
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot approach to Pd-loaded porous polymers with properties tunable by the oxidation state of the phosphorus core†
Xiaoyu
Jiang‡
a,
Wuxue
Zhao‡
b,
Wei
Wang
*a,
Fan
Zhang
*b,
Xiaodong
Zhuang
b,
Sheng
Han
c and
Xinliang
Feng
d
aSchool of Perfume and Aroma Technology, Shanghai Institute of Technology, Shanghai, 200235, P. R. China. E-mail: wangweittg@sit.edu.cn
bSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: fan-zhang@sjtu.edu.cn
cSchool of Chemical and Environmental Engineering, Shanghai Institute of Technology, Haiquan Road 100, Shanghai, P. R. China
dCenter for Advancing Electronics Dresden & Department of Chemistry and Food Chemistry, Technische Universitaet Dresden, 01062 Dresden, Germany
First published on 14th July 2015
Abstract
Two novel Pd-loaded heteroatom-linked microporous polymers Pd@N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P and Pd@NPO were effectively synthesized by one-pot Pd-catalyzed Heck coupling reactions of tris(4-vinylphenyl)amine with tris(4-bromophenyl)phosphine and tris(4-bromophenyl)phosphine oxide, respectively. The Pd atoms loaded into the networks of the resulting porous polymers originated from the Pd-catalyst used in the polymerization, which was achieved under moderate reaction conditions. Besides the nitrogen atoms, the trivalent phosphorus atoms bearing one lone pair of electrons and the pentavalent phosphorus atoms of the phosphoryl groups were used as the linkers for the formation of the frameworks of the porous polymers Pd@NP and Pd@NPO, respectively. The phosphorus atoms with different oxidation states in the networks caused a dramatic variation in the physical and catalytic properties of the as-prepared porous polymers. Pd@NP and Pd@NPO exhibit surface areas of 381 m2 g−1 and 684 m2 g−1, respectively. Both Pd-loaded porous polymers enable efficient Suzuki–Miyaura coupling reactions featuring short reaction times and good yields, with the catalysts being highly stable and easy to recycle. The catalytic activity of Pd@NPO is higher than that of Pd@NP.
P and Pd@NPO were effectively synthesized by one-pot Pd-catalyzed Heck coupling reactions of tris(4-vinylphenyl)amine with tris(4-bromophenyl)phosphine and tris(4-bromophenyl)phosphine oxide, respectively. The Pd atoms loaded into the networks of the resulting porous polymers originated from the Pd-catalyst used in the polymerization, which was achieved under moderate reaction conditions. Besides the nitrogen atoms, the trivalent phosphorus atoms bearing one lone pair of electrons and the pentavalent phosphorus atoms of the phosphoryl groups were used as the linkers for the formation of the frameworks of the porous polymers Pd@NP and Pd@NPO, respectively. The phosphorus atoms with different oxidation states in the networks caused a dramatic variation in the physical and catalytic properties of the as-prepared porous polymers. Pd@NP and Pd@NPO exhibit surface areas of 381 m2 g−1 and 684 m2 g−1, respectively. Both Pd-loaded porous polymers enable efficient Suzuki–Miyaura coupling reactions featuring short reaction times and good yields, with the catalysts being highly stable and easy to recycle. The catalytic activity of Pd@NPO is higher than that of Pd@NP.
Introduction
During the past decades, carbon–carbon and carbon–heteroatom coupling reactions have made great progress in synthetic organic chemistry. Among the varieties of coupling methods, the Suzuki–Miyaura reaction is one of the most common and efficient methods for the application of carbon–carbon bond formation and it has been extensively investigated.1 However, the practical application of these reactions on a large scale remains a big challenge, typically due to the following three reasons:2,3 (1) expensive catalysts with a requirement for a large excess of ligands; (2) sensitivity of the catalytic systems to air; (3) the difficult recovery of the product as well as the low reusability and recycling of the catalysts. As one of the available strategies of addressing these issues, researchers have successfully combined Pd nanoparticles with various supports, including porous silicon dioxide,4 metal oxides,5 carbon structures,6 covalent organic frameworks (COFs)7 and microporous organic polymers (MOPs),8 to develop heterogeneous catalysts.Microporous organic polymers (MOPs), composed of C, H, O, N and other main group elements, are a new class of porous material with nano-scale porosity and they have attracted tremendous attention due to their porous structures associated with prominent physical properties and potential applications, such as in light harvesting,9 sensing,10 gas separation and storage,11,12 catalysis,8,13 and energy storage and conversion.14 According to different design strategies, MOPs can be separated into the following four kinds: (1) conjugated microporous polymers (CMPs);15,16 (2) hyper-cross-linked polymers (HCPs);17,18 (3) covalent microporous organic frameworks (COFs);19,20 (4) polymers of intrinsic microporosity (PIMs).21,22 Conjugated microporous polymers (CMPs) represent one of the fastest developing types of porous materials because of their outstanding properties of good thermal and chemical stability, high surface area and well-defined porosity.15 In particular, some CMPs which contain functional units such as bipyridines,23 metalloporphyrins,24,25 and triazine rings26,27 have been used to construct heterogeneous catalytic systems by loading Pd nanoparticles onto the networks in one or two steps, which is much more effective and controllable than those of traditional methods in the other porous supports.28–30 Notably, phosphorus (P), as a light element with facile chemical modification, has been widely used for tuning the photophysical and electrochemical properties of functional materials, such as, organic light-emitting diodes (OLEDs)31 and electron transport materials (ETMs),32 and also constructing various chelated ligands.33 Very recently, several P-containing porous polymers have been successfully achieved.34,35 In these reports, the phosphorus atom acts as either a linker for the formation of the cross-linked network, a functional moiety to achieve phosphorescent emitters36 or as a ligand of heterogeneous catalysts.37 The phosphorus atom, with different valence states, has been used for tuning the luminescence properties of a functional material. However, as far as we know, it has been used less for affecting the structural characteristics of a porous material by changing the valence states of the heteroatoms (e.g. P) in the network. In this paper, we have successfully developed an effective approach to synthesize two novel Pd-loaded microporous polymers by a one-pot palladium-catalyzed Heck cross-coupling reaction of tris(4-vinylphenyl)amine with either tris(4-bromophenyl)phosphine or tris(4-bromophenyl)phosphine oxide under mild experimental conditions. This method of combining the palladium catalyst with the in situ catalyzed polymerization, enables the confinement of the nascent Pd particles in the polymer networks. The two kinds of porous polymers exhibit remarkably different thermal stabilities and porous structures. They exhibit high activities for catalyzing Suzuki–Miyaura coupling reactions, even in aerobic conditions, and they can be recycled and reused for at least three cycles without significant deactivation. Their porous structures also offer a steric influence for size-selectively catalyzing substances with different molecular sizes. The differences in the properties of the as-prepared polymers can mainly be attributed to the phosphorus atoms with different valance states in the networks.
Experimental section
Reagents and chemicals
Triphenylamine, 1,4-dibromobenzene, phosphorus oxychloride, methyl phosphonium iodide, NaH, and n-butyllithium were purchased from Aladdin and Aldrich. All solvents were dried before use. N,N-Dimethylformamide (DMF) was dried with CaH2 and then distilled under reduced pressure before use. Tetrahydrofuran (THF) was refluxed with sodium. All air-sensitive reactions were carried out in a nitrogen atmosphere and performed with Schlenk techniques. The key building blocks of tris(4-bromophenyl)phosphine,31 tris(4-formylphenyl)amine,38 tris(4-vinylphenyl)amine,38 and tris(4-bromophenyl)phosphine oxide39 were synthesized by modified protocols of the previous reports.The Pd-loaded porous polymers were prepared via a typical Pd-catalyzed Heck cross-coupling polymerization.
Synthesis of the porous polymer Pd@N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) P
P
Tris(4-vinylphenyl)amine (126.0 mg, 0.39 mmol), tris(4-bromophenyl)phosphine (193.4 mg, 0.39 mmol), and K2CO3 (357.2 mg, 2.73 mmol) were used in a mixture of anhydrous DMF (10 mL). The reaction mixture was treated three times by freezing and thawing, and then tetrakis(triphenylphosphine)palladium(0) (16.5 mg, 0.015 mmol) was added. The combined mixture was treated two times by freezing and thawing again, and then it was heated up and stirred at 120 °C for 72 h under a nitrogen atmosphere. After the reaction mixture was cooled to room temperature, the resulting precipitate was filtered and collected to afford the crude product which had a yellow colour. According to a typical workup for a porous polymer, the crude product was sequently washed with water, chloroform, methanol, and acetone several times and then treated by Soxhlet extraction with THF for 24 h and dried under vacuum at 80 °C for 4 h to give Pd@NP as a yellow solid (145 mg, 83%).
Synthesis of the porous polymer Pd@NPO
Using the reactants tris(4-vinylphenyl)amine (126.0 mg, 0.39 mmol), tris(4-bromophenyl)phosphine oxide (199.6 mg, 0.39 mmol), K2CO3 (357.2 mg, 2.73 mmol) and tetrakis(triphenylphosphine)palladium(0) (16.5 mg, 0.015 mmol) in 10 mL of DMF, a similar procedure as mentioned above was performed to give Pd@NPO as a yellow solid (156 mg, 88%).
Catalytic properties of the as-prepared porous polymers
As an example, a typical procedure is presented: to a reaction tube boronic acid (1.2 mmol, 1.2 equiv.), potassium carbonate (276.0 mg, 2 mmol, and 2.0 equiv.), aryl bromide or aryl chloride (1.0 mmol, 1.0 equiv.), Pd@NP (0.1 mol% Pd based on arylbromide), ethanol (6 mL) and water (2 mL) were added and then tube was sealed with a Teflon screw cap. All these additions were done in air, and the reaction system did not undergo any extra treatment, such as degassing. The reaction mixture was stirred in a preheated oil bath (70 °C). The reaction progress was monitored by TLC analysis. After cooling down to room temperature, the product was extracted into dichloromethane and washed with water to remove any potassium carbonate. Further purification was done via column chromatography over silica gel to afford the desired cross-coupling products. In recycling tests, the catalyst was simply washed with dichloromethane, water, and ethanol and then dried, it was then used again directly after each cycle.
Characterization
Fourier transform infrared (FTIR) spectra were recorded with a Spectrum 100 spectrometer (Perkin Elmer, Spectrum 100). X-ray powder diffraction patterns were recorded with a transmission geometry using a Rigaku X-ray diffractometer with Cu–Kα irradiation (λ = 0.15406 nm) at 40 kV, 20 mA over the 2θ range from 5 to 60°. Thermogravimetric analysis (TGA) was measured by a TAQ5000IR with a heating rate of 20 °C min−1 under flowing N2. X-ray photoelectron spectroscopy (XPS) experiments were carried out on an AXIS Ultra DLD system from Kratos with Al Kα radiation as the X-ray source for the radiation. Solid-state 13C CPMAS NMR was conducted on a Bruker AVANCE III 300 Spectrometer. Samples were spun at 5 kHz in 4 mm zirconium rotor within a MAS probe. An acquisition time of 20 ms, a contact time of 1 ms and a 6.5 μs pre-scan delay were used. The recycle time was 2 s to obtain fully relaxed spectra. Chemical shifts were externally referenced to adamantane at 38.48 ppm. Elemental analysis was carried out using inductively coupled plasma emission spectrometer (ICP) analyses on an iCAP 6000 Radial. The fluorescence spectroscopy emission spectra were obtained with a FluoroMax-4 spectrophotometer. SEM measurements were performed on a FEI Sirion-200 field emission scanning electron microscope. Transmission electron microscopy (TEM) characterizations were conducted using a JEM-2100 (JEOL Ltd, Japan) with an accelerating voltage of 200 KV. Nitrogen sorption isotherm measurements were performed on a Micromeritics ASAP 2010 surface area and pore size analyzer at 77 K. Prior to the measurement, the samples were degassed in a vacuum at 200 °C for 12 h.Results and discussion
The synthetic routes are shown in Scheme 1. Tris(4-vinylphenyl)amine was polymerized with either tris(4-bromophenyl)phosphine or tris(4-bromophenyl)phosphine oxide using a typical Pd-catalyzed Heck cross-coupling reaction to form a yellow coloured precipitate, which was washed with water, chloroform, methanol, and acetone and further purified with THF to afford the porous polymers Pd@NP (yield: 83%) and Pd@NPO, (yield: 88%), respectively. The components of the resulting porous polymer samples were analyzed by inductively coupled plasma (ICP) analysis, showing the presence of the Pd element in the networks, with a content of 1.0 wt% and 0.89 wt% for Pd@NP and Pd@NPO, respectively. The as-prepared polymer samples were almost insoluble in common organic solvents, e.g. toluene, tetrahydrofuran, dimethylformamide, dichloromethane, methanol, acetone etc.
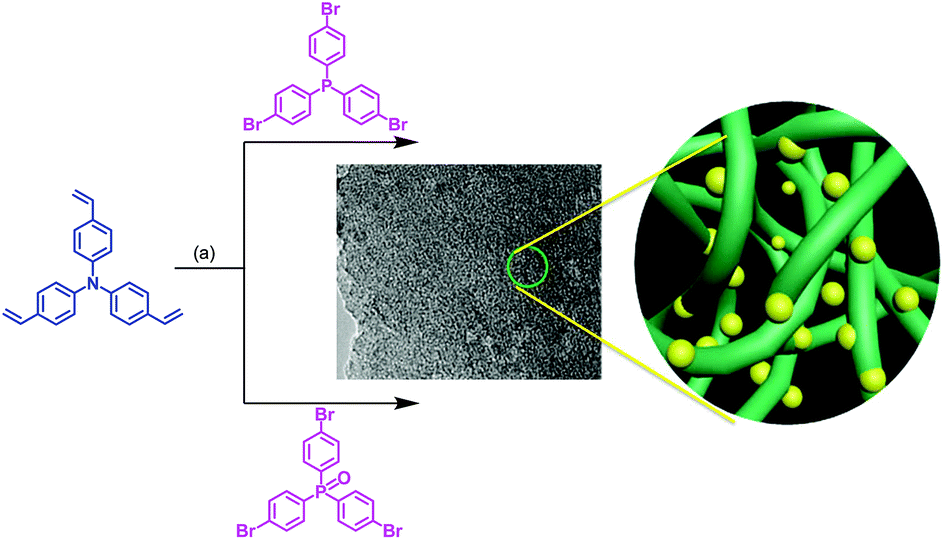 | ||
Scheme 1 Synthetic procedure for the porous polymers Pd@NP and Pd@NPO where (a) is K2CO3, Pd(PPh3)4, DMF, 120 °C, and 72 h ( : porous framework; : porous framework;  : Pd particle). : Pd particle). | ||
The chemical structures of the polymers were confirmed by 13C solid-state and 31P solid-state nuclear magnetic resonance (NMR) spectroscopy. In the solid-state 13C CP/MAS NMR spectra (Fig. 1a), the resonance of the terminal vinyl carbon atom of the tris(4-vinylphenyl)amine monomer at about 114 ppm was not detected for Pd@NP or Pd@NPO. The peak at approximately 148.0 ppm was attributed to the quaternary N–Car sites for both porous polymers.40 Two signals at approximately 130.0 ppm and 147 ppm for the two polymers corresponded to the carbon atoms of the ethylene Car–CHCH–Car units and the Car–P sites, respectively.41 In the solid-state 31P CP/MAS NMR spectra (Fig. 1b), for Pd@NPO, a signal at 28.5 ppm arose from the –PO moiety. While, in the case of Pd@NP, two signals at about 25.3 ppm and −7.0 ppm were observed, corresponding to quaternary phosphonium and tertiary phosphine atoms, respectively.42,43 These results indicate that a high degree of polymerization has been achieved by using the Pd-catalyzed Heck cross-coupling reaction.
 | ||
| Fig. 1 (a) 13C CP-MAS NMR spectra of the Pd@NP and Pd@NPO. (b) 31P CP-MAS NMR spectra of the Pd@NP and Pd@NPO (* attributed to side bands43). | ||
For comparison, the Fourier transform infrared (FTIR) spectra of tris(4-vinylphenyl)amine, Pd@NP and Pd@NPO were collected and are shown in Fig. 2a. The characteristic mono-substituted terminal CC vibration peak appears at about 910 cm−1 for the tris(4-vinylphenyl)amine.9 While for Pd@NP and Pd@NPO, the peak at about 910 cm−1 disappeared, and a low-intensity peak at around 962 cm−1 was observed, which is the characteristic vibration peak of the bis-substituted CC, indicating the complete consumption of the tris(4-vinylphenyl)amine monomer after the Heck cross-coupling reaction. The structures and thermal stabilities of the polymers were examined using X-ray diffraction (XRD) and thermogravimetric analysis, respectively. The XRD of Pd@NP and Pd@NPO indicates amorphous morphologies, which are typical for porous polymers formed through metal-catalyzed coupling reactions (Fig. 2b). Thermogravimetric analysis (TGA) (Fig. 2c) under a nitrogen atmosphere disclosed that both Pd@NP and Pd@NPO exhibit a high thermal stability with only a 5% weight loss at 400 and 550 °C, respectively, which is the same as the previously reported P-containing porous polymers.37 The good thermal stabilities for these types of porous polymers could be reasonably assigned to their conjugated aromatic frameworks. The elemental components of the Pd@NP and Pd@NPO samples were further examined by mapping. Of note, the elemental mapping images show uniform distributions of carbon, phosphorus, and palladium elements in the network of Pd@NP (Fig. 3 and S2†) and Pd@NPO (Fig. S3 and S4†).
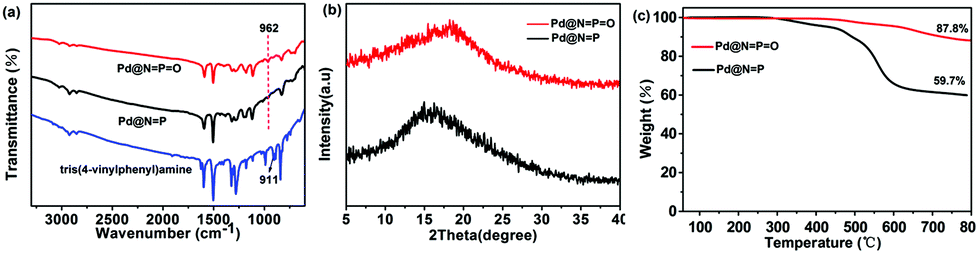 | ||
| Fig. 2 Characterization of Pd@NP and Pd@NPO: (a) FTIR spectra compared with tris(4-vinylphenyl)amine; (b) XRD patterns and (c) TGA curves. | ||
 | ||
| Fig. 3 (a) Typical scanning electron microscopy (SEM) image of Pd@NP and the corresponding elemental mapping images of (b) carbon, (c) phosphorus, and (d) palladium in the selected area. | ||
The porous structures of Pd@NP and Pd@NPO were measured by nitrogen adsorption–desorption isotherm measurements at 77 K (Fig. 4a). The nitrogen adsorption and desorption isotherms revealed that both porous polymers exhibit type II isotherms according to the IUPAC classification. Pd@NP possesses a much lower surface area (Brunauer–Emmett–Teller (BET) value: 381 m2 g−1; Langmuir surface area: 481 m2 g−1) and a smaller total pore volume (0.239 cm3 g−1) than Pd@NPO (BET value: 684 m2 g−1; Langmuir surface area: 702 m2 g−1; total pore volume: 0.342 cm3 g−1). The pore size distributions of Pd@NP and Pd@NPO were calculated using nonlocal density functional theory (NLDFT). As shown in Fig. 4b, the pore size distribution of Pd@NP mainly appears at 0.98 nm and, approximately, at 0.52 nm. Besides the peak at ∼0.91 nm, remarkably intensive peaks were also recorded at 0.45 nm and 1.67 nm for Pd@NPO. These compare with most of the reported CMPs, COFs, and MOFs which have small pores of less than 2 nm. The phosphorus(III) linker, bearing one lone pair of electrons, projects toward the trigonal pyramid structure, and thus likely endows the network of Pd@NP with less rigidity. Meanwhile, the phosphorus(V) atom of the phosphoryl group in the network of Pd@NPO adopts a tetrahedral geometrical structure with an increased rigidity, which would be favorable for the formation of a much more spacious scaffold.31,39 On the other hand, in comparison with the building block tris(4-bromophenyl)phosphine, tris(4-bromophenyl)phosphine oxide, with a stronger electron-withdrawing phosphoryl group, offers an enhanced polarity.44 These structural and electronic characteristics of the building blocks might benefit an increased degree of polymerization for Pd@NPO. Hence, a much better thermal stability and a pronounced increase in the surface area and pore size were achieved for Pd@NPO in comparison to those of Pd@NP. Moreover, the morphologies and microstructures of Pd@NP and Pd@NPO were investigated by transmission electron microscopy (TEM) (Fig. 5). The alternating areas of light and dark contrast in the TEM images revealed their disordered porous structural natures.
 | ||
| Fig. 4 (a) Nitrogen adsorption–desorption analysis of the porous polymers; (b) pore size distribution of the porous polymers. | ||
 | ||
| Fig. 5 TEM images of (a) Pd@NP and (b) Pd@NPO. | ||
In order to gain insight into the structural information, in particular the state of the palladium element in the networks of these polymers, X-ray photoelectron spectroscopy (XPS) measurements were performed. They revealed two broad peaks at 336.8 eV and 342.1 eV, as displayed in Fig. S4,† suggesting the existence of both Pd(0) and Pd(II) in Pd@NP and Pd@NPO.45 On the basis of XPS analysis, the content of the Pd element in both of porous polymers can be evaluated as 1.10 wt% and 0.97 wt% for Pd@NP and Pd@NPO, respectively, which are consistent with those values from the aforementioned ICP analysis.
Pd-based catalysts have been extensively used in homogeneous systems for the formation of carbon–carbon or carbon–heteroatom bonds.46,47 Owing to Pd being a high-cost, scarce resource, the development of reusable and recyclable Pd catalysts, for example, for heterogeneous Pd-based catalysis, is desirable when taking environmental, economic and safety factors into account. In this respect, Pd@NP and Pd@NPO were examined as catalysts for the Suzuki–Miyaura coupling reaction. All reactions were directly performed under aerobic conditions (in water/EtOH, with a base of K2CO3 at 70 °C, without the addition of extra ligands and degassing treatment of the reaction system, and exposed to air). As an example, with only 0.1 mol% of the Pd@NP catalyst (the content is calculated relative to the aryl bromides), the coupling reaction of phenylboronic acid with substituted aryl bromides can smoothly occur in air, affording coupling products in a nearly quantitative yield within a very short time after a simple workup procedure (entries 1–3 in Table 1) (for the experimental details, see the Experimental section). It only took 1.5 hours for most of the reactions to achieve the coupling products in good yields, making such kinds of Pd-loaded porous polymers among the most effective catalysts for Suzuki cross-couplings.30 The superior catalytic performance of Pd@NP is presumably due to the triarylphosphine units in the microporous structure, which might provide effective binding sites for Pd atoms, and the adsorption of the reactants into the porous channels by capillary condensation of the micropores, which is where the catalytic sites are located. As a comparison, Pd(PPh3)4 (1.3 mg, equal to the amount of the Pd in 10 mg of the Pd@NP above) was seen to catalyze the reaction of phenyl bromide and phenylboronic acid under the same reaction conditions without the addition of extra ligands, affording the cross-coupling product in a yield of only 55.4%. This result might be attributed to the deactivation of the traditional catalyst [Pd(PPh3)4], as a reduced amount of the assistant ligand was used, which seems to be avoided in the heterogeneous catalytic system of the as-prepared Pd-loaded porous polymer. Such a phenomenon was also observed in the other phosphorus-containing porous polymers for catalyzing hydroformylations, which were just reported during the preparation of our manuscript.48 On the other hand, the catalytic properties of the as-prepared porous polymers highly depend on the porous structures, which were elucidated by their catalytic behaviors after using varying substrates. Taking Pd@NP as the catalyst under similar reaction conditions as those aforementioned, the different substrates p-methyl-bromobenzene, p-ethyl-bromobenzene and p-propyl-bromophenyl were reacted with phenylboronic acid, affording the coupling products p-methylbiphenyl, p-ethylbiphenyl and p-propylbiphenyl in yields of 80.4%, 55.2% and 18.9%, respectively (entries 6–8, Table 1). Obviously, increasing the size of the substituent group in the p-position of the substrate directly led to a decline in the yield of the corresponding cross-coupling product as a consequence of the steric effect of the porous structure of Pd@NP. Further, using a phenyl derivative with the substituent group at the m- or o-position as the substrate also resulted in remarkably different yields (entries 4 and 5, Table 1), verifying the spacial effect of the porous network of the as-prepared polymers on the catalytic behaviors. Pd@NPO, with a larger average pore size than Pd@NP, exhibits higher catalytic activities, indicating the space-confinement effect of the porous structure on the reactants. Additionally, the coupling of phenylboronic acid with chlorobenzene only gave a 35.5% yield (entry 14, Table 1), probably due to the sluggish activity of the aryl chloride with respect to that of the aryl bromide under the experimental conditions, which might be improved by optimizing the reaction conditions or performance under an inert atmosphere. In the case of using 4-methoxy-bromobenzene and phenylboronic acid as substrates, the yield of the coupling product is still very high (98%, entry 9, Table 1). However, the coupling reaction between p-bromobenzaldehyde and phenylboronic acid only gave the coupling product with a poor yield of 11%. The reason can probably be attributed to the deactivation of the Pd atoms by the strong polar substrates in the polymer matrix.
P and Pd@NPOa
|
|
|||
|---|---|---|---|
| Entry | R | Catalyst | Yield (%) |
a Reaction conditions: 1 mmol aryl bromide, 1.5 mmol arylboronic acid, 2 mmol K2CO3, 10 mg catalyst, and H2O/EtOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v) at 70 °C for 1–2 h. :3 v/v) at 70 °C for 1–2 h.
|
|||
| 1 | H | Pd@NP/Pd@NPO |
99.2/99.5 |
| 2 | H | Pd@NP/Pd@NPO |
99.0/99.3 |
| 3 | H | Pd@NP/Pd@NPO |
98.6/99.0 |
| 4 | o-CH3 | Pd@NP/Pd@NPO |
60.3/65.3 |
| 5 | m-CH3 | Pd@NP/Pd@NPO |
71.575.5 |
| 6 | p-CH3 | Pd@NP/Pd@NPO |
80.4/89.2 |
| 7 | p-CH2CH3 | Pd@NP/Pd@NPO |
53.2/55.6 |
| 8 | p-CH2CH2CH3 | Pd@NP/Pd@NPO |
18.9/20.7 |
| 9 | p-OCH3 | Pd@NP/Pd@NPO |
97.0/98.0 |
| 10 | o-F | Pd@NP/Pd@NPO |
80.5/82.5 |
| 11 | m-F | Pd@NP/Pd@NPO |
69.7/72.5 |
| 12 | p-F | Pd@NP/Pd@NPO |
85.0/86.0 |
| 13 | p-CHO | Pd@NP/Pd@NPO |
11.0/14.0 |
| 14 | H/XCl |
Pd@NP/Pd@NPO |
35.5/33.8 |
| 15 | H | Pd(PPh3)4 | 55.2 |

The recyclability and reusability of these porous polymers were examined by using phenyl bromide and phenylboronic acid as substrates. The results demonstrated that the yields of the cross-coupling products remained at more than 98% after three continuous runs, and the catalyst could be readily recovered by a simple treatment after each cycle and directly reused in the next run, indicating that such kinds of Pd-loaded porous polymers represent efficient and economic catalytic systems.
To consider if the amount of Pd(PPh3)4 used in the polymerization would have a significant effect on the content of the Pd element in the networks and the porosities of the resulting porous polymers, we also examined the polymerization of tris(4-vinylphenyl)amine with tris(4-bromophenyl)phosphine under 1% mmol and 8% mmol of the Pd(PPh3)4 catalyst, comparing with that of 4% mmol of Pd(PPh3)4. The loading amount of Pd in the resulting polymer samples, confirmed by the inductively coupled plasma (ICP) analysis, was 0.64 wt%, 1.0 wt% and 4.67 wt% with respect to the usage of 1% mmol, 4% mmol and 8% mmol of the Pd(PPh3)4 catalyst for the polymerization. The yields of the porous polymers are also increased when a greater amount of the Pd catalyst was used, which was likely due to the enhancement of the degree of the polymerization. Accordingly, the porous structures of the resulting porous polymers also varied, and the polymer product with the largest surface area was found in the case of 4% mmol of Pd(PPh3)4. These results indicate that the amount of the Pd(PPh3)4 catalyst used for the polymerization has a significant effect on the characters of the resulting polymers, including the content of Pd element loaded into the networks (Fig. S5 and Table S1†).
Conclusions
In summary, the Pd-loaded microporous polymers Pd@NP and Pd@NPO, with P(III) atoms bearing one lone pair of electrons and the P(V) atoms of the phorsphoryl groups as linkers in the networks, respectively, were effectively synthesized by one-pot Pd-catalyzed Heck coupling reactions. The Pd-catalyst used for the in situ polymerization of the two porous polymers serves as the source of Pd atom which is uniformly embedded in the network of these polymers. Pd@NPO exhibits a much higher thermal stability, larger surface area and pore size than Pd@NP. These polymers enable the effective catalysis of the Suzuki cross-coupling reaction under aerobic conditions. The activities of the as-prepared porous polymers exhibit pronounced size-selection properties, confined by the spacial effects of the porous structures of the networks. They showed excellent catalyst recyclability and reusability. The catalytic activity of Pd@NPO is much higher than that of Pd@NP. The phosphorus linkers with different valence states in the networks play a crucial role in the different structures and properties of the two porous polymers, which will be explored for building new material systems applicable for transforming organic substrates to much more complex structures with a wide scope.
Acknowledgements
This work was financially supported by the National Basic Research Program of China (973 Program: 2013CBA01602, 2012CB933404), the Natural Science Foundation of China (21174083, 51403126 and 21102091), and the Shanghai Committee of Science and Technology (15JC1490500).Notes and references
- F. S. Han, Chem. Soc. Rev., 2013, 42, 5270 RSC.
- Y. Xiong, B. J. Wiley and Y. Xia, Angew. Chem., Int. Ed., 2007, 46, 7157 CrossRef CAS PubMed.
- M. Lamblin, L. Nassar-Hardy, J. C. Hierso, E. Fouquet and F.-X. Felpin, Adv. Synth. Catal., 2010, 352, 33 CrossRef CAS PubMed.
- R. B. Bedford, U. G. Singh, R. I. Walton, R. T. Williams and S. A. Davis, Chem. Mater., 2005, 17, 701 CrossRef CAS.
- B. M. Choudary, S. Madhi, N. S. Chowdari, M. L. Kantam and B. Sreedhar, J. Am. Chem. Soc., 2002, 124, 14127 CrossRef CAS PubMed.
- Y. Kitamura, S. Sako, T. Udzu, A. Tsutsui, T. Maegawa, Y. Monguchi and H. Sajiki, Chem. Commun., 2007, 5069, 10.1039/B712207A.
- S. Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816 CrossRef CAS PubMed.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819 CrossRef CAS.
- L. Sun, Z. Liang, J. Yu and R. Xu, Polym. Chem., 2013, 4, 1932 RSC.
- L. Sun, Y. Zou, Z. Liang, J. Yu and R. Xu, Polym. Chem., 2014, 5, 471 RSC.
- R. Dawson, E. Stockel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239 CAS.
- W. C. Song, X. K. Xu, Q. Chen, Z. Z. Zhuang and X. H. Bu, Polym. Chem., 2013, 4, 4690 RSC.
- Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083 RSC.
- F. Vilela, K. Zhang and M. Antonietti, Energy Environ. Sci., 2012, 5, 7819 CAS.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012 RSC.
- S. Ren, R. Dawson, A. Laybourn, J. X. Jiang, Y. Khimyak, D. J. Adams and A. I. Cooper, Polym. Chem., 2012, 3, 928 RSC.
- L. J. Abbott and C. M. Colina, Macromolecules, 2014, 47, 5409 CrossRef CAS.
- M. Seo, S. Kim, J. Oh, S. J. Kim and M. A. Hillmyer, J. Am. Chem. Soc., 2015, 137, 600 CrossRef CAS PubMed.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548 RSC.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010 RSC.
- B. S. Ghanem, Polym. Chem., 2012, 3, 96 RSC.
- J. Vile, M. Carta, C. G. Bezzu and N. B. McKeown, Polym. Chem., 2011, 2, 2257 RSC.
- P. Arab, A. Verlander and H. M. El-Kaderi, J. Phys. Chem. C, 2015, 119, 8174 CAS.
- A. M. Shultz, O. K. Farha, J. T. Hupp and S. T. Nguyen, Chem. Sci., 2011, 2, 686 RSC.
- X. Liu, H. Li, Y. Zhang, B. Xu, S. A. H. Xia and Y. Mu, Polym. Chem., 2013, 4, 2445 RSC.
- A. Modak, M. Pramanik, S. Inagaki and A. Bhaumik, J. Mater. Chem. A, 2014, 2, 11642 CAS.
- A. Bhunia, V. Vasylyeva and C. Janiak, Chem. Commun., 2013, 49, 3961 RSC.
- S. Kidambi and M. L. Bruening, Chem. Mater., 2004, 17, 301 CrossRef.
- F. Wang, J. Mielby, F. H. Richter, G. Wang, G. Prieto, T. Kasama, C. Weidenthaler, H. J. Bongard, S. Kegnaes, A. Furstner and F. Schuth, Angew. Chem., Int. Ed., 2014, 53, 8645 CrossRef CAS PubMed.
- Q. Song, Y. Jia, B. Luo, H. He and L. Zhi, Small, 2013, 9, 2460 CrossRef CAS PubMed.
- C. Liu, Y. Li, Y. Li, C. Yang, H. Wu, J. Qin and Y. Cao, Chem. Mater., 2013, 25, 3320 CrossRef CAS.
- S. O. Jeon and J. Y. Lee, J. Mater. Chem., 2012, 22, 4233 RSC.
- T. Li, S. Kaercher and P. W. Roesky, Chem. Soc. Rev., 2014, 43, 42 RSC.
- P. J. C. Hausoul, T. M. Eggenhuisen, D. Nand, M. Baldus, B. M. Weckhuysen, R. J. M. Klein Gebbink and P. C. A. Bruijnincx, Catal. Sci. Technol., 2013, 3, 2571 CAS.
- S. Qiao, W. Huang, Z. Du, X. Chen, F. K. Shieh and R. Yang, New J. Chem., 2015, 39, 136 RSC.
- Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943 RSC.
- Q. Zhang, Y. Yang and S. Zhang, Chem. – Eur. J., 2013, 19, 10024 CrossRef CAS PubMed.
- P. Taranekar, Q. Qiao, H. Jiang, I. Ghiviriga, K. S. Schanze and J. R. Reynolds, J. Am. Chem. Soc., 2007, 129, 8958 CrossRef CAS PubMed.
- Y. Wang, M. I. Ranasinghe and T. Goodson, J. Am. Chem. Soc., 2003, 125, 9562 CrossRef CAS PubMed.
- J. X. Jiang, A. Trewin, F. Su, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, Macromolecules, 2009, 42, 2658 CrossRef CAS.
- Q. Zhang, S. Zhang and S. Li, Macromolecules, 2012, 45, 2981 CrossRef CAS.
- S. L. Qiao, W. Huang, Z. K. Du, X. H. Chen, F. K. Shiehc and R. Q. Yang, New J. Chem., 2015, 39, 136 RSC.
- P. J. C. Hausoul, T. M. Eggenhuisen, D. Nand, M. Baldus, B. M. Weckhuysen, R. J. M. K. Gebbink and P. C. A. Bruijnincx, Catal. Sci. Technol., 2013, 3, 2571 CAS.
- X. Chen, S. Qiao, Z. Du, Y. Zhou and R. Yang, Macromol. Rapid Commun., 2013, 34, 1181 CrossRef CAS PubMed.
- Y. Dai, S. Liu and N. Zheng, J. Am. Chem. Soc., 2014, 136, 5583 CrossRef CAS PubMed.
- H. Li, C. C. C. Johansson Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147 CrossRef CAS.
- N. Kambe, T. Iwasaki and J. Terao, Chem. Soc. Rev., 2011, 40, 4937 RSC.
- Q. Sun, Z. Dai, X. Liu, N. Sheng, F. Deng, X. Meng and F. S. Xiao, J. Am. Chem. Soc., 2015, 137, 5204 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py00576k |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |