TMA:AIBN was 1:100:0.1 and PMMA with a DP of 70 was used as the macro-CTA, 2, (the polymers/polymerizations were named as 4–X; where X denotes the composition of chloroform in a solvent mixture), see Table S1.†
Under these different solvent conditions a range of assembly morphologies were obtained for the targeted block copolymer PMMA70-b-PTMA100. When pure chloroform was used as the polymerization solvent, a bimodal molecular weight distribution was observed by SEC and only well-defined aggregates (ca. 200 nm) were observed by light scattering (Table S1 4–100 and Fig. S3†). Examination of the reaction mixture by dry state TEM indicated that vesicles were formed during the dispersion polymerization (Fig. 1a, 4–100). To eliminate the possibility of imaging artefacts caused by imaging particles in the dry-state, SLS was also conducted on the sample to further determine the morphology (Fig. S3†). A Zimm analysis revealed that the P-ratio (Rg/Rh, ratio between radius of gyration and hydrodynamic radius) of the nanostructures was close to 1 (Rg/Rh = 1.17), indicating the formation of hollow structures.57,58 SANS was also conducted on a more concentrated sample (20 mg mL−1), which was obtained directly from the polymerization solution without dilution (Fig. S4†). The Guinier-Porod fit was firstly applied to confirm a spherical shape for these assemblies.59 Furthermore, the thickness of the vesicle membrane was determined from a plot of q2I(q) versus q.60 The first minimum of this plot q corresponds to the first zero of the membrane factor given by:
| d = 2π/q |
 | ||
| Fig. 1 Representative dry state TEM images of self-assemblies prepared by RAFT dispersion polymerization for the target polymers PMMA70-b-PTMA100, 4–X in different solvents (from a to g: volume compositions of CHCl3 in a CHCl3 and 1,4-dioxane mixture, 100%, 75%, 50%, 33%, 25%, 12.5% and 0%) and their corresponding size and size distributions as determined by DLS (h). Scale bar: 100 nm (1000 nm for c, d, and e). | ||
When a mixture of chloroform and 1,4-dioxane (volume composition of chloroform: 75%) was used as the solvent (Table S1 4–75, Fig. S5†), once again a bimodal polymer formed and a more complex morphology was observed. In this case lamellae with tentacles were observed by TEM analysis (Fig. 1b, 4–75). These assemblies appeared well defined by DLS analysis and had a hydrodynamic diameter of ca. 300 nm. AFM analysis was conducted on this sample, which further confirmed the formation of lamellae with tentacles morphology. Moreover, it showed the height of the lamellae and tentacles were ca. 25 and 35 nm, respectively (Fig. S6†). Further decreasing the volume composition of chloroform during the polymerization to 50% led to the formation of long flexible cylinders (Table S1 4–50, Fig. S7†). From TEM analysis, the cylinders were around 5 μm long (Fig. 1c, 4–50). Even longer and thinner cylinders were observed when the compositions of chloroform were 33% and 25%, respectively (Fig. 1d, 4–33 and 1e, 4–25; Fig. S8 and S9†). These cylinders were more than 10 μm in length and around 55 nm in width by TEM analysis. However, when the amount of chloroform used in the dispersion polymerization was dropped to 12.5%, much shorter worm-like structures were obtained (Fig. 1f, 4–12.5; Fig. S10†) and the width of cylinders slightly increased to 75 nm. DLS was also used to analyze the resultant morphologies. However, due to the highly elongated structures obtained in these systems, the sizes obtained from DLS are relative values.62
In the case of pure 1,4-dioxane as the polymerization solvent, low contrast disk-like structures with a diameter of ca. 100 nm were observed by TEM analysis (Fig. 1g, 4–0; Fig. S11†). AFM was then conducted on this sample and the heights of the structures were ca. 3–5 nm (Fig. S12†), which confirmed the disc-like morphology. SAXS was used to further analyze the morphology in solution (Fig. S13†). Kratky plots for spheres and cylinders were plotted with the raw data to emphasize the deviation of the scattering intensity at high-q values. The sphere plot (q2I(q) versus q) showed a horizontal asymptote at high q values which suggests the presence of spheres. In the meantime the cylinder plot (qI(q) versus q) also tended to a horizontal asymptote at high-q values thus confirming the presence of cylinders in solution. Such plots can suggest the assemblies have both spherical and cylindrical features. Slopes of −1 and −2 were observed in the log(I(q)) versus log(q) plot, which suggests the presence of rods with a very short length. Finally different models were applied and attempts to analyze this profile with a disperse spherical uniform model,61 uniform convex lens,63,64 or uniform cylindrical models (with short length to account for a disk-like morphology)59 yielded poor quality data fits. This suggests that the morphologies were not convex lenses, simple spheres or disks. Indeed, a core–shell cylindrical model with dispersity in the core radius provided the best results (Fig. S13†).59 This fit suggested that structures were formed with a core radius of ca. 9 nm, a corona thickness of ca. 8 nm and a core length or depth of ca. 3 nm, suggesting the formation of disk-like structures. Whilst this fit was the best of those examined, the quality was not as good as expected. Moreover, the large difference of dimensions obtained between the structure models utilized in the SAXS analysis and the TEM analysis suggests that the former did not reflect the entire sample. This may be due to a solvation effect or large sample dispersity in either diameter or height. Given the observations made by TEM analysis it is suggested that the dispersity is in the height of the disk rather than the diameter.
Overall, these results show that various morphologies can be accessed by varying the composition of solvent during the RAFT dispersion polymerization of TMA. However, the polymers obtained under the range of polymerization solvents were not monodisperse (Fig. S14†). Indeed bimodal chromatograms were observed by SEC analysis in each case. To eliminate the possibility that TMA monomer had undergone side-reactions during the polymerization, mixtures of TMA and CPDT were stirred at 60 °C in chloroform under N2 atmosphere for 24 hours. Monitoring by 1H NMR spectroscopy analysis, there was no obvious change observed before and after heating, which indicated that both TMA and the chain transfer agent were stable under the polymerization conditions (Fig. S15†). Therefore, it was concluded that the observed bimodal distributions of the polymers were induced by the RAFT dispersion polymerization process.
Effect of TMA block length
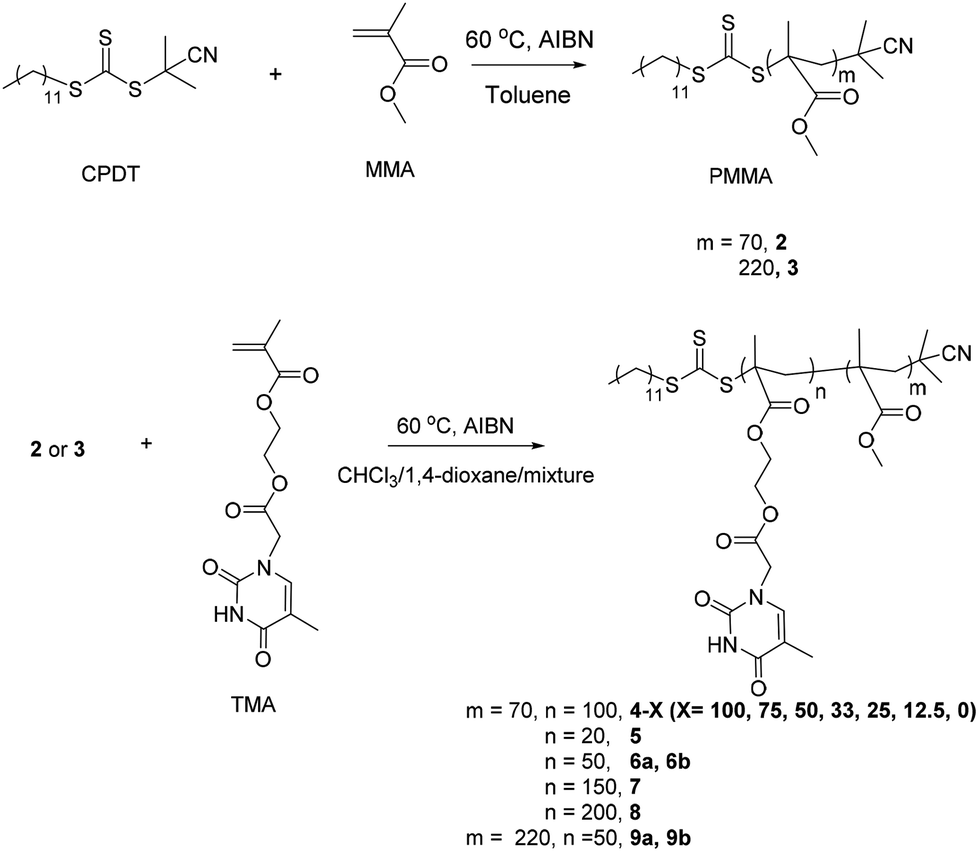
In addition to the effect of the solvent used for the polymerizations, the effect of varying TMA block length was also investigated using macro-CTA 2, using both chloroform and 1,4-dioxane as the polymerization solvents. In chloroform the target DPs of the thymine-containing block were 20, 50, and 100 and different morphologies were obtained for each polymerization in this series. When the DP of the thymine-containing block was 20, spherical micelles were observed by TEM analysis (Fig. 2a, 5) and they were confirmed as solid spheres by SLS (Rg/Rh = 0.79, Fig. S16†). Further increasing the DP of the thymine-containing block to 50 resulted in the formation of cylinders (Fig. 2b, 6a). The length of cylinders was more than 500 nm by TEM analysis, however, some spherical micelles were also observed. To further determine the morphologies in solution, SANS was also conducted on sample 6a. The Guinier-Porod model was used firstly to provide information on the shape of the scattering objects (Rg and anisotropy). A dimension parameter of 1.32 was obtained, indicating the formation of elongated objects. However, as the minimum q value is 0.004 Å−1 and the structures are relatively big (Rg > 100 nm), it is difficult to have full confidence in this fit (Fig. S18†). As a mixture of spheres and cylinders were observed by TEM analysis, a linear combination (sum model fit) of a model for spheres with some dispersity (sphere fit)65 and a model for cylinders with some dispersity (cylinder fit) was created to analyze the structures in solution (Fig. S18†). This confirmed the observation by TEM analysis that both cylinders and spheres coexist in solution. As discussed above for polymer 4–100, when the DP of the thymine-containing block was increased to 100, only vesicles were observed by TEM analysis (Fig. 1a).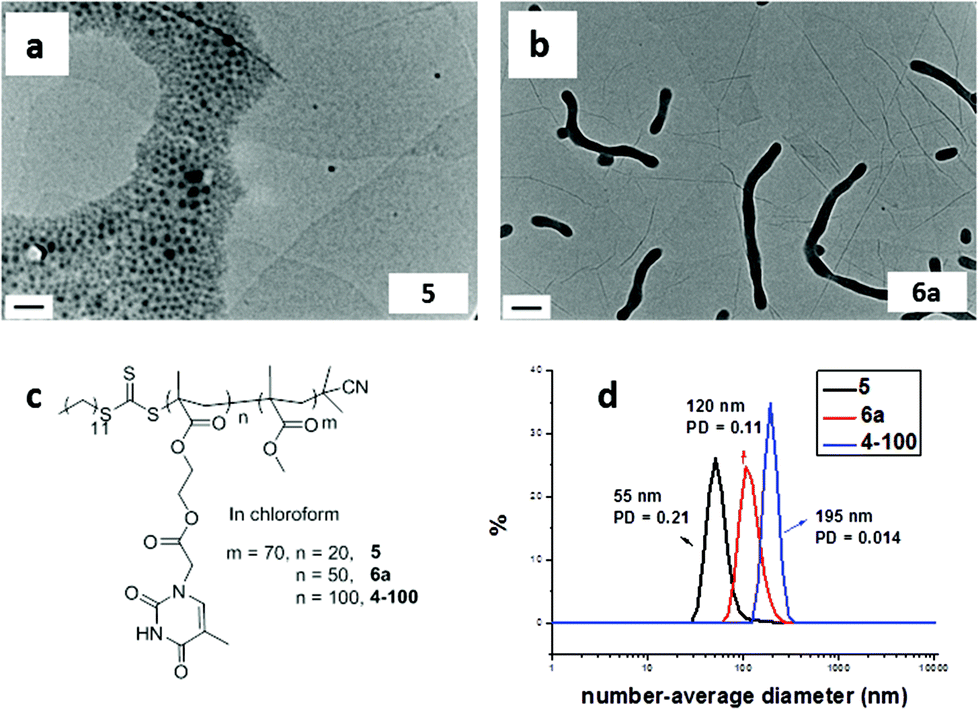 | ||
| Fig. 2 (a) and (b) Representative TEM images of self-assemblies prepared by RAFT dispersion polymerization in chloroform for a target copolymer PMMA70-b-PTMAn, their corresponding chemical structures (c) and DLS particle size distributions (d) with increasing TMA block length. Scale bar: 100 nm. | ||
Thus, with decreasing TMA block length and hence increasing weight fraction of PMMA from 54 to 32 to 19%, as expected a morphology transition, from sphere to cylinder to vesicle was observed in the dispersion polymerization performed in chloroform. This observation is consistent with the previous observation in the literature for RAFT dispersion polymerizations, however it is interesting as the polymers in this work have much broader polydispersities which does not seem to affect the self-assembly process.13
In comparison, when 1,4-dioxane was used as the polymerization solvent, a sphere-cylinder-vesicle transition was not observed when increasing the block length of TMA from 50 to 200 (Table S1, 6b, 4–0, 7, 8; Fig. S19–S24†). Instead across the PMMA composition range (from 32 to 19 to 14 to 11 weight%, for polymers 6b, 4–0, 7, and 8, respectively) only larger micelles were observed which increased in size (from 21 to 341 nm) as the DP of the PTMA block increased. The possible reason for this observation is that the nucleobase polymers have better solubility and weaker intra- and intermolecular chain interactions in 1,4-dioxane than chloroform (Ka = 12 versus 20 M−1 at 60 °C). It has been shown by van Hest, for related nucleobase systems, that solvents such as water lead to a higher than calculated lypophilic fraction, which in turns leads to the formation of spherical micelles across a broader composition window.38
Effect of adenine-containing mediator
A mediator is commonly used in nucleobase-containing chemistry for a number of reasons, including to avoid the precipitation of polymer, to protect the activity of a catalyst66 or control polymer tacticity.29 Inspired by a previous study,66 an adenine-containing mediator was used in this work to investigate its effect on the resultant self-assembly morphologies as well as the dispersity of the resultant copolymers. The adenine-containing mediator, 9-hexyladenine (Scheme 2) was synthesized according to a previous literature report (Scheme S1 and Fig. S25†).52 | ||
| Scheme 2 Synthetic route for the thymine-containing diblock copolymers using RAFT dispersion polymerization in the presence of adenine-mediator. | ||
RAFT dispersion polymerization was carried out in the presence of the adenine-containing mediator (amount of TMA:mediator = 1:1) using PMMA70 as the macro-CTA and chloroform as the solvent. The studied DPs of the thymine-containing block were 20, 50, and 100 (Scheme 2, to afford polymers 5′, 6a′, and 4–100′ respectively). When the DP of the thymine-containing block was 20, small spherical micelles were obtained (Fig. 3a, 5′), which was very close to the morphology obtained in the absence of the mediator (Fig. 2a, 5). Moreover, the dispersity of the resultant copolymer obtained in the presence of the mediator was much narrower compared to the copolymer obtained in the absence of mediator (Fig. 3d and S26†). However, the monomer conversion was also lower (66%) perhaps due to the improved solubility of TMA in the presence of mediator, which changes the kinetics from a dispersion polymerization to a solution polymerization.
 | ||
| Fig. 3 Representative TEM images (a–c) of self-assemblies prepared by RAFT dispersion polymerization in chloroform with adenine-containing mediator for a target copolymer PMMA70-b-PTMAn and SEC traces (d) of resultant copolymers obtained with or without mediator for DP 20 thymine block. Scale bar: 100 nm. | ||
In the case of the DP 50 TMA block, spherical micelles were observed in the presence of the mediator (Fig. 3b, 6a′), which is different from the polymerization performed, under identical conditions, without the addition of the mediator (Fig. 2b, 6a where cylinders were formed). In the presence of the adenine-containing mediator, the solubility of PTMA in chloroform increases, which decreases the apparent packing parameter of the assembly,67 therefore spherical micelles were formed instead of cylinders.
Further increasing the DP to 100 resulted in the formation of vesicles (Fig. 3c, 4–100′; Fig. S28†) similar to that observed for the polymerization without mediator (Fig. 1a, 4–100). However, in this case the vesicles possessed wrinkled membranes, which are thought to be due to the complementary interactions between the PTMA and adenine-containing mediator disrupting the assembly process. Overall, addition of an adenine-containing mediator does not significantly change the observed morphology for the series of PTMA block copolymers. Moreover, a narrowing of the dispersity of the resultant copolymer is only observed for the short thymine-containing block and for the longer thymine-containing blocks, little effect on dispersity is observed.
Effect of macro-CTA
When PMMA with a DP of 70 was used as the macro-CTA above, all the polymers obtained were poorly defined and bimodal distributions were observed by SEC analysis in the majority of polymerization conditions. In order to improve the control of polymerization, a macro-CTA with a higher DP was studied as it should be better able to stabilize the growing solvent-insoluble thymine blocks. To explore this, PMMA with a DP of 220 (PMMA220, 3) was used as the macro-CTA for comparison with PMMA70, 2 and the target DP of the thymine-containing block was kept constant at 50. In chloroform, spherical micelles (9a) were observed by TEM analysis when 3 was used as the macro-CTA (Fig. 4a). As expected this is different from the morphology formed when using 2 as the macro-CTA (Fig. 2b) as the longer PMMA block leads to a higher solvophilic weight fraction (59 versus 32%, polymers 9a and 6).39 | ||
| Fig. 4 Representative TEM images of self-assemblies prepared by RAFT dispersion polymerization for a target copolymer PMMA220-b-PTMA50 in (a) 9a, chloroform; (b) 9b, 1,4-dioxane. Scale bar: 100 nm. | ||
In contrast, when 1,4-dioxane was used as the solvent the resultant self-assembly, 9b, was not well-defined as determined by both TEM and DLS analysis (Fig. 4b and S30†). From TEM analysis, a phase-separated pattern was observed and by DLS a broad and fluctuating distribution suggested that the resultant structures were not stable in solution. In this case it was observed that the resultant polymer, 9b, has a significantly narrower dispersity than the related polymers prepared in chloroform (9a) or with the shorter macro-CTA (4–0) due to the improved solubility of the growing diblock in 1,4-dioxane (Fig. S30†).
In this study, nearly all the block copolymers obtained were bimodal by SEC analysis, despite this the resultant self-assemblies were surprisingly well-defined and predictable. In a dispersion polymerization, polymerization proceeds in the polymer particles as they absorb monomers from the continuous phase,68 however, in our system as the pyrimidine–pyrimidine interactions are relatively weak (compared to purine–purine interactions) and the monomer has good solubility in the polymerization solvent, we hypothesize that the monomer absorption into the core of the particles is modest and thus the aggregates can readily establish a thermodynamic equilibrium, which leads to the formation of well-defined structures despite the broad dispersity of the constituent copolymers.
Conclusions
In conclusion, RAFT dispersion polymerizations have been utilized to polymerize thymine-containing monomers using PMMA as a macro-CTA. Effects of various factors including choice of polymerization solvent, the targeted degree of polymerization, the length of macro-CTA and the presence of an adenine-containing mediator were investigated. In general, the resultant aggregates induced by the polymerizations are well-defined despite the high dispersity of the resultant polymers. Furthermore, by changing the polymerization solvent a wide variety of morphologies could be targeted. This study demonstrates that RAFT dispersion polymerization is a facile method to prepare a variety of thymine-containing nanoobjects which may find interest in further assembly applications or recognition.Acknowledgements
The authors thank the University of Warwick, NSF in ERC and EPSRC for funding. The SECs used in this research were obtained from Birmingham Science City, with support from Advantage West Midlands (AWM), and partly funded by the European regional development fund (ERDF). Stephen King (Rutherford Appleton Laboratory, Oxford, U.K.) is acknowledged for carrying out the SANS measurements. Dr Rebecca Williams is thanked for corrections and suggestions.Notes and references
- J. K. Oh, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6983–7001 CrossRef CAS PubMed.
- M. F. Cunningham, Prog. Polym. Sci., 2008, 33, 365–398 CrossRef CAS PubMed.
- J. Jennings, M. Beija, J. T. Kennon, H. Willcock, R. K. O'Reilly, S. Rimmer and S. M. Howdle, Macromolecules, 2013, 46, 6843–6851 CrossRef CAS.
- V. Ladmiral, M. Semsarilar, I. Canton and S. P. Armes, J. Am. Chem. Soc., 2013, 135, 13574–13581 CrossRef CAS PubMed.
- X. W. Zhang, F. Boisson, O. Colombani, C. Chassenieux and B. Charleux, Macromolecules, 2014, 47, 51–60 CrossRef CAS.
- Y. Kitayama, Y. Kagawa, H. Minami and M. Okubo, Langmuir, 2010, 26, 7029–7034 CrossRef CAS PubMed.
- Y. Kagawa, H. Minami, M. Okubo and J. Zhou, Polymer, 2005, 46, 1045–1049 CrossRef CAS PubMed.
- J. Nicolas, A.-V. Ruzette, C. Farcet, P. Gérard, S. Magnet and B. Charleux, Polymer, 2007, 48, 7029–7040 CrossRef CAS PubMed.
- C. Farcet, B. Charleux and R. Pirri, Macromolecules, 2001, 34, 3823–3826 CrossRef CAS.
- Y. Kitayama, K. Kishida, H. Minami and M. Okubo, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1991–1996 CrossRef CAS PubMed.
- Y. Kitayama, M. Yorizane, H. Minami and M. Okubo, Polym. Chem., 2012, 3, 1394–1398 RSC.
- J. Tonnar, P. Lacroix-Desmazes and B. Boutevin, Macromolecules, 2007, 40, 6076–6081 CrossRef CAS.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Soft Matter, 2012, 8, 7753–7767 RSC.
- X. Zhang, S. p. Boissé, W. Zhang, P. Beaunier, F. D'Agosto, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 4149–4158 CrossRef CAS.
- Z. Jia, V. A. Bobrin, N. P. Truong, M. Gillard and M. J. Monteiro, J. Am. Chem. Soc., 2014, 136, 5824–5827 CrossRef CAS PubMed.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 RSC.
- N. J. Warren, O. O. Mykhaylyk, A. J. Ryan, M. Williams, T. Doussineau, P. Dugourd, R. Antoine, G. Portale and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 1929–1937 CrossRef CAS PubMed.
- M. Semsarilar, N. J. W. Penfold, E. R. Jones and S. P. Armes, Polym. Chem., 2015, 6, 1751–1757 RSC.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- J. Rieger, Macromol. Rapid Commun., 2015 DOI:10.1002/marc.201500028.
- C. A. Figg, A. Simula, K. A. Gebre, B. S. Tucker, D. M. Haddleton and B. S. Sumerlin, Chem. Sci., 2015, 6, 1230–1236 RSC.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polym. Chem., 2015, 6, 3054–3062 RSC.
- S. Sugihara, S. P. Armes, A. Blanazs and A. L. Lewis, Soft Matter, 2011, 7, 10787–10793 RSC.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Macromolecules, 2012, 45, 5081–5090 CrossRef CAS.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Macromolecules, 2014, 47, 1664–1671 CrossRef CAS.
- Y. Kang, A. Pitto-Barry, H. Willcock, W.-D. Quan, N. Kirby, A. M. Sanchez and R. K. O'Reilly, Polym. Chem., 2015, 6, 106–107 RSC.
- Y. Tao, K. Satoh and M. Kamigaito, Macromol. Rapid Commun., 2011, 32, 226–232 CrossRef CAS PubMed.
- C. R. South and M. Weck, Macromolecules, 2007, 40, 1386–1394 CrossRef CAS.
- R. McHale, J. P. Patterson, P. B. Zetterlund and R. K. O'Reilly, Nat. Chem., 2012, 4, 491–497 CrossRef CAS PubMed.
- P. K. Lo and H. F. Sleiman, J. Am. Chem. Soc., 2009, 131, 4182–4183 CrossRef CAS PubMed.
- Y. Tao, Y. Yang, D. Shi, M. Chen, C. Yang and X. Liu, Polymer, 2012, 53, 1551–1557 CrossRef CAS PubMed.
- R. J. Thibault, P. J. Hotchkiss, M. Gray and V. M. Rotello, J. Am. Chem. Soc., 2003, 125, 11249–11252 CrossRef CAS PubMed.
- F. Ilhan, T. H. Galow, M. Gray, G. Clavier and V. M. Rotello, J. Am. Chem. Soc., 2000, 122, 5895–5896 CrossRef CAS.
- A. K. Boal, F. Ilhan, J. E. DeRouchey, T. Thurn-Albrecht, T. P. Russell and V. M. Rotello, Nature, 2000, 404, 746–748 CrossRef CAS PubMed.
- L. P. Stubbs and M. Weck, Chem. – Eur. J., 2003, 9, 992–999 CrossRef CAS PubMed.
- H. J. Spijker, A. J. Dirks and J. C. M. van Hest, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4242–4250 CrossRef CAS PubMed.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- A. L. Schmitt, M. H. Repollet-Pedrosa and M. K. Mahanthappa, ACS Macro Lett., 2012, 1, 300–304 CrossRef CAS.
- P. Lim Soo and A. Eisenberg, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 923–938 CrossRef PubMed.
- X. Wang, J. L. Davis, J. P. Hinestrosa, J. W. Mays and S. M. Kilbey, Macromolecules, 2014, 47, 7138–7150 CrossRef CAS.
- O. Terreau, L. Luo and A. Eisenberg, Langmuir, 2003, 19, 5601–5607 CrossRef CAS.
- W. Batsberg, S. Ndoni, C. Trandum and S. Hvidt, Macromolecules, 2004, 37, 2965–2971 CrossRef CAS.
- S. Hvidt, C. Trandum and W. Batsberg, J. Colloid Interface Sci., 2002, 250, 243–250 CrossRef CAS PubMed.
- P. Linse, Macromolecules, 1994, 27, 6404–6417 CrossRef CAS.
- D. J. Adams, M. F. Butler and A. C. Weaver, Langmuir, 2006, 22, 4534–4540 CrossRef CAS PubMed.
- L. Chen, T. Ci, T. Li, L. Yu and J. Ding, Macromolecules, 2014, 47, 5895–5903 CrossRef CAS.
- S. Jain and F. S. Bates, Macromolecules, 2004, 37, 1511–1523 CrossRef CAS.
- Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2007, 40, 4446–4455 CrossRef CAS.
- Y. Kang, A. Lu, A. Ellington, M. C. Jewett and R. K. O'Reilly, ACS Macro Lett., 2013, 2, 581–586 CrossRef CAS.
- Z. Wang, Y. Yu, D. Zhang and D. Zhu, Chin. Sci. Bull., 2006, 51, 1947–1954 CrossRef CAS PubMed.
- J. P. Patterson, A. M. Sanchez, N. Petzetakis, T. P. Smart, T. H. Epps III, I. Portman, N. R. Wilson and R. K. O'Reilly, Soft Matter, 2012, 8, 3322–3328 RSC.
- T. M. Reed and T. E. Taylor, J. Phys. Chem., 1959, 63, 58–67 CrossRef CAS.
- S. Kline, J. Appl. Crystallogr., 2006, 39, 895–900 CrossRef CAS.
- N. S. L. D. Calculator, http://www.ncnr.nist.gov/resources/sldcalc.html.
- H. Peng, D. Chen and M. Jiang, J. Phys. Chem. B, 2003, 107, 12461–12464 CrossRef CAS.
- J. P. Patterson, M. P. Robin, C. Chassenieux, O. Colombani and R. K. O'Reilly, Chem. Soc. Rev., 2014, 43, 2412–2425 RSC.
- A. Guinier and G. Fournet, Small-angle scattering of X-rays, Wiley, 1955 Search PubMed.
- S. Hocine, A. Brulet, L. Jia, J. Yang, A. Di Cicco, L. Bouteiller and M.-H. Li, Soft Matter, 2011, 7, 2613–2623 RSC.
- J. B. Hayter, Physics of amphiphiles–micelles, vesicles, and microemulsions, Varenna on Lake Como, Villa Monastero, 1983 Search PubMed.
- N. Petzetakis, D. Walker, A. P. Dove and R. K. O'Reilly, Soft Matter, 2012, 8, 7408–7414 RSC.
- H. Kaya, J. Appl. Crystallogr., 2004, 37, 223–230 CrossRef CAS.
- H. Kaya and N.-R. De Souza, J. Appl. Crystallogr., 2004, 37, 508–509 CrossRef CAS.
- P. Bartlett and R. H. Ottewill, J. Chem. Phys., 1992, 96, 3306–3318 CrossRef CAS PubMed.
- J. Romulus and M. Weck, Macromol. Rapid Commun., 2013, 34, 1518–1523 CrossRef CAS PubMed.
- J. Rodríguez-Hernández, F. Chécot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef PubMed.
- G. Odian, Principles of Polymerization, John Wiley & Sons, Inc., Hoboken, 4th edn, 2004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra, SEC, DLS, SANS, SAXS, SLS data. See DOI: 10.1039/c5py00617a |
| This journal is © The Royal Society of Chemistry 2015 |