
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the structure–control relationship of amide-functionalized SG1-based alkoxyamines for nitroxide-mediated polymerization and conjugation†
Elise
Guégain
a,
Vianney
Delplace
a,
Thomas
Trimaille
b,
Didier
Gigmes
b,
Didier
Siri
b,
Sylvain R. A.
Marque
b,
Yohann
Guillaneuf
b and
Julien
Nicolas
*a
aInstitut Galien Paris-Sud, CNRS UMR 8612, Univ. Paris-Sud, Faculté de Pharmacie, 5 rue Jean-Baptiste Clément, F-92296 Châtenay-Malabry cedex, France. E-mail: julien.nicolas@u-psud.fr; Web: https://twitter.com/julnicolas Fax: +33 1 46 83 55 11; Tel: +33 1 46 83 58 53
bAix-Marseille University CNRS UMR 7273, Institut de Chimie Radicalaire, Avenue Escadrille Normandie-Niemen, 13397 Marseille Cedex 20, France
First published on 19th March 2015
Abstract
The functionalization of alkoxyamines prior to nitroxide-mediated polymerization (NMP) induces important structural variations when compared to the parent molecules. This may have important consequences on the design of functionalized materials by pre-functionalization. In this context, a wide range of amide-functionalized alkoxyamines (a functionality often obtained after conjugation from COOH– and N-succinimidyl-containing alkoxyamines) based on the nitroxide SG1 (N-tert-butyl-N-(1-diethyl phosphono-2,2-dimethylpropyl) nitroxide) have been synthesized and their dissociation rate constants (kd) have been determined. To rationalize their reactivity, a multi-parameter procedure was applied and enabled to discriminate disubstituted amide-functionalized alkoxyamines from monosubstituted ones. Monosubstituted alkoxyamines exhibited lower kd than their disubstituted counterparts (Ea increase of ∼7–10 kJ mol−1) because of the occurrence of intramolecular hydrogen bonding (IHB) between the alkyl and the nitroxide fragments. NMP of styrene, n-butyl acrylate and methyl methacrylate with a small amount of acrylonitrile was then successfully performed from two representative secondary SG1-based alkoxyamines employed for conjugation: namely AMA (COOH-containing) and AMA-NHS (N-succinimidyl derivative), and compared to polymerizations initiated with AMA-Gem, an AMA-based alkoxyamine pre-functionalized with the anticancer drug Gemcitabine (Gem) and subjected to IHB. Although AMA-NHS showed the best results due to its lower Ea, the strong polarity of the Gem moiety that counter-balanced the detrimental effect of IHB over its kd still allowed for a reasonable control.
Introduction
Nitroxide-mediated polymerization (NMP),1,2 atom-transfer radical polymerization (ATRP)3,4 and reversible addition–fragmentation chain transfer (RAFT)5,6 polymerization – to mention only the most popular – are very efficient reversible deactivation radical polymerization (RDRP) techniques. Over the last few decades, the field of macromolecular synthesis has been revolutionized as designing well-defined, complex and functional architectures is now possible with great ease. The use of a preformed alkoxyamine,1 a 2-in-1 molecule which undergoes reversible thermal homolysis to produce an initiating radical and a persistent nitroxide, is definitely the most efficient procedure to perform NMP. The NMP mechanism is governed by a reversible activation–deactivation equilibrium where the nitroxide reversibly deactivates the growing radicals into dormant alkoxyamine functionalities (Scheme 1a).1,7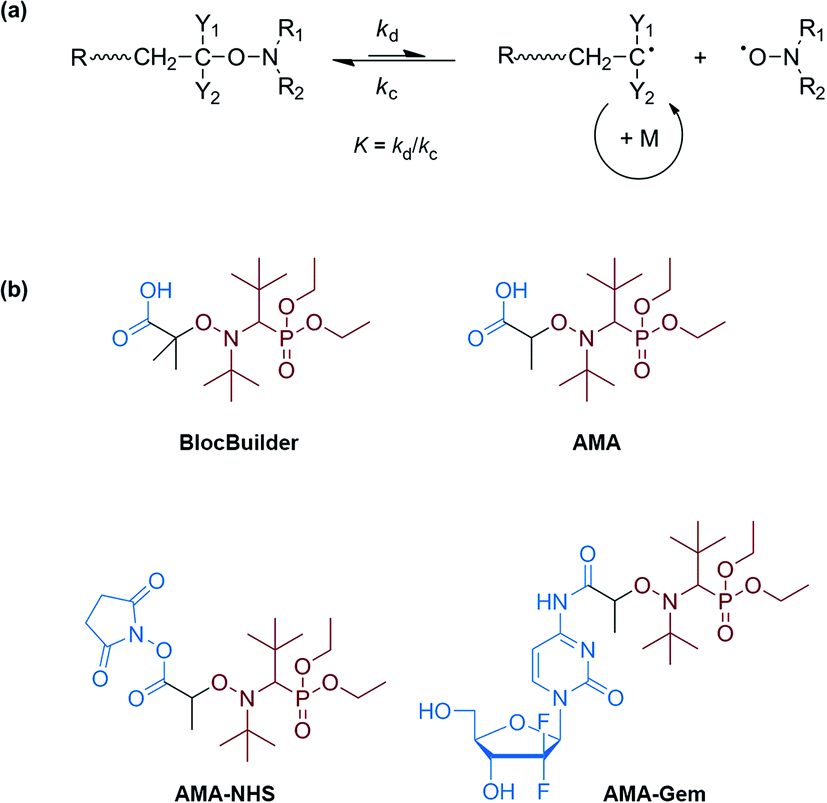 | ||
| Scheme 1 General scheme of the NMP mechanism with kc = combination rate constant, kd = dissociation rate constant and K = activation–deactivation equilibrium constant (a). Structures of the BlocBuilder, AMA, AMA-NHS and AMA-Gem SG1-based alkoxyamines (b). | ||
Among the features of NMP that benefitted from recent developments is the ease of access to functionalized alkoxyamines for (bio)conjugation purposes.1 For instance, carboxylic acid-containing SG1-based alkoxyamines, such as the BlocBuilder alkoxyamine (2-methyl-2-(N-tert-butyl-N-(1-diethoxyphosphoryl-2,2-dimethylpropyl)aminoxy) propionic acid) or its secondary counterpart, the AMA-SG1 (2-(N-tert-butyl-N-(1-diethoxyphosphoryl-2,2-dimethylpropyl)aminoxy) propionic acid) alkoxyamine (Scheme 1b), enabled coupling with primary amines. This is achieved either after their conversion into the corresponding N-succinimidyl (NHS) ester derivative8–10 or through their direct use in combination with benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) as a coupling agent (Scheme 2).11,12
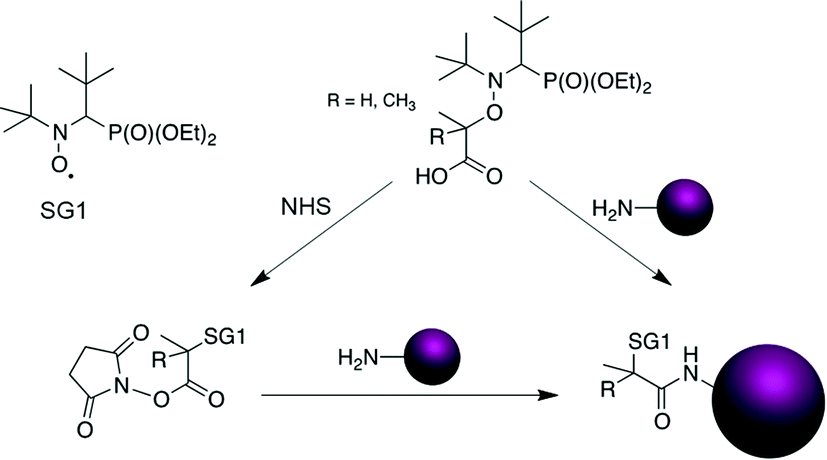 | ||
| Scheme 2 Functionalization of SG1-based alkoxyamines via an amide linkage. | ||
In particular, these NHS-based alkoxyamines were employed for the linkage of preformed NHS-functional PEG-based polymers to a protein9 whereas PyBOP was used for the direct coupling of the alkoxyamine to the terminal amine group of a peptide through solid-phase peptide synthesis11 or to an anticancer drug,12 prior to performing NMP. This last strategy, which can be assimilated to a “grafting from” approach in the case of bulky substrates, exhibits two main benefits compared to the reverse strategy involving the coupling of preformed α-functional polymers (generally termed “grafting to”): (i) the efficiency of the conjugation is generally higher, especially for bulky substrates, due to a lower steric hindrance and (ii) the purification of the resulting conjugates is facilitated as only the unreacted monomer has to be removed. However, pre-functionalizing alkoxyamines induces important structural changes when compared to the parent molecules. In particular, all pre-functionalized SG1-based alkoxyamines employed for (bio)conjugation so far have amide functionalities whereas the parent BlocBuilder and AMA alkoxyamines have a carboxylic acid group (see Scheme 2). Since the dissociation rate constant (kd) of the alkoxyamine is governed by the structure of the alkyl moiety, with a combination of polar, steric and stabilization effects,13 such structural modifications are likely to impact their dissociation. This is of high importance since, for a given monomer and nitroxide, the kd value determines the degree of control over the polymerization.14 Therefore, pre-functionalization of alkoxyamines could alter their controlling ability compared to that of non-functionalized counterparts.
This context prompted us to first investigate the reactivity of a series of functionalized amide-containing alkoxyamines based on the SG1 nitroxide and deriving from the BlocBuilder or the AMA alkoxyamines to determine whether their radical reactivity was influenced by the functionalization. In a second step, NMP of representative monomers (i.e., styrene, n-butyl acrylate and methyl methacrylate with a small amount of acrylonitrile) was achieved from a selection of alkoxyamines to illustrate our findings. We selected three secondary SG1-based alkoxyamines (they usually give higher coupling yields due to their less hindered structures compared to tertiary analogs),9,15 which can find application in the “grafting from” strategy (Scheme 2): (i) the AMA alkoxyamine, bearing a carboxylic group; (ii) its NHS derivative (AMA-NHS); and (iii) an amide-functionalized alkoxyamine bearing the anticancer drug Gemcitabine (AMA-Gem, Scheme 1), synthesized by the direct coupling between AMA and Gemcitabine. The main idea was to perform point-by-point comparisons between these three alkoxyamines to highlight the potential influence of the functionalization over the polymerization. Comparing AMA, AMA-NHS and AMA-Gem gave indications about structure–control relationship for each class of monomer and helped to predict their behavior for the design of functional materials depending on the grafting strategy (i.e., “grafting from” vs. “grafting to”). Note that AMA-Gem is of high interest as we recently showed that the controlled growth of short polyisoprene chains from this alkoxyamine enabled the resulting amphiphilic species to self-assemble into stable, narrowly dispersed nanoparticles of 130–160 nm in diameter with significant in vivo anticancer activity on human pancreatic carcinoma-bearing mice.12 Considering the great potential of this strategy, an in-depth investigation of the behavior and the controlling ability of AMA-Gem towards different monomer families is of high importance to design well-defined functionalized materials for anticancer applications.
Experimental part
Materials
2-Methoxyethylamine (99%), tert-butylamine (98%), N-isopropylacrylamide (>99%), CuBr (98%), copper powder (<10 microns, 99%), 2-bromo propionic acid (99%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%), N,N′-dicyclohexylcarbodiimide (DCC, 99%), N,N-diisopropylethylamine (DIPEA, 99%) and (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP, 98%) were purchased from Aldrich. N-Hydroxysuccinimide (NHS, 98%) was supplied by Acros and Gemcitabine hydrochloride (Gem HCl) from Sequoia Research Products Limited. Styrene (S, 99%), n-butyl acrylate (nBA, 99%), methyl methacrylate (MMA, 99%), and acrylonitrile (AN, 99+%) were purchased from Aldrich and were used as received (except for MMA which was distilled). Deuterated chloroform (CDCl3) was obtained from Euristop. Dry dimethylformamide (DMF) and dry toluene were obtained from Aldrich and all other solvents were purchased from Carlo Erba. N-tert-Butyl-N-(1-diethyl phosphono-2,2-dimethylpropyl) nitroxide (SG1, 86%) and BlocBuilder MA 1 were kindly supplied by Arkema. The O-acetylated-glucosamine was synthesized as described in ref. 16. 2-Bromo-N,N-dimethylpropanamide and 2-bromo-N-isopropylpropanamide were prepared as described in ref. 17. 2-Bromo-2-methyl-N,N-dimethylpropanamide was prepared as described in ref. 18. Alkoxyamine 2 was prepared as described in ref. 19. Alkoxyamine 3b was prepared as described in ref. 20. Alkoxyamine 4 was prepared as described in ref. 11. Alkoxyamines 13–15 were prepared as described in ref. 21. 2-[N-tert-Butyl-N-(1-diethoxyphosphoryl-2,2-dimethylpropyl)aminoxy]-propionic acid (AMA, 8),22 2-[N-tert-butyl-N-(1-diethoxyphosphoryl-2,2-dimethylpropyl)aminoxy]-N-propionyloxysuccinimide (AMA-NHS, 11),9 and AMA-Gem 1212 were synthesized according to previously reported methods. Note that for AMA, an enhancement of crystallization is performed by dissolving the colorless oil in 1 mL of ethyl acetate followed by coevaporation of both ethyl acetate and residual dichloromethane (DCM) under vacuum. For AMA-NHS alkoxyamine, only the major diastereoisomer was obtained.Analytical techniques
1H NMR spectroscopy was performed in 5 mm diameter tubes in CDCl3 on a Bruker Avance-300 (300 MHz) spectrometer. The chemical shift scale was calibrated on the basis of the solvent peak (δ = 7.26 ppm). 13C NMR spectroscopy was performed in 5 mm diameter tubes in CDCl3 on a Bruker Avance-300 (75 MHz) spectrometer. The chemical shift scale was calibrated on the basis of the solvent peak (δ = 77.0 ppm). Size exclusion chromatography (SEC) was performed at 30 °C with two columns from Polymer Laboratories (PL-gel MIXED-D; 300 × 7.5 mm; bead diameter, 5 μm; linear part, 400–4 × 105 g mol−1) and a differential refractive index detector (Spectrasystem RI-150 from Thermo Electron Corp.), using chloroform (CHCl3) as an eluent, a Waters 515 pump at a flow rate of 1 mL min−1, and toluene as a flow-rate marker. The conventional calibration curve was based on poly(methyl methacrylate) (PMMA) standards (peak molar masses, Mp = 625–625![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 g mol−1) or polystyrene (PS) standards23 (peak molar masses, Mp = 162–523000 g mol−1) from Polymer Laboratories. This technique allowed Mn (number-average molar mass), Mw (weight-average molar mass), and Mw/Mn (dispersity, Đ) to be determined.
500 g mol−1) or polystyrene (PS) standards23 (peak molar masses, Mp = 162–523000 g mol−1) from Polymer Laboratories. This technique allowed Mn (number-average molar mass), Mw (weight-average molar mass), and Mw/Mn (dispersity, Đ) to be determined.
Methods
Synthesis of SG1-based alkoxyamines
Determination of the dissociation rate constants (kd)
Polymerization reactions
In all cases, the targeted Mn at 100% monomer conversion was 20000 g mol−1.
Results and discussion
Synthesis and dissociation behavior of amide-functionalized SG1-based alkoxyamines
A broad library of different SG1-based alkoxyamines was prepared by varying the structure of the amide group and the stabilization of the alkyl moiety (i.e., tertiary or secondary alkyl moiety) (1–16, Scheme 3). Alkoxyamine 8 (AMA) was prepared by copper metal-mediated synthesis22 whereas its N-succinimidyl derivative 11 (AMA-NHS) was obtained from AMA by a DCC-assisted coupling reaction with N-hydroxysuccinimide.9 Alkoxyamines 3a and 3b were prepared by the reaction of the corresponding N-succinimidyl derivatives of the BlocBuilder alkoxyamine 1.8 For hindered alkoxyamines 4–6 and 12, a direct coupling between the parent molecule and the corresponding amine was performed using PyBOP as a coupling agent.11 The reaction proceeded through the formation of an activated benzatriazole ester of the BlocBuilder under basic conditions, followed by the nucleophilic attack of the amino compound, leading to the desired alkoxyamine in nearly quantitative yields after only 20–30 min (or 24 h for alkoxyamine 12). Tertiary alkoxyamines 2 and 7 with disubstituted amine moieties were prepared by atom transfer radical addition (ATRA) from the corresponding alkyl bromides.24,25 Alkoxyamines 13–16 were prepared by 1,2 intermolecular radical addition onto the corresponding olefins.26 This synthetic route was shown to be very efficient for the preparation of secondary functionalized SG1-based alkoxyamines that can also act as precursors for macromolecular engineering. The penultimate unit effect (PUE) is known to drastically increase the dissociation rate constant of alkoxyamines 13–16 due to the presence of the hindered 1-carboxy-1-methyl-ethyl fragment.27 Therefore, the corresponding secondary SG1-based alkoxyamines 9 and 10 were also prepared with a simple methyl group in place of the 1-carboxy-1-methyl-ethyl fragment from the alkyl halide derivatives by ATRA.25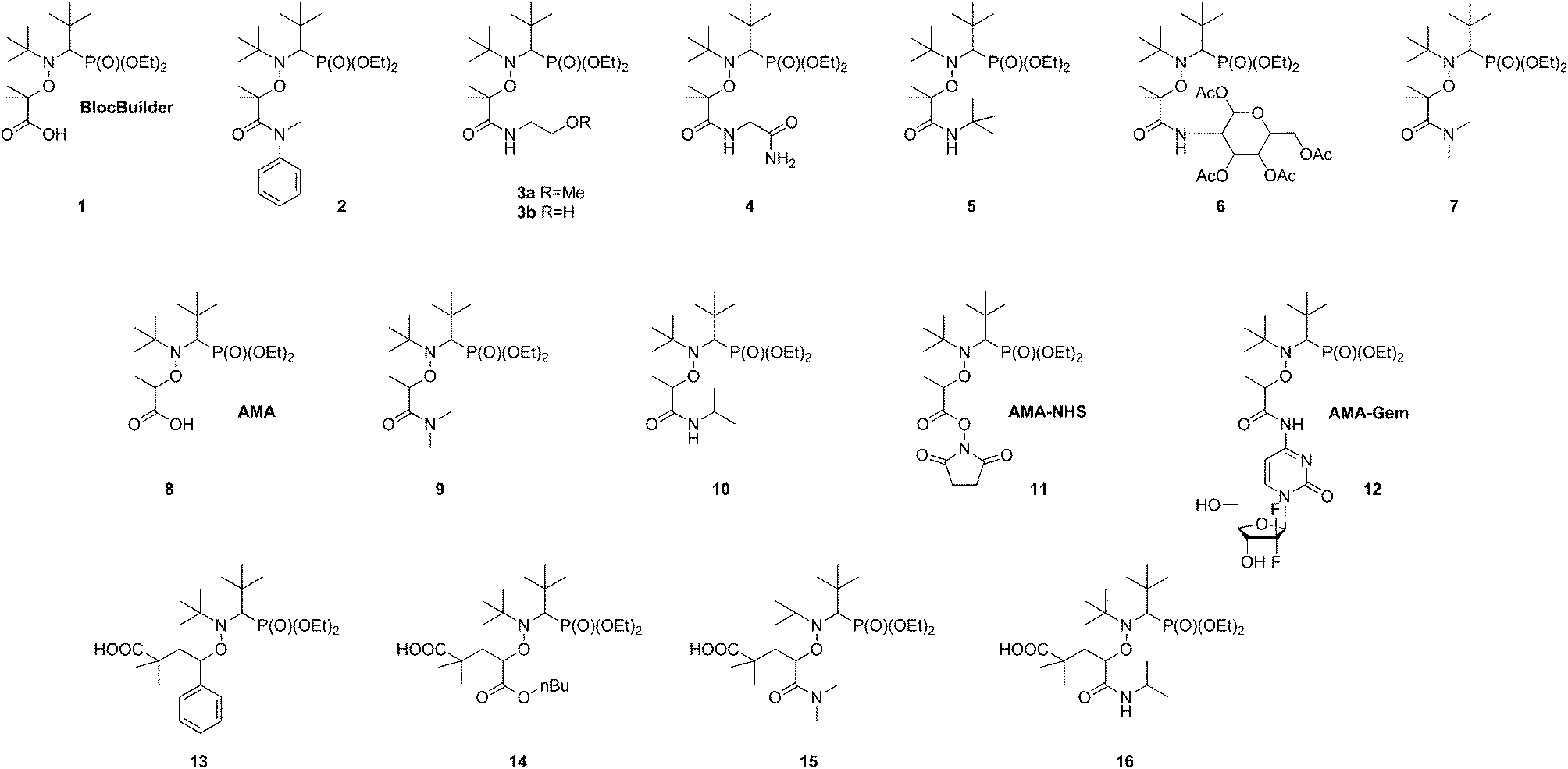 | ||
| Scheme 3 Structure of the different amide-functionalized SG1-based alkoxyamines synthesized and evaluated in this study. | ||
The dissociation rate constant (kd) measurements were performed either by monitoring the alkoxyamine concentration decay by means of 31P NMR in the presence of an excess of thiophenol as an alkyl radical and a nitroxide scavenger or by monitoring the increase of nitroxide concentration using ESR with oxygen as an alkyl radical scavenger. It has already been shown that kd values were not significantly different whatever be the techniques used.28 All experiments were carried out two times and activation energies Ea were estimated using the averaged frequency factor A = 2.4 × 1014 s−1.29 The results are summarized in Table 1.
| Alkoxyamine | E a (kJ mol−1) | Ref. |
|---|---|---|
| a Measured using 31P NMR. b Measured using ESR. | ||
| 1 (BlocBuilder) | 112 | 13 |
| 2 | 105.5a | This work |
| 3a | 122b | This work |
| 3b | 125 | 20 |
| 4 | 117.9a | This work |
| 5 | 123.5b | This work |
| 6 | 121.6b | This work |
| 7 | 112.3b | This work |
| 8 (AMA) | 130.7/132.8 | 28 |
| 9 | 124.5b | This work |
| 10 | 135b | This work |
| 11 (AMA-NHS) | 127.2b | This work |
| 12 (AMA-Gem) | 131b | This work |
| 13 | 115/116 | 27 |
| 14 | 124.5 | 27 |
| 15 | 122.5/123.4 | 27 |
| 16 | 130b | This work |
To rationalize the reactivity observed with molecules 1–16, a multi-parameter procedure was applied. This approach was shown to be very robust to describe and emphasize the various effects involved in the C–ON bond homolysis of alkoxyamines. Considering that parameters given in the literature cannot fully describe many alkyl moieties, they were estimated to characterize steric, polar and stabilization effects (see ESI† for details). Experimental kd values were then plotted against the steric Charton constant ν, the electrical Hammett constants σI, and the radical stabilization constants σRS, representing the steric, polar and stabilization effects, respectively (Fig. 1).
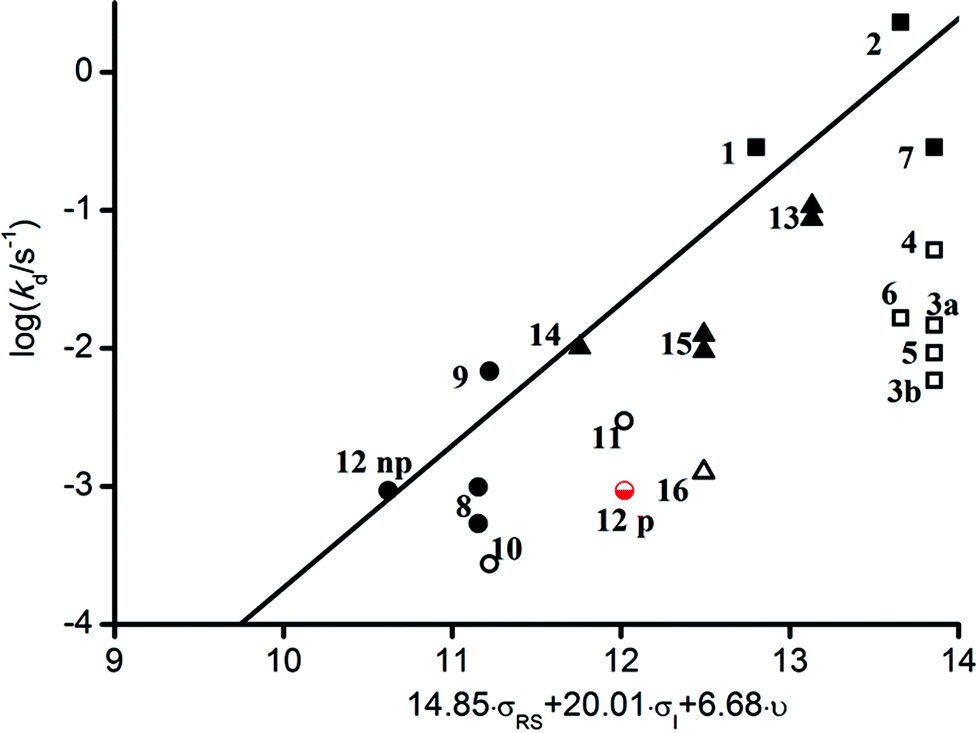 | ||
| Fig. 1 Plot of log (kd/s−1) vs. the steric Charton constant ν, the electrical Hammett constants σI, and the radical stabilization constants σRS parameters. (■ and □) Tertiary alkoxyamines, (○ and ●) alkoxyamine exhibiting the penultimate unit effect (PUE), and (▲ and △) secondary alkoxyamines. Empty symbols denote outliers. For alkoxyamine 12, 2 data sets are presented: one with a low polar parameter (12np) and one with a high polar parameter (12p); see text and ESI† for details. | ||
Each experimental logkd at 120 °C of all alkoxyamines has then been correlated to the steric Charton constant ν, the electrical Hammett constants σI, and the radical stabilization constants σRS parameters to give the best linear relationship (see ESI†). The obtained parameters leading to a linear relationship did not significantly differ from those already reported29 showing that alkoxyamines 2, 7, 9 and 15 behave as expected for SG1-based alkoxyamines. In contrast, alkoxyamines 3–6, 10 and 16 exhibited a higher Ea than expected (ca. 7–10 kJ mol−1). The main difference between these 2 groups of alkoxyamines comes from the nature of the nitrogen in the amide function (i.e., disubstituted for the former and monosubstituted for the latter). It is known that long-range steric or polar effects arose for SG1-based alkoxyamines bearing different ester chains. Nevertheless in the case of amide bonds, similar unusual behaviours reported for alkoxyamines 3–6 denote that the type of alkyl groups has a marginal influence on Ea, as opposed to ester-functionalized analogues. This reactivity could be ascribed to the predominance of the Z diastereoisomer configuration which is favoured due to the relief of steric strain (Fig. 2).
 | ||
| Fig. 2 Structure of the Z and E diastereoisomer configurations for amide-functionalized SG1-based alkoxyamines. The E diastereoisomer is discarded due to the relief of the steric strain. | ||
The Z configuration with the hydrogen atom pointing towards the nitroxide moiety could also explain the decreased lability of alkoxyamines bearing a monosubstituted nitrogen on the amide function. It might indeed induce some intramolecular hydrogen-bonding (IHB) between the alkyl and the nitroxide fragments, which generates a new bond to cleave, hence increasing the activation energy. The predominance of the Z isomer configuration was assessed by X-ray analysis of alkoxyamine 3b (see ESI†). As for alkoxyamines 3–6, only one peak was observed by 31P NMR in the crude materials. Based on 3b, it was assumed that all these alkoxyamines exhibited the Z configuration. The IHB interaction was then studied by DFT calculations on a selection of representative model alkoxyamines 17 and 19 (IHB capable) and 18 and 20 (not IHB capable) (Scheme 4). DFT optimized structures of 3b and 1 (BlocBuilder) were very close to their corresponding X-ray structures (see ESI† for details), demonstrating that the level of theory applied was suitable. The calculated bond distances are summarized in ESI† and showed that, whatever the alkyl moieties, all nitroxide fragments exhibited close conformations. Multi-IHB between the hydrogen on the amide group and the oxygens of the nitroxide was highlighted by the bond distances in alkoxyamine 17, giving dH16⋯O5 = 2.101 Å and dH16⋯O1 = 2.586 Å. In both cases, these values were smaller than the sum of the van der Waals radii of H atoms and oxygen atoms (rH = 1.34 Å, rO = 1.68 Å, and ΣvdW = 3.02 Å). The value for the valence angle <N8H16O1> (larger than 170°) highlighted a strong IHB whereas the value of 106° for <N8H16O5> gave a weak to moderate IHB (Scheme 4). Note that a weak to moderate IHB has already been reported for BlocBuilder MA alkoxyamine30,31 as well as for a SG1-based alkoxyamine carrying hydroxyl group on the alkyl moiety.32,33
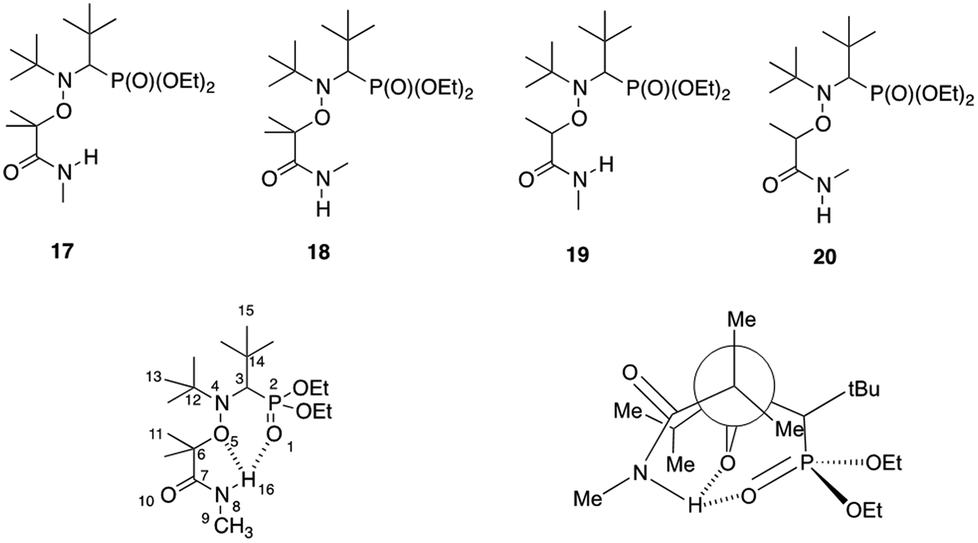 | ||
| Scheme 4 Structures of the calculated amide-functionalized alkoxyamines and representation of the multiple intramolecular hydrogen-bonding interactions. | ||
In conclusion, the establishment of IHB increasing the dissociation energy by ca. 7–10 kJ mol−1 for a series of monosubstituted amide-functionalized SG1-based alkoxyamines has been evidenced. The situation for AMA-Gem 12 is however more complex since the estimation of its polar parameter cannot be performed (the value for the cytosine group is not reported). We used the polarity value of the model phenyl ring in the first approximation and the predicted dissociation behavior fulfilled the linear relationship already reported for SG1-based alkoxyamines (12np in Fig. 1). Nevertheless, as the cytosine moiety should be prone to IHB, a higher activation was expected. A better estimation was then performed using an oxopyrimidinyl group (12p in Fig. 1). As expected for such an alkoxyamine, the activation energy was increased by ca. 10 kJ mol−1, due to the occurrence of IHB.
The experimental activation energy of 12 (Ea = 131 kJ mol−1) was similar to that of the acid-functionalized analogue (Ea = 130/132 kJ mol−1 for the diastereoisomers). This value was lower than that of alkoxyamine 10 (Ea = 135 kJ mol−1), a model of amide-functionalized SG1-based alkoxyamine, and was similar to the reference MONAMS alkoxyamine.34 In the case of AMA-Gem 12, the influence of IHB over the dissociation was entirely counter-balanced by the presence of the polar cytosine ring on the amide function that promoted the dissociation.
The aim of the next section was therefore to estimate its performance in NMP in comparison with two precursors of amide-functionalized alkoxyamines, namely the acid-functionalized AMA alkoxyamine and its NHS derivative (AMA-NHS).
NMP of vinyl monomers initiated by different SG1-based alkoxyamines
 | ||
| Fig. 3 Bulk NMP of S at 120 °C initiated by different SG1-based alkoxyamines: ■, expt. 1 (AMA); ●, expt. 2 (AMA-NHS); ▲, expt. 3 (AMA-Gem). (a) Evolution of ln[1/(1 − conv.)] with time (t); (b) evolution of the number-average molar mass (Mn) and dispersity (Đ) with conv. The full lines represent the theoretical Mn and lines connecting data points are only guide for the eye. | ||
The situation was however less marked by adding 10 mol% of free SG1 at the onset of the polymerizations (expts 4–6, Fig. 4). This drastically reduced the differences of behavior between the three alkoxyamines as: (i) kinetics rather exhibited the same slope (Fig. 4a); (ii) the initiation efficiency was greatly improved for AMA (expt. 4, Fig. 4b) with Mn values almost overlapping with those obtained from AMA-Gem and AMA-NHS and (iii) dispersities reached lower values for AMA and AMA-Gem (Đ ∼1.4), and so was for AMA-NHS (Đ ∼1.15). The presence of 10 mol% of free SG1 at the very beginning of the polymerization helped in reducing the occurrence of irreversible terminations and therefore reduced the differences between the dissociation abilities of these three alkoxyamines.
 | ||
| Fig. 4 Bulk NMP of S at 120 °C initiated by different SG1-based alkoxyamines in the presence of 10 mol% free SG1: ■, expt. 4 (AMA); ●, expt. 5 (AMA-NHS); ▲, expt. 6 (AMA-Gem). (a) Evolution of ln[1/(1 − conv.)] with time (t); (b) evolution of the number-average molar mass (Mn) and dispersity (Đ) with conv. The full lines represent the theoretical Mn and lines connecting data points are only guide for the eye. | ||
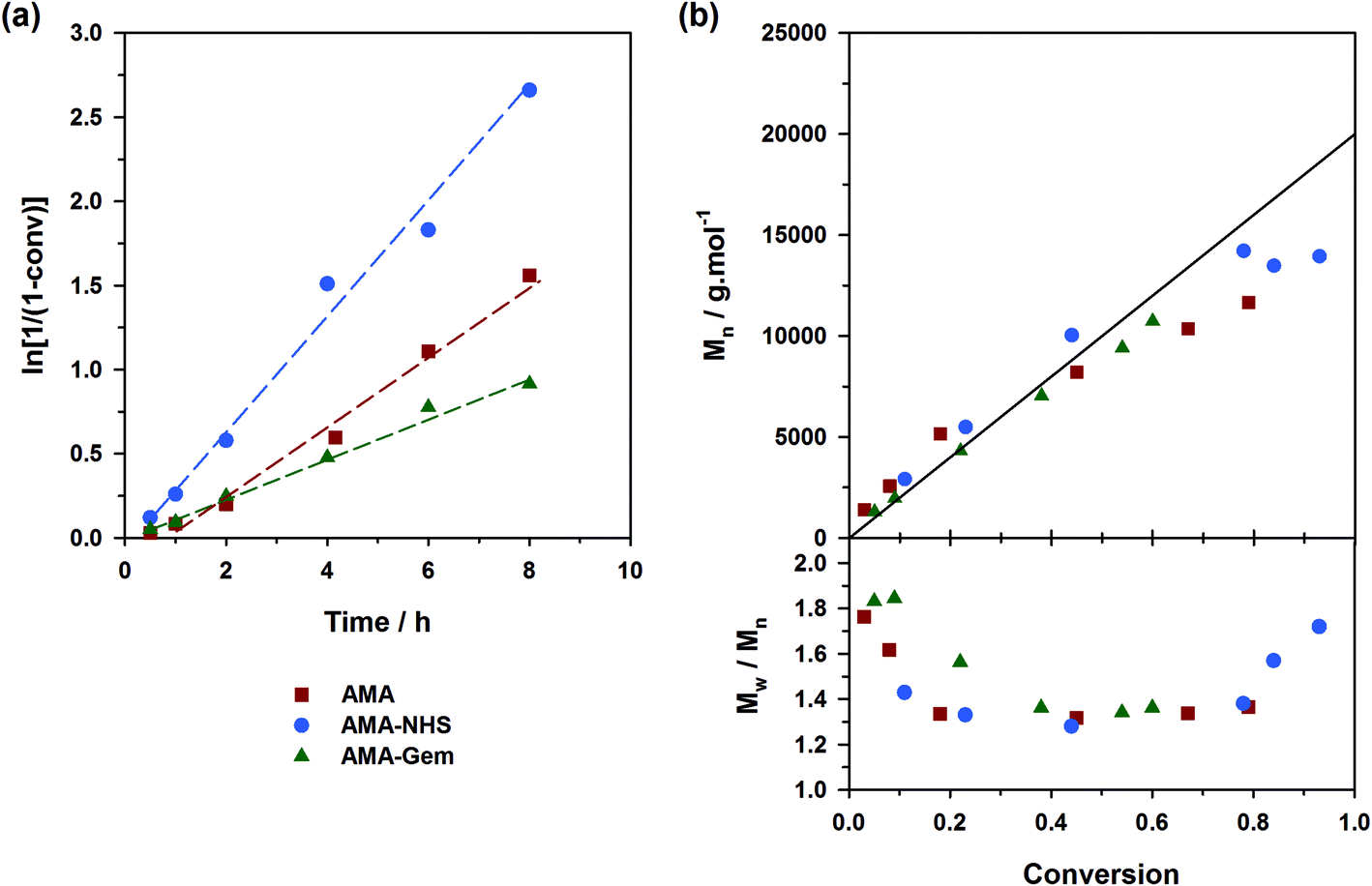 | ||
| Fig. 5 Solution NMP of nBA at 120 °C initiated by different SG1-based alkoxyamines: ■, expt. 7 (AMA); ●, expt. 8 (AMA-NHS); ▲, expt. 9 (AMA-Gem). (a) Evolution of ln[1/(1 − conv.)] with time (t); (b) evolution of the number-average molar mass (Mn) and dispersity (Đ) with conv. The full lines represent the theoretical Mn and lines connecting data points are only guide for the eye. | ||
Controlling the NMP of MMA with secondary alkoxyamines is therefore not straightforward due to their slower decomposition rates compared to their tertiary analogues. Polymerizations were performed at 100 °C (and not at 90 °C to alleviate the detrimental effect of their relatively high Ea) in the presence of 9 mol% of AN (expts 10–12, Fig. 6). Results showed a substantially better control with AMA-NHS compared to AMA and AMA-Gem. With AMA-NHS, Mn values were closer to the predicted ones (indicating a higher initiation efficiency, although quite modest as f = 60%) and dispersities progressively decreased to ∼ 1.4. In contrast, although Mn values were still increasing linearly with MMA conversion for AMA and AMA-Gem up to ca. 30% conversion, significantly higher dispersities (Đ ∼1.6–1.7) were obtained during the polymerization. This particular case, for which a fast-dissociating alkoxyamine is necessary, highlighted the pivotal role of the kd value in the control of the NMP process and exalted the difference between the behaviors of the three alkoxyamines.
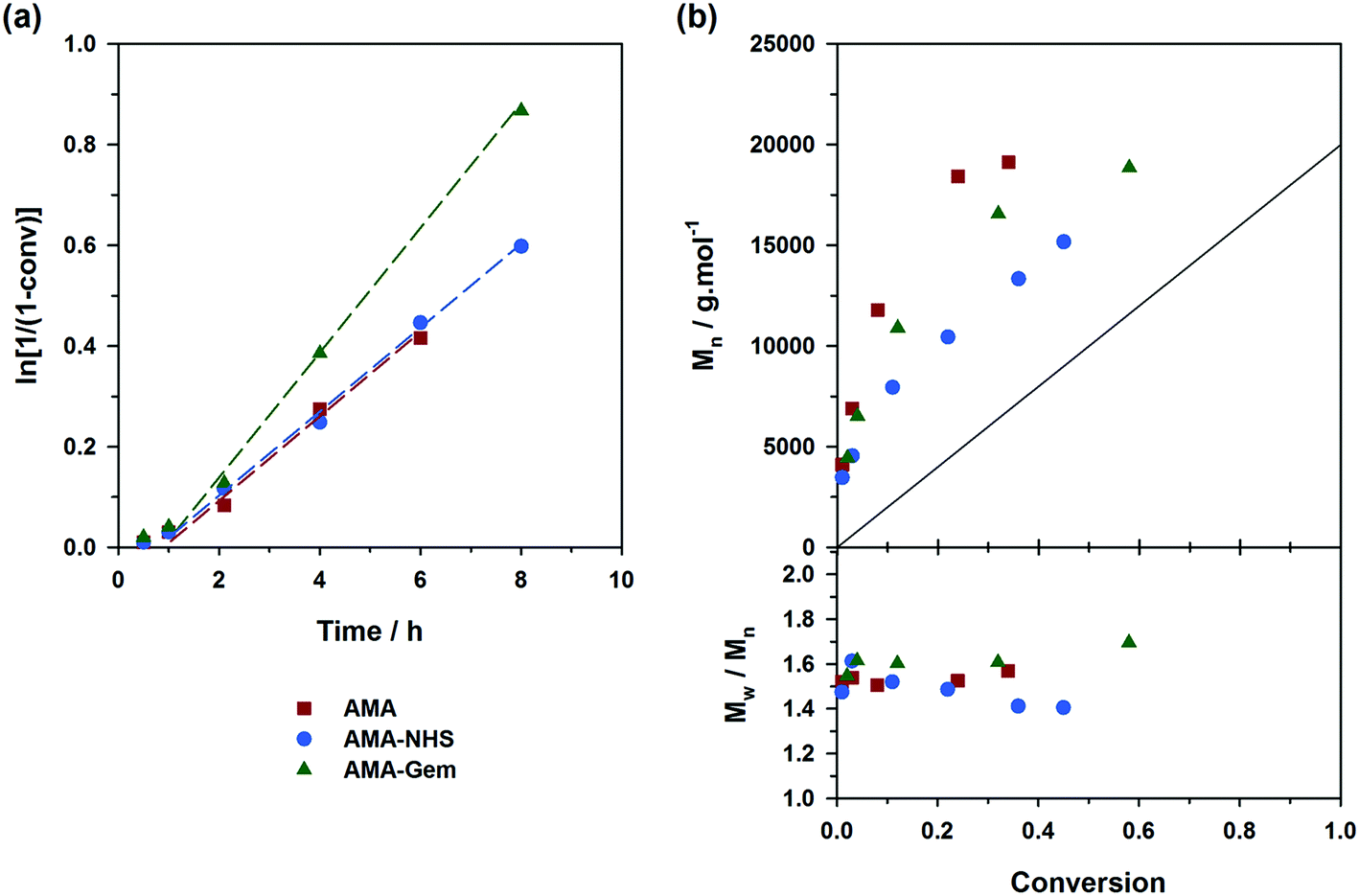 | ||
| Fig. 6 Solution NMP of methyl methacrylate (MMA) with a small amount of acrylonitrile (AN, fAN0 = 9 mol%) at 100 °C initiated by different SG1-based alkoxyamines: ■, expt. 10 (AMA); ●, expt. 11 (AMA-NHS); ▲, expt. 12 (AMA-Gem). (a) Evolution of ln[1/(1 − conv.)] with time (t); (b) evolution of the number-average molar mass (Mn) and dispersity (Đ) with conv. The full lines represent the theoretical Mn and lines connecting data points are only guide for the eye. | ||
A strategy to improve the control is to slightly increase the initial amount of the ‘controlling’ comonomer, while still targeting low values to maintain a high amount of methacrylic units in the resulting copolymer. By using fAN0 = 15 mol%, a significant improvement was indeed witnessed (expts 13–15, Fig. 7). Whereas similar first-order kinetics were observed in all cases, AMA-NHS and AMA-Gem led to higher initiation efficiencies (f = 89% and 75%, respectively) whereas that of AMA was rather unchanged and still poor. Also, dispersities for AMA and AMA-Gem were slightly improved and reached lower values at the end of the polymerization (Đ ∼1.5), compared to similar experiments with 9 mol% AN (see Fig. 6b). However, no beneficial effect on the dispersity was observed with AMA-NHS (expt. 14).
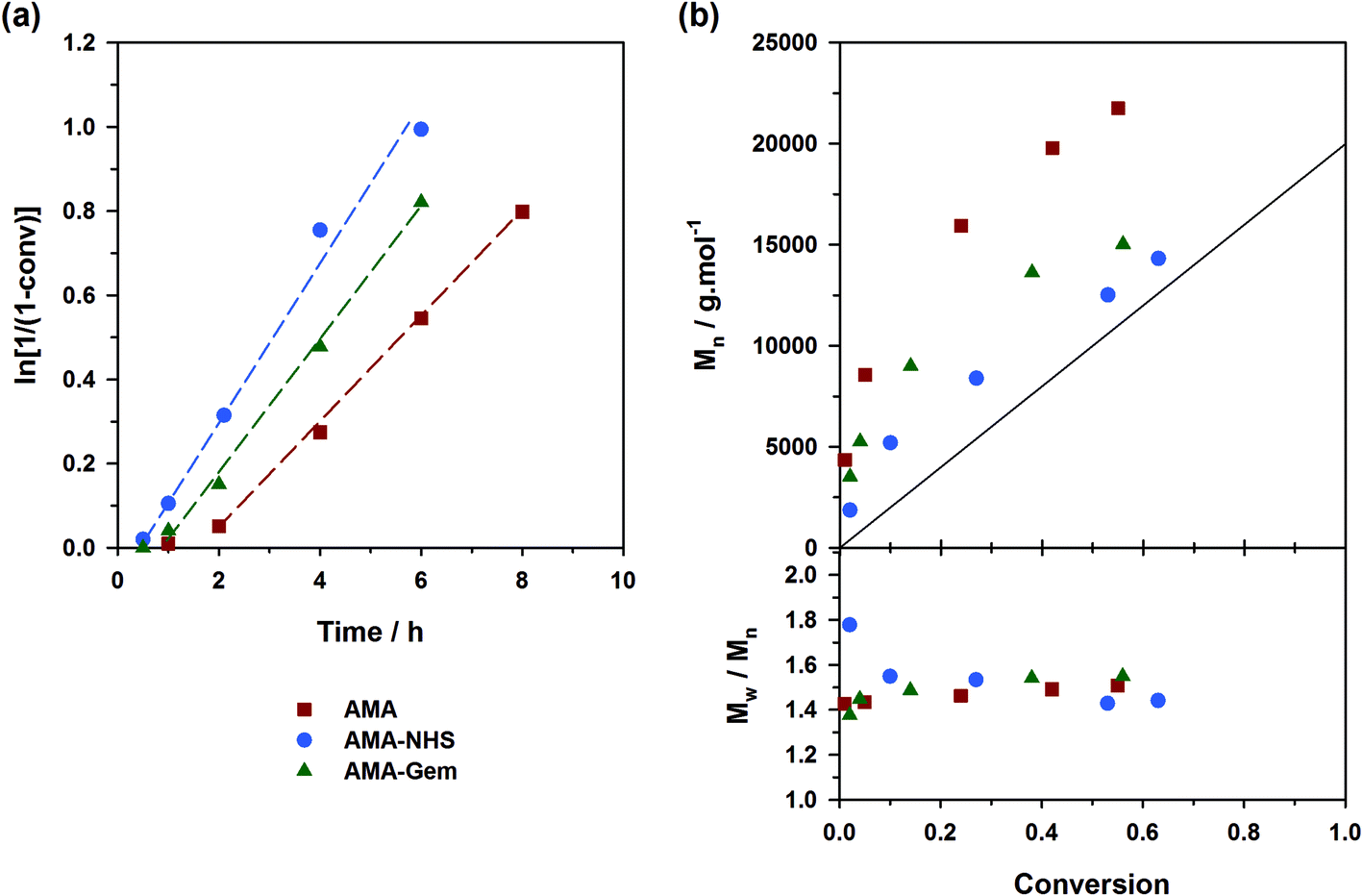 | ||
| Fig. 7 Solution NMP of methyl methacrylate (MMA) with a small amount of acrylonitrile (AN, fAN0 = 15 mol%) at 100 °C initiated by different SG1-based alkoxyamines in the presence of 10 mol% free SG1: ■, expt. 13 (AMA); ●, expt. 14 (AMA-NHS); ▲, expt. 15 (AMA-Gem). (a) Evolution of ln[1/(1 − conv.)] with time (t); (b) evolution of the number-average molar mass (Mn) and dispersity (Đ) with conv. The full lines represent the theoretical Mn and lines connecting data points are only guide for the eye. | ||
General discussion
What resulted from this series of polymerizations with different monomers is overall good agreement between the controlling ability of each alkoxyamine tested (i.e., AMA, AMA-NHS and AMA-Gem) and the value of their activation energy (and implicitly of their dissociation rate constant). AMA-NHS, which exhibits the lowest Ea by ca. 4–5 kJ mol−1, indeed led to a better control than AMA and AMA-Gem in terms of initiation efficiency and dispersity, although differences were quite moderate. The addition of a small amount of free SG1 (10 mol%) had the beneficial effect of reducing these differences and improving the control. A similar effect was observed on the MMA polymerization by increasing the amount of controlling comonomer (fAN0 = 15 mol% vs. 9 mol%).Despite the establishment of IHB for AMA-Gem, the presence of the polar cytosine ring on the amide function, which favors the dissociation, enabled a fair control of different monomer families. In fact, the initiation efficiency of AMA-Gem was slightly better than the AMA alkoxyamine, although they exhibited very close Ea. This result could be explained by a higher rate of decomposition during the polymerization process. The occurrence of IHB is indeed likely prone to vary with the polarity of the solvent (without IHB, the dissociation of AMA-Gem would be dramatically increased with a theoretical Ea = 121 kJ mol−1, see ESI† for details), as recently observed for the NMP of isoprene.31 Note that the nature of the solvent can also impact the alkoxyamine reactivity.57,58
Importantly, it appears that when the ‘grafting from’ strategy is envisioned from NHS- or COOH-containing AMA-based alkoxyamines, the coupling with a polar amine moiety may still allow the synthesis of well-defined polymers (i.e., IHB will be counter-balanced). In contrast, when a non-polar amine group is envisioned, IHB might reach its maximum value and lead to a significantly higher Ea, thus impacting the quality of control. In this case, the coupling of a preformed polymer could be the preferable pathway toward well-defined functionalized materials. Further investigations are therefore necessary to assess this hypothesis by probing the influence of the polarity of the amine group on the quality of the control.
Conclusion
In this article, a series of different amide-functionalized alkoxyamines (which is a functionality often obtained after conjugation from COOH- or NHS-containing alkoxyamines) based on the nitroxide SG1 have been synthesized and their kd have been determined. A multi-parameter procedure was applied to rationalize their reactivity. It enabled to discriminate disubstituted alkoxyamines from monosubstituted ones. Whereas disubstituted alkoxyamines displayed expected dissociation abilities for SG1-based alkoxyamines, monosubstituted counterparts exhibited lower kd (Ea increase of ∼7–10 kJ mol−1) because of the occurrence of IHB between the alkyl and the nitroxide fragments. To probe potential consequences on the synthesis of functionalized materials, S, nBA and MMA with a small amount of AN were successfully polymerized by NMP from AMA and AMA-NHS, which are two representative secondary SG1-based alkoxyamines employed for conjugation, displaying a carboxylic acid and a NHS group, respectively. Their performances were compared to those of AMA-Gem, a pre-functionalized alkoxyamine subjected to IHB bearing an anticancer drug as a functional moiety via an amide bond. It resulted that the strong polarity of the Gem moiety still enabled a fair control despite the detrimental effect of IHB over its kd. Overall, this work may have important consequences on the design of polymer conjugates from functional alkoxyamines and help in determining the most suitable approach for their design.Acknowledgements
We thank the French Ministry of Research for the financial support of the PhD thesis of EG and the French National Research Agency (ANR-11-JS08-0005) for the financial support of the PhD thesis of VD. Arkema is warmly acknowledged for kindly providing the BlocBuilder MA™ alkoxyamine and the SG1 nitroxide. The CNRS is also acknowledged for financial support.Notes and references
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- R. B. Grubbs, Polym. Rev., 2011, 51, 104–137 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3745 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- A. Goto and T. Fukuda, Prog. Polym. Sci., 2004, 29, 329–385 CrossRef CAS.
- J. Vinas, N. Chagneux, D. Gigmes, T. Trimaille, A. Favier and D. Bertin, Polymer, 2008, 49, 3639–3647 CrossRef CAS.
- M. Chenal, C. Boursier, Y. Guillaneuf, M. Taverna, P. Couvreur and J. Nicolas, Polym. Chem., 2011, 2, 1523–1530 RSC.
- J. Parvole, L. Ahrens, H. Blas, J. Vinas, C. Boissiere, C. Sanchez, M. Save and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 173–185 CrossRef CAS.
- T. Trimaille, K. Mabrouk, V. Monnier, L. Charles, D. Bertin and D. Gigmes, Macromolecules, 2010, 43, 4864–4870 CrossRef CAS.
- S. Harrisson, J. Nicolas, A. Maksimenko, D. T. Bui, J. Mougin and P. Couvreur, Angew. Chem., Int. Ed., 2013, 52, 1678–1682 CrossRef CAS PubMed.
- D. Bertin, D. Gigmes, S. R. A. Marque and P. Tordo, Macromolecules, 2005, 38, 2638–2650 CrossRef CAS.
- F. Chauvin, P.-E. Dufils, D. Gigmes, Y. Guillaneuf, S. R. A. Marque, P. Tordo and D. Bertin, Macromolecules, 2006, 39, 5238–5250 CrossRef CAS.
- D. Gigmes, J. Vinas, N. Chagneux, C. Lefay, T. N. T. Phan, T. Trimaille, P.-E. Dufils, Y. Guillaneuf, G. Carrot, F. Boue and D. Bertin, ACS Symp. Ser., 2009, 1024, 245–262 CrossRef CAS.
- J. Klein and D. Herzog, Makromol. Chem., 1987, 188, 1217–1232 CrossRef CAS.
- W. E. Weaver and W. M. Whaley, J. Am. Chem. Soc., 1947, 69, 1144–1145 CrossRef CAS PubMed.
- T. Hama, D. A. Culkin and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 4976–4985 CrossRef CAS PubMed.
- C. Leroi, D. Bertin, P.-E. Dufils, D. Gigmes, S. Marque, P. Tordo, J.-L. Couturier, O. Guerret and M. A. Ciufolini, Org. Lett., 2003, 5, 4943–4945 CrossRef CAS PubMed.
- N. Chagneux, T. Trimaille, M. Rollet, E. Beaudoin, P. Gerard, D. Bertin and D. Gigmes, Macromolecules, 2009, 42, 9435–9442 CrossRef CAS.
- P.-E. Dufils, N. Chagneux, D. Gigmes, T. Trimaille, S. R. A. Marque, D. Bertin and P. Tordo, Polymer, 2007, 48, 5219–5225 CrossRef CAS.
- S. Harrisson, P. Couvreur and J. Nicolas, Polym. Chem., 2011, 2, 1859–1865 RSC.
- The PS calibration is appropriate for PnBA samples as shown by the Mark–Houwink–Sakurada parameters: actually, it leads to an error of about 3–5%, which is within the accepted range for SEC analysis. Indeed, the MHS parameters in THF at 30 °C are the following: KPS = 11.4 × 10−5 dL g−1 and αPS = 0.716 for PS [see: R. A. Hutchinson, D. A. Paquet Jr., J. H. McMinn, S. Beuermann, R. E. Fuller and C. Jackson, Dechema Monogr., 1995, 131, 467 CAS ]; KPnBA = 12.2 × 10−5 dL g−1 and αPnBA = 0.700 for PnBA [see: S. Beuermann, D. A. Paquet Jr., J. H. McMinn and R. A. Hutchinson, Macromolecules, 1997, 29, 1918 Search PubMed ].
- K. Matyjaszewski, B. E. Woodworth, X. Zhang, S. G. Gaynor and Z. Metzner, Macromolecules, 1998, 31, 5955 CrossRef CAS.
- D. Bertin, D. Gigmes, C. LeMercier, S. R. A. Marque and P. Tordo, J. Org. Chem., 2004, 69, 4925–4930 CrossRef CAS PubMed.
- D. Gigmes, P.-E. Dufils, D. Gle, D. Bertin, C. Lefay and Y. Guillaneuf, Polym. Chem., 2011, 2, 1624–1631 RSC.
- D. Bertin, P.-E. Dufils, I. Durand, D. Gigmes, B. Giovanetti, Y. Guillaneuf, S. R. A. Marque, T. Phan and P. Tordo, Macromol. Chem. Phys., 2008, 209, 220–224 CrossRef CAS.
- D. Bertin, D. Gigmes, S. Marque and P. Tordo, e-Polymers, 2003, 002, 1–9 Search PubMed.
- D. Bertin, D. Gigmes, S. R. A. Marque and P. Tordo, Chem. Soc. Rev., 2011, 40, 2189–2198 RSC.
- S. Harrisson, P. Couvreur and J. Nicolas, Macromolecules, 2011, 44, 9230–9238 CrossRef CAS.
- S. Harrisson, P. Couvreur and J. Nicolas, Macromol. Rapid Commun., 2012, 33, 805–810 CrossRef CAS PubMed.
- E. G. Bagryanskaya, P. Brémond, T. Butscher, S. R. A. Marque, D. Parkhomenko, V. Roubaud, D. Siri and S. Viel, Macromol. Chem. Phys., 2014, 216, 475–488 CrossRef.
- P. Brémond, T. Butscher, V. Roubaud, D. Siri and S. Viel, J. Org. Chem., 2013, 78, 10524–10529 CrossRef PubMed.
- S. Marque, C. Le Mercier, P. Tordo and H. Fischer, Macromolecules, 2000, 33, 4403–4410 CrossRef CAS.
- C. Farcet, J. Nicolas and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4410–4420 CrossRef CAS.
- P. Lacroix-Desmazes, J.-F. Lutz, F. Chauvin, R. Severac and B. Boutevin, Macromolecules, 2001, 34, 8866–8871 CrossRef CAS.
- J. Nicolas, B. Charleux, O. Guerret and S. Magnet, Angew. Chem., Int. Ed., 2004, 43, 6186–6189 CrossRef CAS PubMed.
- J. Nicolas, B. Charleux and S. Magnet, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4142–4153 CrossRef CAS.
- C. Farcet, J. Belleney, B. Charleux and R. Pirri, Macromolecules, 2002, 35, 4912–4918 CrossRef CAS.
- Y. Guillaneuf, D. Gigmes and T. Junkers, Macromolecules, 2012, 45, 5371–5378 CrossRef CAS.
- E. Guégain, Y. Guillaneuf and J. Nicolas, Macromol. Rapid Commun., 2015 Search PubMed , accepted.
- G. S. Ananchenko, M. Souaille, H. Fischer, C. Le Mercier and P. Tordo, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3264–3283 CrossRef CAS.
- Y. Guillaneuf, D. Gigmes, S. R. A. Marque, P. Tordo and D. Bertin, Macromol. Chem. Phys., 2006, 207, 1278–1288 CrossRef CAS.
- C. Dire, J. Belleney, J. Nicolas, D. Bertin, S. Magnet and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6333–6345 CrossRef CAS.
- B. Charleux, J. Nicolas and O. Guerret, Macromolecules, 2005, 38, 5485–5492 CrossRef CAS.
- R. McHale, F. Aldabbagh and P. B. Zetterlund, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2194–2203 CrossRef CAS.
- J. Nicolas, C. Dire, L. Mueller, J. Belleney, B. Charleux, S. R. A. Marque, D. Bertin, S. Magnet and L. Couvreur, Macromolecules, 2006, 39, 8274–8282 CrossRef CAS.
- J. Nicolas, L. Mueller, C. Dire, K. Matyjaszewski and B. Charleux, Macromolecules, 2009, 42, 4470–4478 CrossRef CAS.
- J. Nicolas, P. Couvreur and B. Charleux, Macromolecules, 2008, 41, 3758–3761 CrossRef CAS.
- B. Lessard and M. Maric, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2574–2588 CrossRef CAS.
- B. Lessard and M. Marić, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5270–5283 CrossRef CAS.
- B. Lessard, C. Tervo, S. De Wahl, F. J. Clerveaux, K. K. Tang, S. Yasmine, S. Andjelic, A. D'Alessandro and M. Maric, Macromolecules, 2010, 43, 868–878 CrossRef CAS.
- B. H. Lessard, E. J. Y. Ling and M. Marić, Macromolecules, 2012, 45, 1879–1891 CrossRef CAS.
- S. R. S. Ting, E.-H. Min, P. Escale, M. Save, L. Billon and M. H. Stenzel, Macromolecules, 2009, 42, 9422–9434 CrossRef CAS.
- J. Nicolas, S. Brusseau and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 34–47 CrossRef CAS.
- B. Lessard, E. J. Y. Ling, M. S. T. Morin and M. Marić, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1033–1045 CrossRef CAS.
- G. Audran, P. Bremond, S. R. A. Marque and G. Obame, Polym. Chem., 2012, 3, 2901–2908 RSC.
- G. Audran, P. Brémond, S. R. A. Marque and G. Obame, J. Org. Chem., 2012, 77, 9634–9640 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py00283d |
| This journal is © The Royal Society of Chemistry 2015 |