
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pd-complex driven formation of single-chain nanoparticles†
Johannes
Willenbacher
ab,
Ozcan
Altintas
ab,
Vanessa
Trouillet
c,
Nicolai
Knöfel
d,
Michael J.
Monteiro
e,
Peter W.
Roesky
*d and
Christopher
Barner-Kowollik
*ab
aPreparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76131 Karlsruhe, Germany. E-mail: christopher.barner-kowollik@kit.edu; Fax: +49 721 608-45740; Tel: +49 721 608-45741
bInstitut für Biologische Grenzflächen, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
cInstitute for Applied Materials (IAM) and Karlsruhe Nano Micro-Facility (KMNF), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
dInstitut für Anorganische Chemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 15, 76131 Karlsruhe, Germany. E-mail: peter.roesky@kit.edu; Fax: +49-721-608-44854; Tel: +49-721-608-46117
eAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane QLD 4072, Australia
First published on 23rd April 2015
Abstract
We report the facile synthesis of well-defined palladium(II) cross-linked single-chain nanoparticles (Pd-SCNPs) using the ‘repeating unit approach’. The linear precursor polymer (Mn ≈ 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 g mol−1, Đ ≈ 1.17) was synthesized via nitroxide mediated statistical copolymerization of styrene and 4-(chloromethyl)styrene (CMS) followed by a post-polymerization modification of the resulting copolymer to covalently attach the triarylphosphine ligand moieties. The ligand content along the lateral polymer chain was 12%. Intramolecular crosslinking was performed in diluted solution with a suitable precursor complex (Pd[1,5-cyclooctadiene]Cl2) to afford the well-defined Pd-SCNPs, which feature a hydrodynamic diameter of Dh = 5.4 nm. The palladium(II) containing single-chain nanoparticles were characterized in-depth using 1H NMR spectroscopy, 31P{1H} NMR spectroscopy, dynamic light scattering (DLS), size exclusion chromatography (SEC), 1H spin–spin relaxation time (T2) analysis, X-ray photoelectron spectroscopy (XPS), and log-normal distribution (LND) simulations. Finally, the applicability of the Pd-SCNPs as catalyst in the Sonogashira coupling was exemplified.
200 g mol−1, Đ ≈ 1.17) was synthesized via nitroxide mediated statistical copolymerization of styrene and 4-(chloromethyl)styrene (CMS) followed by a post-polymerization modification of the resulting copolymer to covalently attach the triarylphosphine ligand moieties. The ligand content along the lateral polymer chain was 12%. Intramolecular crosslinking was performed in diluted solution with a suitable precursor complex (Pd[1,5-cyclooctadiene]Cl2) to afford the well-defined Pd-SCNPs, which feature a hydrodynamic diameter of Dh = 5.4 nm. The palladium(II) containing single-chain nanoparticles were characterized in-depth using 1H NMR spectroscopy, 31P{1H} NMR spectroscopy, dynamic light scattering (DLS), size exclusion chromatography (SEC), 1H spin–spin relaxation time (T2) analysis, X-ray photoelectron spectroscopy (XPS), and log-normal distribution (LND) simulations. Finally, the applicability of the Pd-SCNPs as catalyst in the Sonogashira coupling was exemplified.
Introduction
The field of single-chain technology has evolved rapidly and attracted significant interest over the past few years due to possible applications in nano-medicine, chemical sensing, and catalysis.1–4 The research area can be divided into two fundamentally different synthetic strategies.5 On the one hand, the ‘selective point folding’ approach uses mutually orthogonal recognition units at distinct positions along the backbone to fold single polymer chains into defined three-dimensional structures.6–9 Placing these recognition units along the polymer chain is synthetically extremely demanding, but provides a better definition of the folding process. On the other hand, the ‘repeating unit approach’ is less synthetically demanding, since the functional groups can either be directly introduced onto the backbone via the copolymerization of a functional monomer or they can easily be grafted onto the polymer in a postpolymerization modification.10–19 However, the ‘repeating unit approach’ is associated with the drawback that the intramolecular crosslinking process as well as the placement of the functional groups is statistical, leading to random three-dimensional objects, the so-called single-chain nanoparticles (SCNPs).Both synthetic strategies derive their inspiration from nature. In each case, polymer chemists seek to mimic the structure and/or function of proteins at a very basic level. Recent all atom simulations by some of us for a ‘selective point folding’ system have demonstrated that polymers below a defined chain length fold into a near-unique closed conformation with a precision which is normally only reached for well-structured proteins.20 Sophisticated examples of ‘selective point folding’ are the hydrogen bond driven dual self-folding of a diblock copolymer and the reversible host–guest complexation driven folding of a water soluble polymer.5,21 Trigger responsive SCNPs were prepared using the ‘repeating unit approach’ via intramolecular crosslinking of dynamic covalent enamine bonds or hydrazone bonds.22–24 The incorporation of metal ions into the SCNP structure – similar to metallo–proteins – has been exploited only to a lesser extent. For example, Pomposo et al. fabricated Cu(II) containing SCNPs with highly selective catalytic activity for the oxidative coupling of terminal alkynes.25 Water-soluble SCNPs with ruthenium catalysts located in a hydrophobic cavity allow the transfer hydrogenation of ketones as demonstrated by Meijer et al.26 The group around Lemcoff synthesized rhodium(I) containing SCNPs from ROMP-derived polycyclooctadiene (polyCOD) with potential application in catalysis.27 The same group expanded the use of their COD-polymer by incorporating iridium(I) and nickel(II) ions.28 In the same study the authors successfully tested their organometallic nanoparticles in several catalytic applications.
In an earlier publication, we studied the ‘selective point folding’ of polystyrenes bearing a triarylphosphine ligand at each chain end.9 In the current study, we wish to transfer the established concept to the preparation of palladium(II) containing single-chain nanoparticles (see Scheme 1). In a facile, three step synthesis, the functional linear precursor polymers are initially generated in a random copolymerization of styrene and 4-chloromethylstyrene by nitroxide mediated polymerization (NMP), and subsequently post-modified with 4-(diphenylphosphino)benzoic acid. Next, the ligand bearing copolymer is cross-linked in a highly diluted solution to produce the palladium(II) cross-linked SCNPs. The above described procedure to synthesize SCNPs entails the advantage that the crosslinking process directly incorporates the catalytically active sites (Pd(PPh2Ar)2Cl2) within the SCNP structure. The Pd-SCNPs and the linear precursor polymer are characterized in-depth using 1H and 31P{1H} NMR spectroscopy, dynamic light scattering (DLS), size exclusion chromatography (SEC), 1H spin–spin relaxation time analysis, X-ray photoelectron spectroscopy (XPS), and log-normal distribution (LND) simulations.
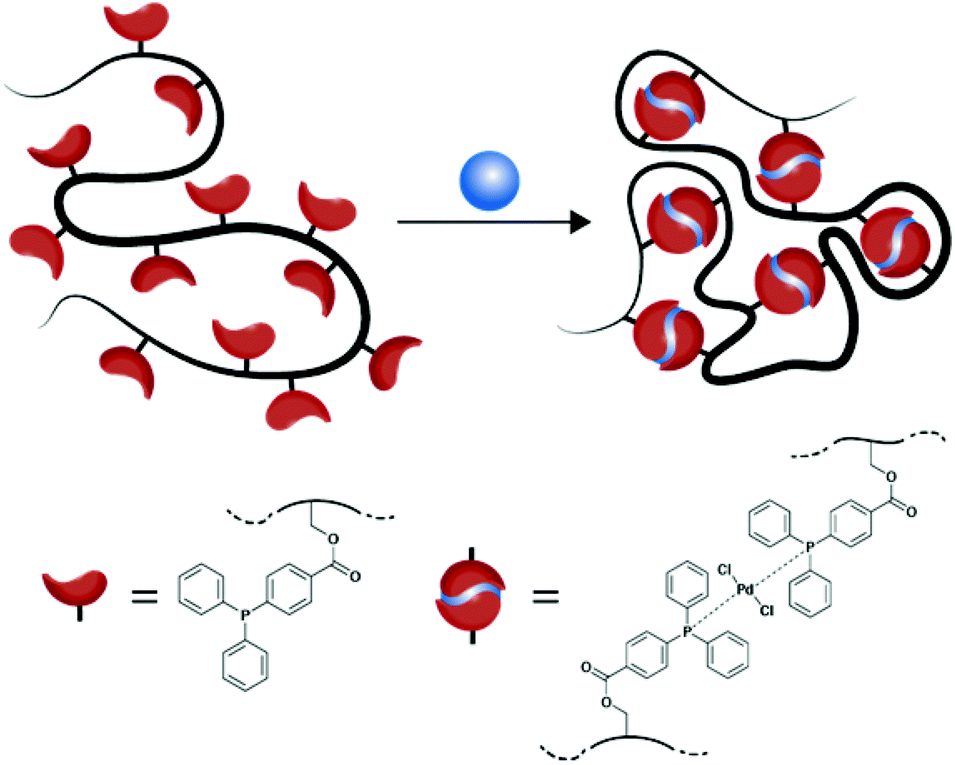 | ||
| Scheme 1 Illustration of the Pd-SCNP formation via the intramolecular crosslinking of functional, linear precursor copolymers by metal–ligand complexation in diluted solution (schematic view, in which we suggest a trans-configuration of the Pd-atoms, see text). | ||
Finally, it is shown that the Pd-SCNPs can be utilized as a catalyst in the Sonogashira coupling of 2-bromopyridine and phenylacetylene.
Experimental section
Materials
2-Bromopyridine (99%, Alfa Aesar), chloroform-d1 (CDCl3, 99.8%, EURISO-TOP), copper(I) iodide (CuI, 99.999%, PURATREM, ABCR), dichloromethane (dry) (DCM, 99.8%, extra dry, over molecular sieve, stabilized, Acros), dichloro(1,5-cyclooctadiene)palladium(II) ([Pd(COD)Cl2], 99%, ABCR) diethylamine (99+%, extra pure, Acros), N,N-dimethylformamide (DMF, 99%, extra pure, Acros), dimethylsulfoxide-d6 (DMSO-d6, 99.8%, EURISO-TOP), 4-(diphenylphosphino)benzoic acid (97%, Acros), K2CO3 (99%, Merck), Na2SO4 (99%, Carl Roth), phenylacetylene (98%, pure, Acros), tetrahydrofuran-d8 (THF-d8, 99.5%, EURISO-TOP) were used as received. Styrene (Sigma-Aldrich) was passed over a column of basic alumina (Acros) and 4-chloromethyl styrene (Acros) was distilled prior to usage and both substances were stored at −19 °C. N,N-Dimethylformamide (DMF), tetrahydrofuran (THF), dichloromethane (DCM), diethylether (Et2O), ethyl acetate and methanol were purchased as analytical grade solvents (Sigma-Aldrich) and used as received.Synthetic procedures
The NMP-initiator 2,2,6,6-tetramethyl-1-(1-phenylethoxy)-piperidine (1, Scheme 2) was synthesized according to a literature procedure.29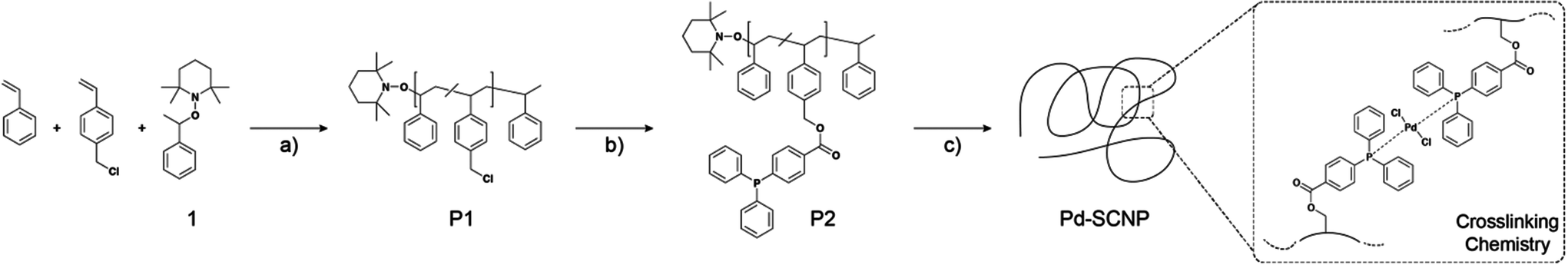 | ||
| Scheme 2 Synthetic strategy for the preparation of palladium(II) crosslinked single-chain nanoparticles (Pd-SCNPs). (a) bulk, 125 °C, 5 h; (b) 4-(diphenylphosphino)benzoic acid, K2CO3, DMF (dry), 50 °C, 16 h; (c) Pd(COD)Cl2, DCM (dry), a.t., 21 h (the crosslinking chemistry presents a schematic view, in which we suggest a trans-configuration of the Pd-atoms, see text). | ||
1H NMR (400 MHz, CDCl3, δ): 7.26–6.20 (m, aromatic protons of PS and initiator), 4.65–4.39 (bs, CH2Cl), 2.50–0.84 (m, aliphatic protons of PS and initiator). SEC (THF, RI): Mn = 10200 g mol−1, Đ = 1.17.
Post modification of poly(styrene-co-chloromethylstyrene) with 4-(diphenylphosphino)benzoic acid
Poly(styrene-co-chloromethylstyrene) (1.0 g, 1 mmol CMS, number of CMS repeating units = 1 equiv.), 4-(diphenylphosphino) benzoic acid (459 mg, 1.5 mmol, 1.5 equiv.) and K2CO3 (415 mg, 3 mmol, 3 equiv.) were dissolved in dry DMF (10 mL) and stirred at 50 °C for 16 h under inert atmosphere. The reaction mixture was diluted with ethyl acetate (300 mL) and washed with brine (3 × 150 mL). The organic phase was separated and dried over Na2SO4. Subsequently, the solvent was removed under reduced pressure. The crude product was diluted with THF (20 mL) and precipitated into cold methanol (200 mL). The functionalized linear precursor polymer P2 was afforded as a white solid by filtration and dried under high vacuum (0.9 g, 0.07 mmol).
1H NMR (400 MHz, CDCl3, δ): 7.99–7.19 (m, aromatic protons of PPh2Ar), 7.26–6.11 (m, aromatic protons of PS and initiator), 5.29–5.00 (bs, CH2OC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 2.50–0.72 (aliphatic protons of PS and initiator). 31P{1H} NMR (162 MHz, CDCl3, δ): −5.06 (s, PPh2Ar). SEC (THF, RI): Mn = 12300 g mol−1, Đ = 1.16.
O), 2.50–0.72 (aliphatic protons of PS and initiator). 31P{1H} NMR (162 MHz, CDCl3, δ): −5.06 (s, PPh2Ar). SEC (THF, RI): Mn = 12300 g mol−1, Đ = 1.16.
Single-chain nanoparticle formation by intramolecular crosslinking with Pd(II)-ions
A solution of the functionalized linear precursor copolymer P2 (50 mg, 0.04 mmol ligand moieties, 1 equiv.) in DCM (dry) (cprecursor = 2.5 mg mL−1) was added dropwise to a stirred solution of Pd(COD)Cl2 (6 mg, 0.02 mmol, 0.5 equiv.) in 20 mL DCM (dry) by syringe pump (1 mL min−1) at ambient temperature in a flame dried Schlenk flask. After the addition the solution was stirred for an additional hour, then it was concentrated in vacuo and the SCNPs were precipitated in cold methanol. Filtration and drying under high vacuum afforded the product as a yellow solid (35 mg).
1H NMR (400 MHz, CDCl3, δ): 8.11–7.19 (m, aromatic protons of Pd-PPh2Ar), 7.26–6.11 (m, aromatic protons of PS and initiator), 5.29–5.00 (bs, CH2OCO), 2.50–0.72 (aliphatic protons of PS and initiator). 31P{1H} NMR (162 MHz, CDCl3, δ): 23.54 (s, PPh2Ar-PdCl2-PPh2Ar).
Sonogashira coupling utilizing Pd-SCNP as catalyst
According to a modified literature procedure,30 2-bromopyridine (476 μL, 5 mmol, 1 equiv.), phenylacetylene (549 μL, 5 mmol, 1 equiv.) and the catalyst Pd-SCNP (see Table 2 for the employed amounts) were dissolved in diethylamine (2 mL) in a flame-dried Schlenk tube. The mixture was degassed by purging the solution with argon for 5 min. Next, CuI (4.8 mg, 0.02 mmol, 0.05 equiv.) was added and the reaction was conducted for 24 h at ambient temperature. Subsequently, the tube was opened to the atmosphere and the mixture was diluted with Et2O (10 mL). The organic layer was filtered and washed with distilled H2O (2 × 20 mL). The organic layer was dried over Na2SO4 and evaporated under reduced pressure to afford the crude product. After 24 h under high vacuum the product was obtained as a brown liquid.Results and discussion
Synthesis of the linear precursor polymer
The linear precursor polymer was synthesized by a combination of nitroxide mediated polymerization and a postpolymerization modification (see Scheme 2). The random copolymerization of styrene and 4-chloromethyl styrene was conducted using a feed ratio of 9:1. The resultant styrene-co-chloromethylstyrene polymer P1 exhibits a narrow molecular weight distribution with a dispersity index of Đ = 1.17 and a number weighted molar mass of Mn = 10200 g mol−1 according to SEC (THF, RI). The 1H NMR spectrum shows the aromatic as well as the aliphatic signals for the polystyrene backbone. In addition, a broad resonance between 4.39 and 4.65 ppm can be assigned to the methylene bridge of the CMS units (see Fig. S1†). The integration ratios of the resonances belonging to the aromatic protons and the methylene unit were utilized to estimate the copolymer composition. A value of 12% CMS units was calculated, which closely resembles the copolymerization feed ratio. In order to attach the ligand moieties onto the linear precursor polymer, P1 was treated with a slight excess of 4-(diphenylphosphino) benzoic acid per CMS unit in the presence of the base K2CO3. In a simple substitution reaction, the triarylphosphine ligands were grafted onto the polymer backbone via an ester bond. The shift of the methylene bridge resonance from 4.54 ppm to 5.25 ppm in the 1H NMR spectrum of P2 evidences full conversion of the CMS to ligand units (see Fig. S1†). New resonances found in the range of 7.19 and 7.99 ppm can be assigned to the aromatic protons of the triarylphosphine moiety. The 31P{1H} NMR spectrum of P2 shows a single resonance at −5.06 ppm representing trivalent uncoordinated triarylphosphine. An additional indication for the successful transformation is the increase in molar mass of P2 to Mn = 12300 g mol−1 according to SEC (see Fig. S2†). The above described design strategy represents a universal approach for the facile introduction of functional groups into the precursor structure.
Preparation of the palladium(II) crosslinked SCNPs
The palladium(II) cross-linked single-chain nanoparticles (Pd-SCNPs) were prepared by intramolecularly crosslinking the linear precursor polymer P2 in diluted solution with a suitable palladium(II) source (see Scheme 2). Dichloro(1,5-cyclooctadiene)palladium(II) ([Pd(COD)Cl2]) has proven to be an ideal candidate, since the COD ligand is an outstanding leaving group in the ligand exchange reaction with the triarylphosphine moieties along the polymer. Intermolecular reactions always pose a problem during the preparation of SCNPs, since they lead to multi-chain aggregates. To circumvent such by-products, highly diluted conditions need to be applied. Feeding the polymer precursor into the Pd solution overcomes the need for dilute conditions as long as the feeding rate is rate determining. A solution of the precursor polymer P2 (cP2 = 0.2 mmol mL−1) was added dropwise (1 mL min−1) to a solution of [Pd(COD)Cl2] via a syringe pump. The ratio of Pd(II) cross-linker to ligand moieties along the polymer chain was chosen to be 1:2 to achieve the highest amount of crosslinking.
During the intermolecular crosslinking of single polymer chains, the hydrodynamic volume decreases due to the transition from a random coil to a more compact structure. The change in hydrodynamic volume can readily be observed and quantified via size exclusion chromatography. The decreased hydrodynamic volume leads to a shift of the SEC trace to longer retention times (see Fig. 2A). Expressed in peak molar masses the shift is ΔMp = 3850 g mol−1, which equals a molecular weight change of ∼21%.
The actual hydrodynamic diameter of the Pd-SCNP can be determined by dynamic light scattering and has a value of Dh = 5.4 nm. Compared with the linear precursor P2 (Dh = 8.8 nm), the difference is ΔDh = 3.3 nm (see Fig. 2B), which equals a change of ∼37%.
Chemical evidence for the crosslinking reaction was obtained from 1H and 31P{1H} NMR spectroscopy. Upon complexation by Pd(II) ions, the proton resonances of the triarylphosphine functional groups are shifted to a lower field (see Fig. 1A). The resonance for the phosphorus atom of the free ligand at δ = −5.06 ppm shifts downfield to δ = 23.54 ppm upon coordination to the metal atom (see Fig. 1B). A complete shift of the phosphorus resonance indicates – within the boundaries of 31P{1H} NMR spectroscopy – a quantitative coordination of the free ligands to the metal atoms. As additional evidence for the correct interpretation of the NMR data, some metal complexes were synthesized as model compounds for Pd-SCNP. As model ligands for the polymer P2 4-methylbenzyl-4-(diphenylphosphino) benzoate (L1) and 4-ethylbenzyl-4-(diphenylphosphino) benzoate (L2) were synthesized. The 31P{1H} NMR spectra of the complexes [Pd(L1)2Cl2] and [Pd(L2)2Cl2] (obtained from [Pd(COD)Cl2] and L1 or L2) showed a chemical shift of δ = 23.5 ppm, which is very similar to the shift observed for Pd-SCNP. [Pd(L1)2Cl2] and [Pd(L2)2Cl2] were fully characterized (refer to the ESI†) and the solid state structures of [Pd(L1)2Cl2] and [Pd(L2)2Cl2] were established by single crystal X-ray diffraction (see Fig. S6 and S7†). Like [Pd(PPh3)2Cl2], [Pd(L1)2Cl2] and [Pd(L2)2Cl2] adopt a trans-configuration in the solid state.31,32 As P2 can form large rings upon cross-linking, we suggest a trans-configuration of the palladium atoms in Pd-SCNP. Since the phosphorus atoms in Pd-SCNP are not in all cases chemically equivalent due to the cross-linking, we next mixed the related ligands L1 and L2 with [Pd(COD)Cl2] in a 1:1:1 ratio. Statistically, a 1:2:1 mixture of [Pd(L1)2Cl2], [Pd(L1)(L2)Cl2], and [Pd(L2)2Cl2] is expected. In comparison to [Pd(L1)2Cl2], a slightly broadened signal but no coupling is observed in the 31P{1H} NMR spectrum of the statistical mixture of [Pd(L1)2Cl2], [Pd(L1)(L2)Cl2], and [Pd(L2)2Cl2] (see Fig. S5†). This data clearly shows that slight differences in the polymer backbone of the triarylphosphine functional group of Pd-SCNP have only a minor influence on the 31P{1H} NMR signals.
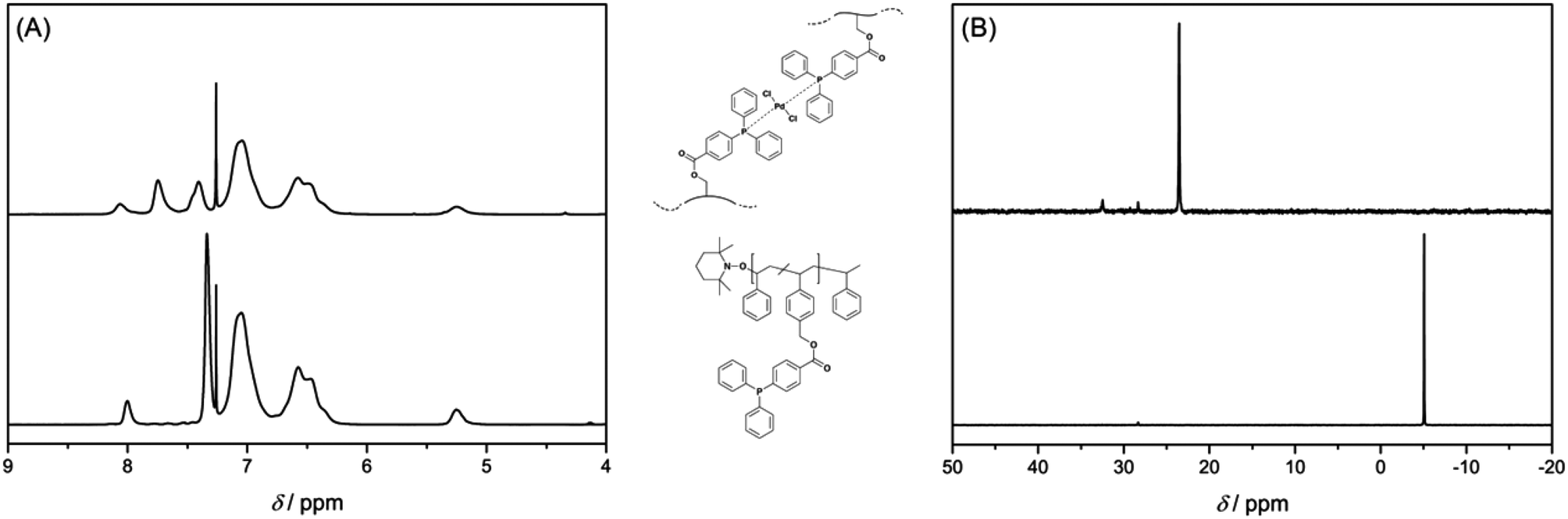 | ||
| Fig. 1 Intramolecular crosslinking by metal–ligand complexation monitored by 1H NMR (A) and 31P{1H} NMR spectroscopy (B) in CDCl3 at 25 °C. The complete shift of the proton and phosphorus resonances indicates quantitative complexation (the crosslinking chemistry presents a schematic view, in which we suggest a trans-configuration of the Pd-atoms, see text). | ||
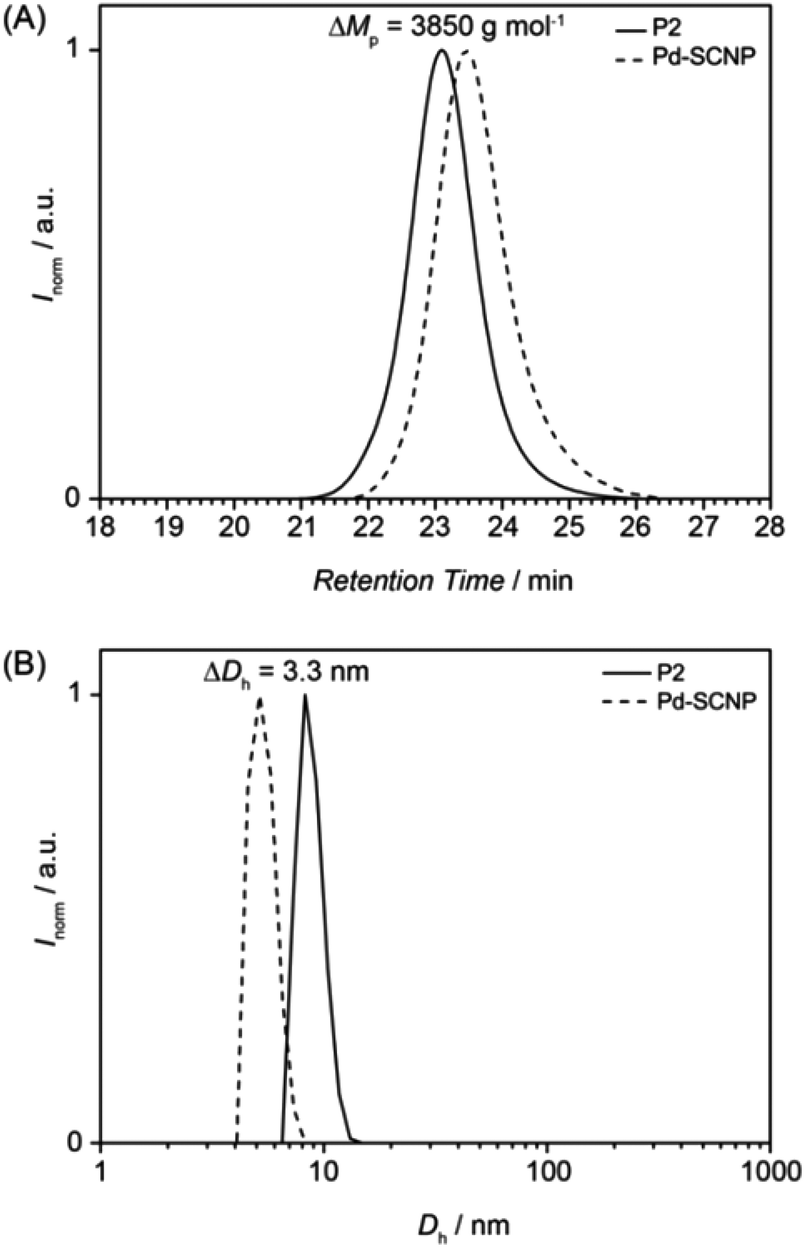 | ||
| Fig. 2 SEC and DLS results of the Pd-SCNP formation. (A) Normalized SEC traces (DMAc, RI) of the linear precursor polymer P2 (solid line) and the Pd-SCNP (dashed line). (B) Number-averaged size distributions of the linear precursor P2 (solid line) and the Pd-SCNP (dashed line) in THF (c = 4 mg mL−1) at 25 °C. | ||
Additional information on the intramolecular crosslinking can be obtained from the 1H spin–spin relaxation time (T2), as T2 is sensitive to molecular motion.33T2 decay curves for the resonance of the methylene bridge (∼5.25 ppm) in P2 and Pd-SCNP (see Fig. 3) were measured via the Car–Purcel–Meiboom–Gill (CPMG) echo train acquisition in THF-d8. Since the intramolecular crosslinking reduces the mobility of the chain-segments, T2 should decrease in Pd-SCNP compared to P2. Analysis of the decay curves via a mono exponential decay function reveals T2 = 147 ms for P2 (see Fig. S3†) and T2 = 70 ms for Pd-SCNP (see Fig. S4†). These results clearly show the transformation from a random coil to a compacted, cross-linked structure.
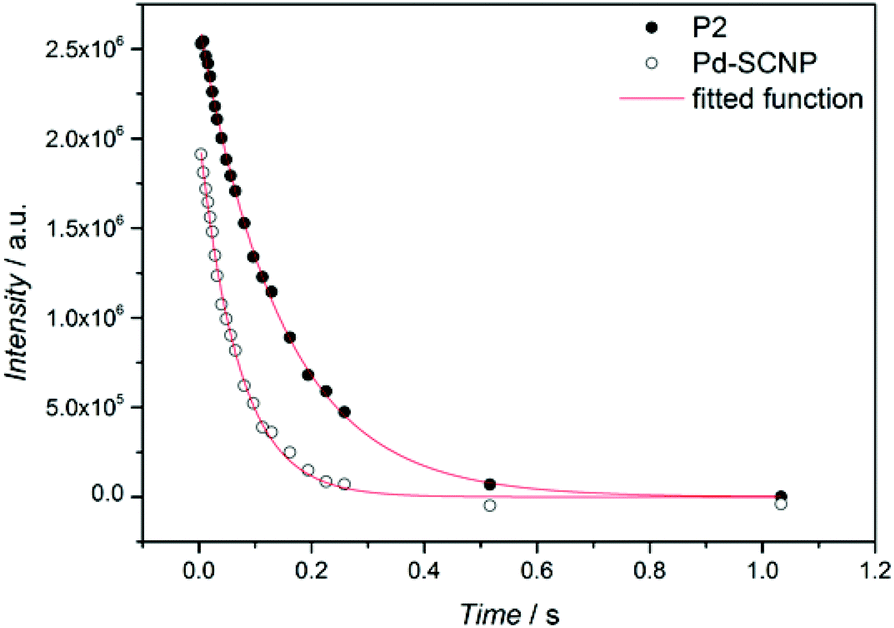 | ||
| Fig. 3 T 2 decay curves for the methylene bridge protons of the linear precursor polymer P2 (dots) and the Pd-SCNP (open dots). The red curves represent mono exponential decay functions that have been fitted to the data points. | ||
Furthermore, the transition from the linear precursor P2 to the more compacted structure in Pd-SCNP was analyzed by log-normal distribution (LND) simulations. Therefore, the log-normal distribution (LND) based on a Gaussian function was used to model the experimental molecular weight distribution (MWD) before and after the intramolecular crosslinking. In particular, the weight distribution was used, since the weight of polymer before and after chain collapse does not change, whereas the number distribution will be very different.34 The following equations for the LND (see ref. 35 for more details) were employed:
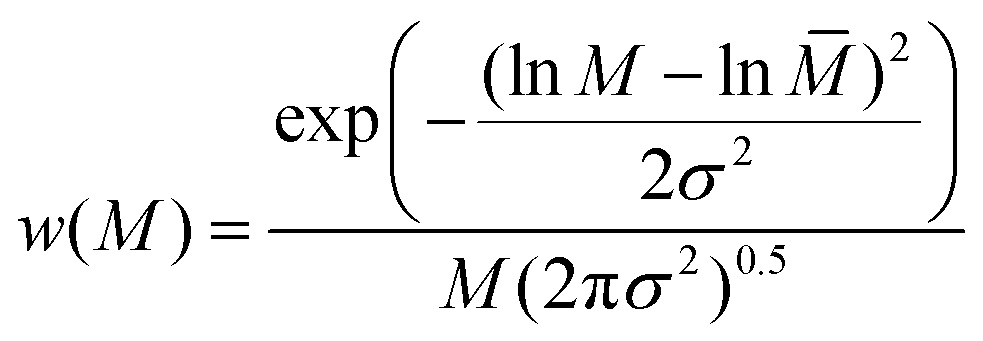 | (1) |
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) = (MnMw)0.5 and σ2 = ln(Đ).
= (MnMw)0.5 and σ2 = ln(Đ).
Eqn (1) is the Gaussian distribution function of w(M) (the weight distribution of the SEC trace), Mn is the number-average molecular weight, Mw is the weight-average molecular weight, and the polydispersity Đ = Mw/Mn. An experimental distribution (e.g. black trace in Fig. 4A) can now be fitted with multiple distributions, each distribution generated from eqn (1) with a pre-determined Mn and Đ. The final simulated distribution can be determined by accounting for the weight fraction of each distribution according to the eqn (2),
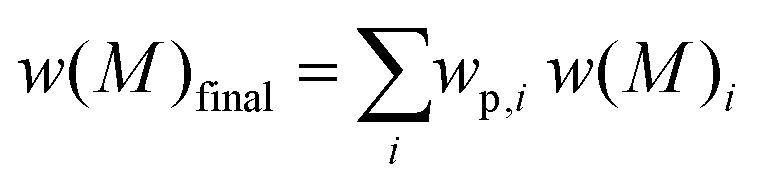 | (2) |
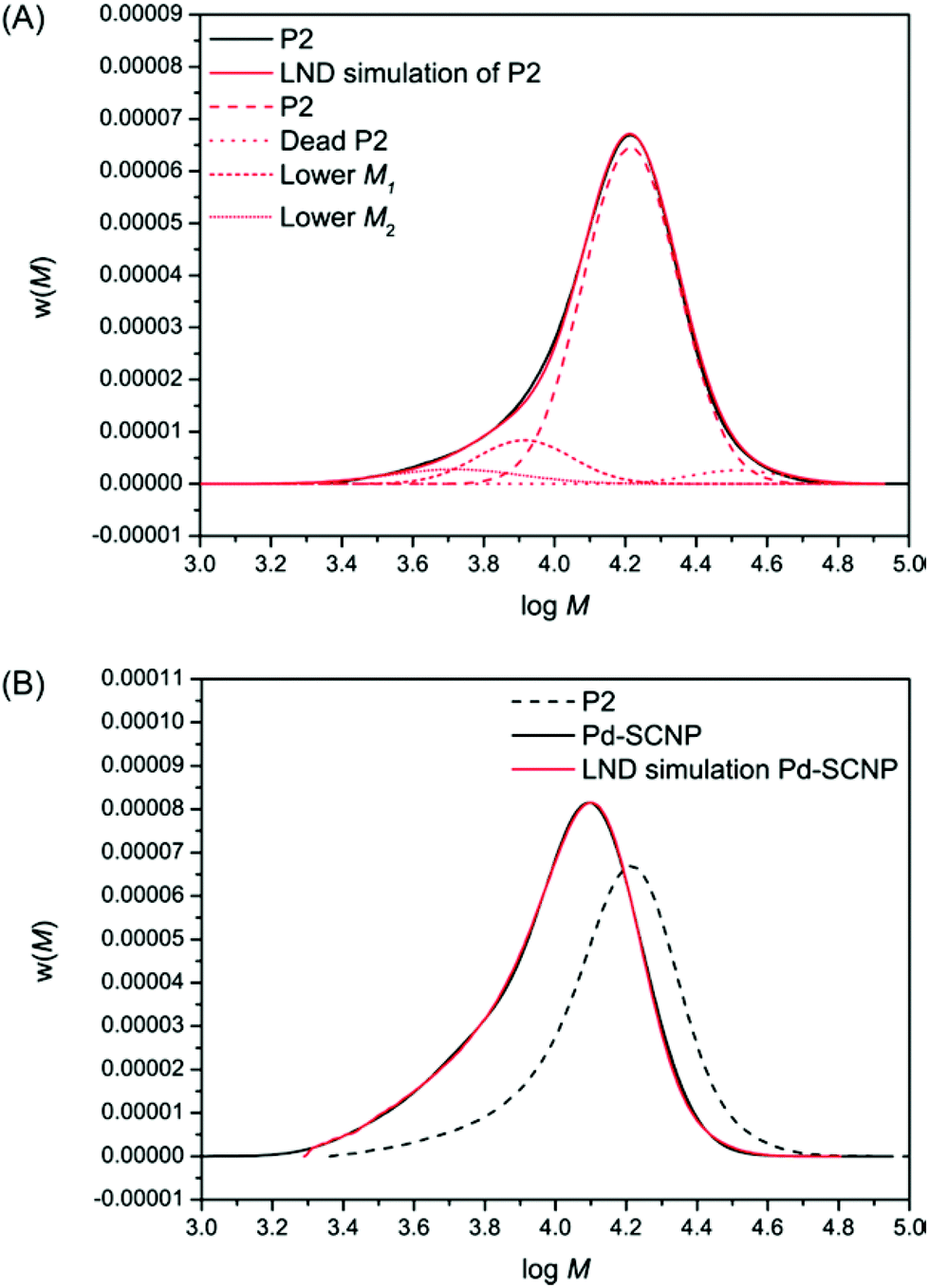 | ||
| Fig. 4 Experimental SEC (DMAc, RI) data and corresponding log-normal distribution (LND) simulations. (A) Black trace: experimental P2, red trace: LND fit of P2 using 4 distributions, namely P2 (red dashed), dead P2 (red dotted), lower M1 (red short dashed), and lower M2 (red short dotted). (B) Black trace: experimental Pd-SCNP, red trace: LND fit of Pd-SCNP using 7 distributions, black dashed trace: experimental P2 for comparison. | ||
The SEC chromatogram for P2 was fitted using the eqn (1) and (2) above (see Fig. 4A). It can be seen that the fit of the chromatogram by the LND model was excellent. To arrive at this fit, the main linear peak (P2, Mn = 17200 g mol−1, Đ = 1.1) consisted of a weight fraction of 0.858, a peak with a weight fraction of 0.069 corresponding for dead polymer formed through bimolecular radical termination (Mn = 34400 g mol−1, Đ = 1.1), and two lower molecular weight peaks with a combined weight fraction of 0.073 (see Table 1 top). Each LND distribution for the polymers in Table 1 is given in Fig. 4A. After metal complexation forming the more compact Pd-SCNPs, we determined that all the linear P2 and the dead polymer formed lower molecular weight species, suggesting ∼100% intramolecular coupling. There are many possibilities for the number of cross-linked products. Here, we used only seven different cross-linked products, but more species cannot be ruled out (see Table 1 bottom). The fit using the LND model is again excellent (see Fig. 4B).
| Distribution | M n/g mol−1 | Đ | w p |
|---|---|---|---|
| P2 | 17200 |
1.1 | 0.858 |
| Dead P2 | 34400 |
1.1 | 0.069 |
| Lower M1 | 8600 | 1.1 | 0.056 |
| Lower M2 | 5730 | 1.2 | 0.017 |
| Distribution | M n/g mol−1 | Đ | w p |
|---|---|---|---|
| P2 | 17200 |
1.1 | 0 |
| Dead P2 | 34400 |
1.1 | 0 |
| Lower M1 | 8600 | 1.1 | 0 |
| Lower M2 | 5730 | 1.2 | 0 |
| Pd-SCNP 1 | 3440 | 1.1 | 0.015 |
| Pd-SCNP 2 | 5160 | 1.1 | 0.024 |
| Pd-SCNP 3 | 6880 | 1.1 | 0.101 |
| Pd-SCNP 4 | 12212 |
1.1 | 0.385 |
| Pd-SCNP 5 | 13416 |
1.1 | 0.179 |
| Pd-SCNP 6 | 14964 |
1.1 | 0.083 |
| Pd-SCNP 7 | 15480 |
1.1 | 0.213 |
The information suggests especially for the low molecular weight species that the change in hydrodynamic volume (HDV) can be as low as 0.2 that of the molecular weight of linear P2. This low HDV suggests a very compact nature, which most probably consists of a single polymer chain.
In order to determine the nature of the Pd-complexes of the SCNPs, P2, Pd-SCNP and the model complex [Pd(PPh3)2Cl2] were subjected to X-ray photoelectron spectroscopy (XPS) analysis. The spectrum of P2 exhibits a peak at 131.1 eV, which is attributed to the triarylphosphine ligand. The signal amounts to 1.1 atomic% (2.7 wt%, 0.0009 mmol mg−1), which is in perfect agreement with the 1H NMR estimations (0.0008 mmol mg−1). The obtained XPS data of all metal containing samples ([Pd(PPh3)2Cl2], and Pd-SCNP) was not fully conclusive. We anticipate some decomposition of the metal complexes during the XPS process, since besides the expected Pd signal (at 338.2 eV) a second Pd-species with a binding energy of 336.4 eV was observed (see Fig. 5). However, the XPS-spectrum of Pd-SCNP shows signals at 131.6 eV, 198.3 eV and 338.2 eV, which are assigned to the phosphorus, chlorine and palladium species, respectively. The peak areas correspond to 0.6 atomic% for phosphorus and chlorine and to 0.3 atomic% for palladium. The ratio P:Cl:Pd of 2:2:1 thus matches the proposed structure of the Pd complexes in the Pd-SCNPs. In addition, the binding energy of the main Pd species in Pd-SCNP (see Fig. 5) and of chlorine and phosphorus (not shown here) are in excellent accordance with the measured binding energy in Pd(PPh3)2Cl2. Furthermore, the found binding energies are comparable with literature values for PdCl2.36,37
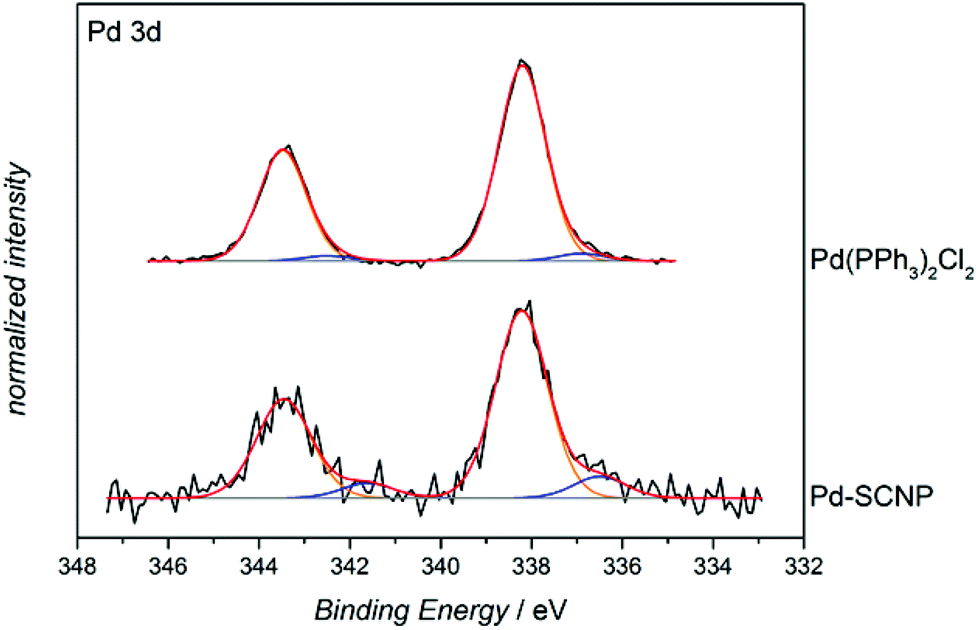 | ||
| Fig. 5 Pd 3d XPS spectra of Pd-SCNP (bottom) and [Pd(PPh3)2Cl2] (top). The comparison of the prepared SCNPs with the model complex shows that the binding energy of the main Pd species at 338.2 eV in Pd-SCNP is in excellent agreement with the binding energy in [Pd(PPh3)2Cl2]. | ||
In order to ascertain the true number of Pd-centers in Pd-SCNP, additional NMR experiments were carried out. For [Pd(PPh3)2Cl2] and Pd-SCNP31P{1H}, NMR samples with an inlet containing an internal standard were prepared. [Pd(PPh3)2Cl2] was chosen as the internal standard for P2, whereas PPh3 was chosen for Pd-SCNP. By utilizing inlets, it was ensured that the internal standards do not interfere with the analyte. These experiments delivered phosphorus contents of 0.0009 mmol mg−1 for Pd-SCNP and 0.0011 mmol mg−1 for P2. The determined values are in good agreement with the estimation from the 1H NMR spectra (refer to the ESI†). As a proof of principle, the Pd-SCNPs were utilized as a novel type of catalyst for the Sonogashira coupling of terminal alkynes and aryl halides. In an exemplary reaction 2-bromopyridine and phenylacetylene were coupled in diethylamine, employing CuI as co-catalyst. The coupling reaction was performed at two ratios of substrate to catalyst. It was observed that an increase in the amount of catalyst improved the yield of the reaction under otherwise identical conditions. To benchmark the Pd-SCNPs against a well-known system, the same coupling reaction was also conducted with the model complex [Pd(PPh3)2Cl2], this time keeping the substrate to catalyst ratio constant. Under these same conditions the model complex delivered a higher conversion of close to 75% (see Table 2).
| Sonogashira coupling | Catalyst | Substrate to catalyst ratio | Conversion after 24 h |
|---|---|---|---|
| 1 | 5 mg Pd-SCNP | 2222:1 |
26% |
| 2 | 10 mg Pd-SCNP | 1111:1 |
45% |
| 3 | 3.2 mg [Pd(PPh3)2Cl2] | 1111:1 |
75% |
Conclusions
Herein, we present the preparation and in-depth characterization of well defined, palladium(II) cross-linked single-chain nanoparticles. The single-chain nanoparticle formation was confirmed by proton nuclear magnetic resonance (1H NMR) spectroscopy, 31P{1H} NMR spectroscopy, dynamic light scattering (DLS), size exclusion chromatography (SEC), 1H spin–spin relaxation time analysis, and X-ray photoelectron spectroscopy (XPS). The design strategy for the linear precursor polymers entails the possibility to easily introduce variable functional groups. In the case at hand, Pd-SCNPs with a hydrodynamic diameter of 5.4 nm were fabricated. The underlying intramolecular crosslinking process directly leads to catalytically active Pd-SCNPs, which was exemplarily demonstrated in a Sonogashira coupling reaction. Employing different precursor complexes (e.g. with Rh-, Ir- or Ni-ion centres) might open the door to various other catalysts. In addition, a novel procedure for interpreting the SEC chromatograms of the SCNPs by log-normal distribution (LND) simulations was introduced. Thus, we were able to show that all linear precursor polymers were converted to the more compact SCNP structure.Acknowledgements
C.B.-K. acknowledges financial support from the Karlsruhe Institute of Technology (KIT) in the context of the Helmholtz BIF program, the German Research Council (DFG) and the Ministry of Science and Arts of the state of Baden-Württemberg. The authors acknowledge Sebastian Bestgen (AK Roesky) for helpful discussions.References
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2012, 33, 958–971 CrossRef CAS PubMed.
- C. K. Lyon, A. Prasher, A. M. Hanlon, B. T. Tuten, C. A. Tooley, P. G. Frank and E. B. Berda, Polym. Chem., 2015, 6, 181–197 RSC.
- M. Ouchi, N. Badi, J.-F. Lutz and M. Sawamoto, Nat. Chem., 2011, 3, 917–924 CrossRef CAS PubMed.
- A. Sanchez-Sanchez and J. A. Pomposo, Part. Part. Syst. Charact., 2014, 31, 11–23 CrossRef CAS PubMed.
- J. Willenbacher, B. V. K. J. Schmidt, D. Schulze-Suenninghausen, O. Altintas, B. Luy, G. Delaittre and C. Barner-Kowollik, Chem. Commun., 2014, 50, 7056–7059 RSC.
- O. Altintas, P. Gerstel, N. Dingenouts and C. Barner-Kowollik, Chem. Commun., 2010, 46, 6291–6293 RSC.
- O. Altintas, E. Lejeune, P. Gerstel and C. Barner-Kowollik, Polym. Chem., 2012, 3, 640–651 RSC.
- O. Shishkan, M. Zamfir, M. A. Gauthier, H. G. Börner and J.-F. Lutz, Chem. Commun., 2014, 50, 1570–1572 RSC.
- J. Willenbacher, O. Altintas, P. W. Roesky and C. Barner-Kowollik, Macromol. Rapid Commun., 2013, 35, 45–51 CrossRef PubMed.
- D. Chao, X. Jia, B. Tuten, C. Wang and E. B. Berda, Chem. Commun., 2013, 49, 4178–4180 RSC.
- P. G. Frank, B. T. Tuten, A. Prasher, D. Chao and E. B. Berda, Macromol. Rapid Commun., 2014, 35, 249–253 CrossRef CAS PubMed.
- B. T. Tuten, D. Chao, C. K. Lyon and E. B. Berda, Polym. Chem., 2012, 3, 3068–3071 RSC.
- E. A. Appel, J. d. Barrio, J. Dyson, L. Isaacs and O. A. Scherman, Chem. Sci., 2012, 3, 2278–2281 RSC.
- C. F. Hansell, A. Lu, J. P. Patterson and R. K. O'Reilly, Nanoscale, 2014, 6, 4102–4107 RSC.
- J. Romulus and M. Weck, Macromol. Rapid Commun., 2013, 34, 1518–1523 CrossRef CAS PubMed.
- J. Lu, N. ten Brummelhuis and M. Weck, Chem. Commun., 2014, 50, 6225–6227 RSC.
- L. Oria, R. Aguado, J. A. Pomposo and J. Colmenero, Adv. Mater., 2010, 22, 3038–3041 CrossRef CAS PubMed.
- D. D. Lane, D. Y. Chiu, F. Y. Su, S. Srinivasan, H. B. Kern, O. W. Press, P. S. Stayton and A. J. Convertine, Polym. Chem., 2015, 6, 1286–1299 RSC.
- P. Wang, H. Pu, J. Ge, M. Jin, H. Pan, Z. Chang and D. Wan, Mater. Lett., 2014, 132, 102–105 CrossRef CAS PubMed.
- D. Danilov, C. Barner-Kowollik and W. Wenzel, Chem. Commun., 2015, 51, 6002–6005 RSC.
- O. Altintas, P. Krolla-Sidenstein, H. Gliemann and C. Barner-Kowollik, Macromolecules, 2014, 47, 5877–5888 CrossRef CAS.
- A. Sanchez-Sanchez, D. A. Fulton and J. A. Pomposo, Chem. Commun., 2014, 50, 1871–1874 RSC.
- B. S. Murray and D. A. Fulton, Macromolecules, 2011, 44, 7242–7252 CrossRef CAS.
- D. E. Whitaker, C. S. Mahon and D. A. Fulton, Angew. Chem., Int. Ed., 2013, 52, 956–959 CrossRef CAS PubMed.
- A. Sanchez-Sanchez, A. Arbe, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2014, 3, 439–443 CrossRef CAS.
- T. Terashima, T. Mes, T. F. A. De Greef, M. A. J. Gillissen, P. Besenius, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 4742–4745 CrossRef CAS PubMed.
- S. Mavila, C. E. Diesendruck, S. Linde, L. Amir, R. Shikler and N. G. Lemcoff, Angew. Chem., Int. Ed., 2013, 52, 5767–5770 CrossRef CAS PubMed.
- S. Mavila, I. Rozenberg and N. G. Lemcoff, Chem. Sci., 2014, 5, 4196–4203 RSC.
- J. Willenbacher, K. N. R. Wuest, J. O. Mueller, M. Kaupp, H.-A. Wagenknecht and C. Barner-Kowollik, ACS Macro Lett., 2014, 3, 574–579 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- G. Ferguson, R. McCrindle, A. J. McAlees and M. Parvez, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 2679–2681 CrossRef.
- S. Otsuka, J. Organomet. Chem., 1980, 200, 191–205 CrossRef CAS.
- J. He, L. Tremblay, S. Lacelle and Y. Zhao, Soft Matter, 2011, 7, 2380–2386 RSC.
- M. Gavrilov and M. J. Monteiro, Eur. Polym. J., 2015, 65, 191–196 CrossRef CAS PubMed.
- M. J. Monteiro, Eur. Polym. J., 2015, 65, 197–201 CrossRef CAS PubMed.
- K. Young-Inn, J. Chan-Kyou, L. Yong-Min and C. Sung-Nak, Bull. Korean Chem. Soc., 2002, 23, 1754–1758 CrossRef.
- M. Ranjbar, S. Fardindoost, S. M. Mahdavi, A. Iraji zad and N. Tahmasebi G, Sol. Energy Mater. Sol. Cells, 2011, 95, 2335–2340 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization methods, additional analytical data as well as additional experimental details. CCDC 1060115 and 1060116. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5py00389j |
| This journal is © The Royal Society of Chemistry 2015 |