
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polysulfobetaine-based diblock copolymer nano-objects via polymerization-induced self-assembly†
Kay E. B.
Doncom
,
Nicholas J.
Warren
and
Steven P.
Armes
*
Department of Chemistry, University of Sheffield, Brook Hill, Sheffield, S3 7HF, UK. E-mail: s.p.armes@sheffield.ac.uk
First published on 13th April 2015
Abstract
A zwitterionic polysulfobetaine-based macromolecular chain transfer agent (PSBMA38) was prepared by reversible addition–fragmentation chain transfer (RAFT) solution polymerization of [2-(methacryloyloxy)ethyl] dimethyl(3-sulfopropyl) ammonium hydroxide (SBMA) in an aqueous solution containing 0.5 M NaCl at 70 °C. This PSBMA38 macro-CTA was then utilized for the RAFT aqueous dispersion polymerization of a water-miscible monomer, 2-hydroxypropyl methacrylate (HPMA). The growing PHPMA block became hydrophobic in situ, leading to polymerization-induced self-assembly. Systematic variation of the mean degree of polymerization of the PHPMA block and the copolymer concentration enabled access to pure phases of spheres, worms or vesicles, as judged by transmission electron microscopy and dynamic light scattering studies. A detailed phase diagram was constructed and the thermo-responsive behavior of selected PSBMA38-PHPMAX nanoparticles was investigated. Finally, the salt tolerance of PSBMA38-PHPMA400 vesicles was compared to that of PGMA71-PHPMA400 vesicles; the former vesicles exhibit much better colloidal stability in the presence of 1 M MgSO4.
Introduction
It is well known that amphiphilic AB diblock copolymers spontaneously undergo self-assembly in aqueous solution in order to minimize the unfavorable interactions between the hydrophobic blocks and the solvent.1 The resulting copolymer morphology depends on the so-called packing parameter, p, which is related to the relative volume fractions of the hydrophilic and hydrophobic blocks.2 A wide range of copolymer morphologies have been reported, including spherical micelles,3 worm-like particles4 and vesicles.5Traditionally, diblock copolymer self-assembly in solution has been achieved via post-polymerization processing techniques such as thin film rehydration6,7 or a solvent switch,8 where the copolymer chains are initially dissolved in a good solvent for both blocks and then a selective solvent for one of the blocks is added in order to induce self-assembly. This approach usually involves additional purification steps to remove the non-selective solvent, e.g. by dialysis or evaporation. This self-assembly route also suffers from a major disadvantage: it is almost invariably conducted in dilute solution (typically <1% w/w copolymer).
Recently, we9–13 and others14–18 have reported that polymerization-induced self-assembly (PISA) of various amphiphilic diblock copolymers can be readily achieved using either aqueous dispersion polymerization10,19 or aqueous emulsion polymerization20–22 based on reversible addition–fragmentation chain transfer (RAFT) chemistry.23–26 This approach allows various copolymer morphologies to be readily prepared in situ at relatively high copolymer concentrations and requires no post-polymerization processing. One early formulation based on RAFT aqueous dispersion polymerization utilized a zwitterionic phosphobetaine homopolymer, poly(2-(methacryloyloxy)ethyl phosphorylcholine) (PMPC), as the stabilizer block and poly(2-hydroxypropyl methacrylate) (PHPMA) as the core-forming block to access a range of copolymer morphologies.10 However, the relatively high mass of the MPC repeat units meant that highly anisotropic copolymer compositions had to be targeted at relatively high copolymer solids (ca. 16–25%) in order to access non-spherical copolymer morphologies (e.g. worms or vesicles).
Polysulfobetaines are closely related to polyphosphobetaines and both classes of polyzwitterions have been shown to be salt-responsive.27–35 The presence of salt generally leads to higher water solubility, which is sometimes known as the ‘anti-polyelectrolyte’ effect.36 Certain polysulfobetaines also display thermo-responsive behavior, with their upper critical solution temperature (UCST) depending on both the copolymer molecular weight and copolymer concentration.29,37–39 Like polyphosphobetaines,40–42 polysulfobetaines have been shown to be highly biocompatible and exhibit anti-fouling properties.41,43–46 Importantly, polysulfobetaines are significantly cheaper than polyphosphobetaines.
In 2014 Pei and Lowe reported the synthesis of a range of diblock copolymer nano-objects comprising a polysulfobetaine stabilizer block and 2-phenylethyl methacrylate (PEMA) as the hydrophobic block. However, these copolymers were prepared via post-polymerization modification. First, a poly(2-(dimethylamino)ethyl methacrylate) (PDMA) macro-CTA was employed for the ethanolic RAFT dispersion polymerization of PEMA.16 These precursor diblock copolymers underwent self-assembly during their PISA synthesis to yield a range of copolymer morphologies, including spheres, worms and vesicles. The PDMA stabilizer block was subsequently quaternized using 1,3-propanesultone to yield polysulfobetaine-based nanoparticles. However, 1,3-propanesultone is known to be carcinogenic and the PISA synthesis was conducted in ethanol rather than water (with the purified copolymer nanoparticles being redispersed in water).
Herein we employ poly(2-(methacryloyloxy)ethyl dimethyl-(3-sulfopropyl)ammonium hydroxide) (PSBMA) as a polysulfobetaine macro-CTA; this is used as a steric stabilizer to conduct the RAFT aqueous dispersion polymerization of 2-hydroxypropyl methacrylate (HPMA) in order to generate a thermo-responsive core-forming block. This formulation yields a range of PSBMA-PHPMA diblock copolymer nano-objects directly via PISA (see Fig. 1). Our approach is both versatile and efficient: it avoids post-polymerization modification with carcinogens such as 1,3-propanesultone and affords the desired nano-objects directly in water (hence subsequent transfer from ethanol to water is not required). Moreover, thermo-responsive behaviour is conferred on the resulting diblock copolymer nano-objects.
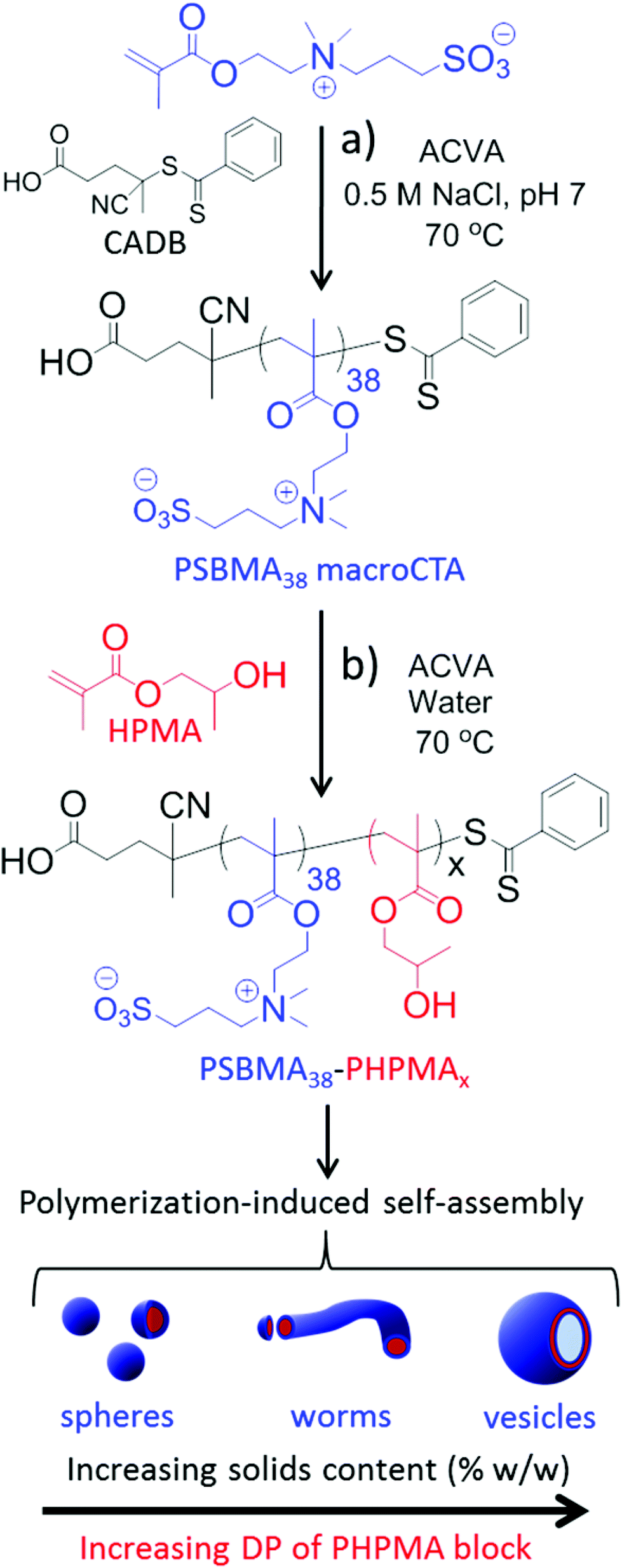 | ||
| Fig. 1 (a) Synthesis of the PSBMA38 macro-CTA via RAFT aqueous solution polymerization of SBMA. (b) RAFT aqueous dispersion polymerization of HPMA using this PSBMA38 macro-CTA at 70 °C to afford various PSBMA-PHPMA diblock copolymer nano-objects via polymerization-induced self-assembly (PISA). | ||
Experimental
Materials
4-Cyanopentanoic acid dithiobenzoate (CADB), [2-(methacryloyloxy)ethyl] dimethyl(3-sulfopropyl) ammonium hydroxide (SBMA) and deuterium oxide (D2O) were obtained from Sigma-Aldrich U.K. and were used as received. 4,4′-Azobis(4-cyanovaleric acid) (ACVA, 99%) and 2-hydroxypropyl methacrylate (HPMA, 98%) were obtained from Alfa Aesar (UK) and were used as received. Deuterated methanol (CD3OD) was obtained from Cambridge Isotope laboratories. Dialysis tubing was received from SpectraPor. Deionized water was used in all experiments.Methods
Polymer characterization
Molecular weight distributions were assessed by aqueous gel permeation chromatography (GPC) at 40 °C using a guard column, a 5 μm PL Aquagel-OH 30 column and a 5 μm PL Aquagel-OH 40 column connected in series to an Agilent Technologies 1260 Infinity refractive index detector, using a phosphate buffer eluent (0.08 M Na2HPO4; adjusted to pH 8.9 using NaOH) at a flow rate of 1.0 ml min−1. The number-average molecular weight (Mn) and polydispersity (Mw/Mn) were calculated using a series of near-monodisperse poly(ethylene oxide) calibration standards.1H NMR spectra were acquired using a Bruker AV1-400 MHz spectrometer in either D2O or CD3OD. At least 64 scans were recorded for each sample. All chemical shifts (δ) are reported in ppm.
DLS measurements were performed using a Zetasizer Nano-ZS instrument (Malvern Instruments, UK) equipped with a 4 mW He–Ne laser operating at 633 nm. Light scattering was detected at 173°. Hydrodynamic diameters were determined using the Stokes–Einstein equation and averaged over three consecutive runs. Copolymer dispersions were diluted to 0.2% w/v prior to analysis at 25 °C. Viscosities (and refractive indices) for aqueous solutions of 1 M NaCl, 2 M NaCl and 1 M MgSO4 at 20 °C were calculated to be 1.096 mPa s (1.343), 1.219 mPa s (1.372) and 1.884 mPa s (1.355) respectively.47
Transmission electron microscopy studies were conducted using a Phillips CM 100 TEM instrument operating at 100 kV, equipped with a Gatan 1k CCD camera. Copper/palladium TEM grids (Agar Scientific) were surfaced-coated in-house to produce a thin film of amorphous carbon. The grids were then plasma glow-discharged for 30 seconds to yield a hydrophilic surface. 10 μL of the aqueous dispersion (diluted to 0.1% w/w) was placed on the grid, then blotted after 1 minute to remove excess sample. The grids were then negatively stained using uranyl formate solution (10 μL, 0.75% w/w) for 20 seconds, followed by blotting to remove excess staining solution and dried using a vacuum hose.
Rheology measurements were performed using an AR-G2 rheometer equipped with a variable temperature Peltier plate, a 40 mL aluminium cone and a solvent trap. Storage (G′) and loss (G′′) moduli were measured as a function of percentage strain and temperature in order to identify the linear viscoelastic region and also to determine the critical degelation temperature. Percentage strain sweeps were conducted at constant temperature (37 °C) using an angular frequency of 1.0 rad s−1. Angular frequency sweeps were carried out at a constant applied strain of 1.0%. Temperature sweeps were conducted at an angular frequency of 1.0 rad s−1 and an applied strain of 1.0%. The temperature was increased by 2 °C, with 5 minutes being allowed for thermal equilibrium between each measurement.
Results and discussion
Synthesis of the PSBMA38 macro-CTA
Synthesis of a PSBMA38 homopolymer using 4,4′-azocyanovaleric acid (ACVA) initiator and 4-cyanopentanoic acid dithiobenzoate (CADB) as the RAFT agent was conducted in aqueous solution containing 0.50 M NaCl at 70 °C. The reaction solution pH was adjusted to 7.2 to facilitate dissolution of the RAFT agent and electrolyte was added because it is well known that this enhances the water solubility of PSBMA.27,30,31 The polymerization kinetics for SBMA exhibited a linear semi-logarithmic plot and reached high conversion (90%) within 3 h (see Fig. S1 in ESI†). The Mn increased monotonically with conversion and the polydispersities remained low throughout the polymerization (Mw/Mn < 1.30). The crude PSBMA was purified by dialysis against water and recovered by lyophilization to yield macro-CTA 1 as a pink powder (Mn = 5 kDa, Mw/Mn = 1.09 vs. poly(ethylene oxide) standards).Analysis of the 1H NMR spectrum of 1 recorded in D2O indicated a mean DP of 38, as calculated by comparing the integrals of the aromatic RAFT end-group signals (k, n and m) between 7.4 ppm and 8.0 ppm with the polymer side-chain signals at 2.98, 3.18, 3.54, 3.78 and 4.46 ppm (assigned as j, f, g, e and d; see Fig. 2).
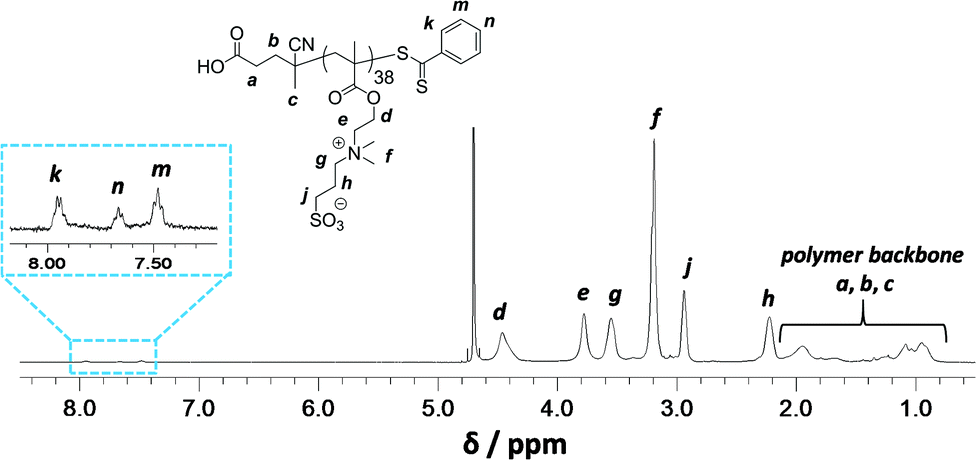 | ||
| Fig. 2 Assigned 1H NMR spectrum recorded for PSBMA macro-CTA 1 in D2O. | ||
Polymerization-induced self-assembly
A series of PSBMA38-PHPMAx (hereafter referred to as S38-Hx) diblock copolymers was prepared using this PSBMA38 macro-CTA for the RAFT aqueous dispersion polymerization of PHPMA in deionized water at 70 °C. It was decided to avoid using an aqueous salt solution to enable direct comparison of the results obtained with this PISA formulation to those previously reported for the PMPC-PHPMA system.10 It is known that the UCST of PSBMA is reduced in the lower molecular weight limit.29,37,38 More specifically, Willcock et al. investigated the effect of chain length at a solution concentration of 1.0 g dm−3: No UCST was observed for PSBMA homopolymers with molecular weights of 20 kDa (DP = 70) or below.39 Therefore, given its relatively low molecular weight and anionic carboxylate end-group (which also suppresses UCST behavior),48 the PSBMA38 macro-CTA used in this work was not expected to exhibit a UCST in the absence of salt. Indeed, control experiments confirmed that a 15% w/w aqueous solution of PSBMA38 remained water-soluble even at 4 °C and all aqueous dispersion polymerization syntheses were performed using an initial PSBMA38 concentration of 12% w/w (or lower). The total solids content of the PISA formulation was varied from 10% w/w to 25% w/w. High HPMA conversions were obtained within 3 h at 70 °C. Three distinct regions can be observed in the semi-logarithmic kinetic plot (see Fig. 3).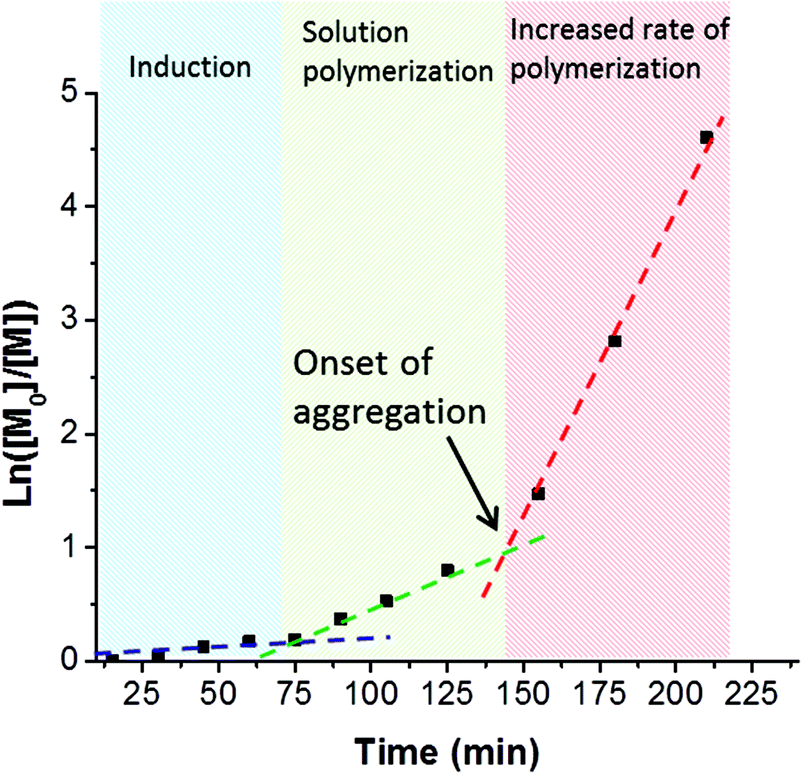 | ||
Fig. 3 First-order kinetic plot for the RAFT aqueous dispersion polymerization of HPMA conducted using a PSBMA38 macro-CTA in deionized water at 70 °C and 20% w/w solids. Reaction conditions: [PSBMA38]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[HPMA]:[ACVA] = 200:1:0.20. Three regimes can be identified: an induction period of approximately 75 min, a relatively slow rate of solution polymerization up to 145 min, and a relatively fast rate of dispersion polymerization after 145 min. The onset of micellization aggregation occurs at 150 min, which corresponds to a HPMA conversion of 67% and equates to a mean PHPMA DP of 133. :[HPMA]:[ACVA] = 200:1:0.20. Three regimes can be identified: an induction period of approximately 75 min, a relatively slow rate of solution polymerization up to 145 min, and a relatively fast rate of dispersion polymerization after 145 min. The onset of micellization aggregation occurs at 150 min, which corresponds to a HPMA conversion of 67% and equates to a mean PHPMA DP of 133. | ||
There is an induction period for the first 75 min, which is commonly observed for RAFT polymerizations of methacrylates conducted with dithiobenzoates,49,50 followed by a significant increase in rate for the next 70 min as solution polymerization occurs and water-soluble diblock copolymer chains are formed. The rate of polymerization further increases after 145 min, since this coincides with micellar nucleation.51 At this point, the HPMA monomer becomes partitioned within the micellar core, thereby increasing the local monomer concentration and hence accounting for the enhanced rate of polymerization.51 The point at which micellar nucleation occurs corresponds to a HPMA conversion of 67% for this particular PISA formulation, which equates to a mean DP of 133 for the growing PHPMA block. This value is significantly higher than the critical degree of polymerization of 92 reported by Blanazs et al. when polymerising HPMA using a poly(glycerol methacrylate) (PGMA47) macro-CTA. This difference can be rationalized by the higher molecular weight of the PSBMA38 block (10.9 kDa) compared to the PGMA47 block (7.5 kDa). Thus the former block occupies a larger volume fraction than the latter and, although the PSBMA38 stabilizer block is shorter, a longer core-forming PHPMA chain is needed to induce micellization.
Systematic variation of the mean DP of the core-forming PHPMA block and the total solids content (Y) enabled a detailed phase diagram to be constructed for the synthesis of S38-Hx-Y nanoparticles (see Fig. 4; full characterization data can be found in Table S1 in the ESI†). The final copolymer compositions were determined by 1H NMR analysis (see Fig. S3 in ESI†). This phase diagram is rather similar to that reported by Sugihara et al. for a zwitterionic PMPC25 stabilizer block10 and by Blanazs et al. for a PGMA78 stabilizer block.11 In each case, the final copolymer morphology is strongly concentration-dependent, with kinetically-trapped spheres being obtained at lower solids (since the probability of sphere–sphere fusion is reduced under these conditions) while equilibrium vesicular morphologies are formed at higher solids. As is usually the case,10–13 the pure worm phase occupies a relatively narrow region and is bounded by mixed phases. There is also a small region corresponding to around 14–16% w/w solids and a PHPMA DP of 250 that contains all three copolymer phases (i.e. spheres, worms and vesicles).
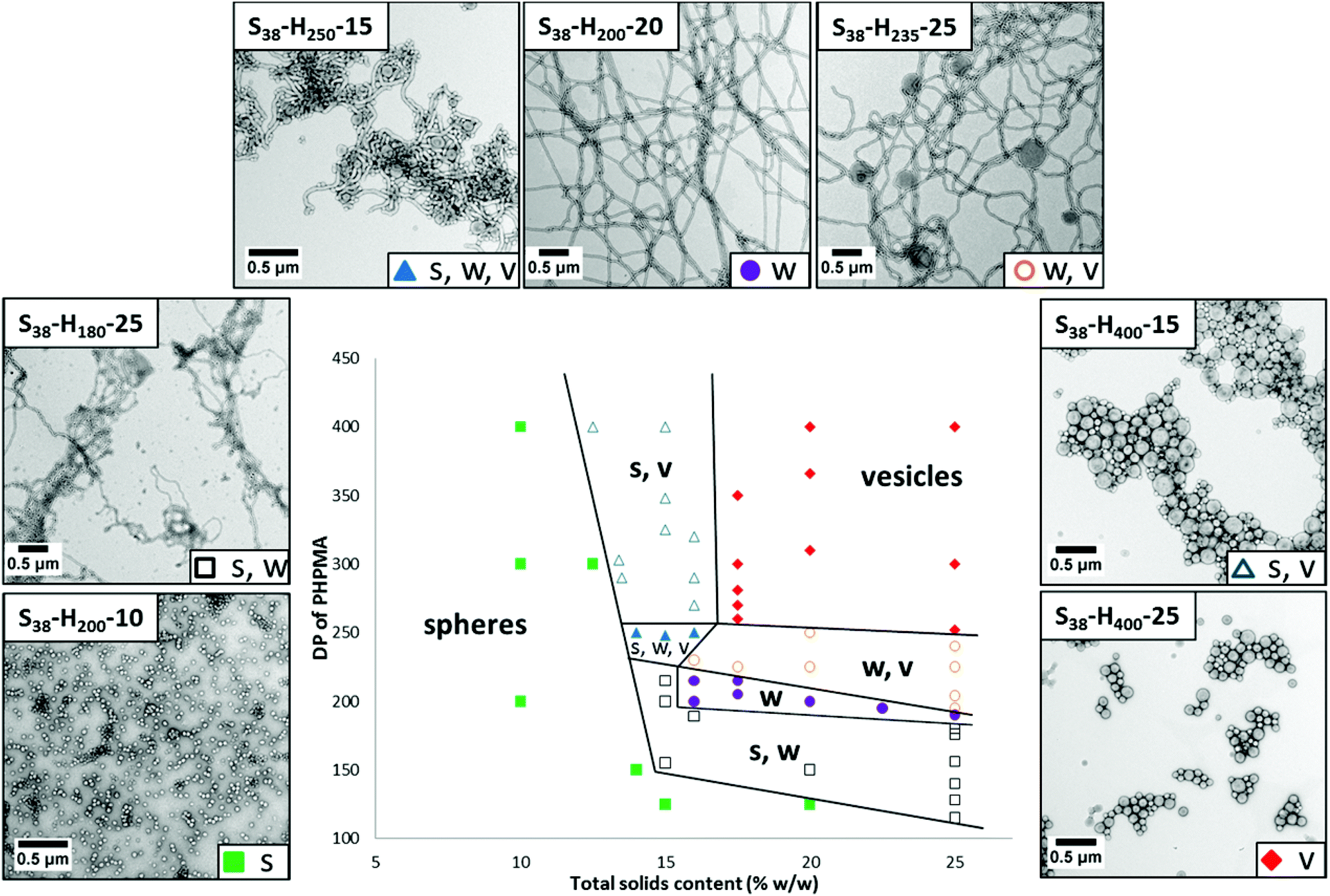 | ||
| Fig. 4 Detailed phase diagram constructed for the preparation of S38-Hx-Y diblock copolymer nano-objects by RAFT aqueous dispersion polymerization of HPMA at 70 °C (for brevity, S denotes PSBMA and H denotes PHPMA). The target DP of the PHPMA block (x) and the total solids content (Y) were systematically varied and the diblock copolymer morphologies were assigned by post mortem TEM analysis of the final copolymer dispersions after appropriate dilution. Also shown are representative TEM images obtained for the various phases; S denotes spheres, W denotes worms and V denotes vesicles. | ||
TEM studies indicate a strong correlation between the mean sphere diameter (DTEM) and the target PHPMA block DP. Thus DTEM = 34 ± 4 nm for PHPMA200, DTEM = 42 ± 3 nm for PHPMA300 and DTEM = 54 ± 6 nm for PHPMA400. TEM consistently undersizes these diblock copolymer nanoparticles relative to DLS. This is because the former technique examines dehydrated nanoparticles with collapsed stabilizer blocks, while the latter interrogates hydrated nanoparticles in aqueous solution. In addition, TEM reports a number-average diameter whereas DLS reports a z-average diameter, which is inherently biased towards larger nanoparticles. If S38-H400 nanoparticles are prepared at higher solids contents (12.5% w/w), then the DLS diameter increases up to 111 nm (polydispersity = 0.05). Along with the greater turbidity of the dispersion, this suggests the presence of larger particles. TEM analysis indicates two distinct populations, corresponding to spheres (DTEM = 57 ± 6 nm) and relatively well-defined vesicle (DTEM = 97 ± 7 nm). The vesicles dimensions observed by TEM correlate quite well with those indicated by DLS. However, the latter technique is insensitive to spheres as the light scattering is dominated by the larger vesicles. Increasing the total solids concentration to 20% w/w for the synthesis of S38-H400 nanoparticles produces a pure vesicle phase (DTEM = 98 ± 13 nm), as indicated by TEM analysis, with DLS studies reporting a z-average diameter of 114 nm (polydispersity = 0.04). Thus the mean size of the pure vesicles is comparable to that of the vesicle population within the spheres plus vesicles mixed phase (Fig. 5 and 6).
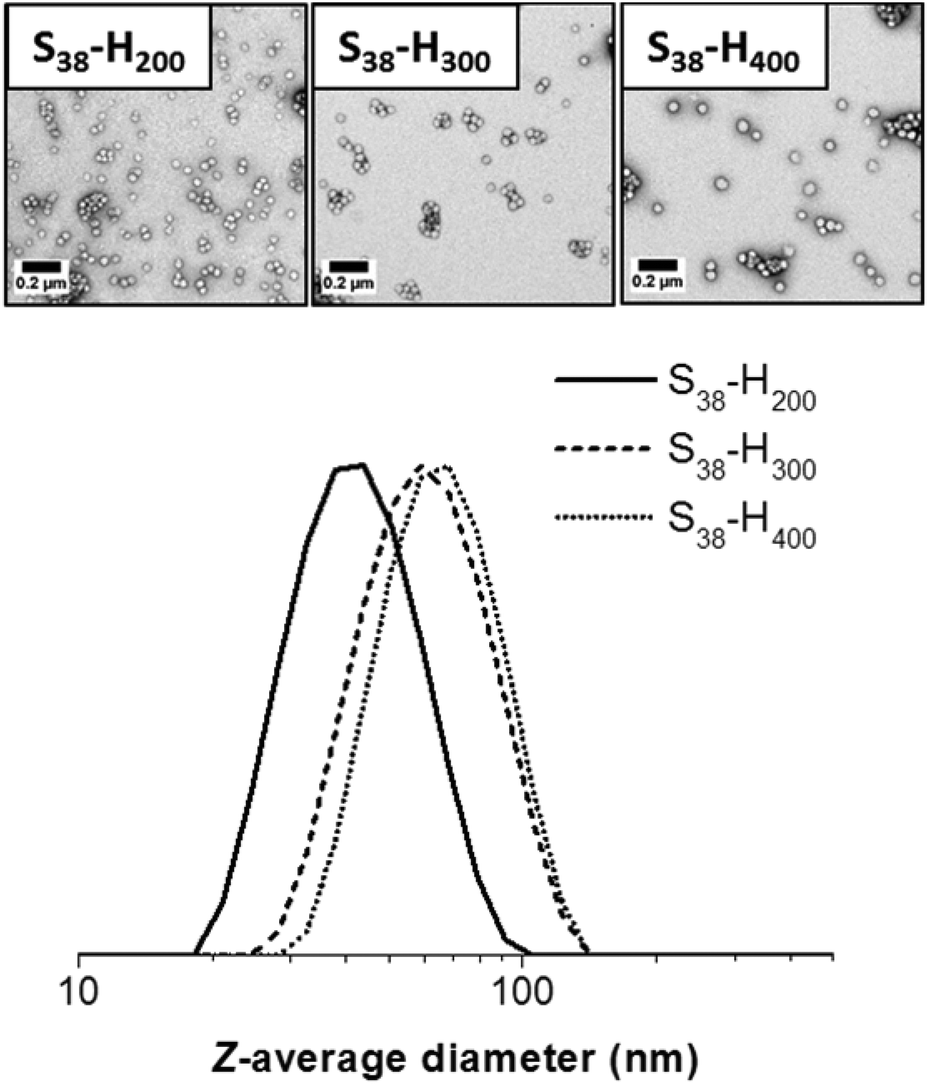 | ||
| Fig. 5 TEM images and corresponding DLS particle size distributions obtained for spherical S38-Hx-10 nanoparticles when x = 200, 300 or 400. | ||
 | ||
| Fig. 6 Representative TEM images obtained for PSBMA38-PHPMA400 nanoparticles prepared at various solids contents. The copolymer morphology evolves from kinetically-trapped spheres (10% w/w) to equilibrium vesicles (20% w/w) via a spheres plus vesicles mixed phase (12.5% w/w). | ||
Varying the target DP of the PHPMA block at a fixed total solids concentration of 20% w/w allows access to pure spheres, worms and vesicles, as well as various mixed phases comprising these two or more morphologies (see Fig. 7). For example, targeting S38-H125 leads to ill-defined spheres (TEM indicates DTEM = 27 ± 5 nm; DLS reports a z-average diameter of 34 nm (polydispersity = 0.14)). Increasing the target DP of the PHPMA block to 150 produces a mixed phase of spheres and worms, as indicated by TEM analysis. Visual inspection of this dispersion confirms an increase in turbidity and formation of a free-standing gel. Targeting a PHPMA block DP of 200 results in the formation of a pure worm phase.
 | ||
| Fig. 7 Representative TEM images obtained for PSBMA38-PHPMAx (denoted as S38-Hx-20 for brevity) nano-objects prepared by RAFT aqueous dispersion polymerization of HPMA at 70 °C at a total solids concentration of 20% w/w. A digital photograph of the physical appearance of each copolymer dispersion is shown as an inset in each TEM image. The scale bar in each case is 0.5 μm. The tube inversion test confirms the formation of free-standing gels in three cases, whereas the other three dispersions remained free-flowing. | ||
Both S38-H225 and S38-H250 formulations produced a mixed phase comprising worms and vesicles, with a higher proportion of vesicles being observed in the latter case as estimated by TEM analysis. The visual appearance and macroscopic behavior of these two dispersions were consistent with these TEM observations. S38-H225 formed an opaque free-standing gel, while S38-H250 comprised an opaque viscous solution. A pure vesicle phase was obtained when targeting a PHPMA block DP of 310 or higher. TEM studies indicate the formation of relatively polydisperse vesicles ranging from approximately 90 nm to 520 nm (DTEM = 214 ± 122 nm), which correspond quite well to the z-average diameter of 202 nm reported by DLS analysis.
Thermo-responsive behavior of S38-Hx-Y dispersions
There is considerable literature precedent for thermo-responsive PHPMA-based nano-objects in aqueous solution.12,13,52,53 Hence the thermo-responsive behavior of selected S38-Hx-Y dispersions were investigated using variable temperature oscillatory rheology. For example, a S38-H200-10 dispersion formed a rather soft worm gel (G′ ∼ 15 Pa) at 20 °C. A temperature sweep from 37 °C to 2 °C led to an increase in gel strength up to approximately 40 Pa, which is consistent with previous observations made by Warren et al.13 It is suggested that the initially short worms become longer upon cooling, hence a larger number of inter-worm contacts leads to a higher gel strength. G′′ exceeded G′ at around 2 °C, indicating degelation.This is a result of greater solvation of the PHPMA core, which lowers the packing parameter1 and results in a morphology transition from a worm gel to free-flowing spheres.52,53 However, this thermal transition proved to be irreversible, since G′′ remained greater than G′ throughout the subsequent heating cycle (see Fig. 8A). This thermo-responsive behavior is in contrast to that reported for PGMA54-PHPMA140 worm gels,52 but is similar to that observed by Warren et al. for a PEG113-PHPMA220 worm gel.13 During such thermal cycling, the temperature was lowered by 2 °C for each measurement, with 5 min being allowed between each data point to ensure thermal equilibrium. The time required to induce degelation at a given copolymer concentration was also examined. The same S38-H200-10 worm gel was held at 2 °C for 200 min (see Fig. 8B). For the first 60 min, the worm gel remained intact since G′ exceeded G′′ (and G′ ∼ 65 Pa). However, G′′ became greater than G′ after 60 min, producing a free-flowing liquid. Degelation was confirmed by visual inspection. TEM studies were undertaken, both for the original worm gel at 20 °C and also 2 h after this dispersion was held at 2 °C. There was a dramatic change in copolymer morphology from worms to spheres, which accounts for the observation of degelation. DLS studies of the diluted copolymer dispersions were also consistent with these TEM observations: the z-average diameter of the original worms at 20 °C was 420 nm (polydispersity = 0.29), whereas the z-average diameter was reduced to 30 nm (polydispersity = 0.25) after cooling to 2 °C for 2 h. The latter dimensions suggest the formation of polydisperse spheres.
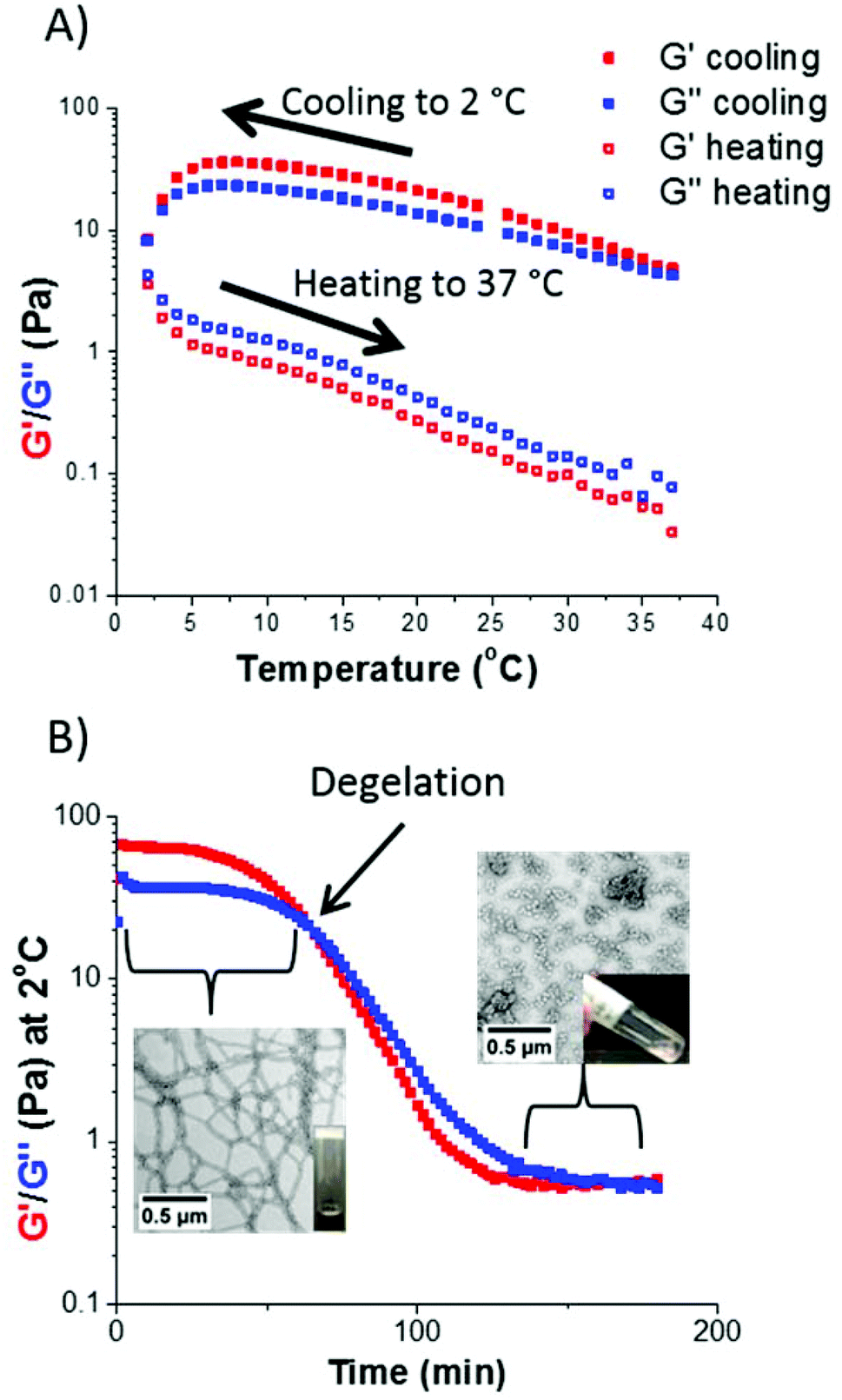 | ||
| Fig. 8 (A) Storage (G′) and loss (G′′) moduli recorded for a 10% w/w S38-H200 worm gel upon cooling from 37 °C to 2 °C followed by heating up to 37 °C. Irreversible degelation occurred at 2 °C. (B) Storage (G′) and loss (G′′) moduli recorded for a 10% w/w S38-H200 worm gel held at 2 °C for 200 min. Degelation occurred after 60 min at this temperature to produce a free-flowing liquid. TEM images recorded before cooling and 2 h after being held at 2 °C confirmed a worm-to-sphere transition. Both sets of measurements were conducted at 1.0% applied strain using an angular frequency of 1.0 rad s−1. | ||
A dispersion of S38-H310-25 vesicles was cooled to 4 °C for 48 h. The original dispersion was an opaque viscous liquid at 20 °C. Visual inspection of the cold dispersion at 4 °C confirmed the formation of a turbid gel, as demonstrated by the tube inversion test. TEM studies indicated that the initial vesicles had transformed into a mixed phase comprising worms and vesicles (see Fig. 9).
 | ||
| Fig. 9 TEM images and physical state of the dispersions obtained for (top) S38-H310-25 vesicles at 20 °C and (middle) a mixed phase of worms and vesicles formed after being held at 4 °C for 48 h and (bottom) a mixed phase of vesicles and short worms obtained after heating to 70 °C for 5 hours. | ||
Formation of the worm population accounts for the observation of macroscopic gelation. This dispersion did not undergo degelation on warming to room temperature, even on a time scale of several weeks. However, heating to 70 °C for 5 hours resulted in degelation. TEM studies indicated the presence of mainly vesicles, but some short worms are also observed. These short worms persist even after heating at 70 °C for 16 h.
Therefore this transition cannot be considered to be fully reversible. Again, this behavior is similar to that of the PEG113-PHPMA220 worms reported by Warren et al.13
The above examples suggest that S38-Hx nano-objects can be transformed into lower-order morphologies upon cooling. This indicates a reduction in the packing parameter under these conditions, which is consistent with a higher degree of plasticization of the PHPMA core-forming block. However, it appears that this thermo-responsive behavior is not fully reversible. This aspect is not yet understood and warrants further studies.
Behavior of S38-Hx nano-objects in the presence of added electrolyte
As previously discussed, polysulfobetaines such as PSBMA are known to exhibit enhanced aqueous solubility in the presence of salt compared to pure water.27,30,31 Therefore the tolerance of PSBMA-stabilized vesicles towards the addition of electrolyte was investigated. S38-H400-25 vesicles were diluted to 0.02% w/v using either water or various salt solutions and their colloidal stabilities were compared to that of PGMA71-PHPMA400-20 (denoted G71-H400-20) vesicles. Each aqueous dispersion was analyzed by DLS immediately after dilution, again after 24 h and finally after ageing for one week at 20 °C. In such experiments, any apparent increase in size is taken to be evidence of particle aggregation caused by colloidal destabilization. The results are summarized in Table 1 (see overleaf).| Nanoparticle type | Aqueous solution | Initial DLS diameter/nm (PDI) | DLS diameter after one week/nm (PDI) |
|---|---|---|---|
| S38-H400-25 vesicles | Water | 140 (0.03) | 140 (0.03) |
| 1 M NaCl | 140 (0.04) | 141 (0.03) | |
| 2 M NaCl | 140 (0.04) | 145 (0.02) | |
| 1 M MgSO4 | 119 (0.05) | 118 (0.09) | |
| G71-H400-20 vesicles | Water | 314 (0.14) | 315 (0.12) |
| 1 M NaCl | 307 (0.19) | 310 (0.22) | |
| 2 M NaCl | 314 (0.19) | 312 (0.15) | |
| 1 M MgSO4 | Precipitate | — |
S38-H400-25 vesicles remained stable in either 1 M or 2 M NaCl solution. In 1 M MgSO4 solution, the vesicles actually appear smaller compared to that observed in water (119 nm vs. 140 nm). TEM studies confirmed that no discernible change in copolymer morphology occurred under these conditions (see Fig. S4 in ESI†). A possible explanation for these DLS observations is that the PSBMA chains expressed at the outer leaflet of the vesicles become less hydrated in the presence of 1 M MgSO4, which in turn reduces the mean hydrodynamic diameter of the vesicles. In contrast, exposure to 2 M MgSO4 solution results in macroscopic precipitation of the S38-H400 vesicles (see Fig. S5 in ESI†). This is commonly known as the “salting out” effect. Dissolution of this 2:2 electrolyte produces a significantly higher ionic strength than 1:1 electrolytes such as NaCl, which explains why the S38-H400 vesicles remain colloidally stable in 2 M NaCl, but become aggregated in the presence of 2 M MgSO4.
As a comparison, G71-H400-20 vesicles were also exposed to various salt solutions. These vesicles also exhibited reasonable colloidal stability in either 1 M or 2 M NaCl for at least a week (see Table 1), although a small degree of aggregation can be detected in the particle size distributions obtained by DLS (see Fig. 10). However, unlike the S38-H400 spheres and vesicles, the G71-H400 vesicles proved to be unstable with respect to particle aggregation in the presence of 1 M MgSO4, with immediate macroscopic precipitation being observed under these conditions (see Fig. S5 in the ESI†). In summary, these ‘added salt’ studies indicate that the zwitterionic PSBMA38 block is a somewhat more salt-tolerant steric stabilizer than the non-ionic PGMA71 block under the same conditions, which is consistent with earlier literature reports.27,37,54–56
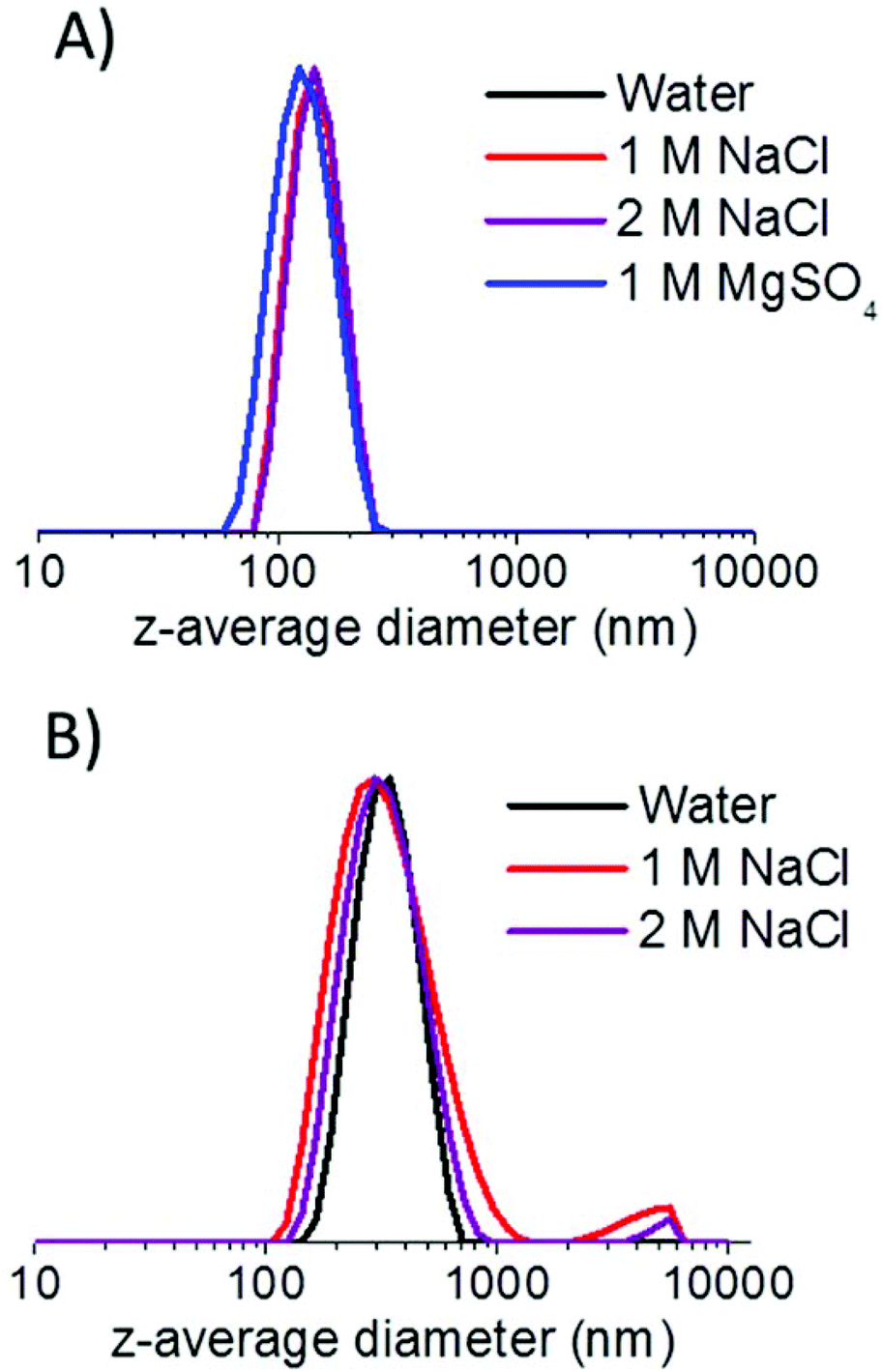 | ||
| Fig. 10 Intensity-average particle size distributions obtained by DLS for (a) S38-H400 vesicles and (b) G71-H400 vesicles after being aged in various aqueous salt solutions for one week. | ||
Conclusions
A near-monodisperse polysulfobetaine macro-CTA with a mean DP of 38 has been utilized as the water-soluble stabilizer block for the RAFT aqueous dispersion polymerization of HPMA to produce a range of copolymer morphologies via polymerization-induced self-assembly (PISA). Systematic variation of the mean DP of the structure-directing hydrophobic PHPMA block and the copolymer concentration enables pure spheres, worms or vesicles to be obtained. However, construction of a detailed phase diagram is required for reproducible targeting of pure worms, since this anisotropic morphology occupies rather narrow phase space. Degelation of a S38-H200 worm gel occurs on cooling: this thermal transition is irreversible as judged by both the tube inversion test and temperature-dependent oscillatory rheology studies. TEM images recorded for dried diluted aqueous dispersions prepared before and after cooling suggests that degelation is the result of a worm-to-sphere transition. In addition, a pure vesicle phase undergoes a sol–gel transition upon cooling as a result of formation of a mixed phase comprising worms and vesicles. Finally, the colloidal stability of S38-H400 vesicles in the presence of added salt was compared to that of a control sample of G71-H400 vesicles. The S38-H400 vesicles remained stable with respect to aggregation in the presence of 1 M MgSO4, whereas G71-H400 vesicles underwent macroscopic precipitation under the same conditions. Thus, the zwitterionic PSBMA stabilizer confers significantly higher salt tolerance than a non-ionic poly(glycerol monomethacrylate) stabilizer.Acknowledgements
SPA acknowledges the European Research Council for a five-year ERC Advanced Investigator grant (PISA 320372) which provided post-doctoral support for KD. EPSRC is also thanked for a Platform grant (EP/J007846/1) to support NJW. Rheanna Perry is acknowledged for providing the PGMA71-PHPMA400 vesicles.Notes and references
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- J. Israelachvili, Intermolecular and Surface Forces, Academic Press, London, 2nd edn, 1991 Search PubMed.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- K. Kita-Tokarczyk, J. Grumelard, T. Haefele and W. Meier, Polymer, 2005, 46, 3540–3563 CrossRef CAS.
- J. Du and R. K. O'Reilly, Soft Matter, 2009, 5, 3544–3561 RSC.
- L. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1996, 118, 3168–3181 CrossRef CAS.
- Y. Li and S. P. Armes, Angew. Chem., Int. Ed., 2010, 49, 4042–4046 CrossRef CAS PubMed.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS PubMed.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- V. Ladmiral, M. Semsarilar, I. Canton and S. P. Armes, J. Am. Chem. Soc., 2013, 135, 13574–13581 CrossRef CAS PubMed.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2013, 136, 1023–1033 CrossRef PubMed.
- Z. An, Q. Shi, W. Tang, C.-K. Tsung, C. J. Hawker and G. D. Stucky, J. Am. Chem. Soc., 2007, 129, 14493–14499 CrossRef CAS PubMed.
- G. Delaittre, M. Save and B. Charleux, Macromol. Rapid Commun., 2007, 28, 1528–1533 CrossRef CAS.
- Y. Pei and A. B. Lowe, Polym. Chem., 2014, 5, 2342–2351 RSC.
- C. A. Figg, A. Simula, K. A. Gebre, B. S. Tucker, D. M. Haddleton and B. S. Sumerlin, Chem. Sci., 2015, 6, 1230–1236 RSC.
- J. Rieger, C. Grazon, B. Charleux, D. Alaimo and C. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2373–2390 CrossRef CAS.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- J. Rieger, F. Stoffelbach, C. Bui, D. Alaimo, C. Jérôme and B. Charleux, Macromolecules, 2008, 41, 4065–4068 CrossRef CAS.
- V. J. Cunningham, A. M. Alswieleh, K. L. Thompson, M. Williams, G. J. Leggett, S. P. Armes and O. M. Musa, Macromolecules, 2014, 47, 5613–5623 CrossRef CAS.
- J. Rieger, G. Osterwinter, C. Bui, F. Stoffelbach and B. Charleux, Macromolecules, 2009, 42, 5518–5525 CrossRef CAS.
- J. Chiefari, Y. Chong, F. Ercole, J. Krstina, J. Jeffery, T. Le, R. Mayadunne, G. Meijs, C. Moad and G. Moad, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- A. B. Lowe, N. C. Billingham and S. P. Armes, Macromolecules, 1999, 32, 2141–2148 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Chem. Rev., 2002, 102, 4177–4190 CrossRef CAS PubMed.
- Y. J. Che, Y. Tan, J. Cao and G. Y. Xu, J. Macromol. Sci., Phys., 2010, 49, 695–710 CrossRef CAS.
- P. Mary, D. D. Bendejacq, M. P. Labeau and P. Dupuis, J. Phys. Chem. B, 2007, 111, 7767–7777 CrossRef CAS PubMed.
- J. C. Salamone, W. Volksen, A. P. Olson and S. C. Israel, Polymer, 1978, 19, 1157–1162 CrossRef CAS.
- J. Virtanen, M. Arotçaréna, B. Heise, S. Ishaya, A. Laschewsky and H. Tenhu, Langmuir, 2002, 18, 5360–5365 CrossRef CAS.
- D. Wang, T. Wu, X. Wan, X. Wang and S. Liu, Langmuir, 2007, 23, 11866–11874 CrossRef CAS PubMed.
- M. Kobayashi, Y. Terayama, M. Kikuchi and A. Takahara, Soft Matter, 2013, 9, 5138–5148 RSC.
- J. Rodríguez-Hernández and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026–2027 CrossRef PubMed.
- W. Schärtl, Light Scattering from Polymer Solutions and Nanoparticle Dispersions, Springer-Verlag, Berlin Heidelberg, Berlin, 2007 Search PubMed.
- D. N. Schulz, D. G. Peiffer, P. K. Agarwal, J. Larabee, J. J. Kaladas, L. Soni, B. Handwerker and R. T. Garner, Polymer, 1986, 27, 1734–1742 CrossRef CAS.
- Z. Dong, J. Mao, D. Wang, M. Yang, W. Wang, S. Bo and X. Ji, Macromol. Chem. Phys., 2014, 215, 111–120 CrossRef CAS.
- H. Willcock, A. Lu, C. F. Hansell, E. Chapman, I. R. Collins and R. K. O'Reilly, Polym. Chem., 2014, 5, 1023–1030 RSC.
- J. Du, Y. Tang, A. L. Lewis and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 17982–17983 CrossRef CAS PubMed.
- S. L. West, J. P. Salvage, E. J. Lobb, S. P. Armes, N. C. Billingham, A. L. Lewis, G. W. Hanlon and A. W. Lloyd, Biomaterials, 2004, 25, 1195–1204 CrossRef CAS PubMed.
- J. P. Salvage, S. F. Rose, G. J. Phillips, G. W. Hanlon, A. W. Lloyd, I. Y. Ma, S. P. Armes, N. C. Billingham and A. L. Lewis, J. Controlled Release, 2005, 104, 259–270 CrossRef CAS PubMed.
- Y. J. Shih, Y. Chang, A. Deratani and D. Quemener, Biomacromolecules, 2012, 13, 2849–2858 CrossRef CAS PubMed.
- Y. J. Shih and Y. Chang, Langmuir, 2010, 26, 17286–17294 CrossRef CAS PubMed.
- Y. Chang, W. Y. Chen, W. Yandi, Y. J. Shih, W. L. Chu, Y. L. Liu, C. W. Chu, R. C. Ruaan and A. Higuchi, Biomacromolecules, 2009, 10, 2092–2100 CrossRef CAS PubMed.
- Y. Chang, S. C. Liao, A. Higuchi, R. C. Ruaan, C. W. Chu and W. Y. Chen, Langmuir, 2008, 24, 5453–5458 CrossRef CAS PubMed.
- F. Lee Rodkey, Clin. Chem., 1966, 12, 517–518 Search PubMed.
- F. Liu, J. Seuring and S. Agarwal, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4920–4928 CrossRef CAS.
- C. Barner-Kowollik, M. Buback, B. Charleux, M. L. Coote, M. Drache, T. Fukuda, A. Goto, B. Klumperman, A. B. Lowe, J. B. McLeary, G. Moad, M. J. Monteiro, R. D. Sanderson, M. P. Tonge and P. Vana, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5809–5831 CrossRef CAS.
- G. Moad, Macromol. Chem. Phys., 2014, 215, 9–26 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
- J. Madsen, S. P. Armes, K. Bertal, S. MacNeil and A. L. Lewis, Biomacromolecules, 2009, 10, 1875–1887 CrossRef CAS PubMed.
- S. F. Lascelles, F. Malet, R. Mayada, N. C. Billingham and S. P. Armes, Macromolecules, 1999, 32, 2462–2471 CrossRef CAS.
- A. B. Lowe, N. C. Billingham and S. P. Armes, Chem. Commun., 1996, 1555–1556 RSC.
- Z. Zhu, J. Xu, Y. Zhou, X. Jiang, S. P. Armes and S. Liu, Macromolecules, 2007, 40, 6393–6400 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional kinetic data; 1H NMR spectra for CTA, macro-CTA and diblock copolymer; TEM images of S38-H400 vesicles dispersed in pure water and in an aqueous solution of 1 M MgSO4; digital photographs of S38-H400 and G71-H400 vesicles in aqueous solutions containing MgSO4. See DOI: 10.1039/c5py00396b |
| This journal is © The Royal Society of Chemistry 2015 |