
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and self-assembly of a fluorine-containing amphiphilic graft copolymer bearing a perfluorocyclobutyl aryl ether-based backbone and poly(acrylic acid) side chains†
Hao
Liu‡
a,
Sen
Zhang‡
a,
Chun
Feng
*a,
Yongjun
Li
ab,
Guolin
Lu
a and
Xiaoyu
Huang
*a
aKey Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules and Key Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, People's Republic of China. E-mail: cfeng@mail.sioc.ac.cn; xyhuang@mail.sioc.ac.cn; Fax: +86-21-64166128; Tel: +86-21-54925520, +86-21-54925310
bState Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, 220 Handan Road, Shanghai 200433, People's Republic of China
First published on 29th April 2015
Abstract
A series of fluorine-containing amphiphilic graft copolymers consisting of a semi-fluorinated poly(2-methyl-1,4-bistrifluorovinyloxybenzene) (PMBTFVB) backbone and hydrophilic poly(acrylic acid) (PAA) side chains was synthesized by the combination of thermal cycloaddition polymerization and atom transfer radical polymerization (ATRP) through the grafting-from strategy. 2-Methyl-1,4-bistrifluorovinyloxybenzene was first homopolymerized via thermal step-growth [2π + 2π] cycloaddition polymerization to form a perfluorocyclobutyl aryl ether-based backbone. This fluoropolymer was transformed into the macroinitiator with the controllable density of the initiating functionality by the mono-bromination of pendant methyls. The target PMBTFVB-g-PAA amphiphilic graft copolymers were achieved by ATRP of tert-butyl acrylate initiated by the macroinitiator followed by the acidolysis of hydrophobic PtBA side chains into hydrophilic PAA segments. Critical micelle concentrations (cmc) of these amphiphilic graft copolymers were determined by fluorescence spectroscopy using N-phenyl-1-naphthylamine as fluorescent probe. Self-assembly behaviors of these fluorine-containing amphiphilic graft copolymers in aqueous media were investigated by transmission electron microscopy (TEM). Diverse micellar morphologies including vesicular, worm-like, and bowl-shaped nanostructures were obtained through tuning the water content and the length of PAA side chain.
Introduction
The self-assembly behavior of amphiphilic copolymers in aqueous media has attracted considerable attention over the past few decades.1,2 This interest is sustained due to their potential applications in a variety of fields including solubilizers,3 drug delivery,4–7 catalysis,8 and microelectronics9,10 for their special physicochemical properties. Morphologies of amphiphilic copolymers are affected not only by the nature and/or length of the chain segment,11 but also by the experimental parameters during the preparation of micelles such as pH value, ionic strength of the solution, micelle preparation conditions, initial copolymer concentration12,13 and so on. As one special class of amphiphilic copolymers, amphiphilic fluorinated copolymers have aroused significant interest since partial fluorination can result in favorable properties such as chemical resistance, low surface energy, and excellent mechanical properties, which can make them to be applied in anti-biofouling surface materials,14,15 proton exchanged films,16 surfactants17 and so on, compared to the non-fluorinated amphiphilic copolymers. Moreover, the very hydrophobic and solvophobic nature of fluoropolymer segments may lead to the formation of unique morphologies during the micellization in aqueous solutions. For instance, Li et al. synthesized μ-[poly(ethylene)][poly(ethylene oxide)][poly(perfluoropropylene oxide)] (μ-EOF) miktoarm star terpolymers, which had a water-soluble poly(ethylene oxide) and two hydrophobic but immiscible components (a polymeric hydrocarbon and a perfluorinated polyether) connected at a common junction.18,19 They systematically studied the aqueous self-assembly behavior and found that upon decreasing the length of the hydrophilic block (O), the resulting micelles evolved from “hamburger” micelles to segmented worms and ultimately to nanostructured bilayers and vesicles. When without the fluoro-segment (F), in comparison with the above-mentioned newly found multicompartment micellar structures, three classical morphologies, i.e. spheres, cylinders, and bilayer vesicles, were generally observed through decreasing the length of the hydrophilic block (O). Tan et al. reported the synthesis and self-assembly of brush-type poly[poly(ethylene glycol)methyl ether methacrylate]-block-poly(pentafluorostyrene) amphiphilic diblock copolymers in aqueous solution, and found the micellar structure comprising of brush-like polymers (hydrophobic main chain and hydrophilic branches) as the coronas and poly(pentafluorostyrene) as the core.20 Mao et al. prepared amphiphilic hyperbranched star-block copolymers containing polycations and the fluoropolymer segment, which could directly self-organize into multicompartment micelles with different diameters.21However, most previous research studies were focused on the synthesis and self-assembly behavior of amphiphilic block copolymers based on fluoropolymers with flexible fluorinated alkane side chains/substituents while trifluorovinyl-containing amphiphilic graft copolymers were rarely studied due to the synthetic difficulties.20–28 Because of their complicated and confined structure, fluorine-containing amphiphilic graft copolymers may form diverse micellar morphologies, which is significant in understanding the structure–property correlation and exploring new materials.29–31 In general, three different strategies, i.e. grafting-through, grafting-onto, and grafting-from, have been employed for synthesizing graft copolymers.32–35 Thanks to the advent of reversible-deactivation radical polymerization (RDRP), including atom transfer radical polymerization (ATRP),36–38 reversible addition–fragmentation chain transfer (RAFT) polymerization,39 nitroxide-mediated polymerization (NMP),40 and single-electron-transfer living radical polymerization (SET-LRP),41 the grafting-from strategy, through which the side chains were formed via RDRP initiated by the pendant initiating groups on the backbone, has become the most popular approach for the synthesis of graft copolymers.42
Perfluorocyclobutyl (PFCB) aryl ether-based polymer is a relatively new class of partially fluorinated polymers, which was first prepared by Babb et al. of Dow Chemical Co. in 1993 through the predominant head-to-head thermal [2π + 2π] step-growth cyclopolymerization of the aryl trifluorovinyl ether (TFVE) monomer at 150–250 °C without any initiator and catalyst.43,44 This kind of semi-fluorinated polymer not only possesses the common properties of fluoropolymers, but shows many other advantages such as optical transparency and improved processability due to the combination of flexible and thermally robust aromatic ethers with a fluorocarbon linkage. However, a very high polymerization temperature (150–250 °C) with an unusual cyclopolymerization mechanism was necessary for the preparation of this class of fluoropolymers in comparison with the commonly used commercial monomers.45 Therefore, our group has developed some approaches to modify PFCB aryl ether-based polymers by combining them with other commercial polymers.46,47 In particular, none has investigated the self-assembly behavior of amphiphilic copolymers bearing a PFCB aryl ether-based segment until now, which may self-assemble into peculiar nanostructures originating from their unique structure by combining flexible and thermally robust aromatic ethers with a fluorocarbon linkage.
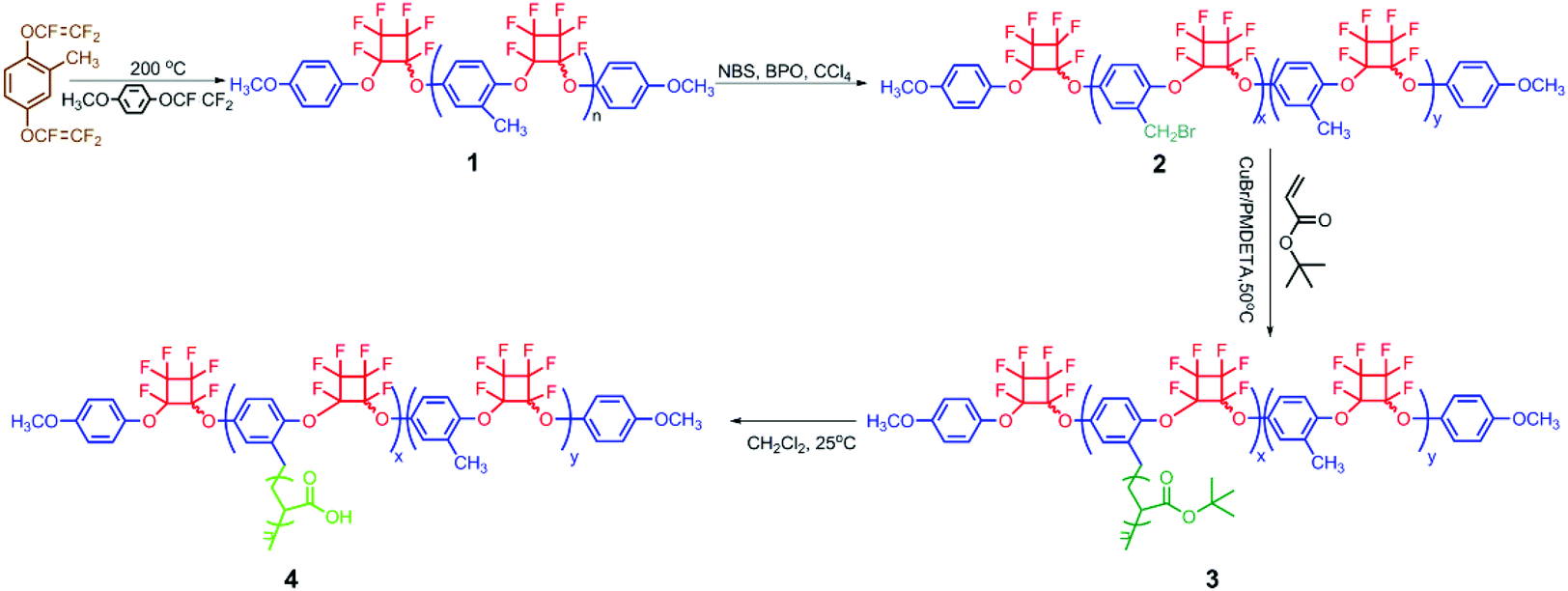
In our previous work, the grafting-from strategy has been utilized to synthesize hydrophobic PFCB aryl ether-based graft copolymers containing the poly(2-methyl-1,4-bistrifluorovinyloxybenzene) (PMBTFVB) backbone via the combination of thermal cyclopolymerization of the MBTFVB monomer and ATRP.48,49 The present work is focused on the synthesis and self-assembly behavior of brush-type amphiphilic PFCB aryl ether-based graft copolymers. The target poly(2-methyl-1,4-bistrifluorovinyloxy-benzene)-g-poly(acrylic acid) (PMBTFVB-g-PAA) amphiphilic graft copolymers were obtained by the acidolysis of hydrophobic poly(tert-butyl acrylate) (PtBA) side chains of poly(2-methyl-1,4-bistrifluorovinyloxybenzene)-g-poly(tert-butyl acrylate) (PMBTFVB-g-PtBA) graft copolymers (Scheme 1), which were prepared by successive thermal cyclopolymerization of MBTFVB and ATRP of tBA. Diverse self-assembled morphologies with the change of the water content were preliminarily explored by transmission electron microscopy (TEM).
 | ||
| Scheme 1 Synthesis of PMBTFVB-g-PAA amphiphilic graft copolymer. | ||
Results and discussion
Synthesis of PMBTFVB-g-PtBA graft copolymers
PMBTFVB-Br 2 macroinitiators with different amounts of benzyl bromide ATRP initiating groups were synthesized on the basis of our previous reports48,49 as shown in Scheme 1 and the results are summarized in Table S1 (see ESI†).PMBTFVB-g-PtBA 3 graft copolymers, aiming to afford PMBTFVB-g-PAA 4 amphiphilic graft copolymers via the acidic hydrolysis of hydrophobic PtBA backbone, were synthesized by ATRP of tBA initiated by PMBTFVB-Br 2 macroinitiators using CuBr/PMDETA as the catalytic system and anisole as the solvent at 80 °C through the grafting-from strategy. The experimental details as well as the molecular weights and molecular weight distributions of the copolymers are summarized in Table 1. It was found that the molecular weights of the resulting copolymers were all much higher than those of PMBTFVB-Br 2 macroinitiators, which indicated that ATRP of tBA was indeed performed. With the increasing polymerization time, the molecular weights of graft copolymers also increased, which accorded with the characteristics of ATRP.37 In the current case, a relatively high feeding ratio of tBA to the benzyl bromide initiating group (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and a relatively low conversion of tBA (<25%) were employed to suppress the intermolecular coupling reactions according to previous reports.50–55 All graft copolymers showed unimodal and symmetrical elution peaks (Fig. S1, see ESI†) with relatively narrow molecular weight distributions (Mw/Mn ≤ 1.37), which confirmed that the intermolecular coupling reaction could be neglected.52
:1) and a relatively low conversion of tBA (<25%) were employed to suppress the intermolecular coupling reactions according to previous reports.50–55 All graft copolymers showed unimodal and symmetrical elution peaks (Fig. S1, see ESI†) with relatively narrow molecular weight distributions (Mw/Mn ≤ 1.37), which confirmed that the intermolecular coupling reaction could be neglected.52
| Sample | Time (min) |
M
n,GPCe (kDa) |
M
w/Mne |
N
tBAf |
n
tBAg |
M
n,NMRh (kDa) |
|---|---|---|---|---|---|---|
|
a Polymerization temperature: 80 °C; feeding ratio: [tBA]:[CH2Br group]:[CuBr]:[PMDETA] = 100:1:1:1; Vanisole:VtBA = 1:2
b Initiated by PMBTFVB-Br 2a.
c Initiated by PMBTFVB-Br 2b.
d Initiated by PMBTFVB-Br 2c.
e Measured by GPC in THF at 35 °C.
f The total number of tBA repeating units obtained from 1H NMR spectra.
g The number of tBA repeating units per PtBA side chain obtained from 1H NMR spectra.
h Obtained from 1H NMR spectra.
|
||||||
| 3a | 30 | 10.5 | 1.32 | 89.7 | 11.8 | 17.3 |
| 3b | 60 | 14.6 | 1.32 | 149.9 | 19.7 | 25.1 |
| 3c | 15 | 11.3 | 1.37 | 147.8 | 10.2 | 24.8 |
| 3d | 30 | 17.3 | 1.31 | 257.4 | 17.8 | 38.9 |
| 3e | 15 | 18.2 | 1.35 | 279.3 | 13.7 | 41.7 |
1H NMR spectrum of the obtained polymer provided further evidence for the successful graft copolymerization of tBA. As shown in Fig. 1A, a characteristic strong peak corresponding to the resonance signal of 9 protons of tert-butyl in PtBA repeating unit appeared at 1.36 ppm after ATRP of tBA. The signal at 1.71 ppm was attributed to 2 protons of CH2CHCO2 moiety of PtBA side chains, which was partly overlapped with the strong peak of tert-butyl. The typical signals of terminal methoxyl and benzene rings of the backbone were still located at 3.66 and 6.92–7.26 ppm after ATRP of tBA. Furthermore, a new strong peak originating from 3 primary carbons in tert-butyl appeared at 27.5 ppm in 13C NMR spectrum after ATRP of tBA, while the resonance signal at 80.5 ppm corresponded to the quaternary carbon in tert-butyl. The typical signal of carbonyl of PtBA side chains was found to be located at 173.3 ppm. A series of peaks ranging from 105.0 ppm to 111.7 ppm represented 4 quaternary carbons in PFCB moiety. The presence of PFCB linkage in PMBTFVB-g-PtBA 3 copolymer was also verified by a series of peaks between −126.6 ppm and −133.0 ppm in 19F NMR spectrum after graft copolymerization of tBA. Therefore, all the aforementioned points confirmed the chemical structure of PMBTFVB-g-PtBA 3 graft copolymer.
 | ||
| Fig. 1 1H NMR spectra of PMBTFVB-g-PtBA 3 in acetone-d6 (A) and PMBTFVB-g-PAA 4 in DMSO-d6 (B). | ||
Importantly, as we can see from Fig. 1A, no signal appeared in the range of 4.00 ppm to 5.00 ppm after ATRP of tBA while the signals originating from 2 protons of –CH2Br initiating group appeared at 4.16 and 4.34 ppm in 1H NMR spectrum before ATRP of tBA. Furthermore, the resonance signal of the carbon of –CH2Br initiating group at 24.5 ppm in 13C NMR spectrum of PMBTFVB-Br 2 was absent in 13C NMR spectrum of PMBTFVB-g-PtBA 3. Both facts clearly showed that all –CH2Br initiating groups in the backbone initiated ATRP of tBA forming PtBA side chains, i.e. the number of grafted PtBA side chains after ATRP of tBA equaled to those of –CH2Br initiating group in the backbone before ATRP of tBA. Therefore, 1H NMR was used to determine the length of PtBA side chains in PMBTFVB-g-PtBA 3 graft copolymer (ntBA), because Mn,GPC obtained by conventional GPC, which is calibrated with linear polystyrene standards, is generally lower than the real value.56 The total number of tBA repeating units (NtBA) and ntBA were calculated according to eqn (1) (Sc, Sd, and SAr–H are the integration areas of peak ‘c’ at 1.71 ppm, peak ‘d’ at 1.36 ppm, and peak ‘Ar–H’ ranging from 6.92 ppm to 7.26 ppm in Fig. 2A, respectively. 28.3 is the number of MBTFVB repeating units in the backbone) and eqn (2) (NATRP is the number of ATRP initiating groups per chain), respectively, and the results are listed in Table 1.
| NtBA = [(Sc + Sd)(28.3 × 3 + 8)]/(11 × SAr–H) | (1) |
| ntBA = NtBA/NATRP | (2) |
 | ||
| Fig. 2 FT-IR spectra of PMBTFVB-g-PtBA 3 (A) and PMBTFVB-g-PAA 4 (B). | ||
Additionally, one can see from Table 1 that the molecular weight of graft copolymers obtained from 1H NMR was much higher than that measured by conventional GPC. This should be attributed to the non-linear architecture of graft copolymers, leading to a lower apparent molecular weight in GPC. This phenomenon was consistent with previous reports.54–56 Although Peng et al. reported that C–F groups of perfluorosulfonic acid-based polymers could serve as initiating groups for ATRP using CuBr/2,2′-bipyridine as the catalytic system,57 we found that trifluorovinyl of PMBTFVB backbone could hardly act as the initiating group for ATRP of tBA employing CuBr/PMDETA as the catalytic system in the current case.
Thus, it can be concluded that a series of PMBTFVB-g-PtBA 3 graft copolymers consisting of a PFCB aryl ether-based backbone (28.3 repeating units) and PtBA side chains (11.8–19.7 repeating units per chain) were successfully synthesized by ATRP of tBA initiated by PMBTFVB-Br 2 macroinitiator.
Synthesis of PMBTFVB-g-PAA amphiphilic graft copolymer
It is well-known that hydrophobic PtBA can be easily converted to hydrophilic PAA via acidic hydrolysis. So, PMBTFVB-g-PAA amphiphilic graft copolymer with PAA side chains can be obtained from the precursor, PMBTFVB-g-PtBA. In the current case, CF3COOH was used to hydrolyze tert-butyoxycarbonyls of PtBA side chains in PMBTFVB-g-PtBA graft copolymer into carboxyls in CH2Cl2 at room temperature according to previous reports.58–60 The structure of the hydrolyzed product was characterized by 1H NMR and FT-IR. 1H NMR spectrum showed the complete hydrolysis of tert-butyoxycarbonyls as indicated by the disappearance of the resonance signal of 9 protons at 1.36 ppm (peak ‘d’ in Fig. 1A) and the appearance of 1 proton in –COOH at 12.30 ppm (peak ‘d’ in Fig. 1B). It also can be seen from Fig. 2A that a new broad peak attributed to carboxyl appeared at 3386 cm−1 after the hydrolysis, which was absent before the hydrolysis (Fig. 2B). Moreover, typical signals of PMBTFVB backbone remained unchanged in both 1H NMR and FT-IR spectra, which illustrated that PMBTFVB segment remained inert during the hydrolysis. Furthermore, the thermal stability of PMBTFVB-g-PtBA 3 and PMBTFVB-g-PAA 4 graft copolymers was investigated by TGA in N2 with a heating rate of 10 °C min−1 and typical TG (thermogravimetry) and DTG (derivative thermogravimetry) curves are shown in Fig. S2 (see ESI†). One can notice that a remarkable weight loss (40%) occurred at around 228 to 246 °C for PMBTFVB-g-PtBA 3 graft copolymer, which was a typical degradation pattern of PtBA,61 while just a 10% weight loss was found for copolymer 4 under the same conditions. Thus, it is clear based on the above-mentioned results that PMBTFVB-g-PAA graft copolymers were successfully obtained from the acidic hydrolysis of PMBTFVB-g-PtBA graft copolymers without affecting the semi-fluorinated backbone.cmc of PMBTFVB-g-PAA amphiphilic graft copolymer
The amphiphilic nature of PMBTFVB-g-PAA 4 graft copolymer, containing a hydrophobic PMBTFVB backbone and hydrophilic PAA side chains, endowed the copolymer with the capability of forming a nanostructure in aqueous media via self-assembly. A fluorescence probe technique, using PNA as the probe, was utilized to determine the critical micelle/aggregation concentration (cmc/cac) of PMBTFVB-g-PAA 4 amphiphilic graft copolymer. The fluorescence spectrum of PNA is sensitively affected by the environment and the polarity of its surrounding. PNA displays higher fluorescence activity in nonpolar environments and its fluorescence can be quenched very easily by polar solvents such as water. In the presence of micelles, PNA is solubilized within the interior of the hydrophobic part and the fluorescence intensity will increase with the rising of the concentration of the polymer. Fluorescence emission spectra of PNA in aqueous solutions of PMBTFVB-g-PAA 4b graft copolymer at different concentrations are shown in Fig. 3A and the relationship of the fluorescence intensity ratio (I/I0) of PNA as a function of the concentration of PMBTFVB-g-PAA 4b graft copolymer is plotted in Fig. 3B. The I/I0 ratios were almost constant while the concentration of graft copolymer 4b was below a certain value. However, the value of I/I0 increased sharply when the concentration of graft copolymer 4b exceeded a certain value, which evidenced the incorporation of PNA probe into the hydrophobic region of micelles. Therefore, cmc/cac of PMBTFVB-g-PAA 4b graft copolymer was determined to be the intersection of two straight lines with a value of 1.80 × 10−5 g mL−1. The cmc/cac values of PMBTFVB-g-PAA 4 graft copolymers are listed in Table 2 and these values were certainly comparable to those of polymeric amphiphiles.8,62 | ||
| Fig. 3 (A) Fluorescence spectra of PNA in aqueous solutions of PMBTFVB-g-PAA 4b and (B) the dependence of the fluorescence intensity ratio of PNA emission band at 418 nm on the concentration of PMBTFVB-g-PAA 4b in pure water. | ||
| PMBTFVB-g-PtBA | PMBTFVB-g-PAA | cmc (g mL−1) |
|---|---|---|
| a Determined by fluorescence spectroscopy using PNA as the probe, excitation wavelength: 340 nm. | ||
| 3a | 4a | 1.26 × 10−5 |
| 3b | 4b | 1.80 × 10−5 |
| 3c | 4c | 6.85 × 10−6 |
| 3d | 4d | 7.29 × 10−6 |
| 3e | 4e | 1.38 × 10−5 |
The ionic strength and pH value have been reported to be important factors to affect the cmc of amphiphilic copolymers. The ionic strength of the solution will increase by adding inorganic salt so that it can influence the hydrophobic segment to make the cmc descend or ascend. In this work, the effect of salt concentration on the cmc of PMBTFVB-g-PAA 4b graft copolymer was explored in the region between 0 and 0.513 M. As shown in Fig. 4A, the value of cmc quickly dropped with the rising of the concentration of NaCl. This phenomenon can be explained that the addition of NaCl resulted in a salting-out effect on the hydrophobic PMBTFVB backbone to increase the hydrophobicity of PMBTFVB segment so that the hydrophobic segments were easier to aggregate to form micelles with the lowering of cmc. A screened electrostatic field of the partially charged PAA segments in the presence of NaCl may also have contributions to this effect. Moreover, it was found that logcmc decreased linearly with the square root of the salt concentration (Fig. 4B), which was similar to a previous report.63
 | ||
| Fig. 4 (A) Relationship between cmc of PMBTFVB-g-PAA 4b and [NaCl]. (B) Linear dependence of logcmc of PMBTFVB-g-PAA 4b on [NaCl]1/2. | ||
Self-assembled nanostructures
Diverse micellar morphologies formed by PMBTFVB-g-PAA 4a copolymer with different water contents when the initial concentration of copolymer 4a was 1 mg mL−1 are shown in Fig. 5. The copolymer first aggregated to form vesicles (ca. 400–1100 nm), not large compound micelles with 25 wt% water content (Fig. 5A) since the edge of the object was much darker and a ring-like image was a typical feature of vesicles in TEM observation. Interestingly, one can note that there was a black dot with a diameter of 120 nm in the center of each vesicle as indicated by red arrows (Fig. 5D). When the water content was raised to 50 wt%, bowl-shaped aggregates were formed with diameters in the range of 250–500 nm and the depths in the range of 120–250 nm (Fig. 5B). At a high water content of 70 wt%, only the aggregates with network morphologies were found in Fig. 5C. Thus, micellar morphologies of PMBTFVB-g-PAA 4 copolymer can be tuned by adjusting the water content in a relatively wide range between 25 wt% and 70 wt%. | ||
| Fig. 5 TEM images of aggregates formed by PMBTFVB-g-PAA 4a graft copolymer with different water contents, the initial content of the copolymer in THF: 1 mg mL−1, (A) 25 wt%, (B) 50 wt%, (C) 70 wt%, and (D) partially enlarged region of (A). | ||
Since the glass transition temperature (Tg) of PMBTFVB backbone was about 53.8 °C much higher than room temperature, which was measured by DSC (Fig. S3, see ESI†), the addition of water would decrease and finally freeze the mobility of PMBTFVB backbone. A higher water content before dialysis would result in a lower mobility of PMBTFVB backbone, thus presumably leading to kinetically trapped morphology. Our previous result on the self-assembly of graft copolymers containing PAA backbone and poly(propylene oxide) side chains in aqueous media showed that the graft copolymer could form vesicles and black dots were also observed at the center of each vesicle.64 Although the exact reason for the formation of black dots was unknown, we speculated that this might result from the contraction of the shell of vesicles during the sample preparation for TEM. During the contraction of the shell, the possibility of the shell of vesicles contracting to the center should be equal, and thus there were more polymer chains at the center and dark dots were then observed in TEM. The formation of a bowl-shaped structure from PMBTFVB-g-PAA 4a copolymer may involve a mechanism similar to that proposed by Riegel.65 As the addition of water progressed, the quality of the solvent for the hydrophobic PMBTFVB backbone worsened, leading to the formation of spherical aggregates. When the water content was increased to 50 wt%, the internal viscosity of spheres with a soft skin was relatively low, resulting in a rapid solvent diffusion in and out of the soft skin and a homogeneous shrinkage of the whole sphere. As a result, large-compound micelles were formed, which were composed of an assembly of reverse micelles (containing islands of hydrophilic PAA chains in the continuous phase of PMBTFVB backbone). Upon continuous addition of water to the assembling system, more solvents (THF) were extracted from the core of spheres, bringing on the hardened external shell due to the increase of Tg of PMBTFVB backbone. Therefore, spaces (bubbles) devoid of polymer aggregates were formed when the solvent was extracted through the shell because this hardened skin prevented the homogeneous shrinkage of the whole precursor sphere. If the internal viscosity was appropriate, solvent/water-filled bubbles within the structure have the chance to coalesce to a single large bubble and bowl-shaped micelles were formed. But when the internal viscosity is too large, the bubbles will not have a chance to coalesce, so porous spheres will be formed. According to the above analysis, the internal viscosity, which must be within a very narrow range, of the aggregates has a crucial effect on the formation of bowl-shaped micelles. Eisenberg et al. successfully prepared bowl-shaped micelles by effectively adjusting the internal viscosity through utilizing the ionic cross-linkers between the amphiphilic triblock copolymer with amino groups.66 The viscosity-control mechanism was also demonstrated by Wang et al.67 who observed the bowl-shaped aggregates from a rigid polyimide homopolymer containing carboxyl end groups. The internal viscosity of LCMs formed from the random copolymers of P(St-co-MAA) could also be adjusted by hydrogen bonding between MAA units to prepare the bowl-shaped micelles.67 For our system, the rigid PMBTFVB backbone together with hydrogen bonding between carboxyls in PAA side chains provided the added viscosity-control mechanism.
We also examined the influence of the initial copolymer concentration and the length of PAA side chains on the self-assembled behaviors of PMBTFVB-g-PAA graft copolymers employing a similar micelle preparation method. As the initial content of the copolymer decreased to 0.2 mg mL−1 with 25 wt% water content, worm-like micelles with a length of several micrometers and a width of about 200 nm were formed by PMBTFVB-g-PAA 4a graft copolymer as shown in Fig. 6A. When more water (50 wt%) was added before dialysis, a network of worm-like micelles was formed by using the graft copolymer 4a (Fig. 6B). A similar tendency was also observed for the self-assembly of poly(γ-benzyl-L-glutamate)-g-poly(ethylene glycol) graft copolymer investigated by Lin et al.68 They found that as the initial concentration of the copolymer decreased from 0.25 mg mL−1 to 0.05 mg mL−1, the morphology of aggregates transformed from vesicles to a mixture of vesicles and spindles. This observation was also consistent with previous reports on the self-assembly of amphiphilic block copolymers and homopolymers.2,69,70 As the polymer concentration decreased, less polymer chains could get into the micelles in the metastable state, when polymer chains of micelle cores were not completely frozen. Thus, a lower polymer concentration would lead to a decrease in the aggregation number of micelles, which facilitated the morphology transformation from bilayers (vesicles) to worm-like micelles. For PMBTFVB-g-PAA 4b graft copolymer with longer PAA side chains, ring-like images were found as shown in Fig. 6C, which was a typical feature of vesicles. The diameter of vesicles was in the range of 800 nm to 1200 nm and there was a darker dot in each vesicle as well. Irregular rod-like network aggregates were formed with a width of about 300 nm as the initial water content was raised to 50 wt%, while bowl-shaped micelles were formed for PMBTFVB-g-PAA 4a graft copolymer under the same conditions. Although the repeating unit of PAA side chains just increased from 12 to 20, the morphology transformed from bowl-shaped micelles to irregular rod-like network aggregates. This observation further indicated that the viscosity control mechanism for the formation of bowl-shaped aggregates was very sensitive,65–67 which seemed to be affected by the length of PAA side chains.
 | ||
| Fig. 6 TEM images of micelles formed by PMBTFVB-g-PAA 4a and 4b graft copolymers with different water contents, the initial copolymer concentration: 0.2 mg mL−1, 4a: (A) 25 wt% and (B) 50 wt%, 4b: (C) 25 wt% and (D) 50 wt%. | ||
It is worth noting that the size distribution of the formed vesicles by PMBTFVB-g-PAA 4a and 4b graft copolymers was relatively narrow, which is not common for the self-assembly of copolymers. Zhou et al. reported the formation of vesicles with narrow size distribution by using Janus hyperbranched polymers containing a hydrophobic hyperbranched poly(3-ethyl-3-oxetanemethanol) segment and hydrophilic hyperbranched polyglycerol.71 The dynamics and mechanism for the formation of vesicles and their bilayer structure were simulated by a dissipative particle dynamics model.70,71 They speculated that the amphiphilic Janus hyperbranched polymers acted as building blocks the same as amphiphilic block copolymers, and formed a bilayer of the vesicle. Thus, in order to decrease the overall steric repulsion between the hyperbranched building blocks, only a specific curvature would match the shape of Janus hyperbranched copolymers and lead to the lowest steric repulsion. In our case, PMBTFVB-g-PAA 4 amphiphilic graft copolymers also serve as building blocks for the formation of nano- or micro-objects in the self-assembly in water as shown in Scheme 2. The amphiphilic graft copolymer first formed unimolecular aggregates in water, and then these aggregates self-assembled into vesicles under certain conditions. Since the graft copolymer might be similar to the amphiphilic hyperbranched copolymer reported by Zhou et al.71,72 and had a specific shape during the formation of vesicles, only a specific curvature match well with this structure and could lead to the lowest steric repulsion between PAA domains. Thus, the size distribution of vesicles formed by PMBTFVB-g-PAA 4 amphiphilic graft copolymer was relatively narrow.
 | ||
| Scheme 2 Schematic illustration of vesicle formation by PMBTFVB-g-PAA 4. | ||
Conclusions
This report provides the synthesis and self-assembly studies on a series of PMBTFVB-based semi-fluorinated amphiphilic graft copolymers, PMBTFVB-g-PAA. Target amphiphilic graft copolymers were prepared by the acidolysis of hydrophobic side chains of PMBTFVB-g-PtBA graft copolymers with relatively narrow molecular weight distributions (Mw/Mn ≤ 1.37), which were obtained by the combination of thermal step-growth cycloaddition of aryl bistrifluorovinyl ether monomer, MBTFVB, and ATRP of tBA into hydrophilic PAA segments. To the best of our knowledge, this is the first report on PFCB aryl ether-based amphiphilic graft copolymer. Self-assembly of PMBTFVB-g-PAA amphiphilic graft copolymer in aqueous solution was investigated by fluorescence spectroscopy and TEM. The cmc of the copolymer in aqueous media decreased with the addition of salt and it was interesting that Log(cmc) decreased linearly with the square root of the salt concentration. Moreover, by tuning the water content, multiple aggregates including vesicular, worm-like and bowl-shaped micelles were observed. It was interesting to note that there was a black dot at the center of each vesicle. Although the reason for the formation of the black dot is unknown, the contraction of the shell of vesicles during the sample preparation for TEM might be one of the possibilities. The formation of bowl-shaped micelles, which were composed of an assembly of reverse micelles (PAA core and PMBTFVB corona) with hydrophilic PAA chains surrounding the structure at the polymer/aqueous solution interface, will only be possible to be observed within a narrow range of values of the internal viscosity of the precursor spheres (LCMs). For our system, the rigid PMBTFVB backbone together with the hydrogen bonding between carboxyls in PAA side chains provided the added viscosity-control mechanism. Due to the special morphology of bowl-shaped micelles, which cannot be easily obtained, they could be employed as templates for preparing bowl-shaped nanostructures.Acknowledgements
The authors acknowledge the financial support from the National Basic Research Program of China (2015CB931900), the National Natural Science Foundation of China (21074145, 21274162, and 21474127), and the Shanghai Scientific and Technological Innovation Project (11ZR1445900, 12JC1410500, and 14JC1493400), and the State Key Laboratory of Molecular Engineering of Polymers (K2014-01).Notes and references
- L. F. Zhang, K. Yu and A. Eisenberg, Science, 1996, 272, 1777–1779 CAS.
- L. F. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1996, 118, 3168–3181 CrossRef CAS.
- R. L. Xu, M. A. Winnik, F. R. Hallett, G. Riess and M. D. Croucher, Macromolecules, 1991, 24, 87–93 CrossRef CAS.
- D. Pan, J. L. Turner and K. L. Wooley, Chem. Commun., 2003, 2400–2401 RSC.
- K. Kataoka, J. Macromol. Sci., Pure Appl. Chem., 1994, A31, 1759–1769 CrossRef CAS PubMed.
- A. Rosler, G. W. M. Vandermeulen and H. A. Klok, Adv. Drug Delivery Rev., 2001, 53, 95–108 CrossRef CAS.
- F. Ahmed and D. E. Discher, J. Controlled Release, 2004, 96, 37–53 CrossRef CAS PubMed.
- S. B. Clendenning, S. Fournier-Bidoz, A. Pietrangelo, G. C. Yang, S. J. Han, P. M. Brodersen, C. M. Yip, Z. H. Lu, G. A. Ozin and I. Manners, J. Mater. Chem., 2004, 14, 1686–1690 RSC.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- J. Ruokolainen, R. Makinen, M. Torkkeli, T. Makela, R. Serimaa, G. T. Brinke and O. Ikkala, Science, 1998, 280, 557–560 CrossRef CAS.
- H. Shen and A. Eisenberg, Macromolecules, 2000, 33, 2561–8572 CrossRef CAS.
- C. Forder, C. S. Patrickios, S. P. Armes and N. C. Billingham, Macromolecules, 1996, 29, 8160–8169 CrossRef CAS.
- K. B. Thurmond, T. Kowalewski and K. L. Wooley, J. Am. Chem. Soc., 1997, 119, 6656–6665 CrossRef CAS.
- J. F. Hester, P. Banerjee and A. M. Mayes, Macromolecules, 1999, 32, 1643–1650 CrossRef CAS.
- S. Perrier, S. P. Armes, X. S. Wang, F. Malet and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1696–1707 CrossRef CAS PubMed.
- L. Gubler, M. Slaski, F. Wallasch, A. Wokaun and G. G. Scherer, J. Membr. Sci., 2009, 339, 68–77 CrossRef CAS PubMed.
- G. Riess, Tetrahedron, 2002, 58, 4113–4131 CrossRef.
- Z. B. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98–101 CrossRef CAS PubMed.
- Z. B. Li, M. A. Hillmyer and T. P. Lodge, Langmuir, 2006, 22, 9409–9417 CrossRef CAS PubMed.
- B. H. Tan, H. Hussain, Y. Liu, C. B. He and T. P. Davis, Langmuir, 2010, 26, 2361–2368 CrossRef CAS PubMed.
- J. Mao, P. H. Ni, Y. Y. Mai and D. Y. Yan, Langmuir, 2007, 23, 5127–5134 CrossRef CAS PubMed.
- J. H. Xia, T. Johnson, S. G. Gaynor, K. Matyjaszewski and J. DeSimone, Macromolecules, 1999, 32, 4802–4805 CrossRef CAS.
- S. Perrier, S. G. Jackson, D. M. Haddleton, B. Ameduri and B. Boutevin, Tetrahedron, 2002, 58, 4053–4059 CrossRef CAS.
- K. Jankova, P. Jannasch and S. Hvilsted, J. Mater. Chem., 2004, 14, 2902–2908 RSC.
- K. T. Lim, M. Y. Lee, M. J. Moon, G. D. Lee, S. S. Hong, J. L. Dickson and K. P. Johnston, Polymer, 2002, 43, 7043–7049 CrossRef CAS.
- S. Qin, H. Li, W. Z. Yuan and Y. Zhang, Soft Matter, 2012, 8, 2471–2476 RSC.
- S. Qin, H. Li, W. Z. Yuan and Y. Zhang, Polymer, 2011, 52, 1191–1196 CrossRef CAS PubMed.
- C. Yang, V. Castelvetro, D. Scalarone, S. Bianchi and Y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4518–4530 CrossRef CAS PubMed.
- S. Forster and T. Plantenberg, Angew. Chem., Int. Ed., 2002, 41, 688–714 CrossRef CAS.
- Y. L. Cai, Y. Q. Tang and S. P. Armes, Macromolecules, 2004, 37, 9728–9737 CrossRef CAS.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- V. Heroguez, Y. Gnanou and M. Fontanille, Macromolecules, 1997, 30, 4791–4798 CrossRef CAS.
- P. Dziezok, S. S. Sheiko, K. Fischer, M. Schmidt and M. Moller, Angew. Chem., Int. Ed., 1997, 36, 2812–2815 CrossRef CAS PubMed.
- M. F. Zhang, T. Breiner, H. Mori and A. H. E. Muller, Polymer, 2003, 44, 1449–1458 CrossRef CAS.
- K. L. Beers, S. G. Gaynor and K. Matyjaszewski, Macromolecules, 1998, 31, 9413–9415 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- J. S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901–7910 CrossRef CAS.
- V. Percec and B. Barboiu, Macromolecules, 1995, 28, 7970–7972 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS PubMed.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- D. A. Babb, B. R. Ezzell, K. S. Clement, W. F. Richey and A. P. Kennedy, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 3465–3477 CrossRef CAS PubMed.
- A. P. Kennedy, D. A. Babb, J. N. Bermmer and A. J. Pasztor, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 1859–1865 CrossRef CAS PubMed.
- S. T. Iacono, S. M. Budy, J. M. Mabry and D. W. Smith, Polymer, 2007, 48, 4637–4645 CrossRef CAS PubMed.
- X. Y. Huang, G. L. Lu, D. Peng, S. Zhang and F. L. Qing, Macromolecules, 2005, 38, 7299–7305 CrossRef CAS.
- G. L. Lu, S. Zhang and X. Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5438–5444 CrossRef CAS PubMed.
- H. Liu, Y. J. Li, S. Zhang, D. Yang, J. H. Hu and X. Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 11–22 CrossRef CAS PubMed.
- H. Liu, S. Zhang, Y. J. Li, D. Yang, J. H. Hu and X. Y. Huang, Polymer, 2010, 51, 5198–5206 CrossRef CAS PubMed.
- S. S. Liu and S. Ayusman, Macromolecules, 2000, 33, 5106–5110 CrossRef CAS.
- H. G. Borner, K. Beers and K. Matyjaszewski, Macromolecules, 2001, 34, 4375–4383 CrossRef.
- G. L. Cheng, A. Boker, M. F. Zhang, G. Krausch and A. H. E. Muller, Macromolecules, 2001, 34, 6883–6888 CrossRef CAS.
- K. Matyjaszewski, S. H. Qin, J. R. Boyce, D. Shirvanyants and S. S. Sheiko, Macromolecules, 2003, 36, 1843–1849 CrossRef CAS.
- Z. Shen, Y. Chen, E. Barriau and H. Frey, Macromol. Chem. Phys., 2006, 207, 57–64 CrossRef CAS PubMed.
- D. Peng, X. H. Zhang and X. Y. Huang, Macromolecules, 2006, 39, 4945–4947 CrossRef CAS.
- Z. F. Jia, Q. Fu and J. L. Huang, Macromolecules, 2006, 39, 5190–5193 CrossRef CAS.
- K. J. Peng, K. H. Wang, K. Y. Hsu and Y. L. Liu, ACS Macro Lett., 2015, 4, 197–201 CrossRef CAS.
- R. K. O'Reilly, M. J. Joralemon, K. L. Wooley and C. Hawker, J. Mater. Chem., 2005, 17, 5976–5988 CrossRef.
- J. Ruehl, A. Nilsen, S. Born, P. Thoniyot, L. P. Xu, S. W. Chen and R. Braslau, Polymer, 2007, 48, 2564–2571 CrossRef CAS PubMed.
- P. P. Li, Z. Y. Li and J. L. Huang, Macromolecules, 2007, 40, 491–498 CrossRef CAS.
- S. M. Ramirez and D. Y. Sogah, Polym. Prepr., 2006, 47, 95–96 CAS.
- Y. Yagci and T. M. Atilla, Prog. Polym. Sci., 2006, 31, 1133–1170 CrossRef CAS PubMed.
- I. Astafieva, K. Khougaz and A. Eisenberg, Macromolecules, 1995, 28, 7127–7134 CrossRef CAS.
- Y. G. Li, Y. Q. Zhang, D. Yang, Y. J. Li, J. H. Hu, C. Feng, S. J. Zhai, G. L. Lu and X. Y. Huang, Macromolecules, 2010, 43, 262–270 CrossRef CAS.
- I. C. Riegel, A. Eisenberg, C. L. Petzhold and D. Samios, Langmuir, 2002, 18, 3358–3363 CrossRef CAS.
- X. Y. Liu, J. S. Kim, J. Wu and A. Eisenberg, Macromolecules, 2005, 38, 6749–6751 CrossRef CAS.
- J. Wang, M. Kuang, H. W. Duan, D. Y. Chen and M. Jiang, Eur. Phys. J. E, 2004, 15, 211–215 CrossRef CAS PubMed.
- C. H. Cai, J. P. Lin, T. Chen and X. H. Tian, Langmuir, 2010, 26, 2791–2797 CrossRef CAS PubMed.
- N. P. Truong, J. F. Quinn, M. V. Dussert, N. P. T. Sousa, M. R. Whittaker and T. P. Davis, ACS Macro Lett., 2015, 4, 381–386 CrossRef CAS.
- C. Feng, G. L. Lu, Y. J. Li and X. Y. Huang, Langmuir, 2013, 29, 10922–10931 CrossRef CAS PubMed.
- Y. Liu, C. Y. Yu, H. B. Jin, B. B. Jiang, X. Y. Zhu, Y. F. Zhou, Z. Y. Lu and D. Y. Yan, J. Am. Chem. Soc., 2013, 135, 4765–4770 CrossRef CAS PubMed.
- Y. L. Wang, B. Li, H. B. Jin, Y. F. Zhou, Z. Y. Lu and D. Y. Yan, Chem. – Asian J., 2014, 9, 2281–2288 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py00452g |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |