
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoparticles of the poly([N-(2-hydroxypropyl)]methacrylamide)-b-poly[2-(diisopropylamino)ethyl methacrylate] diblock copolymer for pH-triggered release of paclitaxel†
Alessandro
Jäger
*ae,
Eliézer
Jäger
*ae,
František
Surman
a,
Anita
Höcherl
a,
Borislav
Angelov
a,
Karel
Ulbrich
a,
Markus
Drechsler
b,
Vasil M.
Garamus
c,
Cesar
Rodriguez-Emmenegger
*a,
Frédéric
Nallet
d and
Petr
Štěpánek
a
aInstitute of Macromolecular Chemistry v.v.i., Academy of Sciences of the Czech Republic, Heyrovsky Sq. 2, 162 06 Prague 6, Czech Republic. E-mail: ajager@imc.cas.cz; jager@imc.cas.cz; rodriguez@imc.cas.cz
bLaboratory for Soft Matter Electron Microscopy, Bayreuth Institute of Macromolecular Research, University of Bayreuth, D-95440 Bayreuth, Germany
cHelmholtz-Zentrum Geesthacht, Centre for Materials and Coastal Research, D-21502 Geesthacht, Germany
dCentre de Recherche Paul-Pascal, CNRS, Université de Bordeaux, 115 Avenue Schweitzer, F-33600 Pessac, France
eDepartment of Physical and Macromolecular Chemistry, Faculty of Science, Charles University in Prague, Albertov 2030, 128 40 Prague 2, Czech Republic
First published on 1st June 2015
Abstract
The potential of self-assembled nanoparticles (NPs) containing the fine tunable pH-responsive properties of the hydrophobic poly[2-(diisopropylamino)ethyl methacrylate] (PDPA) core and the protein repellence of the hydrophilic poly[N-(2-hydroxypropyl)methacrylamide] (PHPMA) shell for in vitro cytostatic activity has been explored on cancer cells. The amphiphilic diblock copolymer poly[N-(2-hydroxypropyl)methacrylamide]-b-poly[2-(diisopropylamino)ethyl methacrylate] (PHPMA-b-PDPA) synthesized by a reversible addition–fragmentation chain transfer (RAFT) technique allows for excellent control of the polymer chain length for methacrylamides. The PHPMA-b-PDPA block copolymer dissolved in an organic solvent (ethanol/dimethylformamide) undergoes nanoprecipitation in phosphate buffer saline (PBS, pH ∼ 7.4) and self-assembles into regular spherical NPs after solvent elimination. The NPs’ structure was characterized in detail by dynamic (DLS), static (SLS) and electrophoretic (ELS) light scattering, small angle X-ray scattering (SAXS), and cryo-transmission electron microscopy (cryo-TEM). The PHPMA chains prevented the fouling of proteins resulting in a remarkable stability of the NPs in serum. On decreasing pH the hydrophobic PDPA block becomes protonated (hydrophilised) in a narrow range of pH (6.51 < pH < 6.85; ΔpH ∼ 0.34) resulting in the fast disassembly of the NPs and chemotherapeutic drug release in a simulated acidic environment in endosomal and lysosomal compartments. A minimal amount of drug was released above the threshold pH of 6.85. The in vitro cytotoxicity studies showed an important increase in the activity of the NPs loaded with drug compared to the free drug. The particle's size below the cut-off size of the leaky pathological vasculature (less than 100 nm), the excellent stability in serum and the ability to release a drug at the endosomal pH with concomitant high cytotoxicity make them suitable candidates for cancer therapy, namely for treatment of solid tumours exhibiting high tumor accumulation of NPs due to the Enhanced Permeability and Retention (EPR) effect.
Introduction
A great deal of attention has been paid to block copolymers that when exposed to an aqueous environment self-assemble into highly organised nanoscale structures.1,2 By changing the solvent selectivity or the insoluble-to-soluble block length ratio, one obtains thermodynamically favoured structures of various morphologies in solution.3 Typical morphologies such as spheres, rods, lamellae and vesicles (polymersomes) can be easily prepared in a controlled manner in aqueous solutions through the self-assembly of asymmetric amphiphilic diblock copolymers.4 Characterised by a bulky core and a relatively thin corona, the particles made by the self-assembly of highly asymmetric diblocks are frozen structures and do not dissociate under dilution.5 Some of these structures such as spheres and vesicles have attracted attention as versatile carriers for drug delivery.6,7 They present improved colloidal stability under physiological conditions when compared to water soluble amphiphilic micelles and possess the ability to encapsulate or integrate a broad range of drugs. However, several problems arise related to the slow drug release if the drug is encapsulated in frozen colloidal particles, especially for applications such as drug release in cancer therapy. For an optimal therapy the hydrophobic guest drug molecules have to be seized into the inner particle core during circulation in the bloodstream and be quickly released at the specific tumour sites.8 Recently, several types of pH-sensitive polymeric NPs have been investigated for such purposes9,10 because the extracellular pH in most solid tumour tissues is more acidic (ranging from pH 6.7 to 7.1),11,12 compared to the normal tissues (buffered at pH 7.4). Furthermore, variations in pH are also encountered when nanoparticles are internalized by cells via endocytosis, where the pH usually drops to as low as 5.0–6.0 in endosomes and 4.0–5.0 in lysosomes.13For such a challenging task, taking into account the small pH window (ΔpH ∼ 0.4) in which the drug must be released, the 2-(diisopropylamino)ethyl methacrylate (DPA) based copolymers have recently proven to be valuable tools capable of fine tuning the release in the desired pH region, both in vitro14 and in vivo15 and quickly release the drug independently of its hydrophobicity16 or hydrophilicity.17 Nevertheless, regardless of the efficiency of the trigger mechanism involved in the drug release towards a successful cancer therapy, the nanocarrier must reach the tumor tissue first. However, nanoparticles in contact with the blood stream are rapidly coated with a corona of proteins which can impair the colloidal stability or can cause the particles to be eliminated via phagocytosis. This can only be circumvented by engineering the nanocarrier system to be “stealth” with a low particle size (between 10 and 100 nm) and having a sufficiently long circulation time to undergo passive specific accumulation in the tumor tissue through the EPR effect.18 Recently published results from our group have demonstrated that unlike poly(ethylene glycol) PEG,19 brushes of poly[N-(2-hydroxypropyl)methacrylamide] (PHPMA) withstand the fouling from blood plasma20 and full blood.21 The unmatched performance of PHPMA was also utilized to improve the circulation of hydrophobic nanoparticles with PHPMA-modified surfaces in vitro22 and in vivo.23
Herein, for the first time, the potential of self-assembled NPs containing the fine tunable pH-responsive properties of the hydrophobic poly[2-(diisopropylamino)ethyl methacrylate] (PDPA) core and protein repellence of the hydrophilic PHPMA shell on in vitro cytostatic activity tested on cancer cells are reported. A diblock copolymer composed of hydrophobic PDPA and hydrophilic PHPMA was prepared by RAFT polymerisation. Poly[N-(2-hydroxypropyl)methacrylamide]-b-poly[2-(diisopropylamino)ethyl methacrylate] (PHPMA25-b-PDPA106) assembled forming nanoparticles. The novel NPs’ self-assembly and structure were characterized in detail by cryo-TEM, SAXS, DLS, SLS and ELS light scattering techniques. Acidic physiological conditions trigger the disassembly of the novel NPs prepared from the block copolymer PHPMA25-b-PDPA106, thus controlling the release of their cargo, the chemotherapeutic paclitaxel (PTX), which results in a high cytostatic effect on HeLa cancer cells in in vitro studies.
Results and discussion
The block copolymer was prepared using RAFT polymerisation. A first block of PHPMA, (Mn = 3600 g mol−1, Mw/Mn = 1.07; Scheme 1, Fig. S1 and S2†) was synthesised and used as a macro chain transfer agent. Subsequently a long second block of PDPA (Mn = 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 g mol−1, Mw/Mn = 1.29) was grown from the macroCTA also by RAFT. RAFT polymerisation accounts for excellent living characteristics in the polymerization of methacrylamides. The block copolymer comprising a hydrophobic block (PDPA) and a hydrophilic block (PHPMA) favours the formation of stable NPs with a bulky core capable of encapsulating and controlling the release of the hydrophobic chemotherapeutic PTX.
200 g mol−1, Mw/Mn = 1.29) was grown from the macroCTA also by RAFT. RAFT polymerisation accounts for excellent living characteristics in the polymerization of methacrylamides. The block copolymer comprising a hydrophobic block (PDPA) and a hydrophilic block (PHPMA) favours the formation of stable NPs with a bulky core capable of encapsulating and controlling the release of the hydrophobic chemotherapeutic PTX.
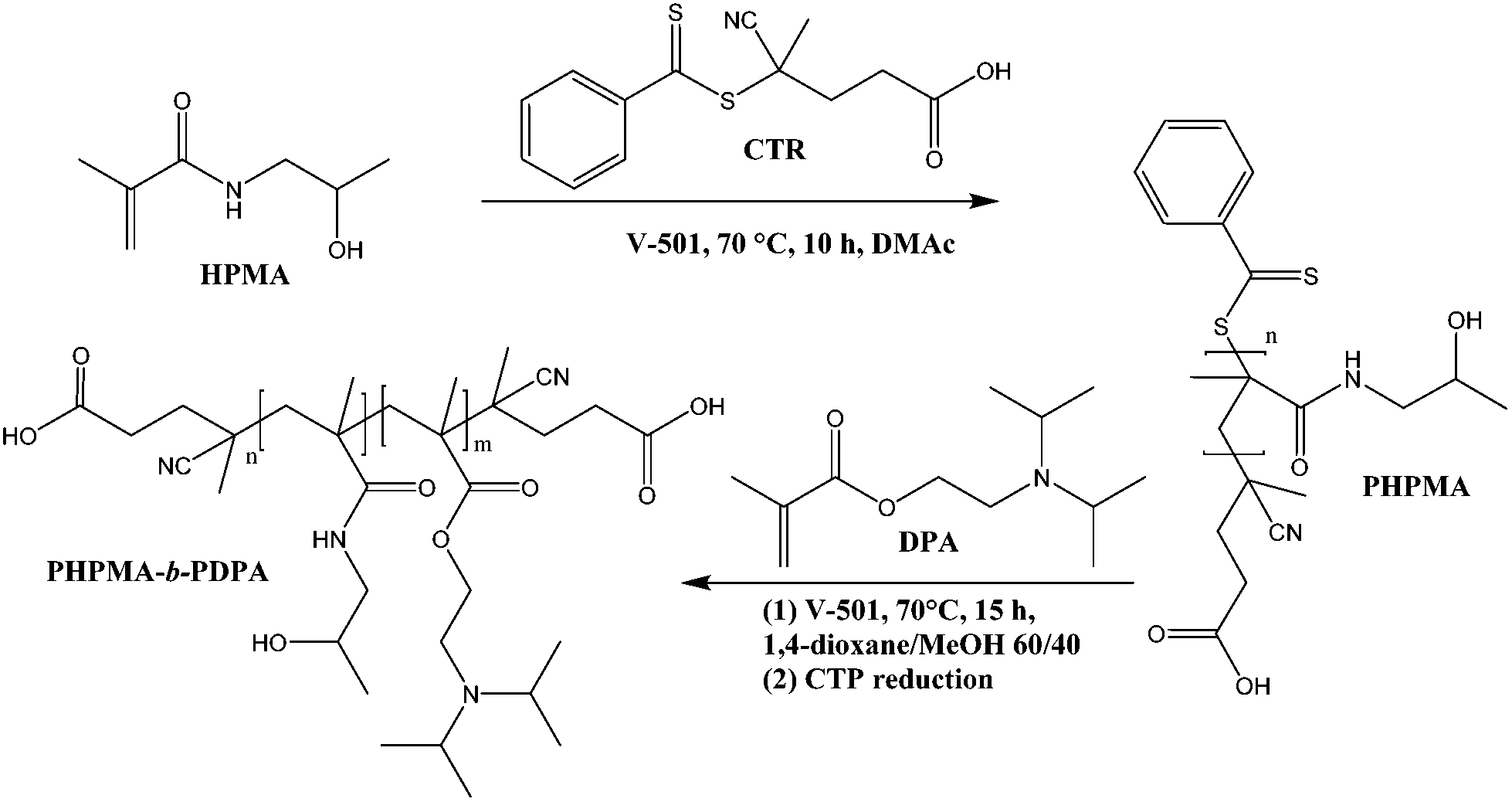 | ||
| Scheme 1 Synthetic route and molecular structure of the PHPMA25-b-PDPA106 block copolymer with pH-triggered assembly/disassembly. | ||
The hydrophilic corona of PHPMA was utilized to protect the hydrophobic core from adsorption of proteins from blood (vide infra). An abrupt change in the light scattering intensity was observed during the titration experiments of the PHPMA25-b-PDPA106 diblock copolymer in aqueous solution simulating physiological conditions. The hydrophobic DPA block is protonated (hydrophilised) in a narrow range of pH (6.51 < pH < 6.85; ΔpH ∼ 0.34, Fig. 1a).
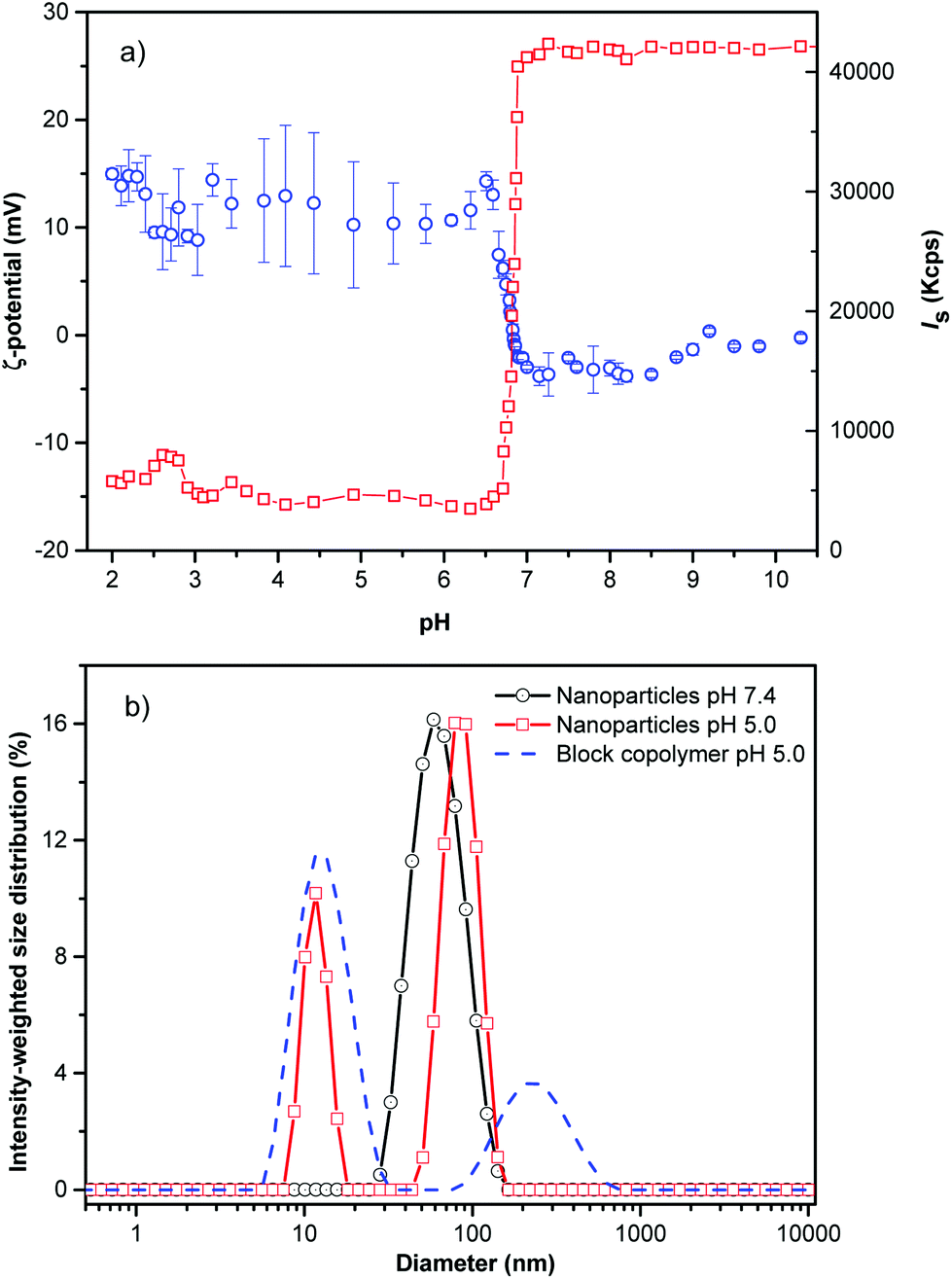 | ||
| Fig. 1 (a) Zeta potential values (black circles) and overall scattering intensity (red squares) as a function of pH for the PHPMA25-b-PDPA106 block copolymer and (b) intensity-weighted size distribution for the PHPMA25-b-PDPA106 NPs at pH 7.4 (black circles), for the nanoparticles at pH 5.0 (red squares) and the single block copolymer at pH 5.0 (blue dashed lines) and 173° angle at a concentration of 1 mg mL−1 diluted in PBS at 37 °C. | ||
The markedly narrow window of pH at which the diblock copolymer (namely DPA block) can be protonated and the large change in scattering intensity and hydrodynamic diameter suggest that pH is a remarkably effective and precise trigger for assembly and disassembly of the system. The NPs were prepared by the solvent-shifting method, whereby the PHPMA25-b-PDPA106 block copolymer was dissolved in ethanol or dimethylformamide (concentration ∼ 1 mg mL−1) and phosphate buffer saline (PBS, pH ∼ 7.4) was added dropwise to the organic solution under stirring until the aqueous weight fraction reached twice the weight of the organic phase. The organic solvent was removed by dialysis or evaporated under reduced pressure. DLS measurement of the diblock copolymer (before NP formation) at pH 5.0 shows that most chains were fully dissolved with an average hydrodynamic diameter of 2RH = 9.0 nm (Fig. 1b).
After PBS addition and solvent evaporation, the DLS shows a monomodal average size distribution referred to as the NPs’ population (2RH = 52 nm, Fig. 1b, black circles) with low dispersity as was estimated by using the cumulant analysis (μ2/Γ2 = 0.120 ± 0.009), and as observed by Cryo-TEM (Fig. 2a). At pH 7.4 (PBS buffer) the average ζ-potential for the studied block copolymer nanoparticles is close to neutrality (∼−2.5 mV) (Fig. 1a). Decreasing the pH below the pKa (DPA) (6.51 < pH < 6.85), the system is rapidly protonated as evidenced by the positive ζ-potential, and mainly composed of molecularly dissolved block copolymer chains, besides a very small number of large aggregates (RH ∼ 125 nm). These aggregates represent about 25% of the intensity-weighted distribution, thus following the relationship Isc ∼ cR3 the number of large particles can be calculated to be 0.0025% of the total number of objects scattering light in the system16 (Fig. 1b). This negligible number of aggregates can be clearly visualized in the volume-weighted distribution of RH shown in Fig. S5.†
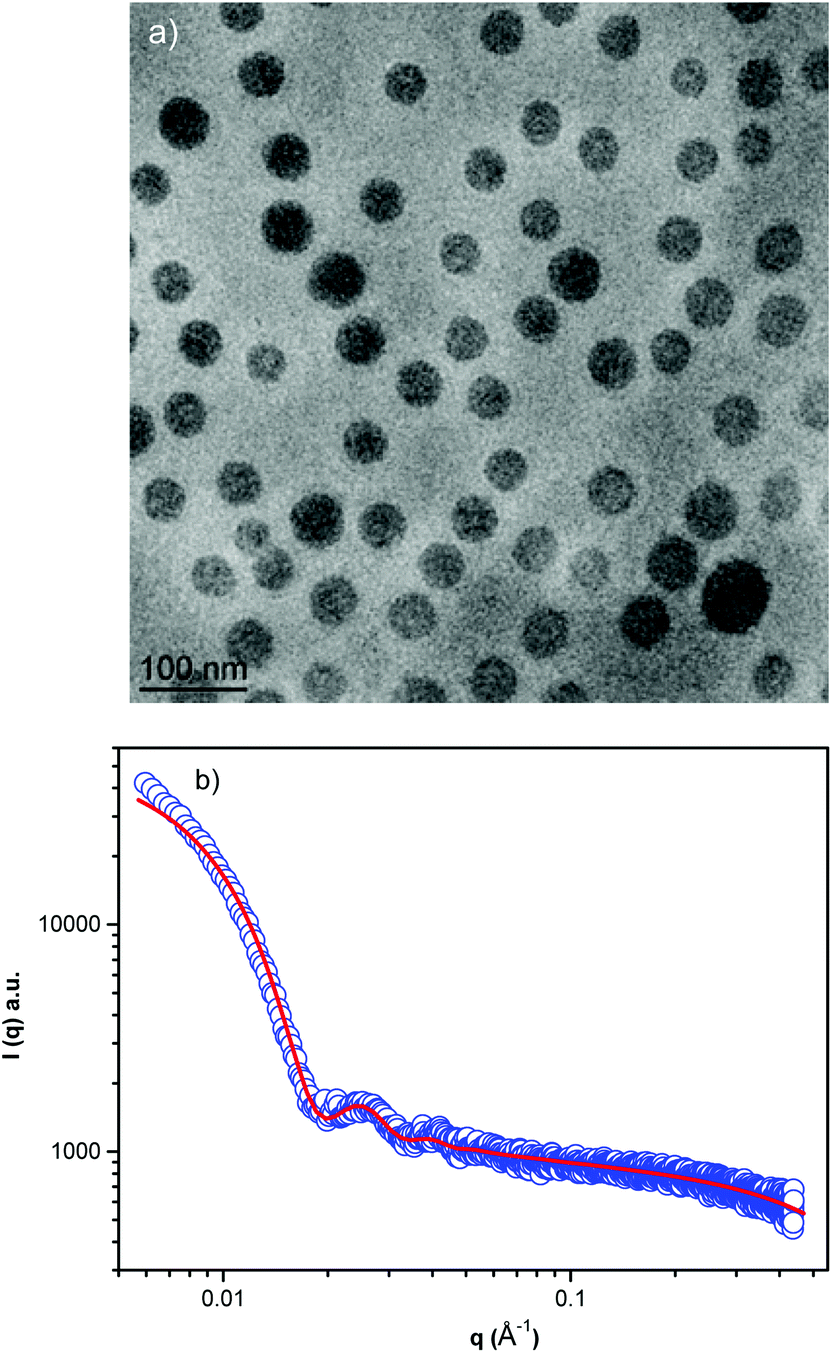 | ||
| Fig. 2 Cryo-TEM image of the block copolymer NPs (a) and (b) small angle X-ray scattering profile in PBS, pH 7.4. | ||
Cryo-TEM images obtained at pH 7.4 reveal well-defined spherical NPs (Fig. 2a). The SAXS profile of the PHPMA25-b-PDPA106 block copolymer NPs (Fig. 2b) indicates the presence of flexible polymer chains described by Gaussian statistics at the outermost layer of the particles as evidenced in the high-q range profile corresponding to the PHPMA chains. These chains are, however, not visible in the Cryo-TEM image (Fig. 2a) due to poor contrast. To model the SAXS profile we utilized a superposition of Gaussian chains as the background and spherical nanoparticles:
| I(q) ∼ K2(q, R, Δη) + background, | (1) |
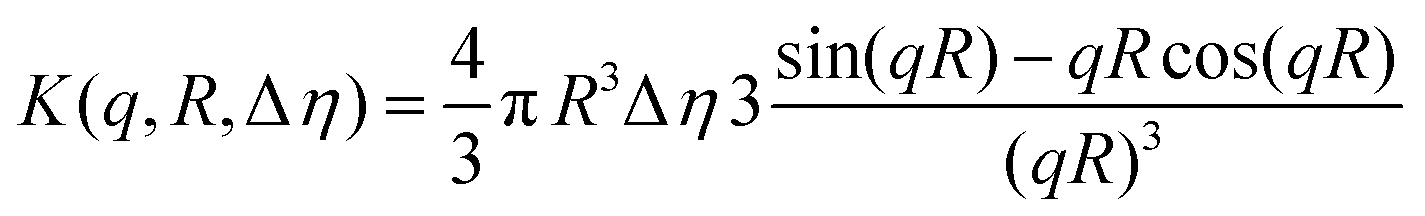 | (2) |
The RG/RH ratio obtained by SLS and DLS data, gave access to the structural characteristics of the particles. It is well established that the NPs’ RG/RH ratio values of 0.775, 1.78, and ≥2 have been reported for hard spheres, random coils and rod-like structures, respectively.16,22,23,26 Furthermore, the RG/RH ratio of spherical objects depends on their inner structure and compactness, being close to 0.775 for compact spheres, 0.8–0.9 for block copolymer micelles due to solvation phenomena and 1.0 for hollow spheres and vesicles.16,23,26 The RG/RH ratio found for the PHPMA25-b-PDPA106 NPs (RG/RH ∼ 0.92) indicates that the particles had a core–shell-like structure.16,27 Therefore, the most probable structural arrangement of the PHPMA25-b-PDPA106 NPs is a particle composed of a PDPA core surrounded by a PHPMA shell. These findings are in agreement with the SAXS measurements, which identify polymer chains with the Gaussian configuration at the outer region of the NPs. Furthermore, aggregation numbers with the same order of magnitude (∼46 chains per particle) were also found for particles with such a structural composition.16,24,27
The efficient NP accumulation in the solid tumor tissue requires an extended circulating capability to enable a time-dependent extravasation of the NPs through the leaky tumor microvasculature (EPR effect). Therefore, long-term stability of the NPs in serum is a pre-requisite for the use of polymer NPs in vivo.23,28,29 As a model of the possible detrimental interaction of the nanoparticles with proteins from blood, we incubated them in human plasma (10% in PBS). The stability of the NPs was monitored by evaluating any possible changes in their hydrodynamic size and scattering intensity over time.23,26,29Fig. 3a shows the temporal stability of the NPs in diluted human blood plasma as a function of the incubation time.
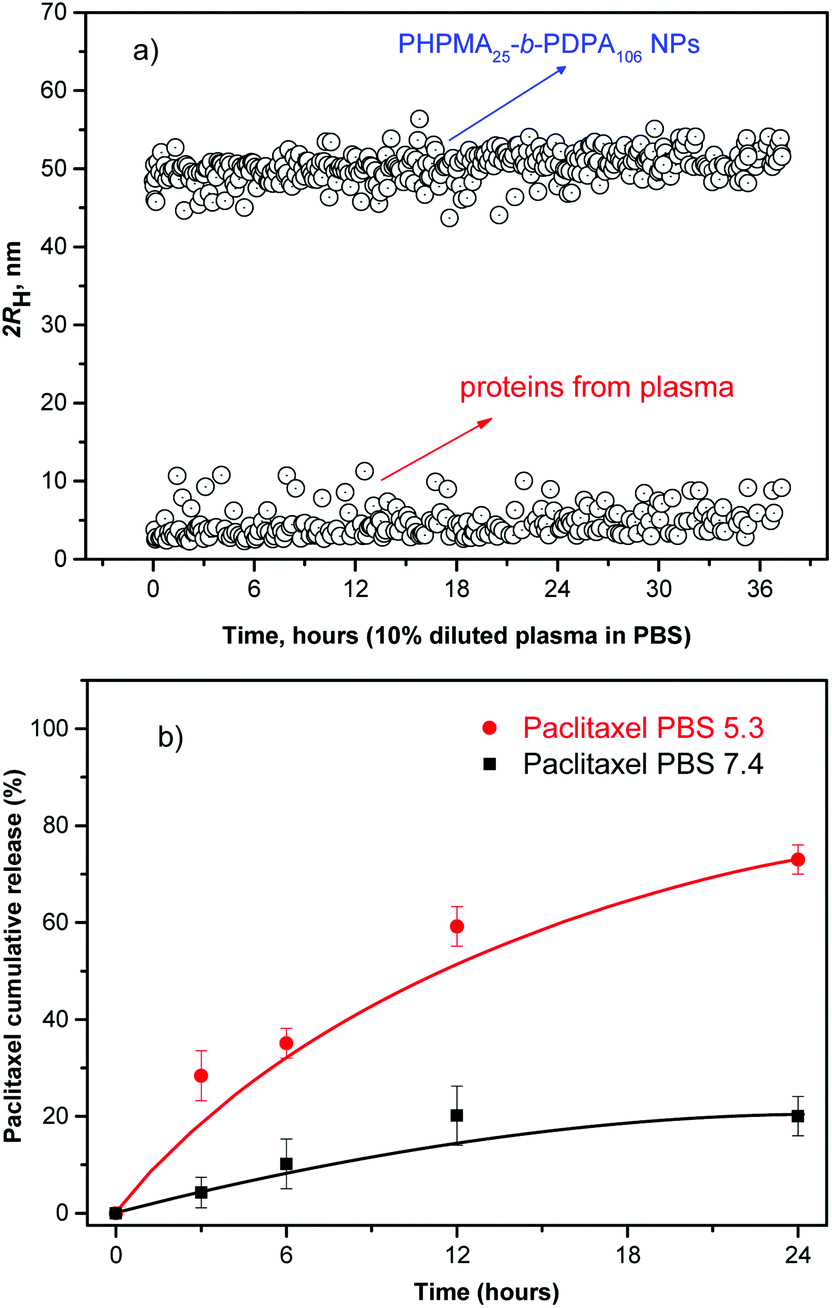 | ||
| Fig. 3 (a) Size distribution in dissolved human plasma (10% v/v in PBS) for the PHPMA25-b-PDPA106 block copolymer NPs as a function of the incubation time and (b) drug release profiles from paclitaxel-loaded PHPMA25-b-PDPA106 block copolymer NPs at pH 7.4 (PBS, closed black squares) simulating transport in blood and at pH 5.3 (PBS, closed red circles) simulating the acidic environment in endosomal and lysosomal compartments at 37 °C. | ||
The size and scattering intensity patterns of the NPs do not change within the studied 36 h suggesting that the NPs are stable in the simulated but highly challenging media. The high stability of the NPs is based on their hydrophilic shell nature, improved colloidal stability and their unmatched resistance to protein adsorption.16,20,29 By preventing any adsorption of proteins the NPs are virtually “invisible” in the blood milieux, which makes them a promising system for in vivo applications.23,29–31
Subsequently, the release profile of the chemotherapeutic PTX was investigated under conditions mimicking the acidic environment in endosomal and lysosomal compartments, pH ∼ 5.0 and 37 °C. The release experiments at 37 °C were also conducted at pH 7.4 to simulate conditions during transport in blood and in normal healthy tissues (Fig. 3b). The drug release profile is clearly pH-sensitive in accordance with the physico-chemical studies presented above.
The results suggest that at pH ∼ 5.0 (acidic environment in endosomal and lysosomal compartments) the block copolymer becomes protonated to form a polycation. The Coulombic repulsion among the chains disrupts the NPs which physically disassemble (pH-triggered disassembly, Fig. 1 and 3b), thus resulting in a 3.5 fold acceleration of the rate of release of the chemotherapeutic (∼70%) within 24 h, (pH ∼ 5.0) compared to physiological conditions (PBS). Moreover, ∼21% of the drug was released from the intact NP cores when kept in PBS at pH 7.4. The release data demonstrate that during the systemic circulation minimal quantity of the drug would be released before reaching the solid tumor environment via the EPR effect. Conversely, the chemotherapeutic is readily released at pH 5.0 with ∼70% of the loaded anticancer drug released within the first 24 h fulfilling the criteria32 for a pH-triggered drug release mechanism exhibiting practical application as nanocarriers in passive tumor-targeted drug delivery.
To investigate the inhibitory effect on tumor cells, the pH-triggered PHPMA25-b-PDPA106 NPs were loaded with the antitumor drug PTX with an overall cargo rate around 2.5 wt% and a loading efficiency of 96% (see the Materials section and Methods section). Given the high loading (negligible free PTX) no further purification was carried out. The cell viability assay using alamarBlue® was used to examine the in vitro cytotoxicity as a classical approach to evaluate the direct effect of the drug-loaded NPs on target cancer cells. The HeLa cell line was selected as a widely used and well-studied cancer cell model system. The drug-loaded NPs were incubated with the HeLa cells for 24 and 48 h. The in vitro cell viability experiments demonstrated that the PTX-loaded-PHPMA25-b-PDPA106 NPs exhibited a similar if not slightly stronger cytotoxic effect compared to the free drug after 24 h of incubation (Fig. 4a). After incubation over 48 h the superior toxicity of the PTX-loaded-PHPMA25-b-PDPA106 NPs compared to free PTX is clearly visible (Fig. 4b). The NPs (IC50 0.1 ng mL−1) exhibited significantly higher cytotoxicity than the free PTX (IC50 1.7 ng mL−1), which corresponds to the values stated in the literature for HeLa cells.33,34 This increased cytotoxicity of the drug-carrying NPs compared to the free drug has been previously reported for analogous systems35–39 and is supposedly the result of enhanced endocytotic uptake of NPs. In particular at low drug concentrations and higher incubation times (below 0.005 μg mL−1, Fig. 4b), the endocytotic uptake of the PTX-loaded NPs is more efficient than the uptake of free drug into the cells.36 A strong toxicity of the drug-loaded NPs after 48 h incubation correlates with the hypothesis that a significant amount of particles is already internalized in endosomal compartments within the first 24 h and that this is closely followed by a fast release of the drug (within 24 h approx. 70% of the drug was released under acidic conditions mimicking the conditions in endosomal and lysosomal compartments, see Fig. 3b). Drug-free PHPMA25-b-PDPA106 NPs were also tested up to the applied maximal concentration (0.4 mg mL−1) with no significant cytotoxic activity (Fig. 4a and b, Fig. S4A and S4B†). After disassembly of the nanoparticles (pH < pKa), the freely dissociated PHPMA25-b-PDPA106 chains present protonated amine groups to the cell. The toxicity of protonated amine groups has been described previously.40 This may contribute to the high toxicity of the presented system. However, the data clearly showed evidence that the drug-free PHPMA25-b-PDPA106 particles have no relevant observable toxic effect on the cells (Fig. 4a and b, Fig. S4A and S4B†). Therefore the high efficiency of the drug delivery system is purely attributed to the efficient transport and release of the drug, and side effects of material toxicity can be excluded. It is important to highlight that the extracellular pH differences between the tumoral tissues and the healthy tissues are not just a characteristic of tumors but also ischemia, inflammation, renal failure or chronic obstructive pulmonary diseases that also share similar pH differences to their healthy counterparts. Therefore the HPMA-b-PDPA copolymer NPs may also find applications as drug carriers for their treatment.
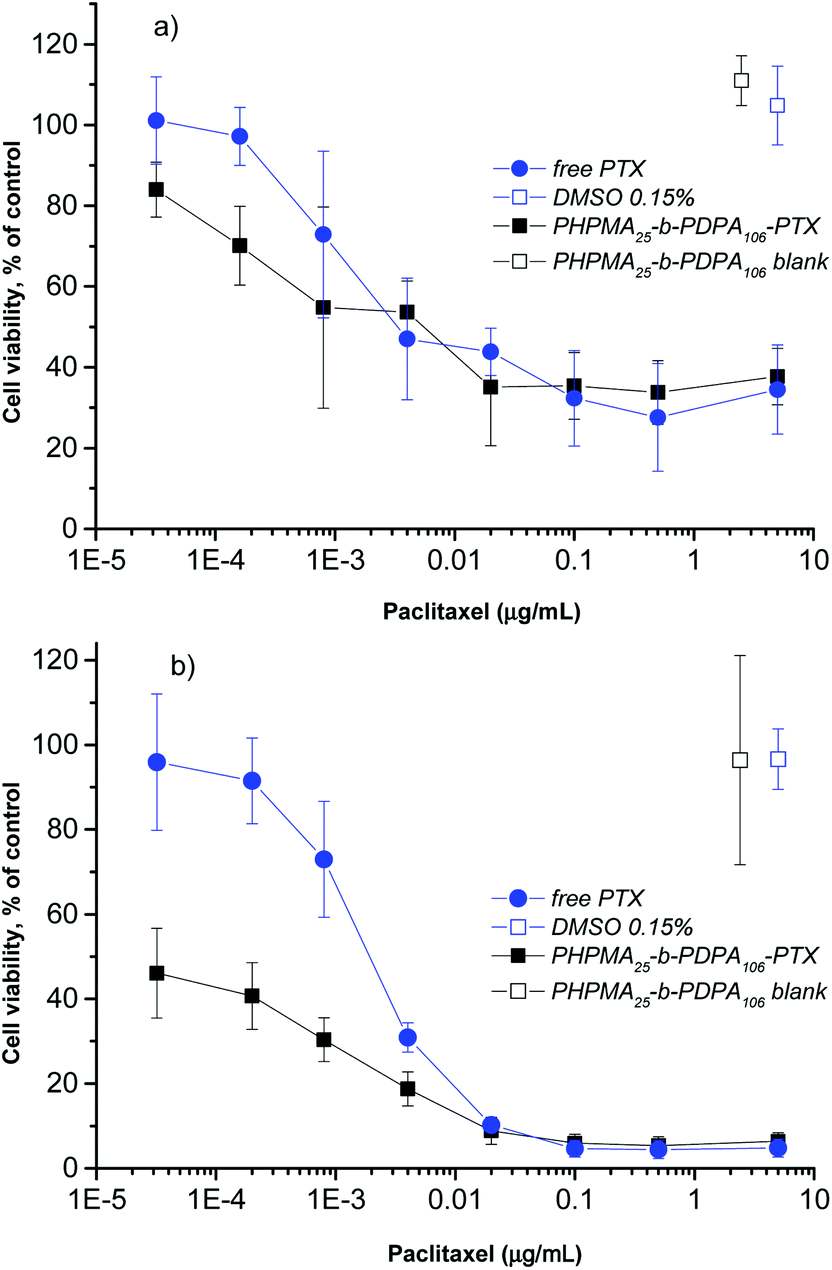 | ||
| Fig. 4 Cell viability of HeLa cell line after 24 h (a) and 48 h (b) incubation with different concentrations of free PTX (closed blue circles) and PTX-loaded PHPMA25-b-PDPA106 (closed black squares) NPs. DMSO 0.15% (open blue squares) and blank PHPMA25-b-PDPA106 NPs (open black squares) were used as controls. | ||
Conclusions
In summary a diblock copolymer of PHPMA and PDPA was prepared by RAFT polymerisation. The diblock copolymer was assembled into spherical NPs consisting of a core of PDPA and a corona of PHPMA as evidenced by SLS, cryo-TEM and SAXS. Presumably, the PHPMA chains prevented the fouling of proteins resulting in a remarkable stability in serum. The reduction of the pH below 6.85 resulted in a rapid increase in the zeta-potential and the fast disassembly of the particles (size decrease in DLS). This was exploited as a trigger for the delivery of hydrophobic drugs into cancer cells. A minimal amount of drug was released above the threshold pH. The in vitro cytotoxicity studies showed an important increase in the activity of the NPs loaded with drug compared to the free drug.The particle's size below the cut-off size of the leaky pathological vasculature (2RH < 100 nm), their excellent stability in serum and the ability to release a drug at the endosomal pH with concomitant high cytotoxicity make them suitable candidates for cancer therapy, namely for treatment of solid tumours exhibiting high tumor accumulation of NPs due to an effective EPR effect.
Experimental section
Materials
Monomer 2-(diisopropylamino)ethyl methacrylate (DPA, 97%) was purified by vacuum distillation prior to use. Monomer N-(2-hydroxypropyl)methacrylamide (HPMA) was synthesized according to the reference.41 Initiator 4,4-azobis(4-cyanopentanoic acid) (V-501, Aldrich) was recrystallized from methanol prior to use. The chain transfer agent, 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CTP, Aldrich), was used as received. Solvents, methanol, dimethylacetamide and 1,4-dioxane were purchased from Lachner, dried over molecular sieves (3 Å) and distilled prior to use.Methods
200 g mol−1, Đ = 1.29. The synthetic parameters and molecular weight data of polymers prepared via RAFT polymerization are described in Table TS1 (ESI†).
Scattering techniques
The dynamic light scattering (DLS) measurements were performed using an ALV CGE laser goniometer consisting of a 22 mW He–Ne linear polarized laser operating at a wavelength (λ = 632.8 nm), an ALV 6010 correlator, and a pair of avalanche photodiodes operating in the pseudo cross-correlation mode. The samples were loaded into 10 mm diameter glass cells and maintained at 25 ± 1 °C. The data were collected using the ALV Correlator Control software and the counting time was 30 s. The measured intensity correlation functions g2(t) were analysed using the algorithm REPES (incorporated in the GENDIST program)43 resulting in the distributions of relaxation times shown in equal area representation as τA(τ). The mean relaxation time or relaxation frequency (Γ = τ−1) is related to the diffusion coefficient (D) of the nanoparticles as: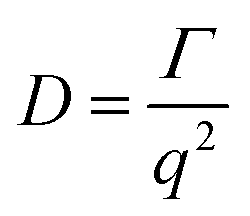 where
where  is the scattering vector, n is the refractive index of the solvent and θ is the scattering angle. The hydrodynamic radius (RH) or the distributions of RH were calculated by using the well-known Stokes–Einstein relation:
is the scattering vector, n is the refractive index of the solvent and θ is the scattering angle. The hydrodynamic radius (RH) or the distributions of RH were calculated by using the well-known Stokes–Einstein relation: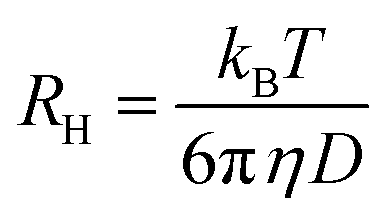 | (3) |
In the static light scattering (SLS), the scattering angle was varied from 30 to 150° with a 10° stepwise increase. The absolute light scattering is related to the weighted-average molar mass (Mw(NP)) and to the radius of gyration (RG) of the nanoparticles by the Zimm formalism represented as:
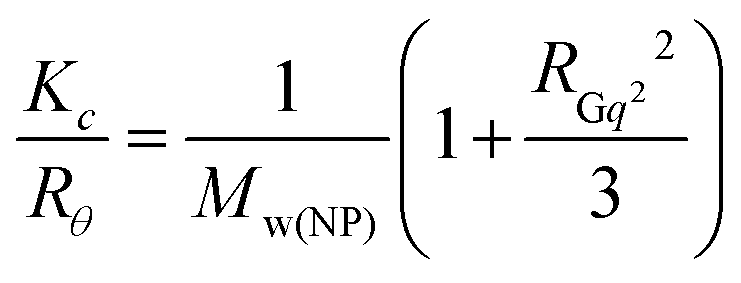 | (4) |
The small angle X-ray scattering (SAXS) experiments were performed on the P12 BioSAXS beamline at the PETRA III storage ring of the Deutsche Elektronen Synchrotron (DESY, Hamburg, Germany) at 20 °C using a Pilatus 2M detector and synchrotron radiation with a wavelength of λ = 0.1 nm. The sample-detector distance was 3 m, allowing for measurements in the q-range interval from 0.11 to 4.4 nm−1. The q range was calibrated using the diffraction patterns of silver behenate. The experimental data were normalized to the incident beam intensity and corrected for the non-homogeneous detector response, and the background scattering of the solvent was subtracted. The solvent scattering was measured before and after the sample scattering to control for possible sample holder contamination. Eight consecutive frames comprising the measurement of the solvent, sample, and solvent were performed. No measurable radiation damage was detected by the comparison of eight successive time frames with 15 s exposures. The final scattering curve was obtained using the PRIMUS program by averaging the scattering data collected from the different frames. The automatic sample changer for sample volume 15 μL and filling cycle of 20 s was used.
pH titration
The pH titration was employed in order to determine the exact pKa of the DPA block in the PHPMA25-b-PDPA106 block copolymer under simulated physiological conditions. The block copolymer sample (c = 0.2 mg mL−1) was previously dissolved in Milli-Q water. The pH was pre-set to pH = 2.0 by adding small amounts of 0.1 mol L−1 HCl solution and the ionic strength was set to 0.15 mol L−1 by adding NaCl to the system. An MPT2 autotitrator connected to a Nano-ZS, Model ZEN3600 (Malvern, UK) zetasizer was used for automated measurement of the pH dependencies of the scattering intensity (Is) and zeta potential (ζ). The Is and ζ-potential were measured at an angle of 173° using a He–Ne 4.0 mW power laser operating at a wavelength of 633 nm. The equipment measures the electrophoretic mobility (UE) of the solution and converts the value to ζ-potential (mV) through Henry's equation. The Henry's function was calculated through the Smoluchowski approximation using the DTS (Nano) program.Cryogenic transmission electron microscopy (Cryo-TEM)
For cryo-TEM studies, a sample droplet of 2 μL was put on a lacey carbon film covered copper grid (Science Services, Munich, Germany), which was hydrophilized by glow discharge for 15 s. Most of the liquid was then removed with blotting paper, leaving a thin film stretched over the lace holes. The specimens were instantly shock frozen by rapid immersion into liquid ethane and cooled to approximately 90 K by liquid nitrogen in a temperature-controlled freezing unit (Zeiss Cryobox, Zeiss NTS GmbH, Oberkochen, Germany). The temperature was monitored and kept constant in the chamber during all the sample preparation steps. After the specimens were frozen, the remaining ethane was removed using blotting paper. The specimen was inserted into a cryo transfer holder (CT3500, Gatan, Munich, Germany) and transferred to a Zeiss EM922 Omega energy-filtered TEM (EFTEM) instrument (Zeiss NTS GmbH, Oberkochen, Germany). Examinations were carried out at temperatures around 90 K. The TEM instrument was operated at an acceleration voltage of 200 kV. Zero-loss-filtered images (ΔE = 0 eV) were taken under reduced dose conditions (100–1000 e nm−2). All images were recorded digitally by a bottom-mounted charge-coupled device (CCD) camera system (Ultra Scan 1000, Gatan, Munich, Germany) and combined and processed with a digital imaging processing system (Digital Micrograph GMS 1.8, Gatan, Munich, Germany). All images were taken very close to focus or slightly under the focus (some nanometers) due to the contrast enhancing capabilities of the in-column filter of the used Zeiss EM922 Omega. In EFTEMs, deep underfocussed images can be totally avoided.Preparation of the nanoparticles
To 2.5 mL of a solution of PHPMA25-b-PDPA106 (20 mg mL−1) and paclitaxel (500 μg mL) in ethanol, 5.0 mL of PBS were added as a precipitant. The increase in the polarity of the solvent led to the aggregation of the polymer chains forming the NPs. The remaining ethanol was removed by evaporation and the final volume of the NPs was concentrated to 5 mg mL−1. Drug-free NPs were prepared by the same way without adding paclitaxel.Paclitaxel (PTX) drug loading and loading efficiency
The total amount of the chemotherapeutic PTX loaded into the nanoparticles was measured by HPLC (Shimadzu, Japan) using a reverse-phase column Chromolith Performance RP-18e (100–4.6 mm, eluent water-acetonitrile with acetonitrile gradient 0–100 vol%, flow rate = 1.0 mL min−1). Firstly 200 μL of the drug loaded NPs were collected from the bulk sample, filtered (0.45 μm) and diluted to 150 μL with HCl (0.05 mol L−1). Such a procedure led to a final pH ∼ 3.0 and therefore a pH-induced NPs disassembly was achieved. Subsequently, the aliquot was diluted to a final volume of 1.0 mL by using acetonitrile (650 μL).15 Afterwards, 20 μL of the final sample was injected through a sample loop. PTX was detected at 227 nm using ultraviolet (UV) detection. The retention time of PTX was 12.20 min under these experimental conditions. An analytical curve with linear response in the range (0.5–100 μg mL−1) was obtained and used to determine the PTX content. The drug-loading content (LC) and the drug-loading efficiency (LE) were calculated by using the following equations: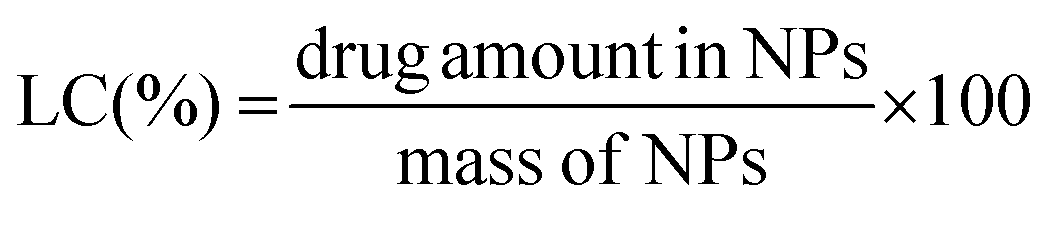 | (5) |
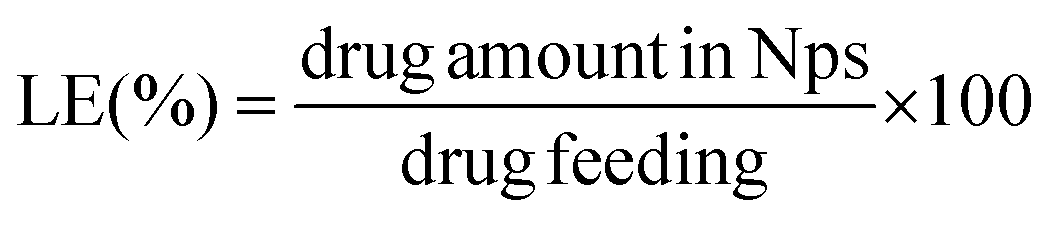 | (6) |
Release experiments
The release experiments were carried out at 37 °C in pH-adjusted release media (pH 7.4 and 5.0). Aliquots (500 μL) of PTX-NPs were loaded into 36 Slide-A-Lyzer MINI dialysis microtubes with MWCO 10 kDa (Pierce, Rockford, IL). These microtubes were dialyzed against 4 L of PBS (pH 7.4 and 5.0).16,23 The release media was changed periodically to reduce the possibility of drug-diffusion equilibrium. The drug release experiments were done in triplicate. At each sampling time, three microtubes were removed from the dialysis system and 0.1 mL from each microtube was sampled and the remaining drug was extracted by using the aforementioned methodologies. The reported data are expressed as the amount of released PTX relative to the total PTX content in the PTX-NPs.Stability of the nanoparticles in blood plasma
The stability of the nanoparticles in blood plasma was monitored in real time while incubating them with diluted human plasma (10% v/v in PBS). Typically, 1 mg mL−1 of the block copolymer nanoparticles was added to a 10% v/v of diluted human plasma in PBS. The dynamic light scattering experiments were performed at intervals for a total incubation time of 36 h.16,22,23,26Cytotoxicity assays
The HeLa cells were cultivated in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal calf serum, 100 units of penicillin and 100 μg mL−1 of streptomycin (Life Technology, CZ). The cells were grown in a humidified incubator at 37 °C with 5% CO2. For the cytotoxicity assay, 5000 cells per well were seeded in duplicates in 96 flat bottom well plates in 100 μL of media 24 h before adding the NPs. For addition of the particles the volume was calibrated to 80 μL, and 20 μL of the 5-times concentrated dilution of PTX or particle dispersion were added per well to obtain a final PTX concentration ranging from 10−5 to 5 μg mL−1. All dilutions were made in full incubation medium with thorough mixing in each dilution step. The sample concentrations of the PTX-loaded particles were adjusted to contain the same total amount of PTX as the samples with free PTX. The cells were incubated with free drug or NPs for 24 h or 48 h. Then 10 μL of alamarBlue® cell viability reagent (Life Technologies, Czech Republic) were added to each well and incubated for a minimum of 3 h at 37 °C. The fluorescence of the reduced marker dye was read with a Synergy H1 plate reader (BioTek Instruments, USA) at excitation 570 and emission 600 nm. The fluorescence intensity of the control samples (with no drug or particles added) was set as a marker of 100% cell viability. The fluorescence signal of “0% viability samples” (all cells were killed by addition of hydrogen peroxide) was used as the background and subtracted from all values prior to calculations. The non-toxic character of the blank particles without the drug was shown by incubation of cells with 0.4 mg L−1 of blank particles. This corresponds to the amount of polymer that is contained in the samples of drug-loaded particles with a total PTX content of 5 μg L−1. For the cell experiments the PTX-dilutions in incubation medium were made from a PTX stock solution of 120 μg mL−1 in PBS/DMSO ((96.5:3.5 v/v).44 Precipitation of the hydrophobic PTX out of the cell culture medium can therefore be excluded because the PTX was previously fully dissolved in a PBS/DMSO solution (96.5:3.5 v/v) and subsequently diluted in the serum-supplied medium under thorough mixing. Consequently, even at a maximal PTX-concentration of 5 μg mL−1, the final DMSO concentration in the incubation medium was below 0.2% and therefore had no effect on cell vitality in the applied setup.45 All the results of the cell experiments are the average of at least 4 measurements (n ≥ 4).
Acknowledgements
This research was supported by the Norwegian Financial Mechanism 2009-2014 under Project contract no 7F14009. C.R.-E. acknowledges the Grant Agency of the Czech Republic (GACR) under contract no. 15-09368Y and the project “BIOCEV – Biotechnology and Biomedicine Centre of the Academy of Sciences and Charles University” (CZ.1.05/1.1.00/02.0109), from the European Regional Development Fund. M. D. acknowledges BIMF (Bayreuth Institute of Macromolecular Research) and BZKG (Bayreuth Center for Colloids and Interfaces) for financial support. F. N. acknowledges support through the CNRS project PICS no. 06130.Notes and references
- J. Rodríguez-Hernández, F. Chécot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691 CrossRef PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC.
- T. Smart, H. Lomas, M. Massignani, M. V. Flores-Merino, L. R. Perez and G. Battaglia, Nano Today, 2008, 3, 38 CrossRef CAS.
- L. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1996, 118, 3168 CrossRef CAS.
- L. Zhang and A. Eisenberg, Polym. Adv. Technol., 1998, 9, 677 CrossRef CAS.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267 CrossRef CAS PubMed.
- G. Gaucher, M.-H. Dufresne, V. P. Sant, N. Kang, D. Maysinger and J.-C. Leroux, J. Controlled Release, 2005, 109, 169 CrossRef CAS PubMed.
- V. P. Chauhan and R. K. Jain, Nat. Mater., 2013, 12, 958 CrossRef CAS PubMed.
- S. Petrova, E. Jäger, R. Konefał, A. Jäger, C. G. Venturini, J. Spěváček, E. Pavlova and P. Štěpánek, Polym. Chem., 2014, 5, 3884 RSC.
- Q. Yin, J. Shen, Z. Zhang, H. Yu and Y. Li, Adv. Drug Delivery Rev., 2013, 65, 1699 CrossRef CAS PubMed.
- B. A. Webb, M. Chimenti, M. P. Jacobson and D. L. Barber, Nat. Rev. Cancer, 2011, 11, 671 CrossRef CAS PubMed.
- E. G. Kelley, J. N. L. Albert, M. O. Sullivan and T. H. Epps III, Chem. Soc. Rev., 2013, 42, 7057 RSC.
- E. S. Lee and Y. H. Bae, J. Controlled Release, 2008, 132, 164 CrossRef CAS PubMed.
- K. Zhou, H. Liu, S. Zhang, X. Huang, Y. Wang, G. Huang, B. D. Sumer and J. Gao, J. Am. Chem. Soc., 2012, 134, 7803 CrossRef CAS PubMed.
- Y. Wang, K. Zhou, G. Huang, C. Hensley, X. Huang, X. Ma, T. Zhao, B. D. Sumer, R. J. DeBerardinis and J. Gao, Nat. Mater., 2014, 13, 204 CrossRef CAS PubMed.
- F. C. Giacomelli, P. Štěpánek, C. Giacomelli, V. Schmidt, E. Jäger, A. Jäger and K. Ulbrich, Soft Matter, 2011, 7, 9316 RSC.
- C. Pegoraro, D. Cecchin, L. S. Gracia, N. Warren, J. Madsen, S. P. Armes, A. Lewis, S. MacNeil and G. Battaglia, Cancer Lett., 2013, 334, 328 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136 CrossRef CAS PubMed.
- T. Riedel, Z. Riedelová-Reicheltová, P. Májek, C. Rodriguez-Emmenegger, M. Houska, J. E. Dyr and E. Brynda, Langmuir, 2013, 29, 3388 CrossRef CAS PubMed.
- C. Rodriguez-Emmenegger, E. Brynda, T. Riedel, M. Houska, V. Šubr, A. B. Alles, E. Hasan, J. E. Gautrot and W. T. S. Huck, Macromol. Rapid Commun., 2011, 32, 952 CrossRef CAS PubMed.
- F. Surman, T. Riedel, M. Bruns, N. Y. Kostina and C. Rodriguez-Emmenegger, Macromol. Biosci., 2015, 15, 636 CrossRef CAS PubMed.
- E. Jäger, A. Jäger, T. Etrych, F. C. Giacomelli, P. Chytil, A. Jigounov, J. L. Putaux, B. Říhová, K. Ulbrich and P. Štěpánek, Soft Matter, 2012, 8, 9563 RSC.
- E. Jäger, A. Jäger, P. Chytil, T. Etrych, F. C. Giacomelli, B. Říhová, P. Štěpánek and K. Ulbrich, J. Controlled Release, 2013, 165, 153 CrossRef PubMed.
- B. Angelov, A. Angelova, S. K. Filippov, T. Naryanan, M. Drechsler, P. Štěpánek, P. Couvreur and S. Lesieur, J. Phys. Chem. Lett., 2013, 4, 1959 CrossRef CAS.
- B. Angelov, A. Angelova, S. K. Filippov, G. Karlsson, N. Terril, S. Lesieur and P. Štěpánek, Soft Matter, 2011, 7, 9714 RSC.
- F. C. Giacomelli, P. Štěpánek, V. Schmidt, E. Jäger, A. Jäger and C. Giacomelli, Nanoscale, 2012, 4, 4504 RSC.
- C. Giacomelli, L. Le Men and R. Borsali, Biomacromolecules, 2006, 7, 817 CrossRef CAS PubMed.
- C. E. de Castro, B. Mattei, K. A. Riske, E. Jäger, A. Jäger, P. Štěpánek and F. C. Giacomelli, Langmuir, 2014, 30, 9770 CrossRef CAS PubMed.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815 CrossRef CAS PubMed.
- K. R. Whiteman, V. Šubr, K. Ulbrich and V. P. Torchilin, J. Liposome Res., 2001, 11, 153 CrossRef CAS PubMed.
- S. Kamei and J. Kopeček, Pharm. Res., 1995, 12, 663 CrossRef CAS.
- K. Cho, X. Wang, S. Nie, Z. Chen and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310 CrossRef CAS PubMed.
- J. E. Liebmann, J. A. Cook, C. Lipschultz, D. Teague, J. Fisher and J. B. Mitchell, Br. J. Cancer, 1993, 68, 1104 CrossRef CAS PubMed.
- X. Peng, F. Gong, Y. Chen, Y. Jiang, J. Liu, M. Yu, S. Zhang, M. Wang, G. Xiao and H. Liao, Cell Death Dis., 2014, 5, e1367 CrossRef CAS PubMed.
- D. Li, Y. Zhang, S. Jin, J. Guo, H. Gao and C. Wang, J. Mater. Chem. B, 2014, 2, 5187 RSC.
- F. Danhier, N. Lecouturier, B. Vroman, C. Jerome, J. Marchand-Brynaert, O. Feron and V. Preat, J. Controlled Release, 2009, 133, 11 CrossRef CAS PubMed.
- J. Panyam, W. Z. Zhou, S. Prabha, S. K. Sahoo and V. Labhasetwar, FASEB J., 2002, 16, 1217 CrossRef CAS PubMed.
- S. K. Sahoo, J. Panyam, S. Prabha and V. Labhasetwar, J. Controlled Release, 2002, 82, 105 CrossRef CAS.
- G. Gaucher, R. H. Marchessault and J.-C. Leroux, J. Controlled Release, 2010, 143, 2–12 CrossRef CAS PubMed.
- W. T. Godbey, K. K. Wu and A. G. Mikos, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 5177 CrossRef CAS.
- K. Ulbrich, V. Šubr, J. Strohalm, D. Plocová, M. Jelínková and B. Říhová, J. Controlled Release, 2000, 64, 63 CrossRef CAS.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033 CrossRef CAS.
- J. Jakes, Czech J. Phys., 1988, 38, 1305 CrossRef.
- E. A. Dubikovskaya, S. H. Thorne, T. H. Pillow, C. H. Contag and P. A. Wender, Proc. Natl. Acad. Sci. U. S. A, 2008, 105, 12128 CrossRef CAS PubMed.
- A. B. Trivedi, N. Kitabatake and E. Do, Agric. Biol. Chem., 1990, 54, 2961 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py00567a |
| This journal is © The Royal Society of Chemistry 2015 |