
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot two polymers: ABB′ melt polycondensation for linear polyesters and hyperbranched poly(ester-urethane)s based on natural L-amino acids†
Rajendra
Aluri
and
Manickam
Jayakannan
*
Department of Chemistry, Indian Institute of Science Education and Research (IISER), Dr. Homi Bhabha Road, Pune 411008, Maharashtra, India. E-mail: jayakannan@iiserpune.ac.in; Fax: +91-20-2590 8186
First published on 6th May 2015
Abstract
We report a novel one-pot ABB′ synthetic route for linear polyester and hyperbranched poly(ester-urethane)s based on multi-functional L-amino acid monomers via a temperature selective melt polycondensation approach. L-Serine, D-serine and L-threonine amino acids were converted into multi-functional ABB′ monomers (A = hydroxyl, B = carboxylic ester and B′ = urethane). At 120 °C, the ABB′ monomer underwent thermo-selective transesterification polycondensation (A reacted with B) to produce linear polyesters with B′ functionality as the pendent functionality in each repeating units. At 150 °C, the ABB′ monomer underwent dual ester-urethane self-polycondensation to produce new classes of hyperbranched poly(ester-urethane)s (A reacted with B and B′). Interestingly, the secondary hydroxyl group in the L-threonine monomer did not react at 120 °C; however, it became active at 150 °C to yield exclusively linear polyesters. The temperature selective polycondensation process was confirmed by appropriate model reactions and 1H and 13C NMR spectroscopic analysis. The role of the macrocyclic formation in the polycondensation process was also investigated by MALDI-TOF MS. The amino acid based new polymers were found to exhibit diverse molecular self-assembly. The linear polyesters adopted a β-sheet conformation which produced a helical nano-fibrous morphology. The hyperbranched polymers underwent a globular coil-like conformation for spherical nano-particular assemblies. Both the secondary structure formation as well as their morphological features were confirmed by circular dichroism spectroscopy and electron and atomic microscopy analyses. The new one-pot synthetic pathway is versatile in making diverse linear and branched polymers based on natural L-amino acids with a nano-fibrous or a spherical morphology for future applications in biomedical and thermoplastic industries.
1. Introduction
Hyperbranched (HB) polymers have recently attained significant importance due to their potential applications in drug delivery,1–3 catalyst development4–6 and self-assembled nanoscale materials.7,8 HB polymers have thermal and visco-elastic properties that are completely different from linear, block and graft macromolecular architectures.9 One-pot synthesis of high molecular weight polymers is an additional advantage associated with the hyperbranched structures.7,8 Over the past two decades, hyperbranched polymers based on polyesters,9 polyurethanes,10,11 polyethers,12–14 polyketals,15 polyamides and poly(ester-amide)s,16 and conducting polymers17 have been reported. Amino acids are important building blocks for polypeptides and their sequence and chain length drive the function and enzymatic activity in the biological system. AB2 or A2B monomers of L-lysine or L-aspartic acid (or L-glutamic acid) were polycondensed to make hyperbranched polyamides18,19 and poly(ester-amides).20–22 To date, no synthetic route has been reported in the literature to make linear polyesters and hyperbranched poly(ester-urethane)s by the identical process from a single amino acid residue. This is partially associated with the non-availability of the synthetic methodologies that could simultaneously activate both carboxylic units (as ester derivatives) and amine units (as urethane derivatives) towards alcohol under the same reaction conditions. Recently, we have reported the development of a dual ester-urethane melt condensation approach for linear poly(ester-urethane)s23 and functional helical polyesters24 based on natural L-amino acids under a solvent free melt polymerization process. This methodology was developed by merging the well-known melt transesterification reaction with melt transurethane process25,26 that was earlier developed in our laboratory. During the course of the development of this methodology, we identified that the carboxylic ester and urethane functionalities had very high selective reactivity towards alcohol (or diols) in the melt condensation reaction. This unique finding was the foundation for the development of a temperature selective one-pot melt polycondensation approach that could produce both linear polyesters and hyperbranched poly(ester-urethane)s from a single multifunctional amino acid source as shown in Fig. 1.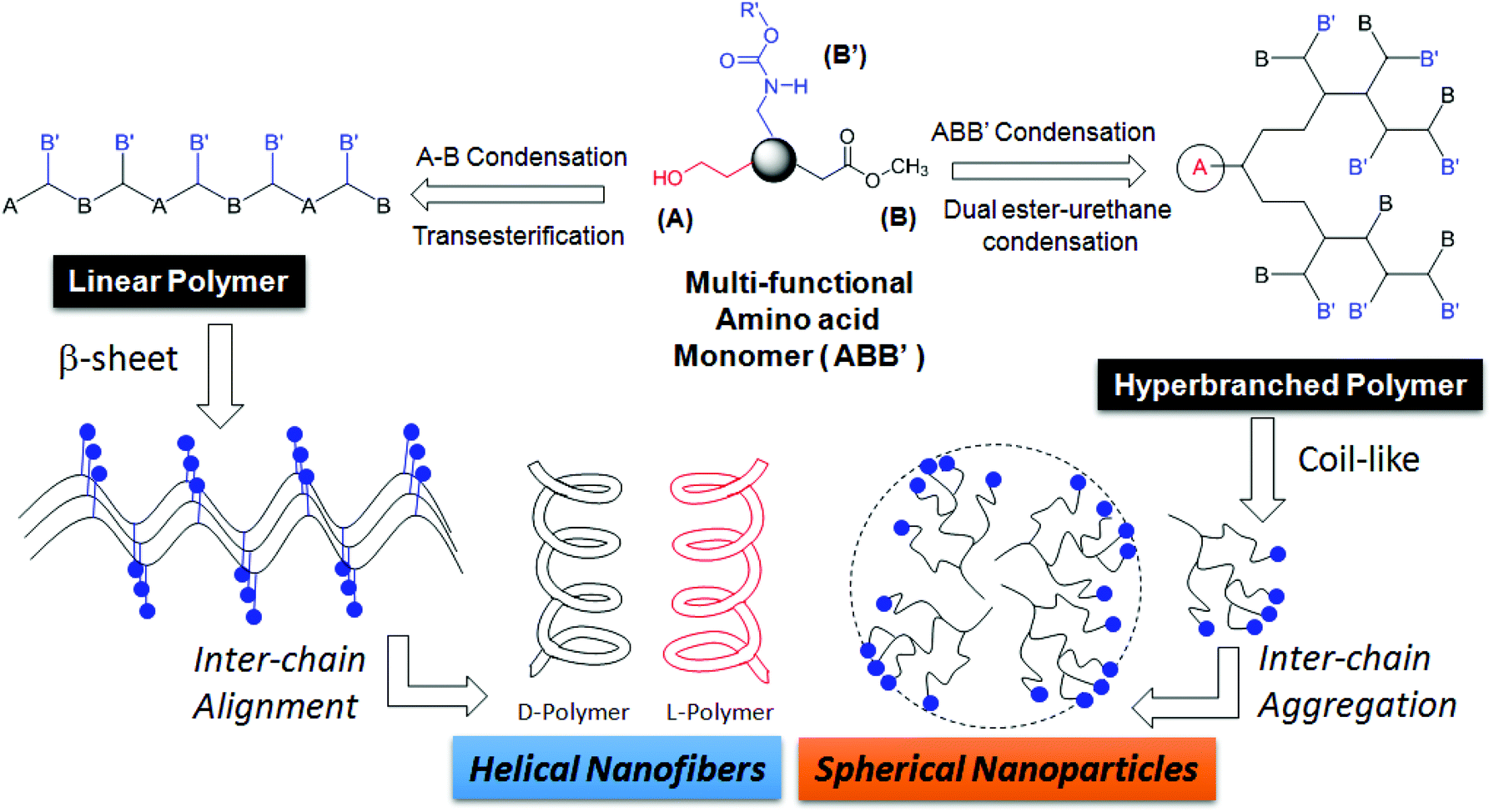 | ||
| Fig. 1 A novel one-pot temperature selective polycondensation approach for linear and hyperbranched polymers based on L-amino acids and their self-assembled nano-structures. | ||
The present investigation reports one of the first melt polycondensation approaches for hyperbranched polymers based on natural L-amino acids. For this purpose, two natural amino acids having carboxylic acid, amines and hydroxyl functional groups are selected. Both L (or) D-serine and L-threonine have fulfilled this requirement with primary and secondary hydroxyl functionalities, respectively. These monomers were readily converted into their corresponding ABB′ monomers, where A, B and B′ represented the hydroxyl, carboxylic ester and urethane functional groups, respectively. Thermo-selective one-pot polycondensation of these monomers produced linear polyesters and hyperbranched poly(ester-urethane)s (see Fig. 1). The reaction of A with B produced A–B linear polyesters with B′ anchored as pendent in each repeating unit. Simultaneous reaction of A towards B and B′ produced hyperbranched poly(ester-urethane)s. Efforts were taken to study the role of cyclization versus linear polymer chains in the molecular weight of the polymers. Further, the ability of the linear and hyperbranched polymers to produce secondary structures was investigated by circular dichroism (CD), electron microscopy and atomic force microscopy. The overall finding revealed that the linear polymers adopted a β-sheet expanded conformation and produced a helical nano-fibrous morphology whereas the hyperbranched polymers produced spherical nanoparticles through coil-like conformations.
2. Experimental
2.1. Materials
L-Serine, D-serine, L-threonine, octyl amine, palmitic acid, cyclohexanol and titanium tetrabutoxide (Ti(OBu)4) were purchased from Aldrich Chemicals and used without further purification. Methyl chloroformate, thionylchloride, and other solvents were purchased locally and purified prior to use.2.2. General procedure
1H and 13C-NMR were recorded using a 400-MHz JEOL NMR spectrophotometer. All NMR spectra were recorded in CDCl3 and DMSO-d6 containing TMS as the internal standard. The mass of the polymers was determined by using an Applied Biosystems 4800 PLUS MALDI TOF/TOF Analyzer. The polymer samples were dissolved in dichloromethane (DCM) at 10 mg mL−1. Dihydroxy benzoic acid (DHB) was used as a matrix. The matrix solution was prepared by dissolving 30 mg in 1 mL. A 1–2 μL aliquot of the polymer/matrix mixture was deposited on top of the MALDI plate and air-dried. High resolution mass spectra (HRMS) of monomers were recorded using a micro mass ESI-TOF MS Spectrometer. Gel permeation chromatographic (GPC) analysis was performed using a Viscotek VE 1122 pump, a Viscotek VE 3580 RI detector and a Viscotek VE 3210 UV/Vis detector in dimethyl formamide (DMF) using polystyrene as the standard. Thermal stability of the polymers was determined using the Perkin Elmer thermal analyzer STA 6000 model at a heating rate of 10 °C min−1 under a nitrogen atmosphere. Thermal analysis of the polymers was performed using a TA Q20 Differential scanning calorimeter. The instrument was calibrated using indium standards. All the polymers were heated to melt before recording their thermograms to remove their previous thermal history; the polymers were heated and cooled at 10 °C min−1 under a nitrogen atmosphere and their thermograms were recorded. Infrared spectra of the samples were recorded using a Perkin Elmer FT-IR spectrophotometer. The absorption studies were done by using a Perkin-Elmer Lambda 45 UV-Visible spectrophotometer in a tetrahydrofuran solvent. Circular dichroism (CD) analysis of the polymer samples was done using a JASCO J-815 spectrometer at 20 °C in THF. FE-SEM images were obtained using a Zeiss ultra plus scanning electron microscope. For FE-SEM the samples were prepared by drop casting the polymer solution on silicon vapors and coated with gold. TEM images were recorded using a Technai-300 instrument by drop casting the sample on a Formvar-coated copper grid. Atomic force microscopy (AFM) images were obtained by drop casting the samples on a freshly cleaved mica surface, using a Veeco Nanoscope IV instrument. The experiment was done in the tapping mode.2.3. Synthesis of L-serine monomer (1)
A typical procedure for carboxylic methyl ester methyl carbamate of amino acids was described for L-serine monomers. To a suspension of L-serine (10.0 g, 95.0 mmol) in methanol (70 mL), thionylchloride (13.7 mL, 190.4 mmol) was added dropwise at 0 °C under a nitrogen atmosphere. The reaction mixture was refluxed for 12 hours under a nitrogen atmosphere. The solvent and excess thionylchloride was removed by distillation. The residue was dried under vacuum to obtain the product as a white solid. The solid mass was stirred in sodium carbonate solution (25 wt%, 54 mL) and dichloromethane (50 mL) at 0 °C. To this ice cold solution, methyl chloroformate (9.82 mL, 128.0 mmol) in dichloromethane (50 mL) was added dropwise and the reaction was continued for 12 h at 25 °C. The reaction mixture was extracted with dichloromethane and the organic layer was dried over anhydrous Na2SO4. The liquid product was further purified by passing through silica gel column using ethyl acetate and pet ether (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 v/v) as an eluent. Yield = 12.6 g (75%). 1H-NMR (400 MHz, CDCl3) δ ppm: 5.87 (s, 1H, –NH), 4.41 (m, 1H, –CH), 3.96–3.88 (dd, 2H, –CH2), 3.76 (S, 3H, COOCH3), 3.68 (S, 3H, NHCOOCH3). 13C-NMR (100 MHz, DMSO-d6) δ ppm: 171.61, 156.91, 61.43, 56.79, 52.17, and 51.90. FT-IR (cm−1): 3389, 2956, 1701, 1526, 1446, 1350, 1268, 1210, and 1066. HRMS (ESI+): m/z [M + Na+] calcd for C6H11NO5 [M+]: 200.0534; found: 200.0537.
:6 v/v) as an eluent. Yield = 12.6 g (75%). 1H-NMR (400 MHz, CDCl3) δ ppm: 5.87 (s, 1H, –NH), 4.41 (m, 1H, –CH), 3.96–3.88 (dd, 2H, –CH2), 3.76 (S, 3H, COOCH3), 3.68 (S, 3H, NHCOOCH3). 13C-NMR (100 MHz, DMSO-d6) δ ppm: 171.61, 156.91, 61.43, 56.79, 52.17, and 51.90. FT-IR (cm−1): 3389, 2956, 1701, 1526, 1446, 1350, 1268, 1210, and 1066. HRMS (ESI+): m/z [M + Na+] calcd for C6H11NO5 [M+]: 200.0534; found: 200.0537.
2.4. Synthesis of L-threonine monomer (2)
L-Threonine (10 g, 84 mmol), SOCl2 (12.21 mL, 168 mmol) methanol (100 mL), methyl chloroformate (8.25 mL, 107 mmol), Na2CO3 (11.42 g, 20 wt%) and dichloromethane (70 mL) were used and the same experimental procedure as for monomer 1 was followed. Yield: 13.0 g (81%). 1H NMR (400 MHz, CDCl3) δ ppm: 5.77 (s, 1H, –NH), 4.29–4.26 (m, 2H, –NH–CH and –CH–OH), 3.74 (s, 3H, –COOCH3), 3.68 (s, 3H, NH–COOCH3), 2.62 (s, 1H, OH), and 1.24–1.21 (d, 3H, OH–CH–CH3). 13C-NMR (100 MHz, CDCl3) δ ppm: 171.84, 157.40, 67.82, 59.15, 52.52 and 19.74. FT-IR (cm−1): 3449, 2975, 2867, 2683, 2362, 2095, 1965, 1729, 1645, 1514, 1457, 1361, 1245, and 1206. HRMS (ESI+): m/z [M + Na+] calcd for C7H13NO5: 214.0687; found: 214.0699.2.5. Synthesis of D-serine monomer (3)
D-Serine (10 g, 95.0 mmol), SOCl2 (13.72 ml, 190.4 mmol), methanol (70 mL), methyl chloroformate (9.82 mL, 128 mmol), Na2CO3 (25 wt%, 54 mL) and dichloromethane (70 mL) were used and the same experimental procedure as for monomer 1 was followed. Yield: 13.31 g (79%). 1H-NMR (400 MHz, CDCl3) δ ppm: 5.86 (s, 1H, –NH), 4.42 (m, 1H, –CH), 3.96–3.90 (dd, 2H, –CH2), 3.77 (S, 3H, COOCH3), 3.69 (S, 3H, NHCOOCH3). 13C-NMR (100 MHz, DMSO-d6) δ ppm: 171.0, 156.71, 61.40, 56.75, 52.17, and 51.90. FT-IR (cm−1): 3380, 2956, 1715, 1520, 1446, 1355, 1260, 1210, and 1066. HRMS (ESI+): m/z [M + Na+] calcd for C6H11NO5 [M+]: 200.0534; found: 200.0497.2.6. Synthesis of L-serine linear polyester (L-SLP)
A typical melt polymerization procedure was explained for L-serine. L-Serine monomer 1 (1.0 g, 5.6 mmol) was taken in a test tube shaped polymerization apparatus and melted by placing the tube in an oil bath at 80 °C with a constant N2 flow and then titaniumtetrabutoxide (0.019 g, 0.06 mmol, 1 mol%) was added into it. The reaction mixture was degassed by purging with nitrogen and subsequent evacuation by vacuum under constant stirring at 80 °C. The oil bath was preheated to 120 °C and the polycondensation was carried out at 120 °C for 4 h with constant stirring under nitrogen purge. During this stage, the methanol was removed along with purge gas and the polymerization mixture became viscous. The viscous melt was further subjected to condensation in a high vacuum (0.01 mbar of Hg) at 120 °C for 2 h. At the end of the polycondensation, the linear polymer was obtained as a solid. It was purified by dissolving in tetrahydrofuran, filtered and precipitated in to diethyl ether. Yield = 0.8 g (98%). 1H NMR (400 MHz, CDCl3) δ ppm: 5.86 (s, 1H, –NH), 4.64–4.42 (m, 3H, –CH2 and –CH), 3.81 (s, 3H, –OCH3), 3.72 (s, 3H, –NHCOOCH3). 13C-NMR (100 MHz, DMSO-d6) δ ppm: 156.58, 107.00, 105.82, 97.20, 66.61, 65.74, 51.80, 33.18, 29.14, 23.33, 23.84. FT-IR (cm−1): 3313, 2956, 1690, 1528, 1449, 1351, 1248, 1205, 1161 and 1055.2.7. Synthesis of L-serine hyperbranched poly(ester-urethane) (L-SHPEU)
Serine monomer 1 (1.30 g, 7.30 mmol) and titanium-tetrabutoxide (0.024 g, 0.073 mmol, 1 mol%) were polymerized at 150 °C for 4 h under nitrogen purge and 2 h under vacuum (0.01 mbar) as described for L-SLP. Yield = 0.77 g (95%). 1H NMR (400 MHz, CDCl3) δ ppm: 6.21–5.70 (m, 1H, NH), 4.63–4.3 (m, 3H, CHCH2OH and CHCH2OH), 3.80 (s, 3H, –OCH3), 3.72(s, 3H, –NHCOOCH3). 13C-NMR (100 MHz, DMSO-d6) δ ppm: 169.13, 156.39, 67.96, 64.92, 61.61, 53.06, 25.59, 14.47, 14.08. FT-IR (cm−1): 3317, 2958, 1521, 1449, 1345, 1205 and 1082.A similar procedure was used to produce L-threonine linear polyester (L-TLP), D-serine linear polyester (D-SLP), and D-serine hyperbranched poly(ester-urethane) (D-SHPEU), and their details are given in the ESI.†
3. Results and discussion
3.1. Serine based linear and HB polymers
Ester-urethane monomers were synthesized starting from naturally available L-amino acids as shown in Scheme 1. L-Serine was converted into its carboxylic acid chloride and subsequently reacted with methanol to yield a methyl ester amine hydrochloride salt. The amine salt was converted into its free amine and further reacted with methyl chloroformate to produce methyl urethane (or carbamate). The resulting molecule represented an ABB′ type monomer in which the hydroxyl (A) unit reacts with ester (B) and urethane (B′) functionalities. A similar procedure was used to obtain monomers based on L-threonine (2) and D-serine (3) (see ESI, SS-1†). Thermogravimetric analysis (TGA) revealed that these monomers were stable up to 170 °C for melt polycondensation (see ESI, SF-1†).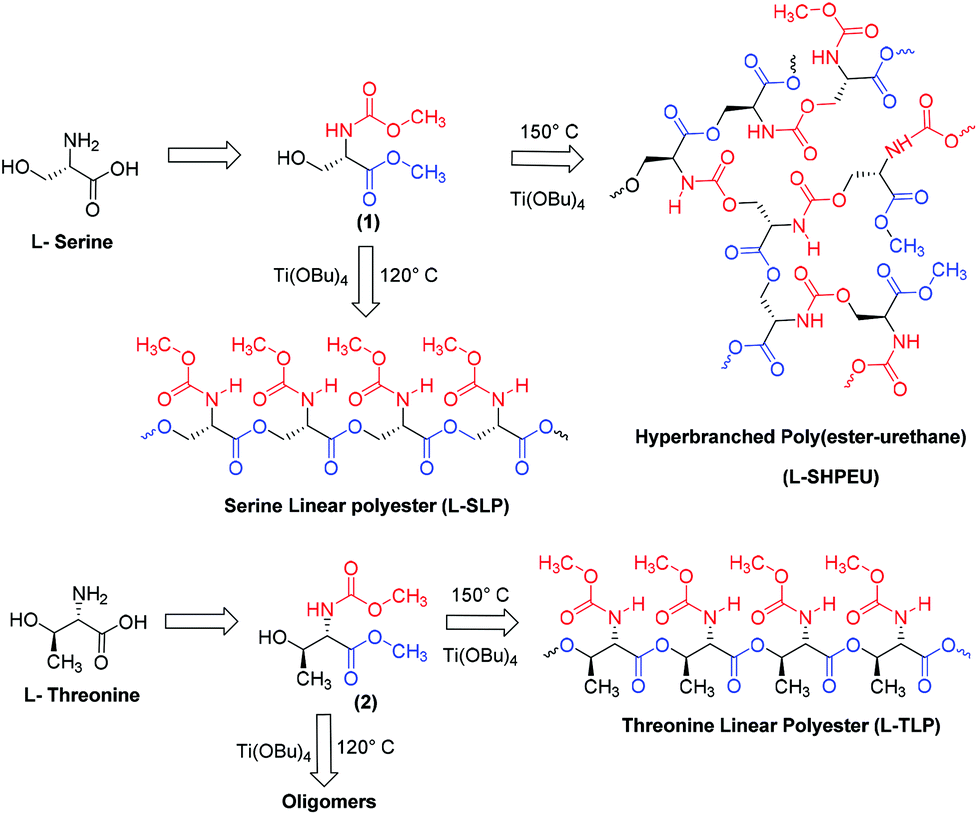 | ||
| Scheme 1 Synthesis of linear and hyperbranched polymers based on L-serine and L-threonine monomers. | ||
The ABB′ monomer was polycondensed using Ti(OBu)4 as a catalyst (1 mol%) at two different temperatures: (i) at 120 °C for selective transesterification of alcohol (A) with ester units (B) to produce linear polyesters and (ii) at 150 °C for dual ester-urethane polycondensation of alcohol (A) with both ester (B) and urethane (B′) to yield new classes of hyperbranched poly(ester-urethane)s. The structures of the linear and hyperbranched polymers are shown in Scheme 1. Prior to the polymerization, the monomer and the catalyst (in a cylindrical polymerization tube) were subjected to evacuation under vacuum and nitrogen purge to make them oxygen and moisture free. Subsequently, the polymerization was conducted under nitrogen purge at the desired temperature (either 120 °C or 150 °C) for 4 hours to obtain the oligomers. These oligomers were further polycondensed for 2 hours under vacuum (0.01 mbar). At the end of the polymerization, the polymers were obtained as white solid mass products.
1H NMR spectra of the L-serine monomer, linear polyester (L-SLP) and HB polymer (L-SHPEU) are shown in Fig. 2. The different peaks in the spectra are assigned with alphabets with respect to their protons in the chemical structures. At 120 °C, the –CH2OH (proton-a) at 3.90 ppm was almost vanished and a new carboxylic ester peak corresponding to –CH2OOCCH– appeared at 4.62 ppm (see Fig. 2b). The integration of the peak intensities confirmed the occurrence of the reaction up to 95%. Thus, the number of the repeating units in the linear polyesters was theoretically estimated to be 15–20 units. Control polymerization was performed at 120 °C and aliquots were collected at regular intervals. 1H NMR spectra of their aliquots (see SF-2†) showed the vanishing of –CH2OH protons and the appearance of a new ester peak. During this process, the urethane methyl protons –CHNHCOOCH3 were completely inert. Hence, at 120 °C in the ABB′ monomer, only the –CH2OH (A) functional group reacted with carboxylic ester B and the urethane functional group B′ was not disturbed. The self-condensation of ABB′ monomers at 150 °C facilitated the simultaneous reaction of –CH2OH (A) with carboxylic ester (B) and urethane (B′) functional groups to produce hyperbranched polymers. In Fig. 2c, the –CH2OH protons completely vanished and a new urethane peak appeared at 4.55 ppm (see peak-a′′) along with the ester peak at 4.62 ppm (see proton-a′). Further, the 1H-NMR spectra of the hyperbranched polymer aliquots (see SF-3†) also supported the fast vanishing of –CH2OH protons and the appearance of new ester and urethane peaks seen as in Fig. 2c. A similar investigation by 13C-NMR further confirmed the temperature selective condensation of the ABB′ monomer to produce linear polyesters and hyperbranched poly(ester-urethane)s (more details in the ESI, see SF-4†).
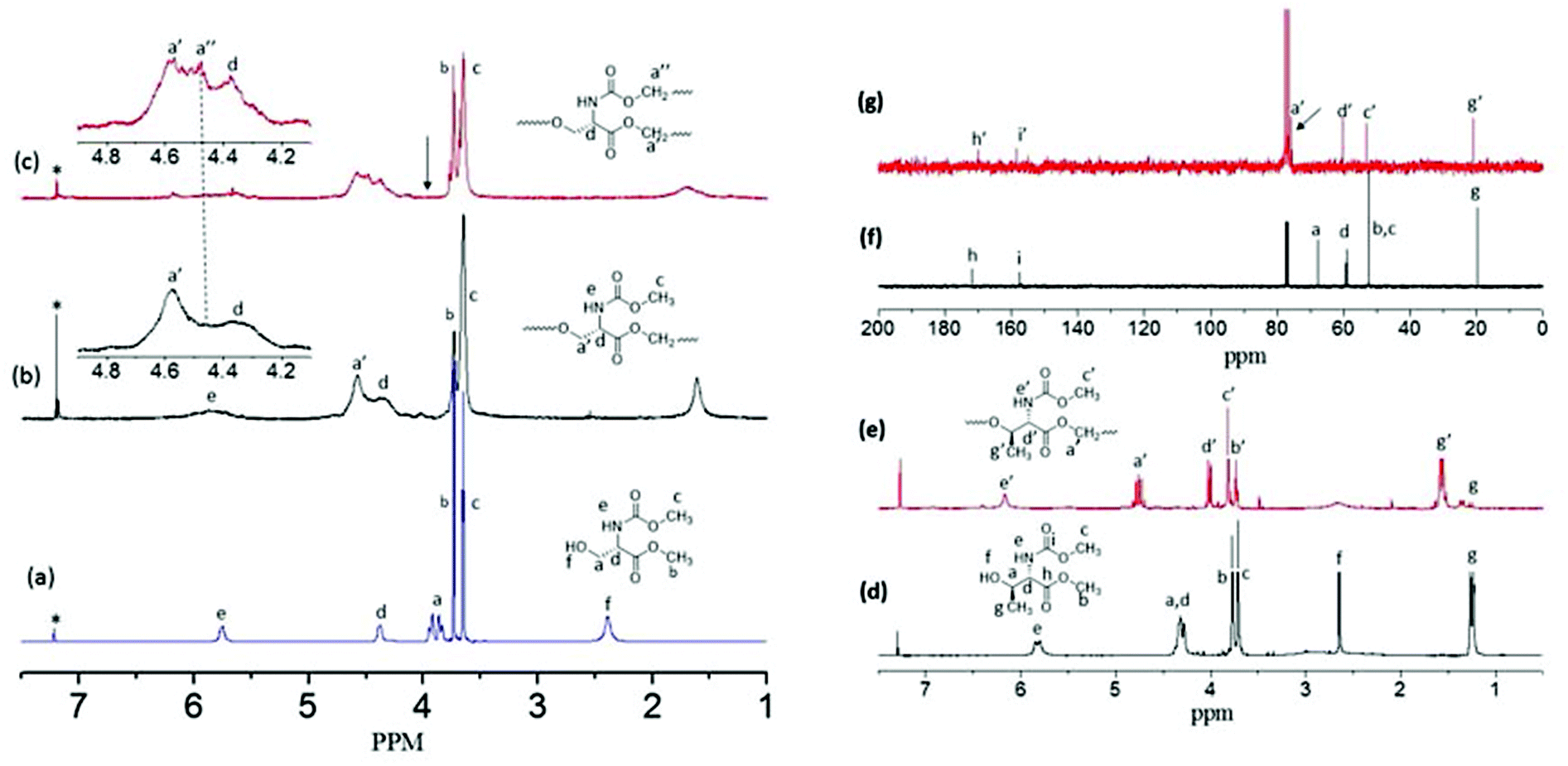 | ||
| Fig. 2 1H-NMR spectra of L-serine monomer 1 (a), its linear polyester (L-SLP) (b) and hyperbranched poly(ester-urethane)s (c). 1H-NMR spectra of L-threonine monomer 2 (d) and its linear polyester (T-LSP) (e). 13C-NMR spectra of L-threonine monomer 2 (f) and T-LSP (g). All the spectra are recorded in CDCl3. | ||
The degree of branching (DB) of the HB polymer was determined using the expression DB = [D + T]/[D + T + L] or DB = [2T]/[D + T + L] or [2D]/[D + T + L] where D, L and T represent the dendritic, linear and terminal units, respectively.12,27,28 The hyperbranched structures produced by AB2 monomers has equal amounts of end groups (B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) B). It is also important to mention that in hyperbranched polymers, the number of dendritic units is always equivalent to the number of terminal units (D = T). In the present case, B and B′ have a difference in reactivity towards A; thus, the difference between B and B′ peak integration values at the terminal units actually represent the amount of dendrite units produced by B in reaction with A (see more details in SF-5†). Thus T = B, T + L = B′ and L = D = B′ − B in the present case. Upon forming the hyperbranched structure the dendritic and linear units appeared together for protons in the polymer-CH2OOCCH and the polymer-CH2-OOCNHCH at 4.6–4.5 ppm (see NMR spectra in SF-5†). The terminal units from both ester and urethane units appeared together at 3.72 and 3.81 ppm, respectively. Based on the NMR integration values, the DB was estimated as 66% (see SF-5†). Earlier reports on HB polyesters and polyethers had shown DB ∼ 60%. Thus, the hyperbranched poly(ester-urethane) has DB similar to literature examples.9
B). It is also important to mention that in hyperbranched polymers, the number of dendritic units is always equivalent to the number of terminal units (D = T). In the present case, B and B′ have a difference in reactivity towards A; thus, the difference between B and B′ peak integration values at the terminal units actually represent the amount of dendrite units produced by B in reaction with A (see more details in SF-5†). Thus T = B, T + L = B′ and L = D = B′ − B in the present case. Upon forming the hyperbranched structure the dendritic and linear units appeared together for protons in the polymer-CH2OOCCH and the polymer-CH2-OOCNHCH at 4.6–4.5 ppm (see NMR spectra in SF-5†). The terminal units from both ester and urethane units appeared together at 3.72 and 3.81 ppm, respectively. Based on the NMR integration values, the DB was estimated as 66% (see SF-5†). Earlier reports on HB polyesters and polyethers had shown DB ∼ 60%. Thus, the hyperbranched poly(ester-urethane) has DB similar to literature examples.9
3.2. Molecular weights and end group analysis
The molecular weights of the newly synthesized linear polyesters and hyperbranched poly(ester-urethane)s were determined by gel permeation chromatography in DMF at 25 °C. The GPC chromatogram of the polymers are shown in the ESI (see SF-6†) and their molecular weights are given in Table ST1 in the ESI.† The molecular weights of the polymers were obtained as Mn ∼ 16000 and Mw ∼ 20000 g mol−1. The molecular weights of the aliquots of the polymer samples corresponding to the polymerization at 120° and 150 °C were also determined by GPC (see SF-6, SF-7 and SF-8†). These molecular weights are plotted against the polymerization time (see SF-9†). In the case of the linear polyesters, the molecular weight increases with an increase in the polymerization time and maximum was achieved toward the end of the reaction. On the other hand, the molecular weights of hyperbranched polymers were enhanced rapidly at the initial stage of the reaction onwards. To further study the enantiomeric effect on the synthesis of the linear and hyperbranched polymers, D-serine monomer (3) was synthesized and polymerized at 120 °C and 150 °C (see SS-1†). The molecular weights of these polymers were also obtained similarly to that of the L-serine counterpart (see Table ST1 in the ESI†). Both natural L-serine monomers and un-natural D-serine monomers followed the thermo-selective polycondensation to produce high molecular weight linear polyesters and hyperbranched poly(ester-urethane)s.
MALDI-TOF mass spectrometry provides direct information on the types of the end groups present in the polymer chains as well as the macrocyclic formation that occurred during the polymerization of ABB′ monomers. MALDI-TOF spectra of the polyester and hyperbranched polymers are shown in Fig. 3a and b, respectively. The polymer showed two sets of intense peaks for every repeating unit with respect to the linear and cyclic units (as both Na+ and K+ ions). Each repeating unit mass was resolved for the repeating unit formula: (145)n + 32 + 23 for sodium ion peaks [(145)n + 32 + 39 for potassium ion peaks]. In Fig. 3a, the mass peaks are visible up to 12 repeating units. The intensity of the linear units became more abundant with increase in the molecular weights of polymers at higher conversion. The hyperbranched polymers (see Fig. 3b) showed peaks up to 11 units. Interestingly, the hyperbranched structures predominantly showed mass peaks with respect to cyclic structures, and the linear units are almost absent at higher conversion. A similar observation was earlier reported in the synthesis of hyperbranched polyethers29 and other hyperbranched polymers.30 This indicated that the macrocyclic formation was one of the major obstacles in building the high molecular weight polymers in hyperbranched polymers. Further, the chain ends have active ester and urethane end groups confirming their thermo-stability towards the melt condensation approach. The TGA-plots of the linear and HB polymers revealed that the newly designed polymers are stable up to 200 to 230 °C (see SF-15†). The thermal properties of the polymers were studied by differential scanning calorimetry (DSC) at 10° min−1 heating and cooling rates. DSC thermograms of the linear polyesters showed glass transition (Tg) temperature at 2 to 4 °C (see SF-16†). The HB polymers showed higher Tg values at 40 to 55 °C (see SF-16†). The higher Tg values in HB polymers are attributed to the presence of H-bonding end groups at the periphery of the HB structure compared to that of linear polyesters.
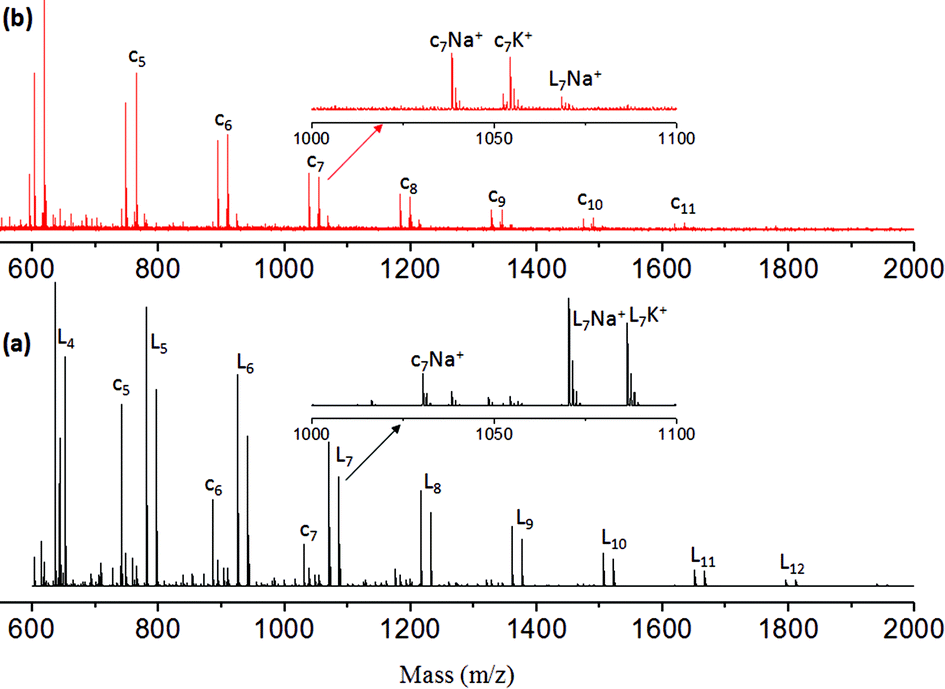 | ||
| Fig. 3 MALDI-TOF Mass spectra of L-SLP (a) and L-SHPEU (b). | ||
3.3. L-Threonine based linear polyesters
L-Threonine amino acid has a secondary hydroxyl group unlike its L-serine counterpart. The L-threonine monomer was subjected to thermo-selective polymerization at 120 °C and 150 °C. At 120 °C, the monomer did not polymerize even after prolonged condensation for more than 10 h (it produced only oligomers of 1–3 units). At 150 °C, the self-condensation of the L-threonine occurred significantly to produce a white solid polymer mass. 1H and 13C NMR spectra of the L-threonine monomer and the polymer (produced at 150 °C) are shown in Fig. 2d–g. The molecular weight of the L-threonine polymer was determined as Mn = 8500 and Mw = 13500 (see SF-6 and Table ST1†). To clarify the role of the secondary hydroxyl group reactivity in L-threonine monomers towards esters and urethanes in the condensation reaction model reactions were carried out using cyclohexanol (see Scheme SS-2 in the ESI†). Cyclohexanol was chosen because of its secondary hydroxyl group and its high boiling point suitable for the melt process. n-Octyl urethane monomer and palmitic carboxylic ester were synthesized (see SS-2†). The reaction of palmitic ester with cyclohexanol produced a new ester linkage R-COOCH-Cy (90% yield) which appeared at 4.72 ppm (see SF-10a†). On the other hand, the n-octyl urethane monomer underwent a slow reaction at 150 °C and the new urethane linkage R-HNCOOCH-Cy (30% yield) was observed at 4.33 ppm (see SF-10b†). The comparison of the model reaction products with the polymer spectrum (see Fig. 2e) revealed that the only new ester linkage was formed in the L-threonine monomer self-condensation at 150 °C. This was further supported by the comparison of 13C NMR spectra of the monomer and the polymer in Fig. 2f and 2g. In Fig. 2g, the carbon atom CH–CH(CH3)–OH in the monomer completely disappeared at 67.82 ppm and a new peak for the ester carbon atom CH–CH(CH3)–O–CO–R appeared at 75.0 ppm (near the solvent carbon). Thus, at 150 °C, L-threonine monomers produced exclusively linear polyesters. The L-threonine linear polyesters showed very low Tg at −25 °C with respect to their steric hindered polymer backbones (see SF-16†). The reason for this trend was attributed to the steric hindrance induced by the α-methyl unit in the L-threonine monomer.
3.4. Self-assembly and morphology of linear and HB polymers
The newly synthesized polymers were derived from enantiomeric L-serine, D-serine and L-threonine monomers; thus, the circular dichroism (CD) spectroscopic analysis was carried out to investigate their secondary structures in solution. Typically, peptides showed a positive band at 192 nm and two negative bands at 208 and 222 nm with respect to α-helical conformation.31 The β-sheet structure exhibited a positive CD band at 205 nm and a negative CD band at 230 nm. Random coil structures are expected to show a single negative CD band at 205 nm.24 CD spectra of the L-serine and D-serine monomers (see Fig. 4a) showed opposite Cotton signals with respect to their stereo-isomeric structures. Similarly, the L-threonine monomer also showed expected CD signals. In Fig. 4b, the CD spectra of the linear polyester from the L-serine and D-serine monomers showed helicity with opposite CD bands. The L-serine linear polyesters showed one positive CD band at 225 nm for π–π* and a negative CD band around 237 nm for n–π* transition.24 These band signatures represent the secondary structures for the polymer chains in the β-sheet conformation.24 Absorption spectra of the polymers were matched with that of the CD signal wavelength region. This proved that the high temperature condensation reaction did not disturb the optical activity of the monomers in the reaction and their optical properties were preserved in the polymer structures as well. Surprisingly, the HB polymers (see Fig. 4c) obtained from L-serine (or D-serine) did not show any CD signal. In Fig. 4d, the L-threonine linear polyester (from secondary alcohol) showed a positive CD signal with respect to the proline type II random coil structure. Thus, based on the above CD signals, it may be summarized that L (or D)-serine linear polyesters exhibited β-sheet conformation whereas random-coil was obtained for the sterically hindered secondary alcohol based polyesters of L-threonine. The highly branched structures did not allow the polymer chains to undergo self-organization in the hyperbranched structure; thus, they did not show CD signals (see Fig. 4c). | ||
| Fig. 4 CD spectra of monomers 1, 2 and 3 (a). CD spectra of linear L and D-serine polyesters (b). CD spectra of L and D-serine hyperbranched poly(ester-urethane)s (c). CD spectrum of L-threonine linear polyester (d). | ||
The polymer samples were subjected to electron microscopy analysis (FE-SEM and HR-TEM) to visualize the size and shape of the self-assembled objects. FE-SEM images of the L-serine linear polyesters showed the formation of a long twisted nano-fibrous morphology (see Fig. 5a) (see SF-11 to SF-13 for more images†). This was further confirmed by the HR-TEM image in Fig. 5b. The thickness and length of the nanofibers were obtained as 30 nm and 2–3 μm, respectively. The formation of nanofibers was attributed to the β-sheet hydrogen bonding interactions facilitated by the pendent urethanes attached to each repeating unit of linear polyesters.24 FE-SEM images of the hyperbranched polymers showed the formation of spherical nanoparticle assemblies (see Fig. 5c). This suggested that the branched macromolecular architectures adapted the globular coil-like conformation and thus produced spherical particles. The larger size of the HB nanoparticles indicated that they were produced by the aggregation of more than one hyperbranched polymer chain together with the spherical shape as shown in Fig. 1. The L-threonine polyester showed the CD signal for a coil-like proline type-II self-assembly that produced spherical particles (see Fig. 5d). FT-IR spectra of all the above three polymers were recorded to trace their morphological differences (see SF-14†). All three polymers showed the existence of hydrogen bonding interactions with respect to a N–H stretching vibrational band at 3350 cm−1 and a CO band at 1722 cm−1.24 Thus, the difference in the morphological features are not due to the variation in their hydrogen bonding ability and it is mainly due to the difference in their topology of the polymer structure. Linear polyesters with urethane hydrogen bonding pendent at each repeating units self-assembled to produce an expanded nano-fibrous morphology. Though the hydrogen bonding interactions are present in the hyperbranched structures in the urethane units, the branched network disturbed their chain alignment. In the case of the L-threonine polymer, the proline type-II did not allow the chain to adopt the expanded conformation.
 | ||
| Fig. 5 FE-SEM (a) and HR-TEM (b) images of L-serine linear polyesters. FE-SEM images of L-serine based HB poly(ester-urethane) (c) and L-threonine linear polyester (d). | ||
Recently, Lederer and co-workers32 reported the molecular self-assembly of dendronized glycopolymers based on maltose shell having lysine amino acid residues. It was found that the molecular weight of the polymers (dendron generation) and pH played vital roles in the aggregation phenomena of these big molecules and their transformation from helical fibres to globular structures. In the present investigation, the newly designed hyperbranched polymers (resembled the dendritic architectures) underwent coil-like conformational changes to produce globular particles. In order to investigate the role of the molecular weights on the globular particle formation by the HB polymers, more than 10 aliquots of the HB samples were collected starting from 30 minutes of the polymerization till the end of the process at 6 h (GPC data and MALDI-TOF of representative aliquots are given in SF-8 and SF-17†). These samples were subjected to FE-SEM and atomic force microscopy, and their images are given in Fig. 6 (for more images see SF-18†). It is very clear from the FE-SEM images that the low molecular weight polymers collected at the initial stage of the polymerization showed larger aggregates of 250 nm (see Fig. 6a–c). With the increase in the molecular weights, the HB polymers produced smaller aggregates of <40 nm. AFM images (see Fig. 6d–f) of the aliquots also confirmed the spherical shape of the nano-aggregates as well as their sizes with that of SEM images.
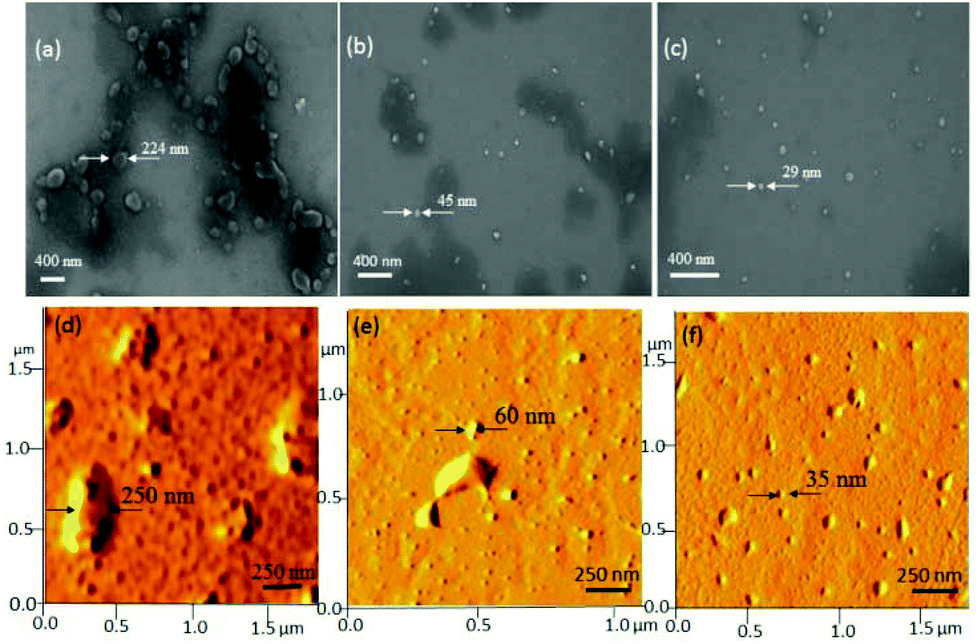 | ||
| Fig. 6 FE-SEM images of HB aliquots having molecular weights: Mn = 10000 g mol−1 (a), 12000 g mol−1 (b) and 16000 g mol−1 (c). (d)–(f) AFM images of HB aliquots having molecular weights: Mn = 10000 g mol−1 (d), 12000 g mol−1 (e) and 16000 g mol−1 (f). | ||
To further analyze the role of the HB polymers, the size of the nanoparticles was plotted against Mn of the HB polymers and shown in Fig. 7a. At low molecular weight samples, the HB polymer chains showed larger size aggregate formation. The size of the particle became <40 nm beyond Mn = 12000 g mol−1. CD signals of these HB polymer aliquots were also recorded and their spectra are shown in Fig. 7b. The CD signals were relatively visible at low molecular weight samples; however, they gradually disappeared with increase in molecular weights. The appearance of CD signals at low molecular weight samples was attributed to the loosely packed HB chains which in turn facilitated the partial alignment of the chains to become CD active. This observation was supported by the larger particles size in FE-SEM and AFM images in Fig. 6a and d. The higher molecular weight HB samples produced tightly packed nanoparticles; thus, the samples became CD inactive. Hence, as observed by Lederer and co-workers32 in dendronized polymers, in the present case, the HB polymers based on natural L-amino acids attained globular spherical conformation at higher molecular weights. It may be concluded that the molecular weights of the hyperbranched structures of amino acid residues are very crucial factors in determining the globular nature of the structures. Higher molecular weight HB polymers adopted globular structures as in the case of dendrite macromolecules. In the present investigation, for the first time, the roles of the polymer topology in the molecular self-assemblies of the amino acid based polymers were resolved. This was accomplished with help of linear and hyperbranched polymers produced from identical starting materials. Though the present approach is demonstrated only for L-serine and L-threonine based polymers, this approach is not restricted only to these examples and it may be expanded to other multifunctional amino acid monomers or other monomeric resources for making diverse polymer structures with appropriate molecular self-assemblies. Further, the present investigation opens up new synthetic pathways for linear or hyperbranched polymers based on multi-functional amino acids.
 | ||
| Fig. 7 (a) Plot of nanoparticle size versus the Mn of the HB polymer aliquots. (b) CD spectra for various molecular weight HB polymer aliquots. | ||
4. Conclusion
In conclusion, a new temperature selective polycondensation approach has been successfully developed to make new classes of linear polyesters and hyperbranched poly(ester-urethane)s based on natural L-amino acid monomers. L-Serine (also D-serine) and L-threonine provided exact structural requirements with hydroxyl, carboxylic acid and amine functionalities that facilitated the synthesis of new ABB′ monomers (A = hydroxyl, B = ester and C = urethane). The ABB′ monomer underwent a temperature selective polycondensation reaction among A and B at 120 °C to produce linear polyesters. In these linear polyesters, the un-reacted functional group B′ remained pendent in each unit. At 150 °C, hydroxyl (A) reacted equally with both B and B′ under the dual ester-urethane melt condensation approach to yield novel hyperbranched poly(ester-urethanes). These optically active polymers showed diverse CD signals with respect to their polymer topology. The linear polyester produced by L-serine and D-serine showed expanded chain conformation through hydrogen bonding interactions that resulted in the self-assembly of a nano-fibrous morphology. Hyperbranched polymers did not show any CD signals and produced spherical morphologies. The branched structures did not allow the chain to pack; thus, they favored coil-like conformation (no CD signal) that assembled the polymer chains as the globular spherical nanoparticles. L-Threonine based polymers adapted random-coil proline type II structures that also produced spherical nanoparticle assemblies. The globular nanoparticle nature of HB polymers were found to be driven by the molecular weights of the samples. Both the vanishing of the CD signal and the formation of tightly packed nanoparticle formation were found to be maxima at higher molecular weight HB samples. The present investigation provides the first insight into the molecular self-assemblies of linear and hyperbranched polymers based on L-amino acids from single starting materials. Both the synthetic methodology and the linear and hyperbranched structures are new entities in the literature and they may be very useful for applications in biomedical and thermoplastic industries.Acknowledgements
The authors are grateful for the research grant from the Department of Science and Technology (DST), New Delhi, for the project SB/S1/OC-37/2013 under SERB scheme. Rajendra Aluri thanks CSIR, New Delhi for his Ph.D. fellowship. The authors thank NCL-Pune for the HR-TEM images. The authors thank the DST-FIST Project for AFM facilities.Notes and references
- H. Jin, W. Huang, X. Zhu, Y. Zhou and D. Yan, Chem. Soc. Rev., 2012, 41, 5986–5997 RSC.
- A. Sousa-Herves, S. Wedepohl and M. Calderon, Chem. Commun., 2015, 51, 5264–5267 RSC.
- J. Chen, C. Wu and D. Oupicky, Biomacromolecules, 2009, 10, 2921–2927 CrossRef CAS PubMed.
- L. Xu and Z. Ye, Chem. Commun., 2013, 49, 8800–8802 RSC.
- J. Sebastian and D. Srinivas, Chem. Commun., 2011, 47, 10449–10451 RSC.
- S. V. de Vyver, J. Gebboers, S. Helsen, F. Yu, J. Thomas, M. Smet, W. Dehaen and B. F. Sels, Chem. Commun., 2012, 48, 3497–3499 RSC.
- Y. Zhou and D. Yan, Chem. Commun., 2009, 1172–1188 RSC.
- A. Zill, A. L. Rutz, R. E. Kohman, A. M. Alkilany, C. J. Murphy, H. Kong and S. C. Zimmerman, Chem. Commun., 2011, 47, 1279–1281 RSC.
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183–275 CrossRef CAS PubMed.
- S. P. Rannard, N. J. Davis and I. Herbert, Macromolecules, 2004, 37, 9418–9430 CrossRef CAS.
- T. Ranganathan, C. Ramesh and A. Kumar, Chem. Commun., 2004, 154–155 RSC.
- M. Jayakannan and S. Ramakrishnan, Chem. Commun., 2000, 1967–1968 RSC.
- M. Jayakannan and S. Ramakrishnan, Macromol. Rapid Commun., 2001, 22, 1463–1473 CrossRef CAS.
- A. Saha and S. Ramakrishnan, Macromolecules, 2009, 42, 4956–4959 CrossRef CAS.
- S. Chatterjee and S. Ramakrishnan, Macromolecules, 2011, 44, 4658–4664 CrossRef CAS.
- M. Scholl, Z. Kadlecova and H. A. Klok, Prog. Polym. Sci., 2009, 34, 24–61 CrossRef CAS PubMed.
- C. R. Yates and W. Hayes, Eur. Polym. J., 2004, 40, 1257–1281 CrossRef CAS PubMed.
- M. Scholl, T. Q. Nguyen, B. Bruchmann and H. A. Klok, Macromolecules, 2007, 40, 5726–5734 CrossRef CAS.
- M. Scholl, T. Q. Nguyen, B. Bruchmann and H. A. Klok, J. Polym. Sci. Part A: Polym. Chem., 2007, 45, 5494–5508 CrossRef CAS PubMed.
- Y. Bao, G. Shen, H. Liu and Y. Lie, Polymer, 2013, 54, 652–660 CrossRef CAS PubMed.
- Y. Bao, J. He and Y. Li, Polymer, 2012, 53, 145–152 CrossRef CAS PubMed.
- Y. M. Bao, X. H. Liu, X. L. Tang and Y. S. Li, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5364–5374 CrossRef CAS PubMed.
- S. Anantharaj and M. Jayakannan, Biomacromolecules, 2012, 13, 2446–2455 CrossRef CAS PubMed.
- S. Anantharaj and M. Jayakannan, Biomacromolecules, 2015, 16, 1009–1020 CrossRef CAS PubMed.
- P. Deepa and M. Jayakannan, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2351–2366 CrossRef CAS PubMed.
- P. Deepa and M. Jayakannan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2445–2458 CrossRef CAS PubMed.
- C. J. Hawker, R. Lee and J. M. J. Frechet, J. Am. Chem. Soc., 1991, 113, 4583–4588 CrossRef CAS.
- D. Holter, A. Burgath and H. Frey, Acta Polym., 1997, 48, 30–35 CrossRef PubMed.
- M. Jayakannan, J. L. J. V. Dongen, G. C.H. Behera and S. Ramakrishnan, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4463–4476 CrossRef CAS PubMed.
- H. R. Kricheldorf, S. Bohme, G. Schwarz and C. L. Schultz, Macromolecules, 2004, 37, 1742–1748 CrossRef CAS.
- A. Sinaga, T. A. Hatton and K. C. Tam, Biomacromolecules, 2007, 8, 2801–2808 CrossRef CAS PubMed.
- S. Boye, D. Appelhans, V. Boyko, S. Zschoche, H. Komber, P. Friedel, P. Formanek, A. Janke, B. I. Voit and A. Lederer, Biomacromolecules, 2012, 13, 4222–4235 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of polymers, model reactions, NMR data, FTIR spectra, HR-MS data, GPC chromatograms, TGA and DSC thermal data, FE-SEM images. See DOI: 10.1039/c5py00602c |
| This journal is © The Royal Society of Chemistry 2015 |