
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poly(2-oxazoline) molecular brushes by grafting through of poly(2-oxazoline)methacrylates with aqueous ATRP
Dan
Gieseler
and
Rainer
Jordan
*
Professur für Makromolekulare Chemie, Department Chemie, Technische Universität Dresden, Mommsenstr. 4, 01069 Dresden, Germany. E-mail: Rainer.Jordan@tu-dresden.de
First published on 14th May 2015
Abstract
Molecular brushes of poly(2-oxazoline)s (POx) are an intriguing class of polymers as they combine a unique architecture with the properties of POx as a biomaterial. Here, the synthesis of several POx macromonomers with methacrylate end groups and consecutive grafting through polymerization by aqueous atom transfer radical polymerization (ATRP) at room temperature is reported. 1H-NMR spectroscopy and size exclusion chromatography (SEC) confirmed the synthesis of POx molecular brushes with maximum side chain grafting densities, narrow molar mass distributions (Đ ≤ 1.16) and final molar masses corresponding to the initial macromonomer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :initiator ratio. Chain extension experiments show high end group fidelity and formation of block copolymer molecular brushes, and kinetic studies revealed a polymerization behavior of oligo(2-methyl-2-oxazoline) methacrylate very similar to the frequently used oligo(ethylene glycol) methacrylate (OEGMA475). Aqueous solutions of POx molecular brushes with poly(2-ethyl- and 2-isopropyl-2-oxazoline) side chains exhibit the typically defined thermoresponsive behavior with a tunable, very narrow and reversible phase transition.
:initiator ratio. Chain extension experiments show high end group fidelity and formation of block copolymer molecular brushes, and kinetic studies revealed a polymerization behavior of oligo(2-methyl-2-oxazoline) methacrylate very similar to the frequently used oligo(ethylene glycol) methacrylate (OEGMA475). Aqueous solutions of POx molecular brushes with poly(2-ethyl- and 2-isopropyl-2-oxazoline) side chains exhibit the typically defined thermoresponsive behavior with a tunable, very narrow and reversible phase transition.
Introduction
Molecular brushes are composed of a macromolecular backbone with a maximum number of pending polymeric or oligomeric side chains. The very high grafting density induces side chain as well as backbone stretching, which results in a characteristic elongated, worm-like shape of the molecular brush.1 The synthesis of molecular brushes is classified into three methods of which every single one has its specific advantages. These methods are grafting onto (synthesis of side chains and the backbone with a subsequent coupling), grafting from (polymerization of side chains starting from a macroinitiator backbone) and grafting through (polymerization of macromonomers).1 The unique structure, considerable size and high aspect ratio of molecular brushes make them suitable for use as single molecule nanomaterials to be used as templates, actuators and sensors in nano(bio)technology2–4 or even in nanomedicine as drug-delivery systems.5,6Molecular brushes based on poly(2-oxazoline)s (POx) came into the focus of recent research because of the advantageous properties of POx as a biomaterial. POxs are pseudo-peptides synthesized by living cationic ring-opening polymerization (LCROP) of 2-oxazolines in a controlled manner (Đ < 1.2, adjustable molar masses).7,8 Similar to poly(ethylene glycol) (PEG), hydrophilic POx is non-toxic9,10 and non-immunogenic (low to none complement activation)10,11 and suppresses biofouling,12,13 POxylated entities display the same “stealth effect” as PEGylated ones,14–16 and hydrophilic as well as amphiphilic POxs show a biodistribution and excretion which is beneficial for medical applications.17 Additionally their solubility and aggregation behavior can be fine-tuned by structural and compositional variation of the poly(2-oxazoline)s by the use of easily accessible monomers.8,18–20 Furthermore, multiple functionalization and structural versatility are possible using respective initiators, monomers and terminating agents.7,21–30 After early approaches31,32 yielding a variety of POx-based comb polymers,33–46 POx molecular brushes have been synthesized recently by several routes. The majority of the applied methods focuses on the polymerization of macromonomers by group transfer polymerization(GTP), free radical polymerization (FRP) or reversible addition/fragmentation chain transfer polymerization (RAFT). GTP and RAFT can result in molecular brushes with narrow molar mass distributions, but are still limited to relatively short backbones and side chains.31,47,48 Unfortunately, GTP is demanding with respect to the strict reaction conditions and the more robust RAFT has to be stopped at rather low conversions (usually ∼50%) in order to suppress side reactions.48 Nevertheless, RAFT has successfully been used for the synthesis of copolymer molecular brushes made from short (Pn = 5) 2-ethyl- and 2-nonyl-2-oxazoline macromonomers.49 FRP of macromonomers results in molecular brushes with a very long backbone (Pw ∼ 1050) but a broad molar mass distribution.50 Alternatively, in different grafting from approaches, 2-isopropenyl-2-oxazoline was polymerized using FRP, anionic polymerization or rare earth metal catalyzed GTP to create a backbone from which different 2-oxazolines were polymerized via living cationic ring-opening polymerization (LCROP) to yield molecular brushes.51,52 The anionic polymerization resulted in defined brushes, even with block copolymer side chains, but again under very demanding reaction conditions. The rare earth metal catalyzed GTP is stated to perform better, but only indicated by AFM-imaging so far.52 In addition, the rare earth metal catalyst is rather difficult to handle. One of the most interesting features of POx molecular brushes is their defined thermoresponsive behavior. The temperature dependent solubility in water can be fine-tuned by varying the length of the backbone, the side chain or the side chain composition.48,50,51,53–56 Thus, POx molecular brushes are interesting materials to be used as sensors or, with cloud points adjustable close to the human body temperature, for biomedical applications such as theragnostics. Furthermore, it is possible to conjugate antibodies to molecular brushes of 2-ethyl- and 2-isopropyl-2-oxazoline, which could be interesting for the development of active targeting in drug-delivery.57
As outlined above, the existing methods for the synthesis of POx molecular brushes are either experimentally demanding or uncontrolled and/or limited to rather short side chain or backbone lengths. Here, the atom transfer radical polymerization (ATRP) should be an attractive alternative to overcome these drawbacks. However, first attempts to use ATRP for the synthesis of POx molecular brushes were made by ATRP of 2-isopropenyl-2-oxazoline and subsequent LCROP of 2-oxazolines, thus a typical grafting from approach. Unfortunately, ATRP of 2-isopropenyl-2-oxazoline yielded only oligomeric products, which has contributed to a strong complexation of the copper species used for the ATRP catalyst by the monomer and the growing oligomers.55 To the best of our knowledge, ATRP of POx macromonomers has not been attempted so far and could be more viable. For example, a similar oligo(ethylene glycol) methacrylate can be polymerized by aqueous ATRP in a fast and well-controlled manner. Even grafting from a protein is possible.58,59 Moreover, the synthesis of suitable POx macromonomers is well known and has been reported in the literature.31,37,48,60–62
Here we report on the well-controlled synthesis of POx molecular brushes by the grafting through of poly(2-oxazoline)methacrylates by aqueous ATRP.
Results and discussion
Synthesis of poly(2-oxazoline)methacrylate macromonomers
Defined POx macromonomers were synthesized according to Kobayashi et al.60 by LCROP of 2-methyl- (MeOx), 2-ethyl- (EtOx) or 2-isopropyl-2-oxazoline (iPrOx) with methyl triflate as the initiator at 90 °C in acetonitrile (Scheme 1). The LCROP was terminated by a mixture of methacrylic acid and triethylamine creating the termination agent, a methacrylate anion, in situ to yield the methacrylate end functionalized POx (MeOxm-MA, EtOxm-MA and iPrOxm-MA). Analogous to oligo(ethylene glycol) macromonomers, short POx macromonomers with m = 7 but also longer ones with m = 21, 23 were synthesized to investigate their consecutive polymerizability by ATRP. The length of the macromonomers was controlled by the initial [M]0:[I]0 ratio. While MeOx yields highly water-soluble polymers, EtOx and especially iPrOx give thermosensitive polymers.63–68
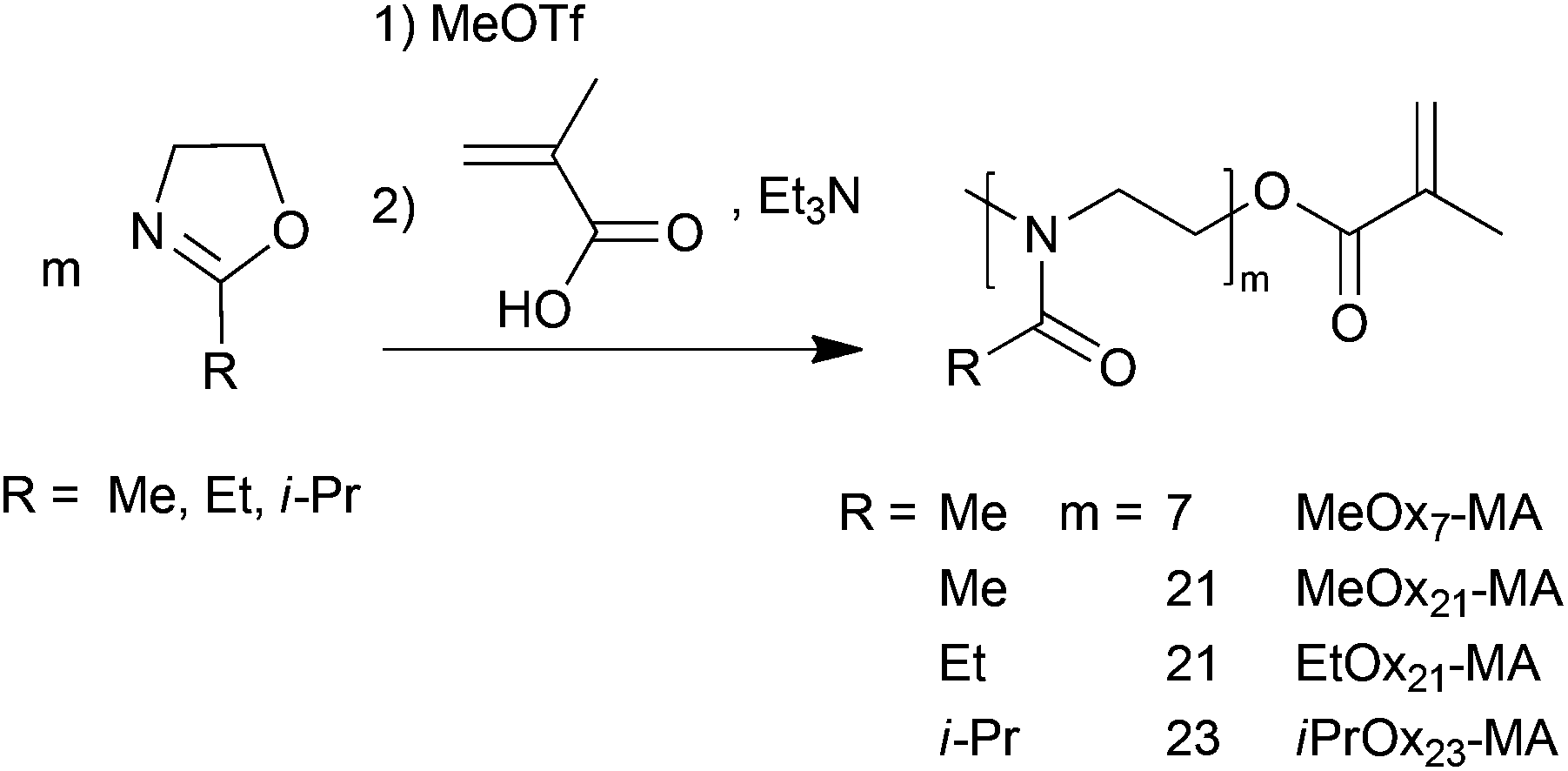 | ||
| Scheme 1 Synthesis of poly(2-oxazoline) macromonomers by LCROP with methyl triflate as the initiator and methacrylic acid/triethylamine termination. | ||
The four POx-MA macromonomers were analyzed by means of SEC, matrix assisted laser desorption ionization–time-of-flight mass spectrometry (MALDI-ToF-MS) and 1H-NMR spectroscopy. With the latter, the chemical composition of all macromonomers could be confirmed, especially the methacrylic end function by the appearance of two singlets around 5.8 ppm, corresponding to the two vinylic protons (Fig. 1). Comparison of the peak integral ratio of the terminal methyl group originating from the initiator (two multiplets between 2.8 and 3.1 ppm) with the methacrylic group indicates a quantitative functionalization by the termination reaction. The 1H-NMR spectrum also shows minor traces of diethyl ether (from precipitation) and triethylammonium ions (from the termination reaction). The impurities could be removed by dialysis, however, because of the relatively low molar mass of the macromonomers; this also caused a significant loss of the product, and since both impurities are not interfering with the consecutive ATRP reaction, the macromonomers were used as such for further experiments. The presence of triethylammonium salt caused obstruction in the SEC trace of the low molar mass MeOx7-MA as both compounds had a similar elution time. This resulted in a relatively high apparent dispersity of ĐSEC = 1.31 while all other macromonomers gave dispersity values of ĐSEC = 1.08–1.09 and, thus, close to the theoretical limit (Fig. 2a, Table 1). This is also corroborated by the MALDI-ToF-MS data, as only molar mass distributions corresponding to the desired macromonomer structure of poly(2-methyl-2-oxazoline) (Δm/z = 85), poly(2-ethyl-2-oxazoline) (Δm/z = 99) or poly(2-isopropyl-2-oxazoline) (Δm/z = 113) with a methyl group at the one and a methacrylic group at the other chain end can be observed in the MALDI-ToF-MS spectra (Fig. 2b and c). A straightforward analysis of the short MeOx7-MA is again obstructed only this time, by the overlap of the mass signals of the macromonomer with matrix components. Nevertheless, close inspection of the mass spectrum indicates a much better dispersity of MeOx7-MA as calculated from the deformed SEC elugram.
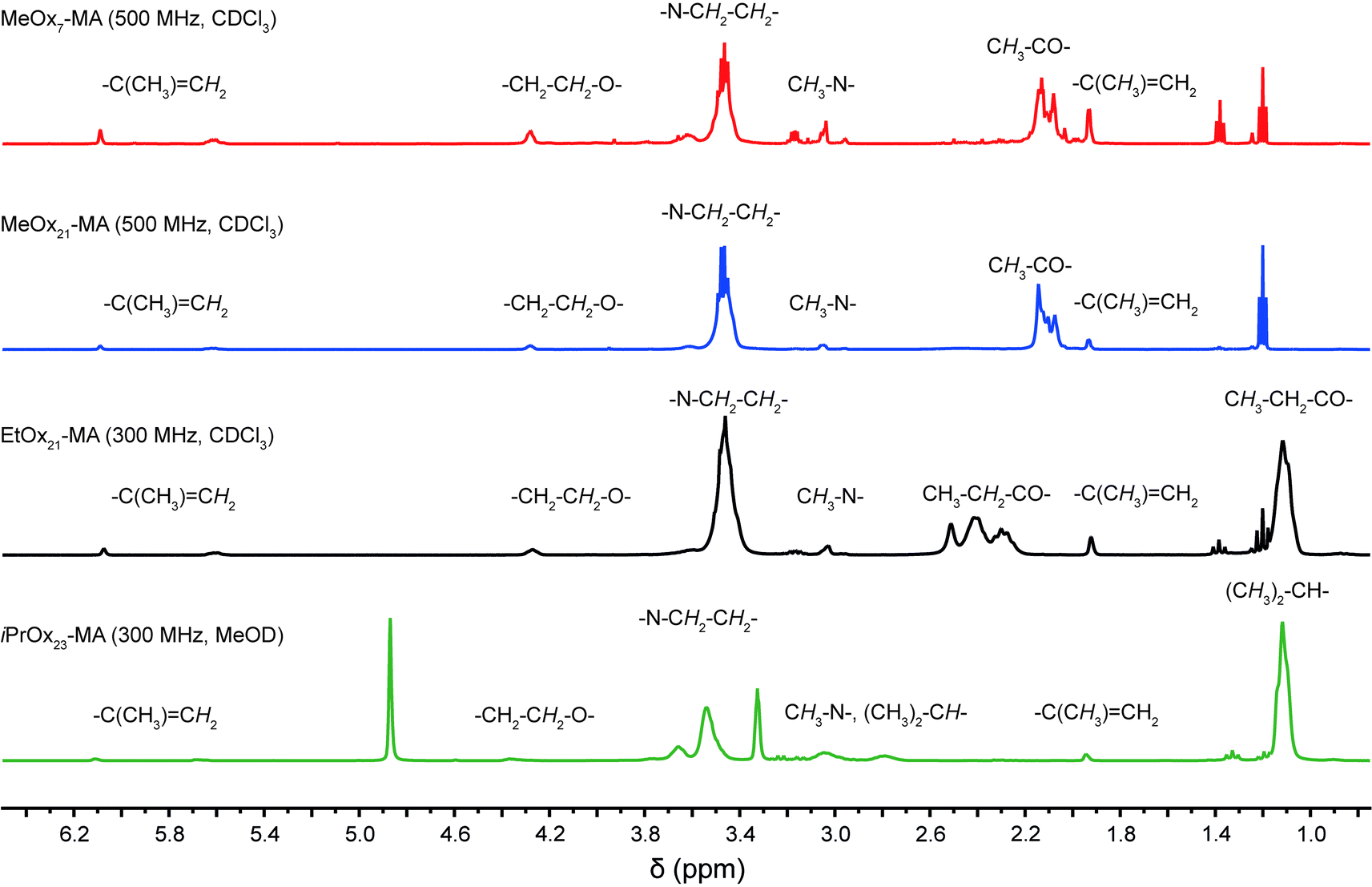 | ||
| Fig. 1 1H-NMR-spectra of the synthesized macromonomers with signals aligned to the respective structural units. | ||
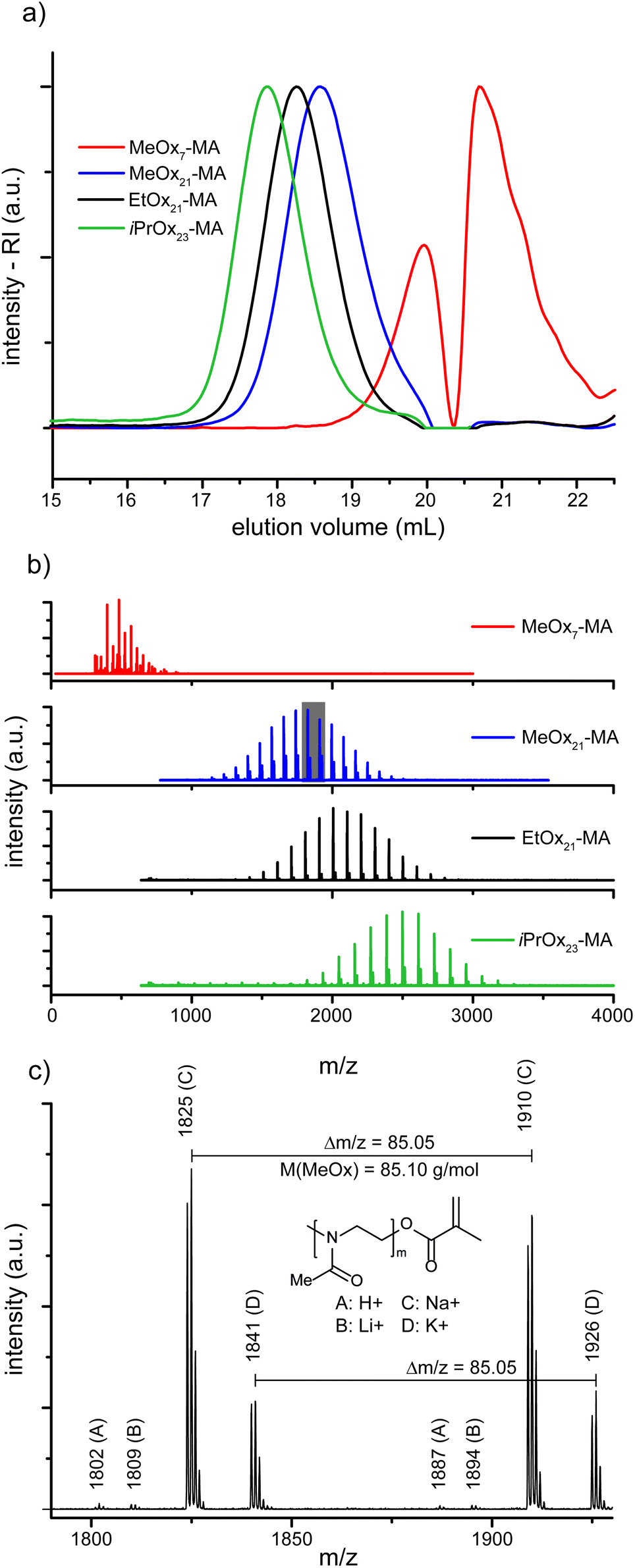 | ||
| Fig. 2 Characterization of the POx-MA macromonomers. (a) SEC elugrams of the macromonomers showing a narrow molar mass distribution and the growth of the molar mass with increasing side chain length and degree of polymerization. (b) MALDI-ToF-MS spectra of the macromonomers demonstrating a narrow molar mass distribution and only the desired macromonomer species. Distribution of MeOx7-MA is overlapped by peaks of the matrix. (c) Detailed section of the spectrum of MeOx21-MA. The chosen part is highlighted (grey area) in (b). | ||
| Name | R | [M]0/[I]0 | M theo. (g mol−1) | P n,NMR | M n,NMR (g mol−1) | M n,MALDI (g mol−1) | M n,SEC (g mol−1) | Đ SEC |
|---|---|---|---|---|---|---|---|---|
|
a As calculated from [M]0:[I]0.
b As determined by end group analysis from 1H-NMR spectroscopy data.
c As determined by MALDI-ToF-MS.
d As determined by SEC (DMAc, 5 g L−1 LiBr, 1 vol% H2O, calibrated with PMMA standards).
e Apparent dispersity obtained from the obstructed elution peak as shown in Fig. 2a. The actual dispersity is significantly lower.
|
||||||||
| MeOx7-MA | Me | 5 | 525 | 7 | 695 | 524 | 424 | 1.31e |
| MeOx21-MA | Me | 20 | 1802 | 21 | 1887 | 1790 | 2010 | 1.09 |
| EtOx21-MA | Et | 20 | 2083 | 21 | 2182 | 2050 | 2484 | 1.08 |
| iPrOx23-MA | i-Pr | 20 | 2363 | 23 | 2703 | 2450 | 3063 | 1.08 |
Synthesis of POx molecular brushes
We tested several ATRP systems with different ligands (N,N,N′,N′,N′′-pentamethyldiethylenetriamine (PMDETA), tris[2-(dimethylamino)ethyl]amine (Me6TREN), 2,2′-bipyridine (bpy)), with or without initial addition of a deactivating copper(II)-complex to polymerize the POx macromonomers to molecular brushes (Table 2).| No. | Ligand | [I] (mM) | Conv.a (%) | [M]:[I]:[Cu(I)]:[Cu(II)]:[L] |
M n,theo (kg mol−1) | M n,SEC (kg mol−1) | Đ SEC |
|---|---|---|---|---|---|---|---|
| a As determined by SEC analysis of the unpurified molecular brush. b As calculated from initial macromonomer–initiator ratio and conversion. c As determined by SEC, DMAc, 5 g L−1 LiBr, 1 vol% H2O, calibrated with PMMA standards. | |||||||
| 1 | PMDETA | 38 | 86.7 | 28:1:1:0:2 |
16.8 | 23.6 | 2.83 |
| 2 | Me6TREN | 35 | 89.5 | 30:1:1:0:1 |
18.8 | 57.8 | 2.23 |
| 3 | bpy | 35 | 93.5 | 30:1:1:0:2 |
20.0 | 164 | 1.41 |
| 4 | bpy | 8 | >99 | 30:1:1:9:22 |
20.7 | 10.2 | 1.15 |
As is obvious from Table 2, entries 1–3, no controlled ATRP reaction was possible using PMDETA, Me6TREN or bpy without additional Cu(II) as indicated by the high values for ĐSEC and apparent molar masses being significantly higher than expected. This is in agreement with earlier literature reports. The highly reactive complexes with PMDETA and especially Me6TREN disproportionate in aqueous solution into a more complex reaction system that result in high radical concentration and loss of polymerization control. Even when using water stable and less reactive bpy complexes, initially added Cu(II) species might be necessary to maintain reaction control.58,69–71 Accordingly, the use of 2,2′-bipyridine as the ligand and copper(I)- as well as copper(II)-bromide (Table 2, entry 4) in aqueous solution proved to be a suitable method and result in the desired molecular brushes in high yields, with narrow molar mass distributions and final molar masses in the expected range (Scheme 2). While the initiator, 2-hydroxyethyl-2-bromoisobutyrate (HEBIB), and copper(I)-complex were used in equimolar amounts, a nine-fold excess of the deactivating copper(II)-complex was necessary to achieve and maintain satisfying reaction control even at high conversions of the macromonomers. A similar recipe was also successfully applied for the polymerization of oligo(ethylene glycol) methacrylate (OEGMA475) by ATRP.58
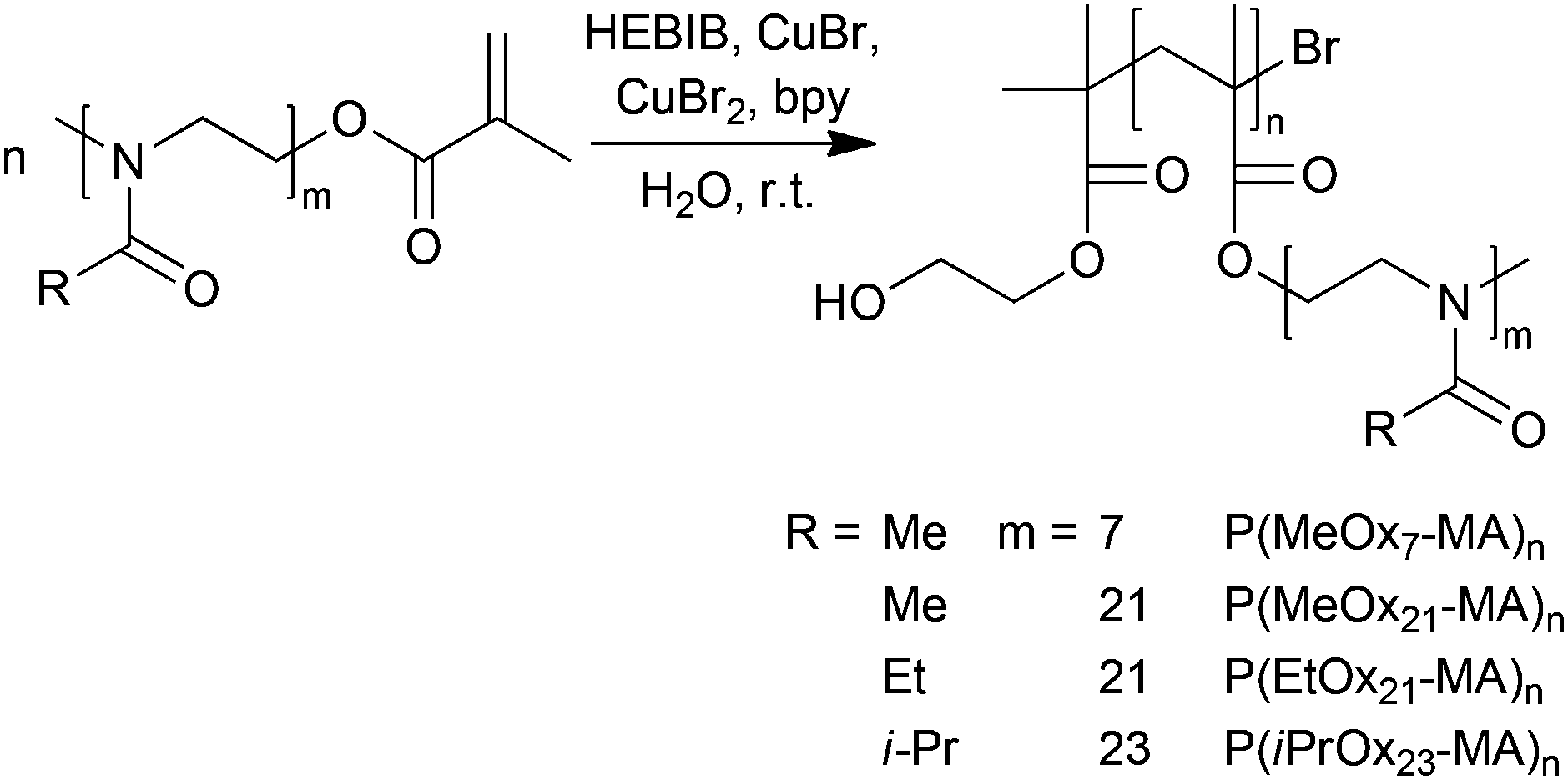 | ||
| Scheme 2 Synthesis of POx molecular brushes by aqueous ATRP at room temperature from POx macromonomers. The components were typically used at a ratio of [I]:[Cu(I)]:[Cu(II)]:[bpy] = 1:1:9:22. | ||
A series of eleven molecular brushes was successfully synthesized by using this approach and analyzed by SEC, NMR and SEC-MALLS (Table 3). In general, conversions >80% could be reached within 6 h or less. The residual macromonomer and catalyst could be removed by column chromatography. Fig. 3a shows the SEC elugrams of a series of molecular brushes P(MeOx7-MA)n with n = 10–820 and the steady increase of the molar masses and low dispersities (Đ = 1.16–1.21) for all brushes. The symmetric SEC traces exhibit neither high nor low molecular subpopulation, which indicates a fast and quantitative initiation, and chain transfer or termination reactions seem to be not significant.
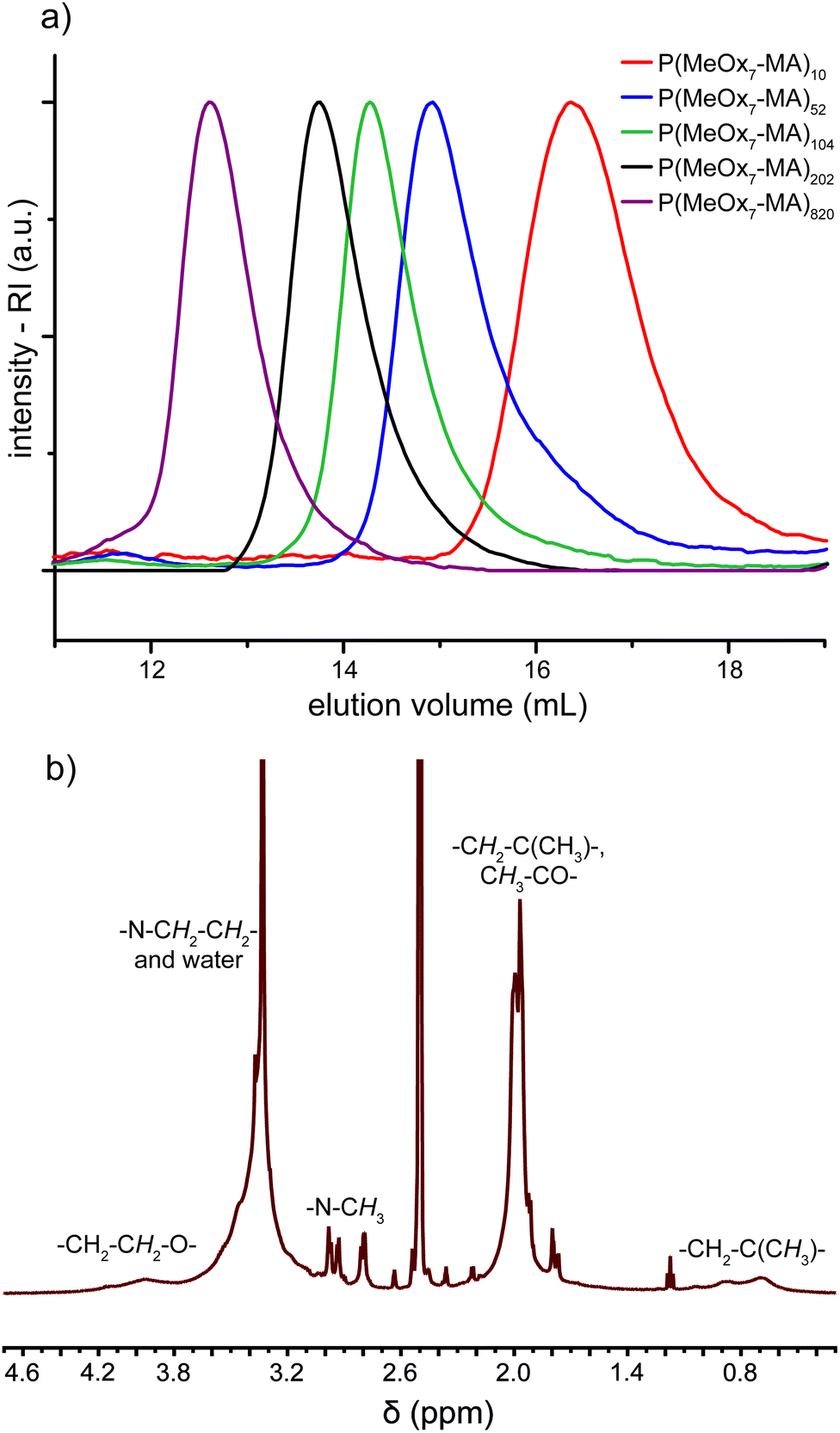 | ||
| Fig. 3 (a) SEC elugrams (DMAc, 5 g L−1 LiBr, 1 vol% H2O, calibrated with PMMA standards) of five molecular brushes from MeOx7-MA with increasing backbone length. (b) 1H-NMR-spectrum (500 MHz, DMSO-d6) of P(MeOx7-MA)52 with assignment of the respective structural units. | ||
:[Cu(I)]:[Cu(II)]:[bpy] = 1:1:9:22; reaction times are as listed
| Name | n | [I] (mM) | t R (h) | Conv.b (%) | M n,theo (kg mol−1) | M n,SEC (kg mol−1) | Đ SEC | M n,SEC-MALLS (kg mol−1) |
|---|---|---|---|---|---|---|---|---|
|
a As calculated from the initial macromonomer–initiator ratio and conversion.
b As determined by SEC analysis of the unpurified molecular brush.
c As determined by SEC, DMAc, 5 g L−1 LiBr, 1 vol% H2O, calibrated with PMMA standards.
d As determined by SEC-MALLS, H2O, 0.72 g L−1 NaN3, 8.5 g L−1 NaNO3, 35 °C.
e [I]:[Cu(I)]:[Cu(II)]:[bpy] = 1:2:9:25.
f Not determined due to the low amount of polymer or because of polymer–column interactions.
|
||||||||
| P(MeOx7-MA)10 | 10 | 5 | 2 | >99 | 7.4 | 6.9 | 1.16 | —f |
| P(MeOx7-MA)52 | 52 | 5 | >99 | 36.1 | 15.5 | 1.16 | 40.0 | |
| P(MeOx7-MA)104 | 104 | 5 | >99 | 72.3 | 24.2 | 1.16 | 65.9 | |
| P(MeOx7-MA)202 | 202 | 5 | >99 | 140.5 | 36.3 | 1.15 | 101.1 | |
| P(MeOx7-MA)820 | 820 | 1 | 3 | 82.1 | 695.0 | 93.5 | 1.21 | —f |
| P(MeOx21-MA)50 | 50 | 5 | 2 | 86.3 | 95.1 | 42.3 | 1.08 | 95.7 |
| P(MeOx21-MA)91 | 91 | 2.5 | 6 | 86.6 | 172.4 | 55.9 | 1.09 | 150.1 |
| P(EtOx21-MA)50 | 50 | 5 | 2 | 89.2 | 108.2 | 48.6 | 1.06 | 118.4 |
| P(EtOx21-MA)92 | 92 | 2.5 | 6 | 86.0 | 200.6 | 71.9 | 1.08 | 212.6 |
| P(EtOx21-MA)183 | 183 | 1e | 5 | 90.9 | 398.7 | 84.4 | 1.10 | 276.6 |
| P(iPrOx23-MA)37 | 37 | 5 | 2 | 65.4 | 99.2 | 46.4 | 1.09 | —f |
1H-NMR spectroscopy verified the anticipated structure of the synthesized brushes. All observable signals can be assigned to the respective structural units as shown exemplarily for P(MeOx7-MA)52 in Fig. 3b. Unfortunately, end group analysis is not possible because of the very low relative intensity of the terminal hydroxyl group and the broad strong proton signals of the high molar mass brush. The two singlets of the vinylic protons of the macromonomers around 5.8 ppm are no longer observable and new broad signals between 0.4 and 1.4 ppm arose, indicating the formation of the methacrylate backbone. Comparing the intensities of these backbone signals with signals originating from the side chain terminal methyl groups around 3.0 ppm (3H), a maximum grafting density can be concluded for the series of P(MeOxm-MA)n. This analysis was not possible for P(EtOxm-MA)n and P(iPrOxm-MA)n because of signal overlapping (data not shown).
Also longer macromonomers (MeOx21-MA and EtOx21-MA) could be successfully polymerized to molecular brushes and for all cases the dispersity as determined by SEC (ĐSEC) is very low (Table 3). As can be expected, the number average molar masses of the molecular brushes as determined by SEC deviate from the expected values especially for longer side chain brushes because of the different macromolecular architecture of the used calibration standard being linear PMMA. Nevertheless, an obvious trend is observable with respect to the increasing molar masses of the brushes with the same side chains but increasing backbone length and for comparable backbones but different side chain lengths. While absolute values differ, these trends at least indicate a controlled polymerization and the possibility of adjustable molar masses by ATRP. For a better analysis, we performed additional characterization by SEC with light scattering detection (SEC-MALLS, see below).
Naturally, the realizable backbone length of molecular brushes by the grafting through of longer macromonomers is limited by the viscosity of the initial reaction solution. We found that a simple dilution resulted in a noticeable loss of control of the ATRP system as the higher dispersity and deviation of calculated vs. determined molar masses for P(MeOx7-MA)820 and P(EtOx21-MA)183 shows (Table 3). The latter could only be obtained with a double amount of the activator copper complex. Here, alternative controlled/living radical polymerization techniques such as Cu0-mediated living radical polymerizations might be more useful.72–75 Related studies are currently ongoing. Nevertheless, aqueous ATRP was found to be a very versatile polymerization to obtain highly defined molecular brushes with considerable side chain and backbone lengths. As the aqueous ATRP can be driven to high conversions it represents a good alternative to other CRP techniques such as RAFT.48 Only the polymerization of iPrOx23-MA turned out to be difficult and stopped at a conversion of around 66%. Already the initial reaction solution was very viscous because of the higher molar mass of iPrOx23-MA and further significant viscosity increase upon conversion of the macromonomer made stirring impossible.
Further analysis of the POx molecular brushes by SEC-MALLS revealed molar masses that corresponded well with the theoretical values in the majority of the cases and corroborates the assumption of a controlled grafting through polymerization. The traces obtained from SEC-MALLS have a symmetrical appearance and again narrow molar mass distributions can be found. Dispersities range even below (Đ ≤ 1.07) the values determined by SEC with refractive index detection, probably because of the lower scattering of the lower molar mass fraction. The molar mass of P(iPrOx23-MA)37 could not be determined because of interaction of the polymer with the stationary phase at 35 °C (standard conditions) as well as 25 °C. Higher temperatures could not be used because of the cloud point of the polymer. Unfortunately, also the longest brush P(MeOx7-MA)820 could not be analyzed by SEC-MALLS due to the low amount of polymer product after the work-up procedure, which had to be performed to quantitatively remove the remaining macromonomer and the catalyst. A typical SEC-MALLS trace along with RI detection of P(MeOx7-MA)52 is shown in Fig. 4. The determined molar masses are in good agreement with the theoretical values. For most cases, the deviation of the determined number average molar masses from the theoretical values is only about 10% and thus within the experimental error of the method (temperature dependence of dn/dc and underestimation of species with low scattering intensity). The determined molar masses for the longest molecular brushes (P(MeOx7-MA)202 and P(EtOx21-MA)183) deviate by about 30%, which cannot be accounted for by an uncontrolled ATRP conversion as the dispersity of the products is still very low. One reason could be the strong hygroscopic nature of the used macromonomers, which made it difficult to adjust the correct [M]0:[I]0 ratio. However, during attempts to additionally freeze-dry the educts prior to use, considerable autopolymerization of the macromonomers was observed which forced us to refrain from additional drying steps. The SEC-MALLS elugram in Fig. 4a shows an additional distribution at very high molar masses which is not appearing in the refractive index detection. Similar elugrams with high molar mass distributions but of strongly varying intensities were also observed for other P(MeOx7-MA)n molecular brushes and are attributed to very large metastable brush aggregates. First dynamic light scattering measurements in methanol (data not shown) revealed only one particle distribution assigned to the molecular brushes, underlining the assumption of aggregates only stabilized by hydrophobic interactions in aqueous solution.
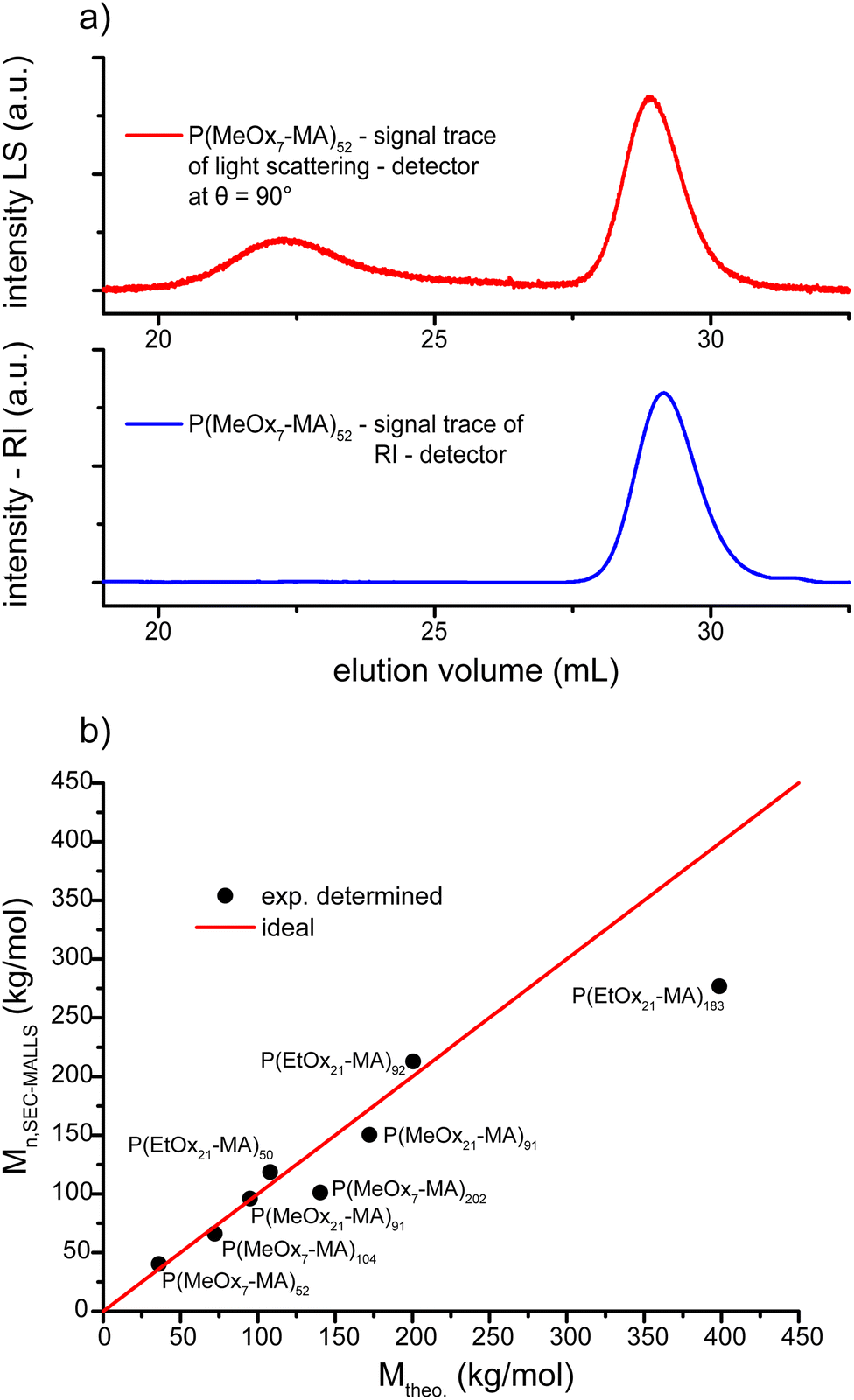 | ||
| Fig. 4 (a) SEC-MALLS elugram of P(MeOx7-MA)52 (H2O, 0.72 g L−1 NaN3 and 8.5 g L−1 NaNO3, 35 °C) in red along with SEC-RI detection (blue). The distribution of the molecular brushes is visible at elution volumes between 27.5 and 31 mL with a narrow and symmetrical appearance. The second high molar mass peak is attributed to metastable brush aggregates. (b) Comparison of the determined number average molar masses with the theoretical values as calculated from the initial macromonomer/initiator ratio and the conversion. Ideal behavior is indicated by the red line. Only P(MeOx7-MA)202 and P(EtOx21-MA)183 deviate noticeably. | ||
Investigation of the polymerization kinetics of MeOx7-MA as the POx analogous to the frequently used OEGMA475 revealed a very similar polymerization behavior of both macromonomers (Fig. 5) under comparable conditions. For MeOx7-MA as well as for OEGMA475 a similar deviation from ideal pseudo-first order kinetics is observable.58 ATRP of MeOx7-MA resulted in a linear increase of the molar mass as a function of the polymerization time at low dispersities (Đ ≤ 1.18) even at high conversions.
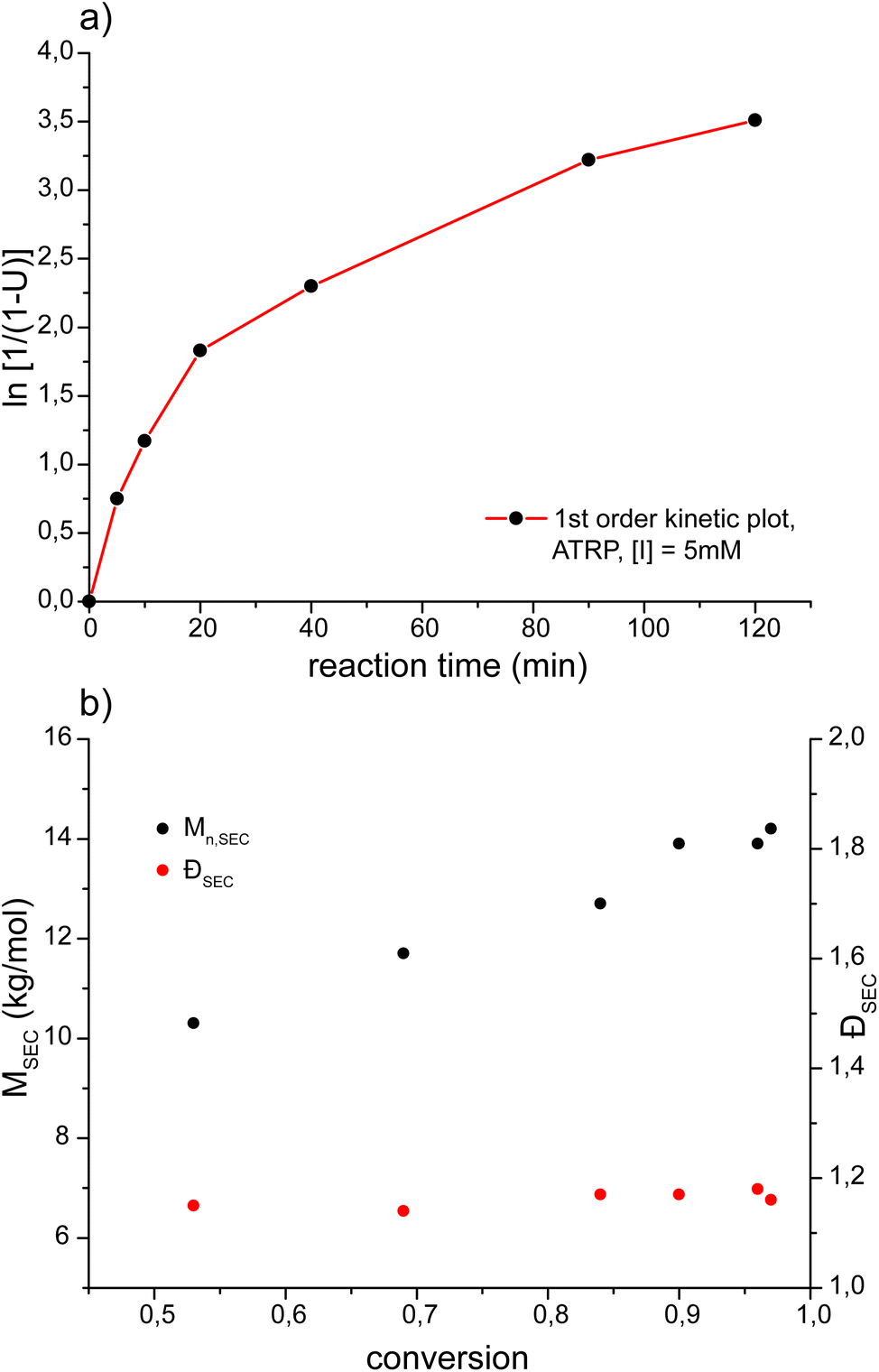 | ||
| Fig. 5 (a) First order kinetic plot for the synthesis of molecular brushes based on poly(2-methyl-2-oxazoline) at room temperature in aqueous solution. Initiator concentration [I] = 5 mM; [macromonomer]:[I]:[CuI]:[CuII]:[bpy] = 50:1:1:9:22. (b) Development of Mn,SEC and ĐSEC with the conversion. | ||
Chain extension of a molecular brush was successfully performed and further corroborates the assumption of a controlled polymerization of POx macromonomers without a significant loss of chain end functionality. First, MeOx21-MA was polymerized to a conversion of >90%, an aliquot of the reaction solution was collected and analyzed and the remaining solution was further converted in situ by the addition of EtOx21-MA to give a P[(MeOx21-MA)-(EtOx21-MA)] block copolymer brush. SEC analysis of the products reveals very low dispersities of both polymers, especially for the block copolymer with a complete shift to a higher molar mass with no indication of the residual homopolymer brush (Fig. 6). Even though the remaining MeOx21-MA participates in the second polymerization, this indicates high end group fidelity and demonstrates the possibility of preparing block copolymer molecular brushes by the grafting through of POx macromonomers by aqueous ATRP.
 | ||
| Fig. 6 SEC elugrams of the two reaction stages of the chain extension. First, the brush was synthesized by aqueous ATRP of MeOx21-MA with initiator concentration [I] = 10 mM; [MM]/[I]/[CuI]/[CuII]/[bpy] = 10/1/1/9/22. After 1.5 h, a part of the reaction mixture was transferred into a solution of EtOx21-MA for in situ chain extension. SEC traces demonstrate a clear shift of the molar mass and the dispersity remains low. | ||
Thermoresponsive behavior
The synthesized molecular brushes based on poly(2-ethyl-2-oxazoline) and poly(2-isopropyl-2-oxazoline) should exhibit a thermoresponsive behavior similar to earlier observations.48,50,51,53–56 The transmittance of aqueous solutions (1 wt%) of the brushes was observed in dependence of the temperature and the cloud point (TCP) was determined at 10% decrease in optical transmittance (Fig. 7). All four brushes showed very sharp transitions that were in the range of only 1–1.5 K. During several consecutive cooling/heating cycles, the materials showed no deviation of the TCP and only a minor heating–cooling hysteresis of approximately 1 K as shown for P(EtOx21-MA)50 and P(iPrOx23-MA)37 in Fig. 7. The determined cloud points appeared in the expected range of TCP = 73 °C for P(EtOx21-MA)50 and TCP = 36 °C for P(iPrOx23-MA)37. An increase of the molar mass of P(EtOx21-MA)n resulted in the expected slight decrease of TCP.48,51,55 The phase transition of P(iPrOx23-MA)37 is intriguingly close to the human body temperature and falls nicely into the row of previously determined TCP's of P(iPrOxm)n molecular brushes, as a brush made by consecutive living anionic polymerization (backbone) and LCROP (grafting from of the side chains) with n = 52, 132, 216 and m = 24 showed TCP = 31, 29.1 and 27 °C, respectively.55 | ||
| Fig. 7 Turbidity measurements for the thermoresponsive brushes in aqueous solution (1 wt%). The heating/cooling rate was 1 K min−1. For optical clarity, cooling traces are only shown for P(EtOx21-MA)50 and P(iPrOx23-MA)37, but heating–cooling hysteresis was small (∼1 K) for all brushes. TCP was determined at 10% decrease in transmittance on heating. | ||
Conclusion
A series of molecular brushes of poly(2-oxazoline)s was synthesized by grafting through polymerization of oligo- and poly(2-oxazoline) macromonomers by aqueous ATRP at room temperature. These molecular brushes feature high grafting densities, narrow molar mass distributions and adjustable molar masses. It was found that surprisingly long and high molar mass molecular brushes are accessible by the grafting through approach; however, the method is limited by the initial viscosity of the reaction solution. Oligo(2-methyl-2-oxazoline)methacrylate showed a very similar polymerization behavior to the commercially available OEGMA475 including the deviation from first order kinetics as well as low dispersities and almost linear increase of the molar mass with the ATRP reaction time. Thus, a versatile POx-based macromonomer to OEGMA475 is now available for the synthesis of structurally complex biomaterials. Chain extension and formation of block copolymer molecular brushes were also successful. Aqueous solutions of POx molecular brushes with poly(2-ethyl- and 2-isopropyl-2-oxazoline) side chains exhibit the typically defined thermoresponsive behavior with a tunable, very narrow and reversible phase transition. The TCP of P(iPrOx23-MA)37 lies at 36 °C close to the human body temperature and falls nicely into the list of previously reported P(iPrOxm)n molecular brushes synthesized by grafting from and living ionic polymerization. In conclusion, aqueous ATRP of oligo- and poly(2-oxazoline) methacrylates was found to be a versatile synthetic route for the preparation of defined POx molecular brushes.Experimental
Materials and methods
All chemicals were purchased from Sigma-Aldrich (Steinheim, Germany) or Acros Organics (Geel, Belgium) and were used as received unless stated otherwise. 2-Methyl-, 2-ethyl-, 2-isopropyl-2-oxazoline, methyltriflate and triethylamine were dried by refluxing over calcium hydride and by distillation under a dry nitrogen atmosphere. 2-Isopropyl-2-oxazoline was synthesized as described elsewhere.64 Methacrylic acid was distilled in vacuo to remove the inhibitor and stored under a dry nitrogen atmosphere at 7 °C. Aqueous solutions were prepared with deionized water (Millipore, 18.2 MΩ cm) unless stated otherwise.
1H-NMR-spectroscopy was performed on a DRX 500 P or an AC 300 P (Bruker, Germany), and spectra were calibrated to the solvent residual signal. MALDI-ToF-MS was performed on a Bruker Biflex (Bruker Daltonics, Bremen, Germany) with an N2-laser (λ = 337 nm) in positive reflector mode. Spectra were recorded in a range of 40–4400 Da and the matrix was suppressed up to 330 Da (MeOx7-MA), 450 Da (EtOx21-MA, IPOx23-MA) or 500 Da (MeOx21-MA), respectively. The device was calibrated from 750 to 3150 Da with Peptide Calibration Standard II (Bruker) prior to every measurement. Samples were prepared by the dried droplet method. Macromonomers were dissolved at 1 g L−1 in methanol with 1% trifluoroacetic acid and subsequently mixed in a ratio of 5:1 (v/v) with the matrix (20 g L−1 sinapinic acid in methanol with 1% trifluoroacetic acid). Turbidity measurements were performed on a Lambda 35 UV/VIS spectrometer equipped with a PTP-1 Peltier System (all from PerkinElmer, Germany) and controlled using Templab software provided by the instrument supplier. The TCP of the molecular brushes were determined by spectrophotometric detection of the changes in transmittance at λ = 500 nm of 1.0 wt% aqueous solutions. The heating/cooling rate was 1.0 K min−1. Given values for the TCP were determined as the temperature corresponding to a 10% decrease in optical transmittance. Size exclusion chromatography (SEC) was done on a PL-GPC 120, equipped with two Gram 100 10 μL 8 × 300 mm columns and dimethylacetamide (DMAc) with 5 g L−1 LiBr and 1 vol% H2O as the mobile phase at 70 °C. The system was calibrated with PMMA standards (PSS, Mainz, Germany) and RI detection. Samples were dissolved in the mobile phase and filtered through 0.2 μm PTFE syringe filters prior to the measurement. SEC-MALLS was performed on a system from Jasco (Gross-Umstadt, Germany) with a PU 2080 HPLC-pump, a Jet-Stream II Plus column oven equipped with one SUPREMA 10 μm 100 Å 8 × 300 mm column and two SUPREMA 10 μm 3000 Å 8 × 300 mm columns (all from PSS). As the mobile phase, double distilled water with 0.72 g L−1 NaN3 and 8.5 g L−1 NaNO3 was used at a temperature of 35 °C. A Dawn DSP laser photometer at λ = 632.8 nm (Wyatt Technology, Dernbach, Germany) and an RI-930 RI detector (Jasco) were used for detection. Samples were dissolved in the mobile phase and filtered through 0.22 μm CME-filters prior to the measurement. Refractive index increments dn/dc were determined on a differential refractometer DR1/b from SLS Systemtechnik (Denzlingen, Germany) in the concentration range of 1–5 g L−1 at a temperature of 25 °C. Polymers were weighed immediately after lyophilization and dissolved in double distilled water containing 0.72 g L−1 NaN3 and 8.5 g L−1 NaNO3. Results of the dn/dc measurement can be found in Table 4.
| Name | dn/dc (mL g−1) |
|---|---|
| P(MeOx7-MA)52 | 0.118 |
| P(MeOx7-MA)104 | 0.116 |
| P(MeOx7-MA)202 | 0.114 |
| P(MeOx21-MA)50 | 0.146 |
| P(MeOx21-MA)91 | 0.153 |
| P(EtOx21-MA)50 | 0.149 |
| P(EtOx21-MA)92 | 0.156 |
| P(EtOx21-MA)183 | 0.153 |
Macromonomer synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)–), 2.10 (br, 21 H, CH3–CO–), 2.95 and 3.05 (m, 3 H, CH3–N–), 3.54 (br, 26 H, –N–CH2–CH2–), 4.27 (br, 2 H, –CH2–CH2–O–), 5.60 (s, 1 H, –CCH2), 6.08 (s, 1 H, –CCH2); Mn,NMR = 695 g mol−1, fNMR = 1. SEC: ĐSEC = 1.31, Mn,SEC = 424 g mol−1. MALDI-ToF-MS: ĐMALDI = 1.04, Mn,MALDI = 524 g mol−1.
CH2)–), 2.09 (br, 63 H, CH3–CO–), 2.94 and 3.04 (m, 3 H, CH3–N–), 3.51 (br, 82 H, –N–CH2–CH2–), 4.27 (br, 2 H, –CH2–CH2–O–), 5.60 (s, 1 H, –CCH2), 6.08 (s, 1 H, –CCH2); Mn,NMR = 1887 g mol−1, fNMR = 1. SEC: ĐSEC = 1.09, Mn,SEC = 2010 g mol−1. MALDI-ToF-MS: ĐMALDI = 1.02, Mn,MALDI = 1790 g mol−1.
CH2)–), 2.36 (br, 42 H, CH3–CH2–CO–), 2.95 and 3.02 (m, 3 H, CH3–N–), 3.50 (br, 82 H, –N–CH2–CH2–), 4.26 (br, 2 H, –CH2–CH2–O–), 5.58 (s, 1 H, –CCH2), 6.06 (s, 1 H, –CCH2); Mn,NMR = 2182 g mol−1, fNMR = 1. SEC: ĐSEC = 1.09, Mn,SEC = 2484 g mol−1. MALDI-ToF-MS: ĐMALDI = 1.02, Mn,MALDI = 2050 g mol−1.
CH2)–), 2.84 and 3.08 (br, 26 H, CH3–N– and (CH3)2–CH–), 3.70 (br, 90 H, –N–CH2–CH2–), 4.42 (br, 2 H, –CH2–CH2–O–), 5.74 (s, 1 H, –CCH2), 6.16 (s, 1 H, –CCH2); Mn,NMR = 2702 g mol−1, fNMR = 1. SEC: ĐSEC = 1.08, Mn,SEC = 3063 g mol−1. MALDI-ToF-MS: ĐMALDI = 1.02, Mn,MALDI = 2450 g mol−1.
CH2)–), 2.10 (br, 21 H, CH3–CO–), 2.95 and 3.05 (m, 3 H, CH3–N–), 3.54 (br, 26 H, –N–CH2–CH2–), 4.27 (br, 2 H, –CH2–CH2–O–), 5.60 (s, 1 H, –CCH2), 6.08 (s, 1 H, –CCH2); Mn,NMR = 695 g mol−1, fNMR = 1. SEC: ĐSEC = 1.31, Mn,SEC = 424 g mol−1. MALDI-ToF-MS: ĐMALDI = 1.04, Mn,MALDI = 524 g mol−1.
CH2)–), 2.09 (br, 63 H, CH3–CO–), 2.94 and 3.04 (m, 3 H, CH3–N–), 3.51 (br, 82 H, –N–CH2–CH2–), 4.27 (br, 2 H, –CH2–CH2–O–), 5.60 (s, 1 H, –CCH2), 6.08 (s, 1 H, –CCH2); Mn,NMR = 1887 g mol−1, fNMR = 1. SEC: ĐSEC = 1.09, Mn,SEC = 2010 g mol−1. MALDI-ToF-MS: ĐMALDI = 1.02, Mn,MALDI = 1790 g mol−1.
CH2)–), 2.36 (br, 42 H, CH3–CH2–CO–), 2.95 and 3.02 (m, 3 H, CH3–N–), 3.50 (br, 82 H, –N–CH2–CH2–), 4.26 (br, 2 H, –CH2–CH2–O–), 5.58 (s, 1 H, –CCH2), 6.06 (s, 1 H, –CCH2); Mn,NMR = 2182 g mol−1, fNMR = 1. SEC: ĐSEC = 1.09, Mn,SEC = 2484 g mol−1. MALDI-ToF-MS: ĐMALDI = 1.02, Mn,MALDI = 2050 g mol−1.
CH2)–), 2.84 and 3.08 (br, 26 H, CH3–N– and (CH3)2–CH–), 3.70 (br, 90 H, –N–CH2–CH2–), 4.42 (br, 2 H, –CH2–CH2–O–), 5.74 (s, 1 H, –CCH2), 6.16 (s, 1 H, –CCH2); Mn,NMR = 2702 g mol−1, fNMR = 1. SEC: ĐSEC = 1.08, Mn,SEC = 3063 g mol−1. MALDI-ToF-MS: ĐMALDI = 1.02, Mn,MALDI = 2450 g mol−1.
ATRP of MeOx7-MA with different ligands and Cu(II) ratios
In general, MeOx7-MA, 2-hydroxyethyl-2-bromoisobutyrate, and the ligand were dissolved in 2 mL of water in a small tube in the desired ratio. The tube was sealed with a rubber septum and the solution was degassed using a nitrogen flow for 30 min. CuBr and CuBr2 were filled into another tube, sealed and evacuated/purged with dry nitrogen (3 times). Subsequently, the macromonomer, initiator, and ligand solution was transferred to the copper salts using a gas-tight syringe. The reaction mixture was stirred for 2 h at r.t. and after that diluted with water and lyophilized. The resulting colored powders were analyzed by SEC. For further details refer to Table 2.Synthesis of molecular brushes (P(Oxm-MA)n)
Interestingly, the work-up procedure for all molecular brushes was facile as the residual macromonomers as well as the remaining catalyst could be easily and quantitatively removed by column chromatography.
Characterization of molecular brushes was performed by SEC, SEC-MALLS and 1H-NMR spectroscopy. For a summary of the experimental conditions and analytical data please refer to Table 3.
Kinetic investigations
The sample for the kinetic investigation was prepared as described above for the synthesis of the molecular brushes. The initiator concentration was 5 mmol L−1, MeOx7-MA was used as a macromonomer and the reaction was conducted at r.t. Samples of 0.05 mL were taken periodically under an inert atmosphere at t = 0, 5, 10, 20, 40, 60, 90 and 120 min and immediately freeze-dried. Conversion, Mn,SEC and ĐSEC were determined for every sample using SEC.Chain extension experiments
In a glass tube, 198.7 mg (0.1053, 10.5 eq.) MeOx21-MA was dissolved in 1 mL of a 10 mM aqueous solution of 2-hydroxyethyl-2-bromoisobutyrate (2.1105 mg, 0.010 mmol, 1 eq.). The tube was sealed with a rubber septum and the solution was purged with nitrogen for 30 min. Meanwhile, 1.4 mg (0.0098 mmol, 1 eq.), CuBr, 20.1 mg (0.0899 mmol, 9.0 eq.), and CuBr2 and 34.7 mg (0.2222 mmol, 22.2 eq.) bpy were filled into a tube, sealed and evacuated/purged with dry nitrogen (3 times). Subsequently, the macromonomer–initiator solution was added to the solid components with a gas-tight syringe under Schlenk conditions. The reaction mixture was stirred for 1.5 h at r.t. An aliquot of 0.1 mL was taken to determine the conversion, molar mass and dispersity of the first reaction step (conv. = 90.8%, Mn,SEC = 18.7 kg mol−1, ĐSEC = 1.09). From the remaining solution, 0.8 mL were transferred to a degassed solution of 199.1 mg (0.0912 mmol, 11.4 eq.) EtOx21-MA in 0.9 mL water. Immediately afterwards a sample of 0.1 mL was taken. The remaining mixture was stirred for another 3 h. The work-up procedure was as described above. The copolymer brush was obtained as a white powder (183.5 mg, 51%). Conv. = 56.3%. 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 0.95 (br, 19 H, CH3–CH2–CO–), 1.91 (30 H, –CH2–C(CH3)– and CH3–CO–), 2.29 (br, 12 H, CH3–CH2–CO–), 2.81 (m) and 2.94 (d) (3 H, CH3–N–), 3.34 (br, 73 H, –N–CH2–CH2– and water). SEC: ĐSEC = 1.07, Mn,SEC = 24.6 kg mol−1.Acknowledgements
Financial support by the “Excellence Initiative by the German Federal and State Governments” (Institutional Strategy, measure “support the best”) is gratefully acknowledged.References
- S. S. Sheiko, B. S. Sumerlin and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 759–785 CrossRef CAS PubMed.
- C. Li, N. Gunari, K. Fischer, A. Janshoff and M. Schmidt, Angew. Chem., Int. Ed., 2004, 43, 1101–1104 CrossRef CAS PubMed.
- C. Tang, B. Dufour, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2007, 40, 6199–6205 CrossRef CAS.
- P. Zhao, L. Liu, X. Feng, C. Wang, X. Shuai and Y. Chen, Macromol. Rapid Commun., 2012, 33, 1351–1355 CrossRef CAS PubMed.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- M. Müllner, S. J. Dodds, T.-H. Nguyen, D. Senyschyn, C. J. H. Porter, B. J. Boyd and F. Caruso, ACS Nano, 2015, 9, 1294–1304 CrossRef PubMed.
- K. Aoi and M. Okada, Prog. Polym. Sci., 1996, 21, 151–208 CrossRef CAS.
- M. Barz, R. Luxenhofer, R. Zentel and M. J. Vicent, Polym. Chem., 2011, 2, 1900–1918 RSC.
- M. Bauer, S. Schroeder, L. Tauhardt, K. Kempe, U. S. Schubert and D. Fischer, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1816–1821 CrossRef CAS PubMed.
- R. Luxenhofer, G. Sahay, A. Schulz, D. Alakhova, T. K. Bronich, R. Jordan and A. V. Kabanov, J. Controlled Release, 2011, 153, 73–82 CrossRef CAS PubMed.
- T. X. Viegas, M. D. Bentley, J. M. Harris, Z. Fang, K. Yoon, B. Dizman, R. Weimer, A. Mero, G. Pasut and F. M. Veronese, Bioconjugate Chem., 2011, 22, 976–986 CrossRef CAS PubMed.
- B. L. Farrugia, K. Kempe, U. S. Schubert, R. Hoogenboom and T. R. Dargaville, Biomacromolecules, 2013, 14, 2724–2732 CrossRef CAS PubMed.
- N. Zhang, T. Pompe, I. Amin, R. Luxenhofer, C. Werner and R. Jordan, Macromol. Biosci., 2012, 12, 926–936 CrossRef CAS PubMed.
- R. Konradi, B. Pidhatika, A. Mühlebach and M. Textor, Langmuir, 2008, 24, 613–616 CrossRef CAS PubMed.
- M. C. Woodle, C. M. Engbers and S. Zalipsky, Bioconjugate Chem., 1994, 5, 493–496 CrossRef CAS.
- S. Zalipsky, C. B. Hansen, J. M. Oaks and T. M. Allen, J. Pharm. Sci., 1996, 85, 133–137 CrossRef CAS PubMed.
- F. C. Gaertner, R. Luxenhofer, B. Blechert, R. Jordan and M. Essler, J. Controlled Release, 2007, 119, 291–300 CrossRef CAS PubMed.
- T. B. Bonné, K. Lüdtke, R. Jordan and C. M. Papadakis, Macromol. Chem. Phys., 2007, 208, 1402–1408 CrossRef PubMed.
- R. Hoogenboom, F. Wiesbrock, M. A. M. Leenen, H. M. L. Thijs, H. Huang, C.-A. Fustin, P. Guillet, J.-F. Gohy and U. S. Schubert, Macromolecules, 2007, 40, 2837–2843 CrossRef CAS.
- R. Luxenhofer, A. Schulz, C. Roques, S. Li, T. K. Bronich, E. V. Batrakova, R. Jordan and A. V. Kabanov, Biomaterials, 2010, 31, 4972–4979 CrossRef CAS PubMed.
- S. Cesana, J. Auernheimer, R. Jordan, H. Kessler and O. Nuyken, Macromol. Chem. Phys., 2006, 207, 183–192 CrossRef CAS PubMed.
- M. W. M. Fijten, C. Haensch, B. M. van Lankvelt, R. Hoogenboom and U. S. Schubert, Macromol. Chem. Phys., 2008, 209, 1887–1895 CrossRef CAS PubMed.
- R. Jordan, K. Martin, H. J. Räder and K. K. Unger, Macromolecules, 2001, 34, 8858–8865 CrossRef CAS.
- S. Kobayashi and H. Uyama, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 192–209 CrossRef CAS PubMed.
- K. Lava, B. Verbraeken and R. Hoogenboom, Eur. Polym. J., 2015, 65, 98–111 CrossRef CAS PubMed.
- K. Lüdtke, R. Jordan, P. Hommes, O. Nuyken and C. A. Naumann, Macromol. Biosci., 2005, 5, 384–393 CrossRef PubMed.
- R. Luxenhofer, M. Bezen and R. Jordan, Macromol. Rapid Commun., 2008, 29, 1509–1513 CrossRef CAS PubMed.
- R. Luxenhofer and R. Jordan, Macromolecules, 2006, 39, 3509–3516 CrossRef CAS.
- M. Reif and R. Jordan, Macromol. Chem. Phys., 2011, 212, 1815–1824 CrossRef CAS PubMed.
- C. Taubmann, R. Luxenhofer, S. Cesana and R. Jordan, Macromol. Biosci., 2005, 5, 603–612 CrossRef CAS PubMed.
- Y. Shimano, K. Sato and S. Kobayashi, Polym. J., 1999, 31, 458–463 CrossRef CAS.
- Y. Shimano, K. Sato and S. Kobayashi, Polym. J., 1999, 31, 219–225 CrossRef CAS.
- G. David, V. Alupei, B. C. Simionescu, S. Dincer and E. Piskin, Eur. Polym. J., 2003, 39, 1209–1213 CrossRef CAS.
- M. Grasmüller, J. C. Rueda-Sanchez, B. I. Voit and O. Nuyken, Macromol. Symp., 1998, 127, 109–114 CrossRef PubMed.
- A. Gross, G. Maier and O. Nuyken, Macromol. Chem. Phys., 1996, 197, 2811–2826 CrossRef CAS PubMed.
- B. Guillerm, V. Darcos, V. Lapinte, S. Monge, J. Coudane and J.-J. Robin, Chem. Commun., 2012, 48, 2879–2881 RSC.
- S. Kobayashi, M. Kaku, S. Sawada and T. Saegusa, Polym. Bull., 1985, 13, 447–451 CrossRef CAS.
- S. Kobayashi, Y. Shimano and T. Saegusa, Polym. J., 1991, 23, 1307–1315 CrossRef CAS.
- O. Nuyken, J. Rueda-Sanchez and B. Voit, Polym. Bull., 1997, 38, 657–664 CrossRef CAS.
- O. Nuyken, J. R. Sanchez and B. Voit, Macromol. Rapid Commun., 1997, 18, 125–131 CrossRef CAS PubMed.
- J. Rueda, S. Zschoche, H. Komber, D. Schmaljohann and B. Voit, Macromolecules, 2005, 38, 7330–7336 CrossRef CAS.
- Y. Shimano, K. Sato, D. Fukui, Y. Onodera and Y. Kimura, Polym. J., 1999, 31, 296–302 CrossRef CAS.
- S.-I. Shoda, E. Masuda, M. Furukawa and S. Kobayashi, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 1489–1494 CrossRef CAS PubMed.
- P. Trivedi and D. Schulz, Polym. Bull., 1980, 3, 37–44 CrossRef CAS.
- H. Uyama, Y. Honda and S. Kobayashi, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 123–128 CrossRef CAS PubMed.
- R. Weberskirch, R. Hettich, O. Nuyken, D. Schmaljohann and B. Voit, Macromol. Chem. Phys., 1999, 200, 863–873 CrossRef CAS.
- M. M. Bloksma, C. Weber, I. Y. Perevyazko, A. Kuse, A. Baumgärtel, A. Vollrath, R. Hoogenboom and U. S. Schubert, Macromolecules, 2011, 44, 4057–4064 CrossRef CAS.
- C. Weber, C. R. Becer, R. Hoogenboom and U. S. Schubert, Macromolecules, 2009, 42, 2965–2971 CrossRef CAS.
- C. Weber, M. Wagner, D. Baykal, S. Hoeppener, R. M. Paulus, G. Festag, E. Altuntas, F. H. Schacher and U. S. Schubert, Macromolecules, 2013, 46, 5107–5116 CrossRef CAS.
- J. Bühler, S. Muth, K. Fischer and M. Schmidt, Macromol. Rapid Commun., 2013, 34, 588–594 CrossRef PubMed.
- N. Zhang, S. Huber, A. Schulz, R. Luxenhofer and R. Jordan, Macromolecules, 2009, 42, 2215–2221 CrossRef CAS.
- N. Zhang, S. Salzinger, B. S. Soller and B. Rieger, J. Am. Chem. Soc., 2013, 135, 8810–8813 CrossRef CAS PubMed.
- C. Weber, A. Krieg, R. M. Paulus, H. M. L. Lambermont-Thijs, C. R. Becer, R. Hoogenboom and U. S. Schubert, Macromol. Symp., 2011, 308, 17–24 CrossRef CAS PubMed.
- C. Weber, S. Rogers, A. Vollrath, S. Hoeppener, T. Rudolph, N. Fritz, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 139–148 CrossRef CAS PubMed.
- N. Zhang, R. Luxenhofer and R. Jordan, Macromol. Chem. Phys., 2012, 213, 973–981 CrossRef CAS PubMed.
- N. Zhang, R. Luxenhofer and R. Jordan, Macromol. Chem. Phys., 2012, 213, 1963–1969 CrossRef CAS PubMed.
- J. Bühler, S. Gietzen, A. Reuter, C. Kappel, K. Fischer, S. Decker, D. Schäffel, K. Koynov, M. Bros and I. Tubbe, et al. , Chem. – Eur. J., 2014, 20, 12405–12410 CrossRef PubMed.
- S. Averick, A. Simakova, S. Park, D. Konkolewicz, A. J. D. Magenau, R. A. Mehl and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 6–10 CrossRef CAS.
- X.-S. Wang and S. P. Armes, Macromolecules, 2000, 33, 6640–6647 CrossRef CAS.
- S. Kobayashi, E. Masuda, S. Shoda and Y. Shimano, Macromolecules, 1989, 22, 2878–2884 CrossRef CAS.
- M. Miyamoto, K. Naka, M. Tokumizu and T. Saegusa, Macromolecules, 1989, 22, 1604–1607 CrossRef CAS.
- C. Weber, R. C. Becer, A. Baumgaertel, R. Hoogenboom and U. S. Schubert, Des. Monomers Polym., 2009, 12, 149–165 CrossRef CAS PubMed.
- R. Hoogenboom, H. M. L. Thijs, M. J. H. C. Jochems, B. M. van Lankvelt, M. W. M. Fijten and U. S. Schubert, Chem. Commun., 2008, 5758–5760 RSC.
- S. Huber and R. Jordan, Colloid Polym. Sci., 2008, 286, 395–402 CAS.
- P. Lin, C. Clash, E. M. Pearce, T. K. Kwei and M. A. Aponte, J. Polym. Sci., Part B: Polym. Phys., 1988, 26, 603–619 CrossRef CAS PubMed.
- J.-S. Park and K. Kataoka, Macromolecules, 2006, 39, 6622–6630 CrossRef CAS.
- F. Rehfeldt, M. Tanaka, L. Pagnoni and R. Jordan, Langmuir, 2002, 18, 4908–4914 CrossRef CAS.
- C. Weber, R. Hoogenboom and U. S. Schubert, Prog. Polym. Sci., 2012, 37, 686–714 CrossRef CAS PubMed.
- H. Bergenudd, G. Coullerez, M. Jonsson and E. Malmström, Macromolecules, 2009, 42, 3302–3308 CrossRef CAS.
- W. Tang and K. Matyjaszewski, Macromolecules, 2006, 39, 4953–4959 CrossRef CAS.
- N. V. Tsarevsky, T. Pintauer and K. Matyjaszewski, Macromolecules, 2004, 37, 9768–9778 CrossRef CAS.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS PubMed.
- W. Wang, J. Zhao, N. Zhou, J. Zhu, W. Zhang, X. Pan, Z. Zhang and X. Zhu, Polym. Chem., 2014, 5, 3533–3546 RSC.
- T. Zhang, Y. Du, F. Müller, I. Amin and R. Jordan, Polym. Chem., 2015, 6, 2726–2733 RSC.
- Y. Zhang, Y. Wang, C. Peng, M. Zhong, W. Zhu, D. Konkolewicz and K. Matyjaszewski, Macromolecules, 2012, 45, 78–86 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |