
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A dinuclear gold(I) complex as a novel photoredox catalyst for light-induced atom transfer radical polymerization†
Frida
Nzulu
a,
Sofia
Telitel
b,
François
Stoffelbach
a,
Bernadette
Graff
b,
Fabrice
Morlet-Savary
b,
Jacques
Lalevée
*b,
Louis
Fensterbank
*a,
Jean-Philippe
Goddard
*c and
Cyril
Ollivier
*a
aUMR CNRS 8232, Institut Parisien de Chimie Moléculaire, Sorbonne Université UPMC Univ Paris 06, 4 place Jussieu, CC 229, F-75252 Paris Cedex 05, France. E-mail: louis.fensterbank@upmc.fr; cyril.ollivier@upmc.fr
bInstitut de Science des Matériaux de Mulhouse IS2M, UMR 7361 CNRS, Université de Haute Alsace, 15 Rue Jean Starcky, 68057 Mulhouse Cedex, France. E-mail: jacques.lalevee@uha.fr
cLaboratoire de Chimie Organique et Bioorganique EA 4566, Université de Haute-Alsace, Ecole Nationale Supérieure de Chimie de Mulhouse, 3 bis rue Alfred Werner, 68093 Mulhouse Cedex, France. E-mail: jean-philippe.goddard@uha.fr
First published on 11th May 2015
Abstract
Controlled/living atom transfer radical polymerization of methacrylates and acrylates initiated by ethyl α-bromophenylacetate (EBPA) as the initiator in the presence of low loadings (1.25 mol% vs. initiator) of a dinuclear gold(I) complex based photocatalyst [Au2(μ-dppm)2]Cl2 has been accomplished in solution and in laminate under UVA and visible-light photoreductive conditions. In solution, the linear increase of molecular weights with methyl methacrylate (MMA) conversion and the low dispersity are consistent with a controlled/living process. In a film, trimethylolpropane triacrylate (TMPTA) was polymerized and the ethyl α-bromophenylacetate (EBPA)/[Au2(μ-dppm)2]Cl2 system resulted in a faster rate of polymerization compared to EBPA/Ir(ppy)3. Chain extensions of polymers were successfully conducted leading to block copolymers, which also confirms the living character of this new system. Photophysical experiments support a conventional photoredox-catalyzed ATRP mechanism. Finally, this approach utilizes a gold catalyst featuring better solubility and lower cost than the well-known Ir(ppy)3 complex.
Introduction
Controlled/living radical polymerization (CRP) is among the most rapidly developing areas of chemistry and polymer science. Over the last few decades, many novel previously inaccessible materials have been prepared using CRP. This process has provided access to nearly all types of macromolecular species exhibiting predetermined molecular weights, low dispersities, and precisely controlled composition and architectures (block copolymers, star-like, comb-like, dendritic etc.).1 The CRP techniques that have been implemented so far consist mainly of reversible addition–fragmentation chain transfer polymerization (RAFT),2 organometallic radical polymerization,3 nitroxide-mediated polymerization (NMP)4 and atom transfer radical polymerization (ATRP).5 In all these CRP methods, control is achieved by the equilibrium maintained between an active and a dormant chain via a reversible deactivation mechanism (RDRP). Currently, these systems are initiated by free-radical initiators and/or metal catalysts and by external stimuli including chemical reducing agents, microwave, heat, electrical current and light.6The use of photochemical stimuli had a tremendous impact not only in organic chemistry,7 molecular biology and electronics but also in polymer chemistry since the pioneer work of Otsu.8 Accordingly some controlled radical photopolymerization (CRP2) methodologies have been recently proposed. The most substantial advantage is that photo-induced processes are extremely fast, and give access to spatially controlled reactions for photografting, photoinduced modification or (micro)patterning of surfaces. They also represent an environmentally friendly alternative to thermal processes allowing reactions to proceed under mild conditions and to use functional groups and materials that decompose at high temperatures. Several CRP2 systems including transition metal-mediated CRP,9 RAFT,10 NMP,4,11 ITP12 and ATRP have been applied. Of all the CRPs, ATRP13 has been the focus of intense research in the polymer community owing to its versatility for the synthesis of well-defined polymers. Quite recently, Matyjaszewski's,14 Yagci's,15 and Hawker's16 groups have successfully devised reactive systems exploiting the redox properties of copper and iridium catalysts respectively in the presence of light. In this field, the development of very efficient photosensitive systems for UVA/visible light remains a huge challenge. Indeed, these types of radiation are safer than the more-energetic UVB and UVC light for the operator, more chemoselective causing less side reactions with reactants, and also eco-friendly (low energy consumption, no release of ozone or harmful UV rays and very little heat emission). Among the few examples of gold catalyzed polymerization,17 the use of a gold photocatalyst to promote photo-ATRP upon UVA/visible light has not been reported so far.
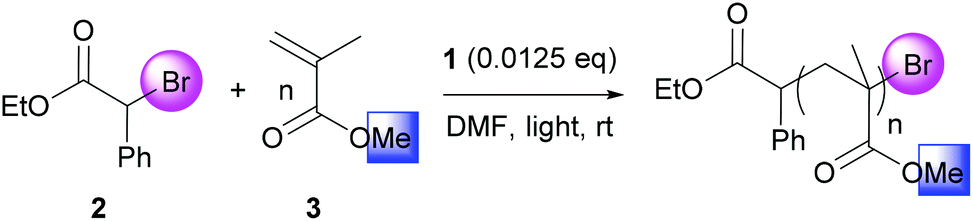
In this context, we showed that photoactivated digold bis(diphenylphosphino)methane dichloride [Au2(μ-dppm)2]Cl21 first reported by Schmidbaur,18 was able to catalyze ATRP processes (Scheme 1). Photoredox properties of the corresponding photoactivated cationic complex [Au2(μ-dppm)2][ClO4] were studied by Che who highlighted its ability to generate carbon-centered radicals by single electron transfer (SET) from alkyl halides.19 In 2013, Barriault nicely demonstrated the synthetic potential of 1 in combination with Hünig's base as a sacrificial electron donor for the photoreduction of unactivated aryl and alkyl bromides.20 Inspired by these seminal studies, we anticipated that the unique properties of 1 would permit SET reduction of activated alkyl bromides, initiate and control ATRP processes. Therefore, we investigated the catalytic properties of 1 in different solvents at rt under light activation to generate a carbon-centered radical from ethyl α-bromophenylacetate (EBPA, 2) and trigger polymerization of rather hydrophilic monomers such as methyl methacrylate (MMA, 3) in solution and trimethylolpropane triacrylate (TMPTA, 5) in laminate. Chain extension experiments with benzyl methacrylate (BnMA, 4) and hydrophobic 2,2,3,3,4,4,4-heptafluorobutyl acrylate (HFBA, 6) were also studied (Scheme 1).
 | ||
| Scheme 1 Atom transfer radical polymerization of various (meth)acrylates catalyzed by digold bis(diphenylphosphino)methane dichloride [Au2(μ-dppm)2]Cl21 as a photoredox catalyst. | ||
Results and discussion
UV-visible and luminescence spectroscopies were used to determine the photophysical properties of 1 and understand its role as a photoredox catalyst (see the ESI, S-8†). The UV-visible absorption spectrum of 1 exhibits two bands at 258 nm (εmax 42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 343 L mol−1 cm−1) and 320 nm (εmax 11343 L mol−1 cm−1). A weak and broad absorption band at 320 nm was visualized while under excitation at the same wavelength, the luminescence spectroscopy of 1 showed a strong emission-band at 390 nm. Thus, the energy of the excited state has been determined as *E(AuII2) ≈ 3.5 eV (*E(AuII2) ≈ 1240/((390 + 320)/2) ≈ 3.5 eV). The frontier molecular orbitals obtained for the optimized geometry of 1 are given in the ESI (Fig. 12†) and suggest a metal to ligand charge transfer transition MLCT for this lowest energy band, the HOMO and the LUMO being located on the metal and ligand moieties, respectively.
343 L mol−1 cm−1) and 320 nm (εmax 11343 L mol−1 cm−1). A weak and broad absorption band at 320 nm was visualized while under excitation at the same wavelength, the luminescence spectroscopy of 1 showed a strong emission-band at 390 nm. Thus, the energy of the excited state has been determined as *E(AuII2) ≈ 3.5 eV (*E(AuII2) ≈ 1240/((390 + 320)/2) ≈ 3.5 eV). The frontier molecular orbitals obtained for the optimized geometry of 1 are given in the ESI (Fig. 12†) and suggest a metal to ligand charge transfer transition MLCT for this lowest energy band, the HOMO and the LUMO being located on the metal and ligand moieties, respectively.
We used laser flash photolysis (LFP) experiments in order to know if the initiator 2 was able to interact with the excited state of complex 1 (AuII*2). The transient absorption spectrum was recorded after laser flash excitation at 355 nm.21 The excited state of 1 is characterized by a transient absorption at about 390 nm in accordance with Che's results.22 Then, LFP allowed the determination of the rate constant of the AuII*2 quenching by 2 (Fig. 1).23 An increasing amount of 2 was added to a solution of 1 under irradiation and a decay of AuII*2 absorption at 390 nm in DCM was observed. When 2 was omitted, the lifetime of 1 was 1.56 μs under a nitrogen atmosphere. This value significantly decreased to 110 ns in the presence of 4 μL of 2 (7.6 × 10−3 M) (Fig. 1). From a Stern–Volmer analysis, we have found that 2 is an excellent oxidative quencher of AuII*2 with a rate constant photolysis of 9.2 × 108 M−1 s−1. In addition, the cyclic voltammogram (CV) of 1 revealed three oxidation waves that appeared at +0.6 V, +1.2 V and +2.0 V in MeCN vs. SCE. The first wave at +0.6 V is assigned to the AuI metal-centered oxidation process while the CV of 2 also gave a reduction potential at −1.3 V (in MeCN vs. SCE). Thus, the oxidation of AuII*2 by 2 would presumably occur as supported by the favorable free energy change ΔG, derived from the Rehm–Weller equation (ΔG° = −1.6 eV) (see the ESI, S-10†). These initial findings suggest that the rate of photoreaction is primarily governed by the homolysis of the C–Br bond of EBPA. These also support that the system 1/2 could be used to initiate radical polymerization with no need of an additional sacrificial donor of electron, as required for the photoreduction of unactivated aryl and alkyl bromides.20
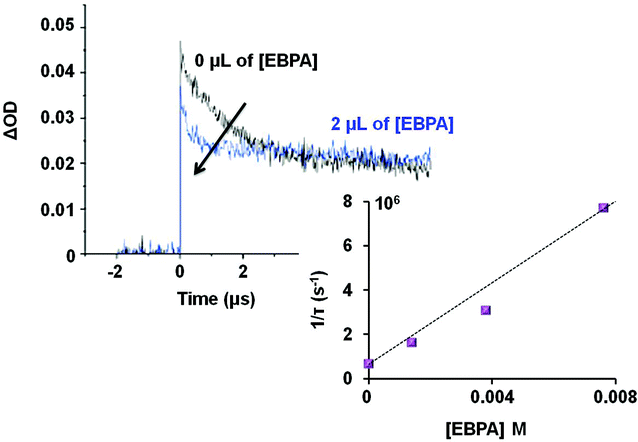 | ||
| Fig. 1 Laser flash photolysis. Decay of the [1]II* signal at 390 nm in dichloromethane with increasing concentrations of 2. The arrow shows that the AuII*2 lifetime decreases as the concentration of 2 increases (from 0 to 7.6 × 10−3 M). ΔOD is the change in optical density associated with the light excitation (λ = 355 nm). | ||
To support this mechanism, we monitored the steady state photolysis of [1] in the presence of 2. We observed a strong decrease of the absorption band at 320 nm upon diode laser exposure concomitantly with the appearance of a band at 420 nm over 10 s of irradiation (Fig. 2). This result confirmed the quenching of the excited state of 1 by the initiator 2 leading to the formation of an ethyl phenylacetyl radical, that could initiate the radical polymerization, and an oxidized gold complex {[Au2(μ-dppm)2Cl2]+, Br−} with an absorption UV signal suspected at 420 nm.
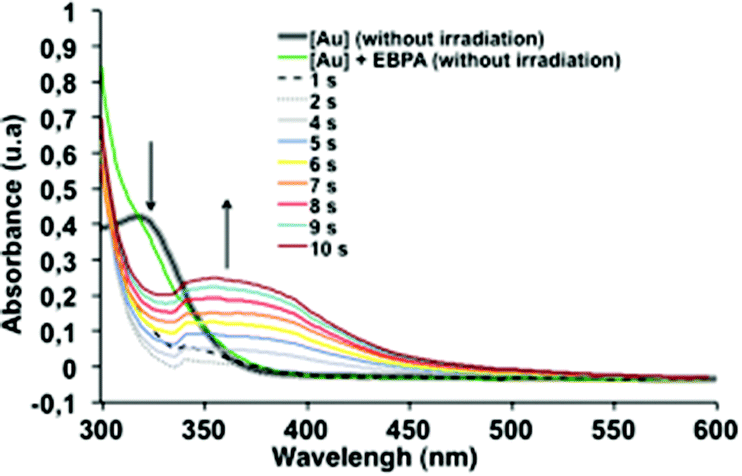 | ||
| Fig. 2 Steady state photolysis of 1 and 2 in dichloromethane upon exposure to the Xe–Hg under air at different irradiation times (from t = 0 to 10 s). 1/2: 1/0.045. UV-vis spectra clearly demonstrate that one species disappears rapidly at a very early stage and one appears. | ||
From this system, the generation of a carbon-centered radical intermediate derived from 2 was also confirmed by ESR spin-trapping experiments (Fig. 3).24 A dichloromethane solution of 2/1 and N-tert-butyl-α-phenylnitrone (PBN) was irradiated (Xe–Hg lamp) for 20 s and monitored by ESR. A characteristic signal of a nitroxide adduct which originates from the addition of the phenylacetate radical to PBN (aN = 14.3 G, aH = 2.5 G) was observed. Similarly, 6 minutes of sunlight exposure to the same solution resulted in an identical ESR signal. This set of experiments confirmed that gold catalyst 1 allows a very responsive photo radical process and 2 is an excellent oxidative quencher. Given the perfect adequacy in terms of photochemical properties and reactivity between catalyst 1 and initiator 2, polymerization of MMA 3 in solution was then carried out.
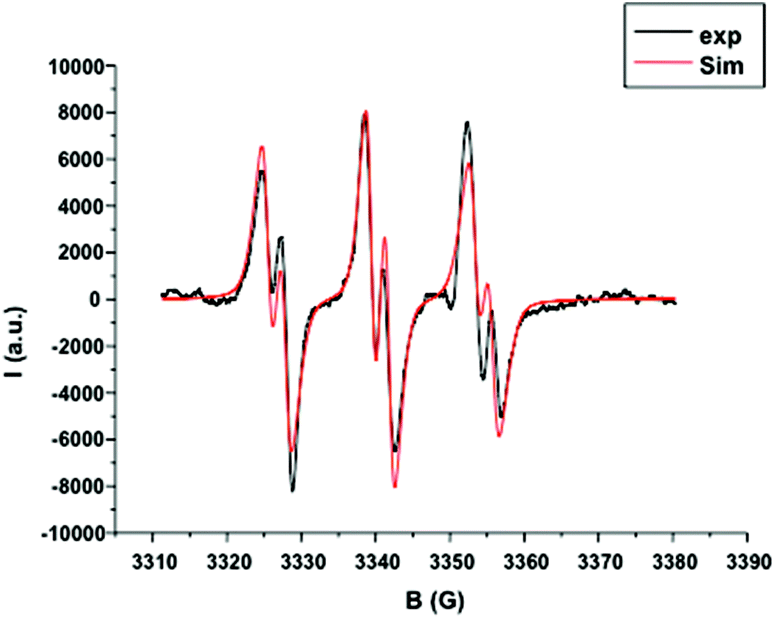 | ||
| Fig. 3 ESR spin trapping (PBN) spectrum. [PBN] = 7.90 × 10−4 M, [1] = 1.57 × 10−4 M, [2] = 1.91 × 10−3 M in 1.5 mL of DCM/tert-butylbenzene 1:2. | ||
The influence of the irradiation wavelength and the solvent was first examined. Typical conditions involved a controlled concentration of MMA (2.08 M) with an initiator/monomer/catalyst (2/3/1) ratio fixed at 1/500/0.0125 (mol/mol). Conversion of 3, molecular weight and dispersity of the so-formed PMMA were measured by gravimetry and GPC analysis and reported in Table 1.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Light (nm) | 2 Conv.b (%) | M n,theo (kg mol−1) | M n,exp (kg mol−1) | Đ |
| a Polymerizations were performed with [MMA] = 2.08 M (vol MMA + vol solvent) at rt with 2/3/1 = 1/500/0.0125 mol/mol. All reactions were degassed before setting the reaction. b Conversion measured by gravimetric analysis. c Determined after one precipitation in MeOH. d Reaction performed in DCM. e Reaction in a mixture 2/3 (1/500) in DMF without the gold catalyst. | |||||
| 1 | 300 | 84 | 42.3 | 34.0 | 1.81 |
| 2 | 350 | 75 | 37.5 | 34.0 | 1.67 |
| 3 | 380 | 54 | 27.4 | 42.0 | 1.77 |
| 4 | 419 | 63 | 31.7 | 48.4 | 1.85 |
| 5 | 350d | 55 | 27.8 | 24.7 | 1.62 |
| 6 | Dark | — | — | — | — |
| 7 | 350e | 100 | 50.0 | 25.0 | 2.1 |
Experiments were performed in N,N′-dimethylformamide (DMF) or dichloromethane (DCM) under UV to visible-light activation at 25 °C. At 300 nm in DMF, 84% of conversion was reached in 24 h (Table 1, entry 1). PMMA with lower number-average molar mass than the theoretical value was obtained with a dispersity of 1.81. Under these reaction conditions, effective initiation proceeded but the physical data of the PMMA can be indicative of side reactions. Lower conversions were observed at 380 and 419 nm (54 and 63% respectively) and the resulting polymers have higher molar mass, revealing a failure of the initiation system (Table 1, entries 3 and 4). A good control of the polymerization was achieved when the irradiation was done at 350 nm (Table 1, entry 2). The conversion of MMA was 75%, the molar mass distribution (Đ) of PMMA decreased to 1.67 with a number-average molar mass closer than the theoretical value. Well controlled polymerization was also obtained in DCM under irradiation at 350 nm (Table 1, entry 5). Molar mass fitted with the theoretical value and the dispersity was 1.62. These last results indicated that the initiation process was efficient and side reactions giving birth to dead chains were very limited. In order to argue in the favor of a controlled radical polymerization, the same mixture of 2/3/1 was placed in the dark and no polymer was formed (Table 1, entry 6). If the gold catalyst was omitted, polymerization occurred but it could not be controlled (Table 1, entry 7).
To evaluate the living character of this polymerization process, chain extension experiments were conducted using MMA or BnMA as the second monomer (Scheme 2). An original sample of PMMA obtained by the present protocol and purified by precipitation in methanol (Mn = 18.0 kg mol−1, Đ = 1.32), was considered as a macroinitiator and placed in DMF with MMA 3 and the gold photocatalyst 1 in a 1/500/0.0125 proportion respectively (Scheme 2A). After 24 h of irradiation at 350 nm, a PMMA homopolymer was isolated showing an important increase of Mn up to 30.9 kg mol−1. From a different PMMA sample (Mn,exp = 14.2 kg mol−1, Đ = 1.50), a block copolymer was obtained with BnMA 4 (PMMA/4/1 1/500/0.0125 mol/mol) (Scheme 2B). Similarly, a considerable Mn,exp increase was observed and has been determined to be 43.8 kg mol−1. The corresponding SEC traces of both experiments showed the evolution of the polymer populations. This clearly demonstrated the possibility of reactivating the bromide terminus of the polymer. Thus, it appears reasonable to postulate a photoATRP pathway for this novel new gold-catalyzed polymerization.
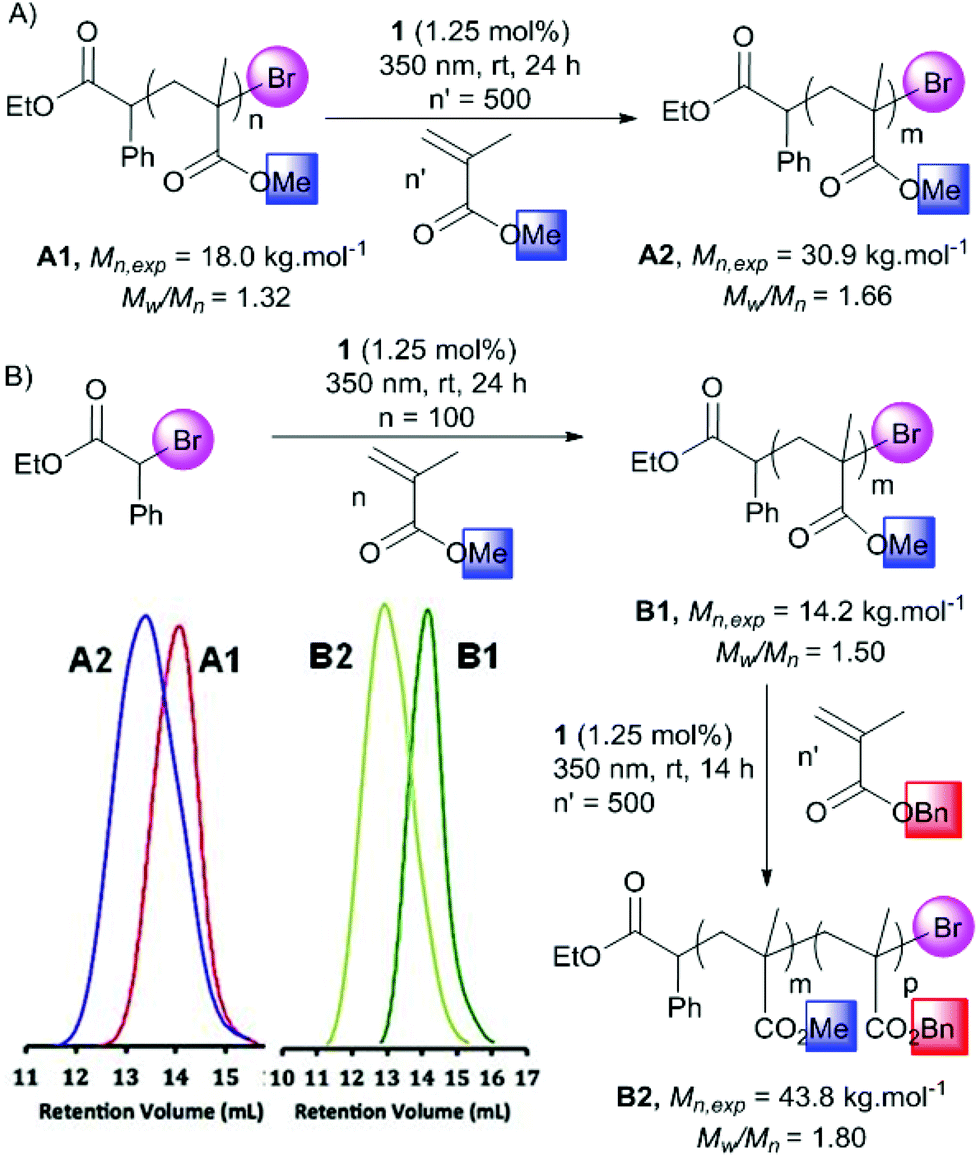 | ||
| Scheme 2 (A) Chain extension of poly(methyl methacrylate) with MMA and SEC chromatogram with the red and blue traces corresponding respectively to A1 and A2. (B) Synthesis of poly(methyl methacrylate)-b-poly(benzyl methacrylate) well defined diblock copolymer and dark green and light green SEC chromatograms corresponding respectively to B1 and B2. | ||
As initial experiments revealed that 1 efficiently catalyzes the polymerization of methacrylates 3 and 4 in solution, comparative kinetic experiments were carried out. A laminate system was chosen to compare the present system (2/1) with the known 2/fac-Ir(ppy)3 (Fig. 4). To address this question, the polymerization of TMPTA 5 in laminate with both systems was monitored by RT-FTIR spectroscopy at 405 nm. The disappearance of a characteristic absorption band of 5 (1600–1650 cm−1) served to measure the conversion of this monomer.25 The 2/fac-Ir(ppy)3 polymerization profile (black curve) showed a rapid initial rate to reach 33% conversion in 800 s. After an activation period of 50 s, the 2/1 system gave a faster polymerization rate than with iridium to reach 38% conversion in 400 s. The iridium catalyst was already known to be very efficient but to our delight, this new gold photocatalyst 1 proved to be even more active for the polymerization of acrylates in bulk.
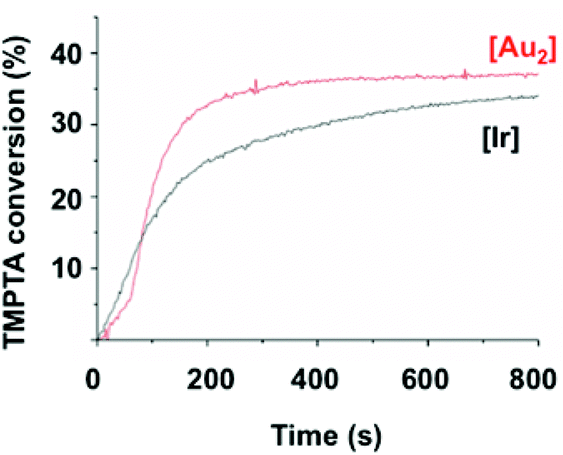 | ||
| Fig. 4 Compared polymerization profiles for TMPTA in the laminate system for different catalysts upon a blue LED bulb irradiation (405 nm). Red curve: 2/5/1 (1/100/0.0125 mol/mol); black curve: 2/5/fac-Ir(ppy)3 (1/100/0.0125 mol/mol). | ||
The dependence of this polymerization process on light was further studied by employing a periodic “on/off” light event during the TMPTA polymerization in laminate 2/5/1 (1/100/0.0125 mol/mol) (Fig. 5). The film was irradiated with LED bulbs (405 nm) with alternating irradiation periods (30 s) and blackout (30 s). The conversion of 5 was monitored by RT-FTIR spectroscopy and is reported in Fig. 5 according to the sequential irradiation. The polymerization rates were dramatically impacted and turning off the light resulted in a significantly low rate. Thus, a clear acceleration of the polymerization was observed under irradiation. This experiment also showed the living character of the polymerization and turning off the light did not induce the interruption of the growing chains.
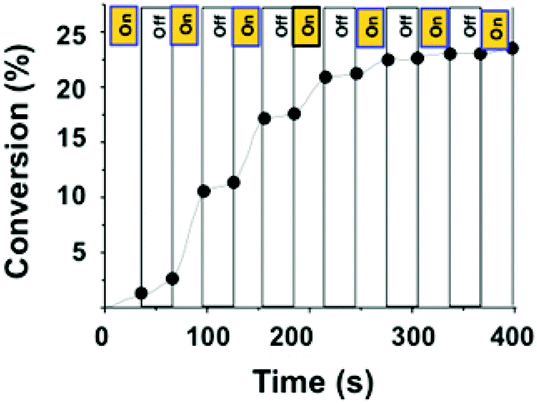 | ||
| Fig. 5 TMPTA conversion as a function of time for intermittent blue LED bulb irradiation. Polymerization was performed from a 2/5/1 (1/100/0.0125 mol/mol) mixture. | ||
An investigation of the living character was extended to surface functionalization. PhotoATRP of TMPTA 5 in laminate with the 2/1 system (2/5/1 1/100/0.0125 mol/mol) allowed obtaining a well defined film after Xe–Hg lamp irradiation for 3 min (Fig. 6A). The residual monomer was removed by washing with acetone and the surface was analyzed by XPS (see the ESI† for the full spectrum). This analysis evidenced the presence of gold and bromine atoms at the surface of the film. A drop of water on the layer made a 55° contact angle in agreement with the wettability character of PMMA. Then, HFBA 6 was deposited on the surface of the first layer and irradiated under the same conditions for 10 minutes (Fig. 6B). The contact angle dramatically changed from 55° to 115°, illustrating the surface modification to a more hydrophobic property due to the presence of perfluoroalkyl chains. This change proved the reactivation of the first layer for the synthesis of a block copolymer. Moreover, XPS of the second layer still showed the presence of gold and bromine atoms and opened the possibility of a third reactivation process (Fig. 6C). TEM (Transmission Electron Microscopy) analysis allowed visualization of the layer surfaces and so helped to exclude the presence of gold nanoparticles which could have been the active catalyst.
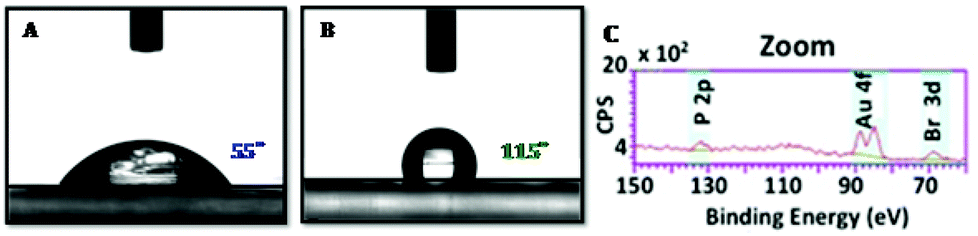 | ||
| Fig. 6 Contact angle experiments: (Picture A) first layer: TPMTA/[Au2]/EBPA: 100/0.0125/1 mol/mol after 3 minutes upon a blue LED bulb irradiation (405 nm). (Picture B) second layer after re-initiation with a fluoromonomer (HFBA 6) after 10 minutes upon a Xe–Hg lamp. (Picture C) represents high resolution XPS spectra after re-initiation and formation of the fluorinated polymer. | ||
All these experiments completed by further mass spectrometry analysis by MALDI-TOF/ESI-TOF and ATRA studies (see the ESI, S17 and S18†) drove us to the following mechanism proposal for this new photoATRP process (Scheme 3). The polymerization relies on the in situ photogeneration of excited state [1]* and its subsequent reaction with alkyl halide (Pn-X) resulting in the formation of the active radical (Pn˙) and [Au2]IIIX. Then, the radical adds onto a monomer and the resulting radical is rapidly deactivated by [Au2]IIIX to form a dormant species and regenerate 1.
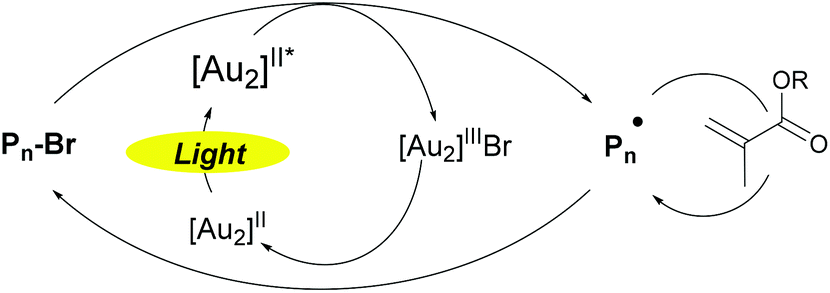 | ||
| Scheme 3 Proposed mechanism for photoATRP of (meth)acrylates using [Au2(μ-dppm)2]Cl2 as a photocatalyst. | ||
Conclusions
In summary, we report here the first gold-catalyzed photoATRP process of methacrylates and acrylates. The novel photocatalyst [Au2(μ-dppm)2]Cl2 in combination with ethyl α-bromophenylacetate (EBPA) as the initiator allows the development of a highly responsive controlled living radical polymerization under UVA, sunlight and visible-light irradiation conditions as evidenced by fluorescence and absorbance measurements, steady-state photolysis, laser flash photolysis (LFP), electron spin resonance (ESR) experiments, Fourier transform infrared (FTIR) spectroscopy and “on–off” switchable experiments. In solution, the linear increase of molecular weights with methyl methacrylate (MMA) conversion and the low dispersity are consistent with a controlled/living process. In the film, trimethylolpropane triacrylate (TMPTA) was polymerized and the ethyl α-bromophenylacetate (EBPA)/[Au2(μ-dppm)2]Cl2 system resulted in a faster rate of polymerization compared to EBPA/Ir(ppy)3. Chain extensions of polymers were successfully conducted leading to block copolymers, which confirmed the living character of this system. Importantly, this approach utilizes a gold catalyst with lower cost and more soluble than conventional iridium complexes. The use of dichloromethane as the solvent without loss of reactivity will open new perspectives in the combination of this photoATRP process with other polymerization modes. Moreover, the reported system is highly sensitive to sunlight and application of photopolymerization using this source of energy will be envisaged.Acknowledgements
This work was supported by the CNRS, UPMC and IUF, which we gratefully acknowledge. This work was supported by the LabEx MiChem part of French state funds managed by the ANR within the Investissements d'Avenir programme under reference ANR-11-IDEX-0004-02. JPG thanks UHA, Fondation pour l'ENSCMu and Actelion for support (Actelion Chair in Innovative Organic Chemistry). The authors acknowledge ANR (NHCX ANR-11-BS07-008) for stimulating fruitful collaborations.Notes and references
- (a) W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93 CrossRef CAS PubMed; (b) A. Goto and T. Fukuda, Prog. Polym. Sci., 2004, 29, 329 CrossRef CAS PubMed.
- (a) C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402 CrossRef CAS PubMed; (b) J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- (a) I. Allaoua, B. E. Goi, M. M. Obadia, A. Debuigne, C. Detrembleur and E. Drockenmuller, Polym. Chem., 2014, 5, 2973 RSC; (b) L. E. N. Allan, M. R. Perry and M. P. Shaver, Prog. Polym. Sci., 2012, 37, 127 CrossRef CAS PubMed; (c) S. Yamago, Chem. Rev., 2009, 109, 5051 CrossRef CAS PubMed.
- (a) J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63 CrossRef CAS PubMed; (b) C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS PubMed.
- (a) J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614 CrossRef CAS; (b) M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721 CrossRef CAS; (c) T. E. Patten, J. Xia, T. Abernathy and K. Matyjaszewski, Science, 1996, 272, 866 CAS; (d) K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS PubMed; (e) K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513 CrossRef CAS PubMed.
- (a) F. A. Leibfarth, K. M. Mattson, B. P. Fors, H. A. Collins and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 199 CrossRef CAS PubMed; (b) Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245 CrossRef CAS; (c) N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. R. de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096 CrossRef CAS PubMed.
- For reviews on photoredox catalysis in organic synthesis, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2010, 40, 102 RSC; (c) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed. For our contributions to this field, see: (d) M. H. Larraufie, R. Pellet, L. Fensterbank, J.-P. Goddard, E. Lacôte, M. Malacria and C. Ollivier, Angew. Chem., Int. Ed., 2011, 50, 4463 CrossRef CAS PubMed; (e) S. Donck, A. Baroudi, L. Fensterbank, J.-P. Goddard and C. Ollivier, Adv. Synth. Catal., 2013, 355, 1477 CrossRef CAS PubMed; (f) A. Baralle, L. Fensterbank, J.-P. Goddard and C. Ollivier, Chem. – Eur. J., 2013, 19, 10809 CAS; (g) M. Daniel, L. Fensterbank, J.-P. Goddard and C. Ollivier, Org. Chem. Front., 2014, 1, 551 RSC; (h) L. Chenneberg, A. Baralle, M. Daniel, L. Fensterbank, J.-P. Goddard and C. Ollivier, Adv. Synth. Catal., 2014, 356, 2756 CrossRef CAS PubMed.
- T. Otsu and A. Kuriyama, Polym. J., 1985, 17, 97 CrossRef CAS.
- (a) S. Yamago, Y. Ukai, A. Matsumoto and Y. Nakamura, J. Am. Chem. Soc., 2009, 131, 2100 CrossRef CAS PubMed; (b) A. Nomura, A. Goto, K. Ohno, E. Kayahara, S. Yamago and Y. Tsujii, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5284 CrossRef CAS PubMed; (c) C. Detrembleur, D.-L. Versace, Y. Piette, M. Hurtgen, C. Jérôme, J. Lalevée and A. Debuigne, Polym. Chem., 2012, 3, 1856 RSC; (d) Y. Nakamura, T. Arima, S. Tomita and S. Yamago, J. Am. Chem. Soc., 2012, 134, 5536 CrossRef CAS PubMed; (e) Y. Nakamura and S. Yamago, Beilstein J. Org. Chem., 2013, 9, 1607 CrossRef PubMed.
- (a) J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo and T. P. Davis, Macromolecules, 2002, 35, 7620 CrossRef CAS; (b) S. Muthukrishnan, E. H. Pan, M. H. Stenzel, C. Barner-Kowollik, T. P. Davis, D. Lewis and L. Barner, Macromolecules, 2007, 40, 2978 CrossRef CAS; (c) M. A. Tasdelen, Y. Y. Durmaz, B. Karagoz, N. Bicak and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3387 CrossRef CAS PubMed; (d) C. Zhang, Y. Yang and J. He, Macromolecules, 2013, 46, 3985 CrossRef CAS; (e) J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508 CrossRef CAS PubMed.
- (a) S. Hu, J. H. Malpert, X. Yang and D. C. Neckers, Polymer, 2000, 41, 445 CrossRef CAS; (b) Y. Guillaneuf, D.-L. Versace, D. Bertin, J. Lalevée, D. Gigmes and J. P. Fouassier, Macromol. Rapid Commun., 2010, 31, 1909 CrossRef CAS PubMed; (c) D.-L. Versace, J. Lalevée, J. P. Fouassier, D. Gigmes, Y. Guillaneuf and D. Bertin, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2910 CrossRef CAS PubMed; (d) D.-L. Versace, J. Lalevée, J. P. Fouassier, Y. Guillaneuf, D. Bertin and D. Gigmes, Macromol. Rapid Commun., 2010, 31, 1383 CrossRef CAS PubMed; (e) Y. Guillaneuf, D. Bertin, D. Gigmes, D.-L. Versace, J. Lalevée and J. P. Fouassier, Macromolecules, 2010, 43, 2204 CrossRef CAS.
- A. Wolpers and P. Vana, Macromolecules, 2014, 47, 954 CrossRef CAS.
- (a) Z. Guan and B. Smart, Macromolecules, 2000, 33, 6904 CrossRef CAS; (b) J. Mosnáček and M. Ilčíková, Macromolecules, 2012, 45, 5859 CrossRef; (c) N. V. Alfredo, N. E. Jalapa, S. L. Morales, A. D. Ryabov, R. Le Lagadec and L. Alexandrova, Macromolecules, 2012, 45, 8135 CrossRef CAS.
- (a) Y. Kwak and K. Matyjaszewski, Macromolecules, 2010, 43, 5180 CrossRef CAS; (b) D. Konkolewicz, K. Schröder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1219 CrossRef CAS.
- (a) M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Rapid Commun., 2011, 32, 58 CrossRef CAS PubMed; (b) M. A. Tasdelen, M. Ciftci and Y. Yagci, Macromol. Chem. Phys., 2012, 213, 1391 CrossRef CAS PubMed.
- (a) B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850 CrossRef CAS PubMed; (b) N. J. Treat, B. P. Fors, J. W. Kramer, M. Christianson, C.-Y. Chiu, J. R. de Alaniz and C. J. Hawker, ACS Macro Lett., 2014, 3, 580 CrossRef CAS.
- (a) L. Ray, V. Katiyar, M. J. Raihan, H. Nanavati, M. M. Shaikh and P. Ghosh, Eur. J. Inorg. Chem., 2006, 3724 CrossRef CAS PubMed; (b) L. Ray, V. Katiyar, S. Barman, M. J. Raihan, H. Nanavati, M. M. Shaikh and P. Ghosh, J. Organomet. Chem., 2007, 692, 4259 CrossRef CAS PubMed; (c) J. Urbano, A. J. Hormigo, P. de Frémont, S. P. Nolan, M. M. Díaz-Requejo and P. J. Pérez, Chem. Commun., 2008, 759 RSC; (d) E. Brulé, S. Gaillard, M.-N. Rager, T. Roisnel, V. Guérineau, S. P. Nolan and C. M. Thomas, Organometallics, 2011, 30, 2650 CrossRef; (e) R. A. Sanguramath, S. K. Patra, M. Green and C. A. Russell, Chem. Commun., 2012, 48, 1060 RSC; (f) F. Nzulu, A. Bontemps, J. Robert, M. Barbazanges, L. Fensterbank, J.-P. Goddard, M. Malacria, C. Ollivier, M. Petit, J. Rieger and F. Stoffelbach, Macromolecules, 2014, 47, 6652 CrossRef CAS.
- H. Schmidbaur, A. Wohlleben, F. Wagner, O. Orama and G. Huttner, Eur. J. Inorg. Chem., 1977, 110, 1748 CAS.
- (a) D. Li, C.-M. Che, H.-L. Kwong and V. W.-W. Yam, J. Chem. Soc., Dalton Trans., 1992, 3325 RSC; (b) C.-M. Che, H.-K. Yip, V. W.-W. Yam, P.-Y. Cheung, T.-F. Lai, S.-J. Shieh and S.-M. Peng, J. Chem. Soc., Dalton Trans., 1992, 427 RSC.
- (a) G. Revol, T. McCallum, M. Morin, F. Gagosz and L. Barriault, Angew. Chem., Int. Ed., 2013, 52, 13342 CrossRef CAS PubMed; (b) T. McCallum, E. Slavko, M. Morin and L. Barriault, Eur. J. Org. Chem., 2015, 81 CrossRef CAS PubMed.
- The wavelength 355 nm corresponds to the third Nd:YAG harmonic.
- C.-M. Che, H.-L. Kwong and C.-K. Poonhieh, J. Chem. Soc., Dalton Trans., 1990, 3215 RSC.
- (a) J. Lalevée, N. Blanchard, C. Fries, M.-A. Tehfe, F. Morlet-Savary and J. P. Fouassier, Polym. Chem., 2011, 2, 1077 RSC; (b) S. Telitel, J. Lalevée, N. Blanchard, T. Kavalli, M.-A. Tehfe, F. Morlet-Savary, B. Graff and J. P. Fouassier, Macromolecules, 2012, 45, 6864 CrossRef CAS.
- (a) N. M. Atherton, M. J. Davies, B. C. Gilbert and P. Tordo, in Spin-Trapping: Recent Developments and Applications, in Electron Paramagnetic Resonance: Volume 16, ed. B. C. Gilbert, N. M. Atherton and M. J. Davies, The Royal Society of Chemistry, Cambridge, U.K., 1998, vol. 16, p. 116 Search PubMed; (b) J. Lalevée, F. Dumur, C. R. Mayer, D. Gigmes, G. Nasr, M. A. Tehfe, S. Telitel, F. Morlet-Savary, B. Graff and J. P. Fouassier, Macromolecules, 2012, 45, 4134 CrossRef.
- S. Telitel, F. Dumur, S. Telitel, O. Soppera, M. Lepeltier, Y. Guillaneuf, J. Poly, F. Morlet-Savary, P. Fioux, J. P. Fouassier, D. Gigmes and J. Lalevée, Polym. Chem., 2015, 6, 613 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data of the catalyst, organic compounds and polymers are provided. Photochemical and photophysical properties of the catalytic system are available. See DOI: 10.1039/c5py00435g |
| This journal is © The Royal Society of Chemistry 2015 |