
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A simple route to deuterated polystyrene block copolymers by reverse iodine transfer polymerisation
Trevor Gavin
Wright
and
Harald
Pasch
*
Department of Chemistry and Polymer Science, University of Stellenbosch, Private Bag X1, Matieland 7602, South Africa. E-mail: hpasch@sun.ac.za; Fax: +27 21 808 4967; Tel: +27 21 808 3173
First published on 17th March 2015
Abstract
Deuterated polystyrene (d-PS), synthesised for the first time using RITP, and hydrogenous polystyrene (h-PS) were characterised using 1H nuclear magnetic resonance (NMR) and size exclusion chromatography (SEC) to compare their polymerisation behaviour. Using h-PS as a macro-initiator, block copolymers of hydrogenous polystyrene-block-deuterated polystyrene (hPS-b-dPS) were prepared for the first time using RITP. These block copolymers were analysed using SEC and liquid chromatography at critical conditions (LCCC) of h-PS to determine possible residual first blocks. At the LCCC conditions for h-PS a separation based on the isotopic effect was observed, where the hPS-b-dPS block copolymers eluted in SEC mode. In order to improve the separation, conditions were established where one component eluted in SEC mode while the other component eluted in liquid adsorption chromatography (LAC) mode. A separation was achieved where the hPS-b-dPS block copolymer was separated from the h-PS precursor. The two-dimensional liquid chromatography (2D-LC) fractionation revealed a third component that was not observed in the one-dimensional fractionations.
Introduction
Controlled/living radical polymerisation (LRP) is an attractive technique for preparing polymers with well-defined architectures in a controlled manner. LRP techniques include atom transfer radical polymerisation (ATRP),1 nitroxide-mediated polymerisation (NMP),2 reversible addition-fragmentation chain transfer polymerisation (RAFT),3 iodine transfer polymerisation (ITP)4 and most recently reverse iodine transfer polymerisation (RITP).5–8 The most popular LRP techniques are reportedly NMP, ATRP and RAFT.9 RITP, a technique that was invented by Lacroix-Desmazes et al.,5,10–14 is a LRP technique where chain transfer agents are generated in situ (Scheme 1), due to the reaction between AIBN and molecular iodine. Molecular iodine inhibits polymerisation, and this inhibitory property is exploited in RITP. There is a period where AIBN and iodine react to form the chain transfer agent, known as the induction period. Once all of the iodine has been consumed, the polymerisation can commence. The resultant polymer has a cyanoisopropyl α-chain end and an iodinated ω-chain end. The living nature of the iodo-terminated polymer can then be used as a macro-initiator to form block copolymers. Several examples of block copolymers prepared using RITP can be found in literature.5,15–19 Deuterated compounds including polymers are typically quite expensive, but these compounds do have some useful applications. The most common use of deuterated polymers is to gain contrast in neutron scattering experiments.20–26 Other applications include the use of deuterated compounds in pharmacology to follow metabolism,27–32 tracing biodegradation of hydrocarbons in water33 and the use of deuterated compounds in nuclear fusion experiments.34–36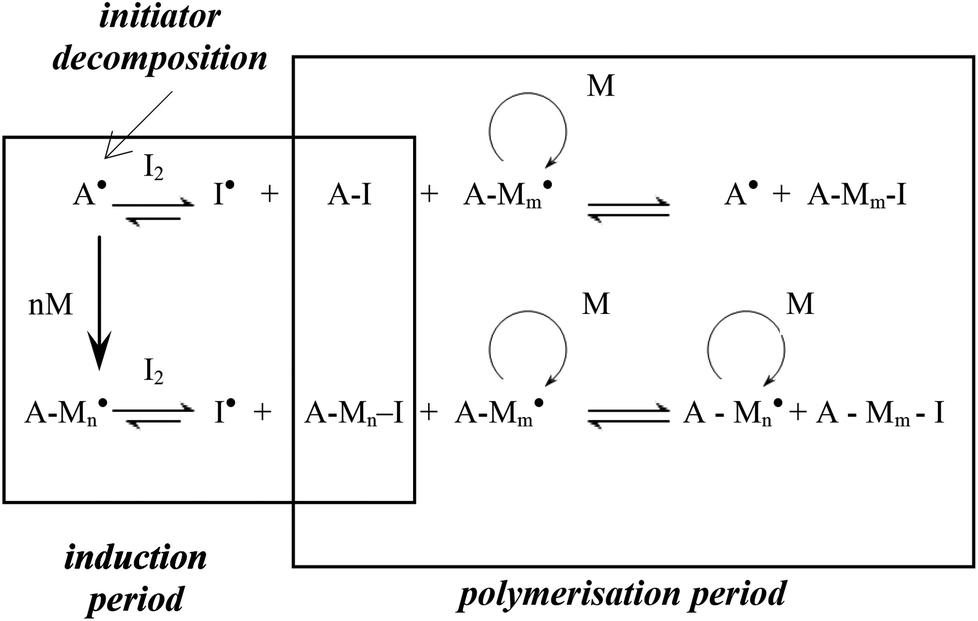 | ||
| Scheme 1 Basic representation of the two stages in the mechanism of RITP.5 | ||
Deuterated polystyrene (d-PS) is usually synthesised using living anionic polymerisation.37,38 In living anionic polymerisation, the reaction typically involves the use of n-butyl lithium as an initiator at a temperature of −78 °C. The polymerisation can be conducted in different solvents including THF and hydrocarbon solvents. In particular in THF, yields are quite high and molar mass dispersities are very low. Hydrogenous PS (h-PS) has also been grafted with d-PS using living anionic polymerisation.39 Deuterated star polymers have been synthesised via RAFT polymerisation at a temperature of 80 °C.40 To the best of our knowledge, the use of RITP to synthesise deuterated polymers has not yet been reported in literature.
This study describes, for the first time, the use of RITP to synthesise d-PS and block copolymers of hydrogenous polystyrene-b-deuterated polystyrene (hPS-b-dPS). Nuclear magnetic resonance spectroscopy (NMR) was used to analyse the homopolymers while size exclusion chromatography (SEC) and high-performance liquid chromatography (HPLC) were used to confirm the formation of the block copolymers. The course of the polymerisation (fate of the initiator and evolution of the chain transfer agent) was followed by 1H NMR to compare the polymerisation behaviour of h-styrene and d-styrene. It was proven that RITP is a simple and direct route to these block copolymers that does not require the complex synthesis of a specific chain transfer agent as e.g. in RAFT.
Experimental section
Chemicals
Styrene (≥99% Sigma-Aldrich) was washed three times with a 0.3 M aqueous solution of sodium hydroxide followed by three washes with distilled de-ionised water. The washed styrene was left to dry overnight using anhydrous magnesium sulphate. The magnesium sulphate was filtered off and the styrene was distilled under vacuum and stored in a refrigerator at –5 °C. Azobis(isobutyronitrile) (AIBN, Riedel de Haën) was recrystallised from methanol, dried under vacuum and stored in a refrigerator at – 5 °C. Deuterated styrene (styrene-d8, Sigma-Aldrich 98%) was passed through a column of alumina to remove inhibitor. Deuterated chloroform (CDCl3, Sigma-Aldrich 99%), deuterated benzene (C6D6, Sigma-Aldrich 99%) and iodine (I2, ACROS Organics, 99.5%, extra pure, resublimed) were used as received.Homopolymerisation of styrene and styrene-d8
The homopolymerisation of styrene (hydrogenous) was performed by adding styrene (4.00 g, 3.84 × 10−2 mol), toluene (4.00 g, 4.34 × 10−2 mol), AIBN (63.6 mg, 3.88 × 10−4 mol) and iodine (51.8 mg, 2.04 × 10−4 mol) into a Schlenk flask. A magnetic stirrer bar was added to the flask to ensure that the reaction mixture was stirred during polymerisation. The Schlenk flask was degassed by three successive freeze–pump–thaw cycles and back filled with UHP argon gas. To commence polymerisation, the flask was submerged in silicone oil heated to 70 °C. The polymerisation reaction proceeded in the dark for 24 hours before stopping the reaction by placing the flask on ice. Finally, the polymer was precipitated in cold methanol and left to dry overnight in a vacuum oven.For the homopolymerisation of styrene-d8, the same reaction conditions were used. However, due to the high cost of deuterated styrene, lower quantities of reagents were used for these reactions. In a typical reaction, styrene-d8 (1.00 g, 8.91 × 10−3 mol), toluene (1.00 g, 1.09 × 10−2 mol), AIBN (3.9 mg, 2.39 × 10−5 mol) and iodine (3.2 mg, 1.26 × 10−5 mol) were added to a Schlenk flask.
In situ 1H NMR monitoring of styrene and styrene-d8 homopolymerisation
The homopolymerisations of styrene and styrene-d8 were also analysed using in situ1H NMR. For hydrogenous styrene polymerisation, styrene (2.00 g, 19.2 × 10−2 mol), AIBN (31.8 mg, 1.94 × 10−4 mol) and iodine (25.9 mg, 1.02 × 10−4 mol) were mixed in a glass vial. A small amount (0.15 g) of this stock solution was injected into a J Young NMR tube and 0.15 g of benzene-d6 was added. The NMR tube was degassed by three successive freeze-thaw pump cycles and filled with UHP argon gas. For styrene-d8 polymerisation, styrene-d8 (0.30 g, 2.67 × 10−3 mol), AIBN (4.80 mg, 2.91 × 10−5 mol) and iodine (3.90 mg, 1.53 × 10−5 mol) were added to the J Young NMR tube.Before starting the in situ1H NMR experiment, a pre-polymerisation spectrum was recorded at 25 °C to be used as a reference. The NMR tube was removed from the NMR magnet and the temperature of the magnet was elevated to 70 °C. After the temperature was stable at 70 °C, the NMR tube was inserted again and the magnet was shimmed at the elevated temperature. Each spectrum was recorded with 15 scans every 15 minutes for 24 hours with a pulse width of 3 μs (40°) and a 4 second acquisition time.
Block copolymerisation of styrene and styrene-d8
The precipitated h-PS was dried in a vacuum oven overnight to ensure that no unreacted monomer was present. h-PS was used as the macro-initiator and typical copolymerisation reaction conditions were as follows; PS-I (1.0 g, 2.78 × 10−5 mol) was mixed with styrene-d8 (1.0 g, 8.91 × 10−3 mol), AIBN (1.37 mg, 8.33 × 10−6 mol) and toluene (2.0 g, 2.17 × 10−2 mol) in a Schlenk flask. The flask was degassed by three successive freeze–pump–thaw cycles and then back filled with UHP argon gas. The flask was then submerged in silicone oil heated to 70 °C and the reaction was run for 24 hours in the dark. The resulting copolymer was precipitated in cold methanol and left to dry in vacuum oven overnight.Analyses
An SEC instrument equipped with a Waters 717plus Autosampler, Waters 600E system controller and Waters 610 fluid unit were used to perform SEC analyses. A Waters 2414 differential refractometer was used for detection. Two PLgel 5 μm Mixed-C columns and a PLgel 5 μm guard column were used. The oven temperature was maintained at 30 °C and 100 μL of 2 mg mL−1 sample was injected into the column set. Tetrahydrofuran (THF) (HPLC grade, BHT stabilised) was used as the eluent for the analyses at a flow rate of 1 mL min−1. Narrow polystyrene standards (h-PS) with molar masses ranging from 800–2 × 106 g mol−1 were used to calibrate the instrument. Molar masses obtained from SEC are reported as h-polystyrene equivalents.1H NMR spectra were recorded on a Varian Unity INOVA 400 MHz spectrometer. Deuterated chloroform (CDCl3) was used to dissolve polymer samples (crude and precipitated).
For in situ1H NMR experiments of hydrogenous styrene, the samples were run in deuterated benzene (C6D6). ACD Labs 10.0 1H NMR processor® was used to process the NMR data. All spectra were phased with automatic phase correction, whilst performing manual baseline correction and integration of proton signals.
For the HPLC analyses, an Agilent 1200 series (Agilent Technologies, Boeblingen, Germany) comprising an auto sampler, vacuum degasser, quaternary pump, column oven, variable wavelength UV detector and Agilent 1260 infinity ELSD was used. The data was recorded and processed using WinGPC Unity (version 7). Critical conditions were established for h-PS using a Phenomenex C18 300 Å (250 × 4.6 mm i.d.) with 5 μm particle size. THF and acetonitrile (ACN) (HPLC grade) were used as the mobile phase and premixed by volume. Samples were dissolved in the premixed solvent with a concentration of 0.5 mg mL−1. The mobile phase was set for a flow rate of 0.5 mL min−1 and sample injection volume was 10 μL. The column oven temperature was kept constant at 35 °C. Conditions where one PS species eluted in SEC mode while the other eluted in liquid adsorption chromatography (LAC) mode were established at a mobile phase composition of THF–ACN 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :52 (v/v). The column oven temperature was kept constant at 46 °C. Samples were dissolved in the premixed solvent with a concentration of 0.5 mg mL−1. In two-dimensional chromatography, the first dimension separated according to chemical composition on a C18 column, whilst the second dimension separated according to molar mass. Samples were dissolved in THF with concentrations of 1 mg mL−1 and 50 μL injected in the first dimension with a flow rate of 0.02 mL min−1. In the two-dimensional liquid chromatography analysis, sample fractions from the first dimension were injected into the second dimension column via an electronically controlled eight port transfer valve (VICI Valco instruments, Texas, USA) consisting of two 50 μL storage loops. The apparatus used in the second dimension consisted of an Agilent 1200 isocratic pump and a 50 mm × 20 mm i.d. PSS Linear M 5 μm styrene-divinylbenzene (SDV) column. THF was the solvent used for analyses in the second dimension, with a flow rate of 3 mL min−1. Detection in the second dimension was done using an ELSD detector. The nebuliser temperature was set to 90 °C and nitrogen gas was used as the carrier gas in the ELSD. The data was recorded and processed using WinGPC Unity (version 7).
:52 (v/v). The column oven temperature was kept constant at 46 °C. Samples were dissolved in the premixed solvent with a concentration of 0.5 mg mL−1. In two-dimensional chromatography, the first dimension separated according to chemical composition on a C18 column, whilst the second dimension separated according to molar mass. Samples were dissolved in THF with concentrations of 1 mg mL−1 and 50 μL injected in the first dimension with a flow rate of 0.02 mL min−1. In the two-dimensional liquid chromatography analysis, sample fractions from the first dimension were injected into the second dimension column via an electronically controlled eight port transfer valve (VICI Valco instruments, Texas, USA) consisting of two 50 μL storage loops. The apparatus used in the second dimension consisted of an Agilent 1200 isocratic pump and a 50 mm × 20 mm i.d. PSS Linear M 5 μm styrene-divinylbenzene (SDV) column. THF was the solvent used for analyses in the second dimension, with a flow rate of 3 mL min−1. Detection in the second dimension was done using an ELSD detector. The nebuliser temperature was set to 90 °C and nitrogen gas was used as the carrier gas in the ELSD. The data was recorded and processed using WinGPC Unity (version 7).
Results and discussion
Homopolymerisation of styrene
H-PS samples were prepared using an [initiator]/[iodine] ratio of 1.7 and targeting molar masses from 10000–100000 g mol−1. The conversion of h-PS could be determined using the 1H NMR spectrum of crude samples. The conversion was then substituted into eqn (1) to determine the molar mass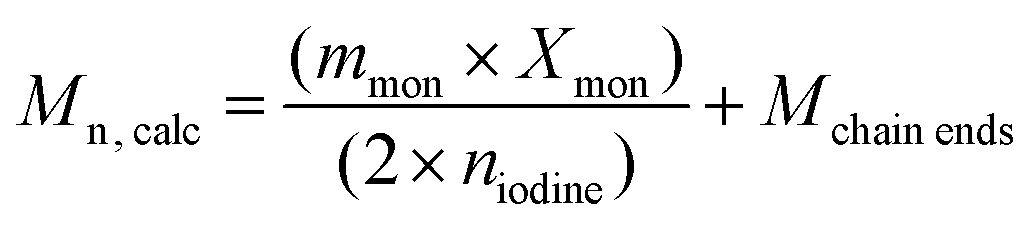 | (1) |
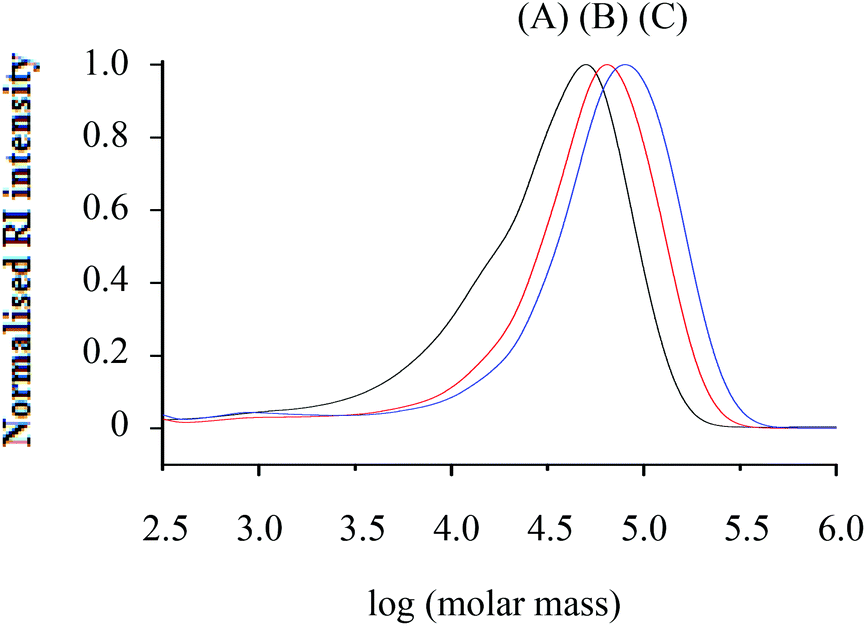 | ||
| Fig. 1 Molar mass distributions from SEC (RI traces) of h-PS (run 3, 5 and 6 in Table 1) synthesised via RITP at 70 °C for 24 hours; (A) Mn, SEC = 24700 g mol−1, (B) Mn, SEC = 43100 g mol−1, (C) Mn, SEC = 55300 g mol−1. | ||
| Run | M n, target (g mol−1) | Conv.b (%) | M n, calc (g mol−1) | M n, SEC (g mol−1) | Đ |
|---|---|---|---|---|---|
| a All reactions run with [AIBN]/[I2] = 1.7. b Determined by 1H NMR of crude sample in CDCl3 by Xmon = (1 − (∫CH2/2/∫C6H5/5)) × 100 where ∫CH2 is the integral of the vinylic protons of residual styrene at 5.1–5.7 ppm, and ∫C6H5 is the integral of the aromatic protons PS. c Calculated by Mn, calc = ((mmon × Xmon)/(2 × niodine)) + Mchain ends. d Calibrated using polystyrene standards (h-PS). | |||||
| 1 | 10000 |
59 | 6000 | 6200 | 1.65 |
| 2 | 30000 |
60 | 17900 |
18400 |
1.77 |
| 3 | 40000 |
62 | 25200 |
24700 |
1.77 |
| 4 | 60000 |
58 | 34600 |
35200 |
1.73 |
| 5 | 70000 |
64 | 44700 |
43100 |
1.70 |
| 6 | 100000 |
60 | 59800 |
55300 |
1.68 |
Homopolymerisation of styrene-d8
Although the synthesis of d-PS via RITP has not been reported in literature, it was expected to follow the same reaction mechanism as h-PS synthesised via RITP. A schematic of the expected polymerisation pathway of styrene-d8via RITP is shown in Scheme 2. The results of polymerisation reactions performed at 70 °C for 24 hours using styrene-d8 are shown in Table 2. At a glance, it is apparent that the monomer conversion for styrene-d8 polymerisation is less than 50%. Regardless of this fact, the dispersity values are between 1.6–1.8, which is typical of styrene polymerised via RITP.7,15,16,41,43 The molar mass distributions from SEC of three d-PS samples (run 2, 3 and 4 Table 2) are shown in Fig. 2.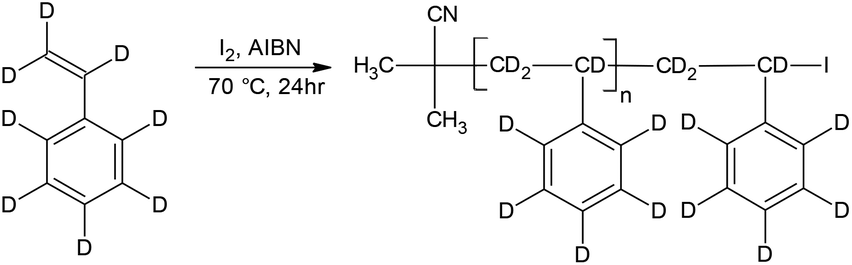 | ||
| Scheme 2 Simplified mechanism of the homopolymerisation of styrene-d8 by RITP. | ||
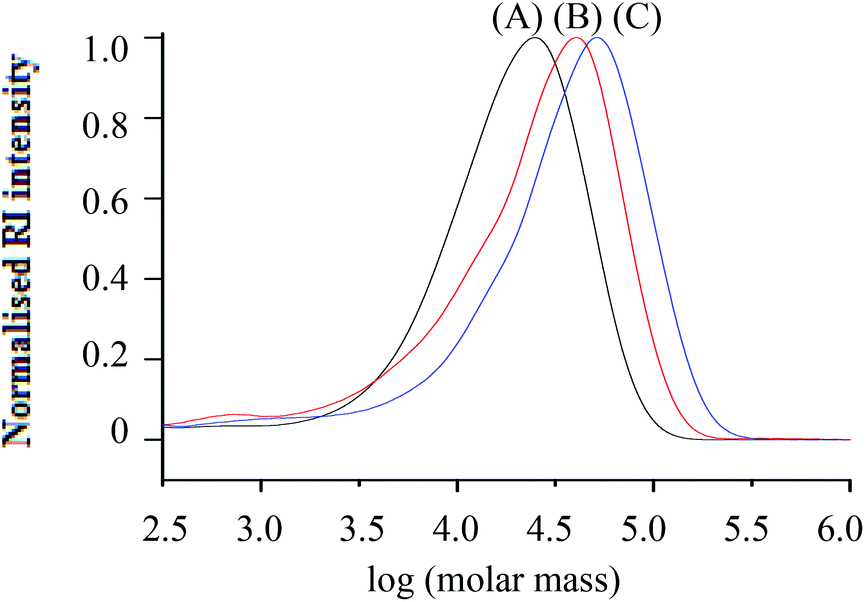 | ||
| Fig. 2 Molar mass distributions from SEC (RI traces) of d-PS (run 2, 3 and 4) synthesised via RITP at 70 °C for 24 hours; (A) Mn, SEC = 12100 g mol−1, (B) Mn, SEC = 19000 g mol−1 and (C) Mn, SEC = 30100 g mol−1. | ||
| Run | M n, target (g mol−1) | Conv.b (%) | M n, calc (g mol−1) | M n, SEC (g mol−1) | Đ |
|---|---|---|---|---|---|
| a All reactions run with [AIBN]/[I2] = 1.7. b Determined gravimetrically. c Calculated by Mn, calc = ((mmon × Xmon)/(2 × niodine)) + Mchain ends. d Calibrated using polystyrene standards (h-PS). | |||||
| 1 | 10000 |
48 | 4800 | 4900 | 1.63 |
| 2 | 25000 |
47 | 11700 |
12100 |
1.72 |
| 3 | 40000 |
47 | 18800 |
19000 |
1.79 |
| 4 | 60000 |
49 | 29600 |
30100 |
1.83 |
| 5 | 100000 |
48 | 48200 |
45500 |
1.77 |
Similar to h-PS, unimodal molar mass distributions with small tailing at lower molar masses were obtained. In RITP there are two stages, an induction period and a polymerisation period. In the induction period, CTAs are generated in situ. These CTAs can either be a result of direct reaction of A˙ with molecular iodine (A–I), or an indirect reaction of A˙ with propagating radicals to form A–Mn–I telomers. The evolution of this CTA has been reported for hydrogenous styrene polymerisation. In the in situ1H NMR spectrum of styrene polymerised via RITP, the proton signal for A–I was observed at δ = 1.65 ppm, which is in agreement with what has been reported previously.5,43 An overlay of the evolution of this A–I transfer agent is shown in Fig. 3 for h-PS and d-PS. The concentration of A–I versus time, the monomer conversion versus time as well as the side reactions due to the formation of 1,2-diiodoethylbenzene have previously been studied.7,43,44 Clearly, the two PS species exhibited similar behaviour with respect to the formation of the transfer agent A–I. The concentration of the A–I transfer agent increased during the induction period until it reached a maximum. The drop in concentration signified the start of the polymerisation period. Fig. 4 shows the evolution of the AIBN concentration for h-PS and d-PS. Again, similar behaviour was observed for the two PS species.
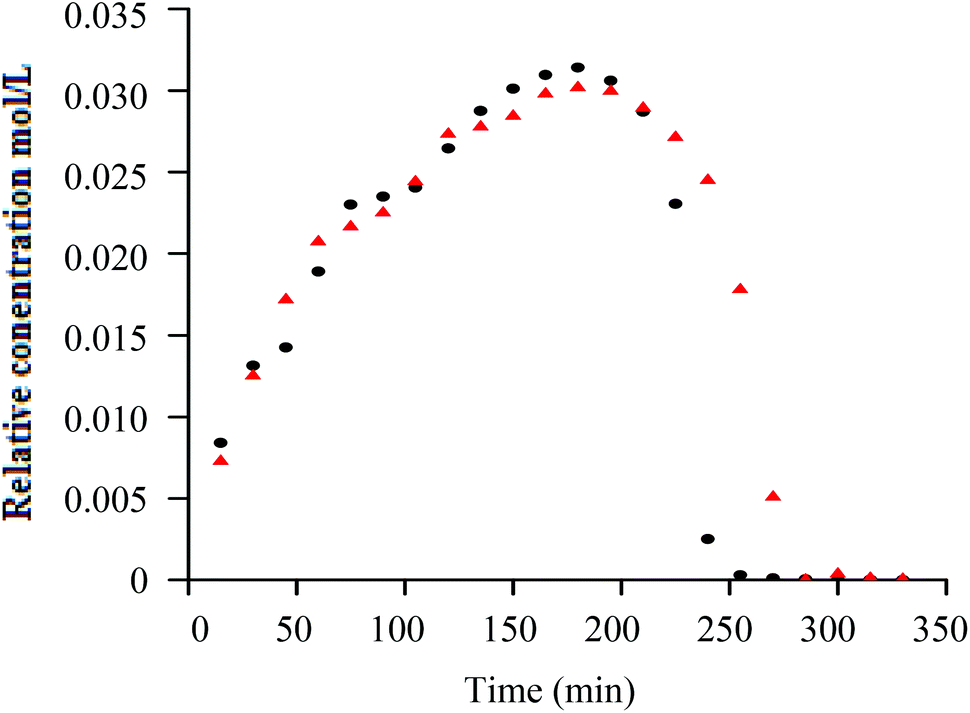 | ||
Fig. 3 Superimposed plots of relative concentration versus time, showing the evolution of the A–I chain transfer agent during the polymerisation of h-PS (●) and d-PS ( ) via RITP at 70 °C for 24 hours. ) via RITP at 70 °C for 24 hours. | ||
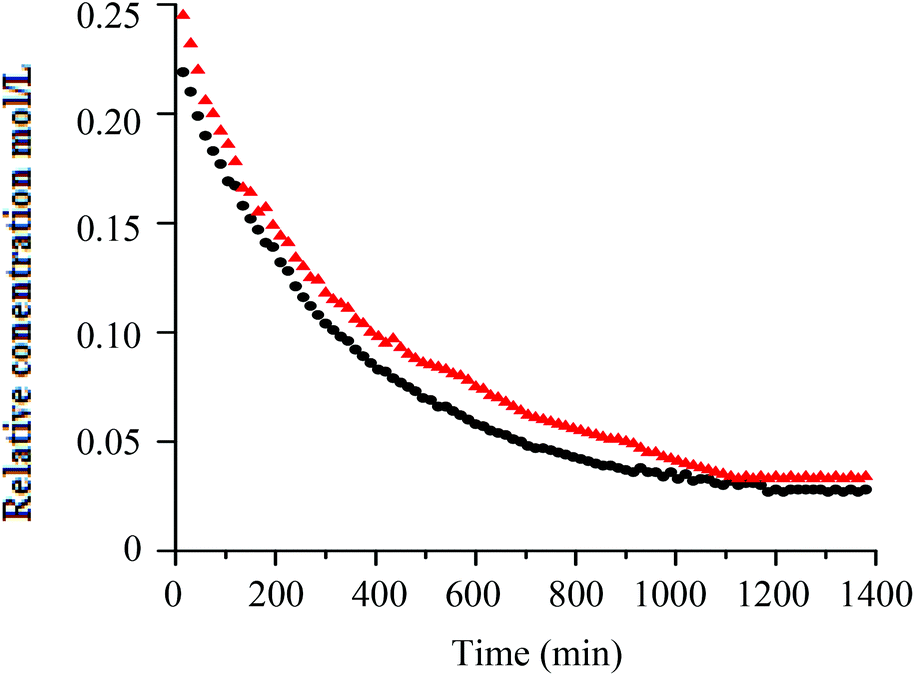 | ||
Fig. 4 Superimposed plots of relative concentration versus time, showing the evolution of the AIBN concentration for h-PS (●) and d-PS ( ) during their respective polymerisation via RITP at 70 °C for 24 hours. ) during their respective polymerisation via RITP at 70 °C for 24 hours. | ||
Synthesis of hPS-b-dPS block copolymers
The synthesis of hPS-b-dPS block copolymers using anionic polymerisation has been reported in literature.45 Withstanding this fact, hPS-b-dPS block copolymers have not, to the best of our knowledge, been prepared via RITP. Due to the high cost of deuterated monomers, RITP offers a relatively cheap and simple method of preparing these rather expensive compounds. It was, therefore, important to understand the mechanism of RITP of deuterated compounds by using a combination of advanced analytical methods. When synthesising the PS homopolymers, it was necessary to add iodine and AIBN to the reaction mixture in order to form chain transfer agents in situ. These homopolymers could then be used as a macro-initiator (PS-I) to synthesise block copolymers.The hPS-b-dPS block copolymers were synthesised using hydrogenous PS-I as a macro-initiator together with styrene-d8. Toluene was added to the mixture to ensure that all PS-I dissolved. Scheme 3 shows a basic representation of the copolymerisation mechanism of hydrogenous PS with styrene-d8. Block copolymers of different molar masses were prepared and analysed by SEC and HPLC. The results for the block copolymerisation reactions are shown in Table 3. The weight percentage of the respective monomer units could be determined by using the molar masses from SEC. This was done using the molar mass values from SEC in eqn (2)
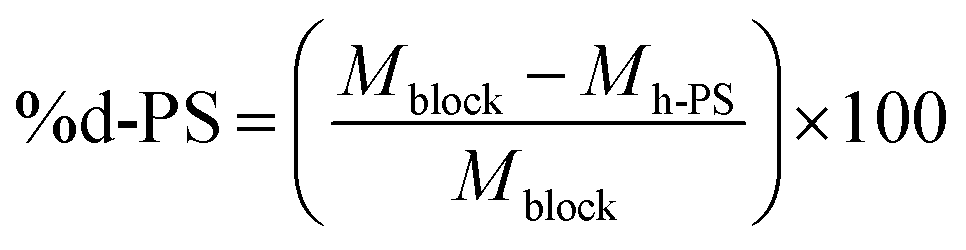 | (2) |
 | ||
| Scheme 3 Simplified mechanism of the copolymerisation of h-PS with styrene-d8via RITP. | ||
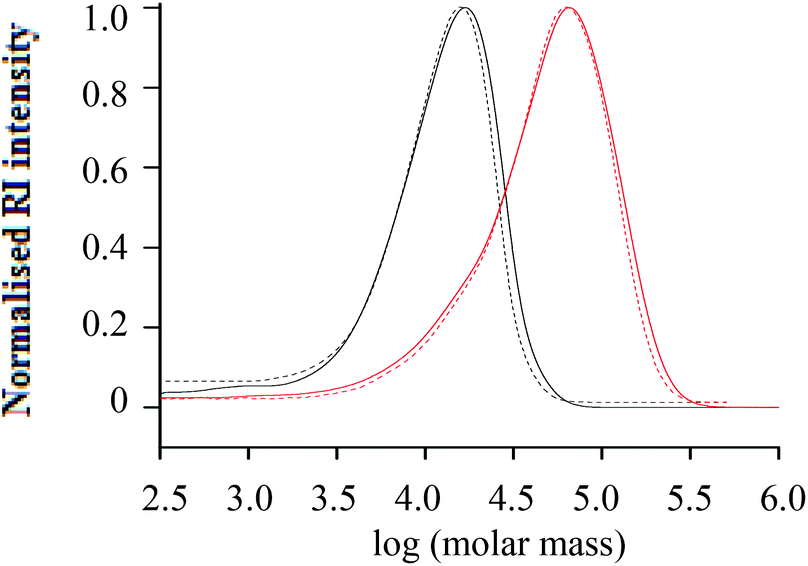 | ||
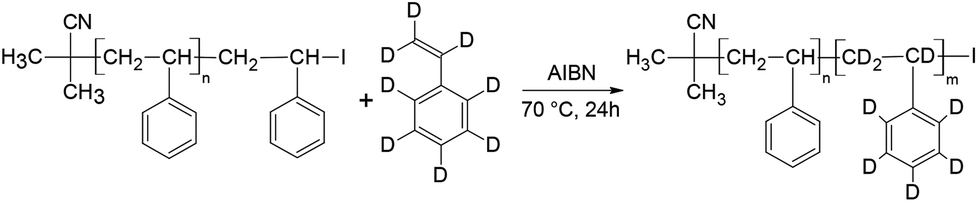
Fig. 5 Molar mass distributions from SEC (RI and UV) of h-PS precursor and hPS-b-dPS block copolymer (run 2 Table 3) synthesised via RITP for 24 hours at 70 °C; (–) = h-PS (RI), (- -) = h-PS (UV), ( ) = hPS-b-dPS (RI), ( ) = hPS-b-dPS (RI), ( ) = hPS-b-dPS (UV). ) = hPS-b-dPS (UV). | ||
| Run | M n, SEC (g mol−1) | Đ | Conv.b (%) | M n, SEC (g mol−1) | Đ | %h-PS:%d-PSc |
|---|---|---|---|---|---|---|
| h-PS | hPS-b-dPS (block copolymer) | |||||
| a Calibrated using PS standards (h-PS). b Determined gravimetrically. c Weight percentage calculated (from Mn, SEC) using %d-PS = ((Mblock − Mh-PS)/Mblock) × 100. | ||||||
| 1 | 18400 |
1.77 | 51 | 31100 |
1.68 | 59:41 |
| 2 | 18400 |
1.77 | 55 | 47600 |
1.63 | 39:61 |
| 3 | 35200 |
1.73 | 49 | 77900 |
1.65 | 45:55 |
Analysis of hPS-b-dPS block copolymers by HPLC
It has been reported that d-PS is more polar than h-PS, due to an enhanced electron donating ability.23 It has also been reported in literature that different isotopes display different chromatographic selectivity in reversed phase liquid chromatography (RPLC).46–48 That is, d-PS interacts less strongly with RP columns than h-PS. This was the basis for selecting a C18 (non-polar) column to analyse the hPS-b-dPS block copolymers.Liquid chromatography at critical conditions (LCCC) was used in this study in order to be as selective as possible with respect to isotopic effects. A Phenomenex C18 300 Å–5 μm stationary phase was used to establish critical conditions at a temperature of 35 °C while varying the composition of the THF–ACN mobile phase. The h-PS and d-PS samples that were prepared via RITP were used to study the hPS-b-dPS block copolymers by LCCC. These samples are listed in Table 4.
| h-PS | M n, SEC (g mol−1) | d-PS | M n, SEC (g mol−1) |
|---|---|---|---|
| a Calibrated using PS standards (h-PS). | |||
| Sample 1 | 6200 | Sample 1 | 4900 |
| Sample 2 | 15600 |
Sample 2 | 12100 |
| Sample 3 | 21600 |
Sample 3 | 19000 |
| Sample 4 | 43100 |
Sample 4 | 30100 |
| Sample 5 | 55300 |
Sample 5 | 45500 |
The critical diagram for the h-PS samples prepared via RITP is shown in Fig. 6. At a mobile phase composition of THF–ACN 55:45 (v/v), h-PS eluted in SEC mode. H-PS eluted in LAC mode when the mobile phase composition was THF–ACN 48:52 (v/v).
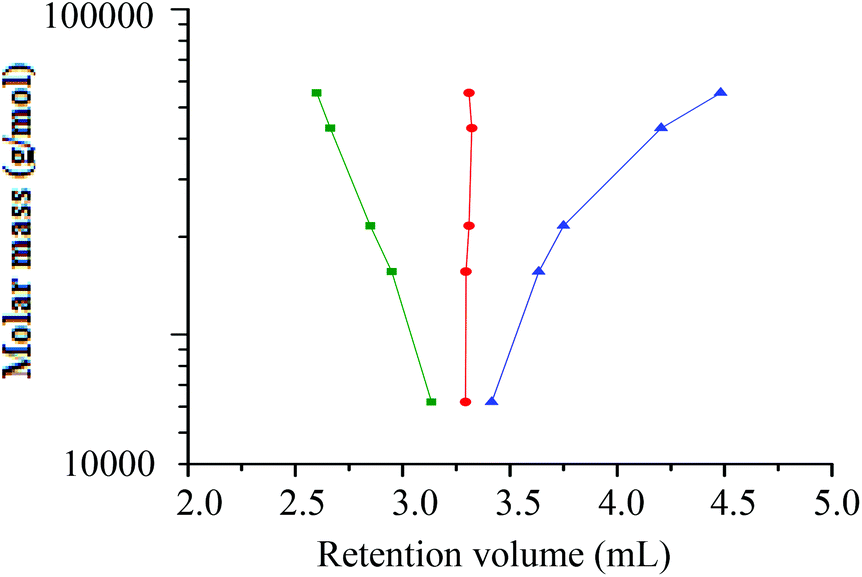 | ||
Fig. 6 Critical diagram of molar mass versus retention volume for h-PS synthesised via RITP; stationary phase: Phenomenex C18 300 Å–5 μm; mobile phase: THF–ACN; ( ) = 55:45 (SEC), ( ) = 55:45 (SEC), ( ) = 50:50 (LCCC) and ( ) = 50:50 (LCCC) and ( ) = 48:52 (LAC) (v/v). ) = 48:52 (LAC) (v/v). | ||
The critical point where all h-PS samples eluted at the same retention volume regardless of their molar mass, was found at a solvent composition of THF–ACN 50:50 (v/v). The critical conditions for h-PS were used to establish where d-PS samples would elute. At the critical conditions of h-PS, the d-PS samples eluted in SEC mode, as seen in Fig. 7.
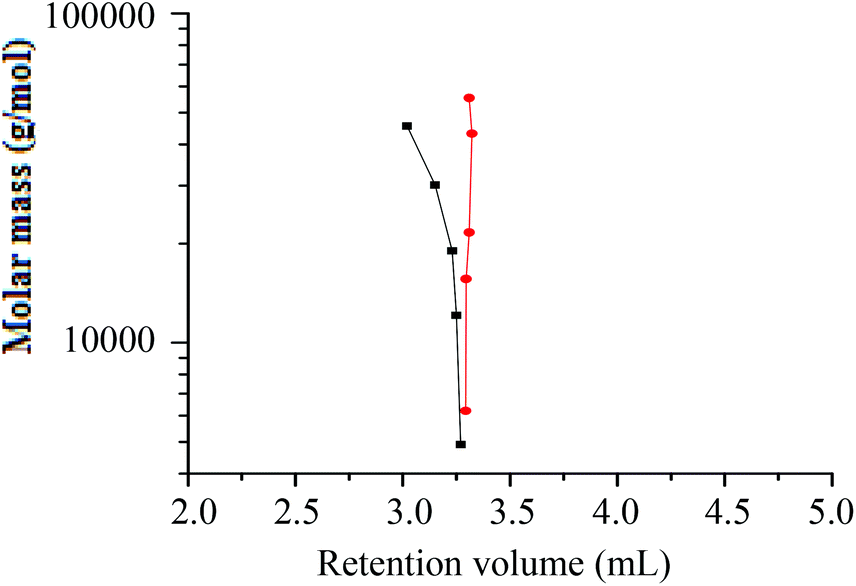 | ||
Fig. 7 Plot of molar mass versus retention volume for h-PS and d-PS samples prepared via RITP. The samples were run at the critical conditions for h-PS; stationary phase: Phenomenex C18 300 Å–5 μm; mobile phase: THF–ACN 50:50; (■) = d-PS (SEC) and ( ) = h-PS (LCCC) (v/v). ) = h-PS (LCCC) (v/v). | ||
At the critical conditions for h-PS, the hPS-b-dPS block copolymers were expected to display the same elution behaviour as the d-PS homopolymers. Fig. 8 shows an overlay of the chromatograms of the h-PS precursor and respective block copolymers (run 1 and 2 Table 3). The hPS-b-dPS block copolymers eluted in SEC mode, as was expected. This type of isotopic separation has been reported in literature before.37,45 It was clear that a higher d-PS content in the block copolymers would result in the block copolymer eluting earlier.
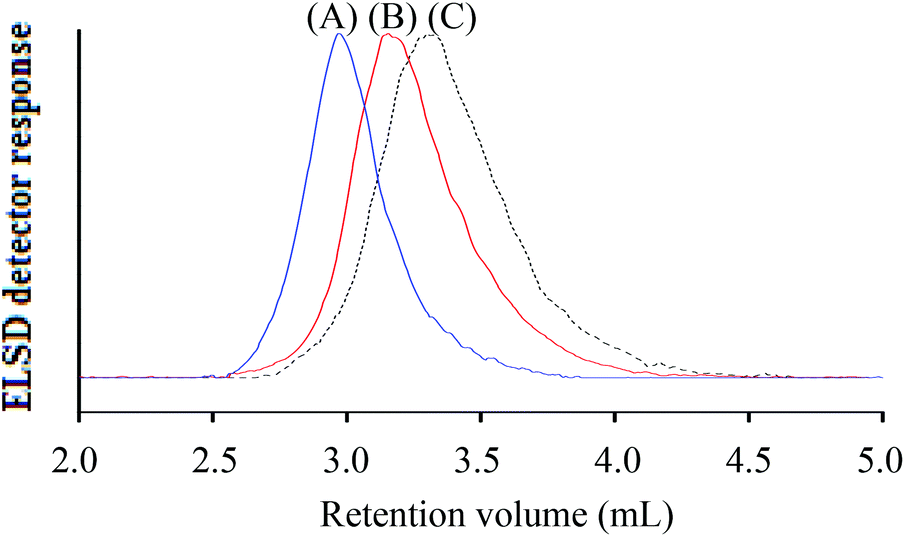 | ||
| Fig. 8 Superimposed chromatograms of the hPS-b-dPS copolymers (solid lines) and the h-PS precursor at the critical conditions of h-PS; stationary phase: Phenomenex C18 300 Å–5 μm; mobile phase: THF–ACN (50:50); (A) hPS-b-dPS (Mn, SEC = 47600 g mol−1), (B) hPS-b-dPS (Mn, SEC = 31100 g mol−1) and (C) h-PS precursor (Mn, SEC = 18400 g mol−1). | ||
A hPS-b-dPS block copolymer of higher molar mass was therefore prepared (run 3 in Table 3). For the high molar mass hPS-b-dPS block copolymer, new separation conditions were established where one PS species eluted in SEC mode while the other eluted in LAC mode. This was done using a solvent composition of THF–ACN 48:52 (v/v).
A plot of molar mass versus retention volume (Fig. 9), for the h-PS and d-PS samples, indicates that the d-PS samples eluted in SEC mode, while the h-PS samples eluted in LAC mode.
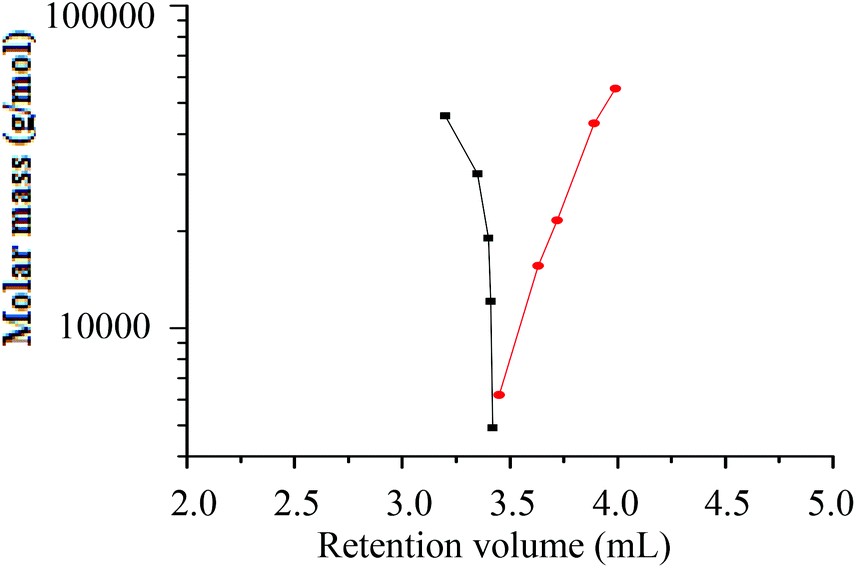 | ||
Fig. 9 Plot of molar mass versus retention volume for h-PS and d-PS samples prepared via RITP at the LAC conditions of h-PS and SEC conditions of d-PS; stationary phase: Phenomenex C18 300 Å–5 μm; mobile phase: THF–ACN (48:52); (■) = d-PS (SEC) and ( ) = h-PS (LAC). ) = h-PS (LAC). | ||
The chromatogram of hPS-b-dPS (run 3), analysed at 46 °C and THF–ACN 48:52 (v/v), is shown in Fig. 10. The chromatogram shows a separation of the hPS-b-dPS block copolymer from the corresponding precursor block. A relatively broad elution pattern is obtained with no baseline separation of the block copolymer and the h-PS homopolymer. This is due to the fact that the block copolymer molecules with longer d-PS blocks shift to SEC elution mode, while the block copolymer molecules with longer h-PS blocks shift to LAC elution mode. Such a separation has been reported in literature for a blend of d-PS and h-PS prepared via living anionic polymerisation.37
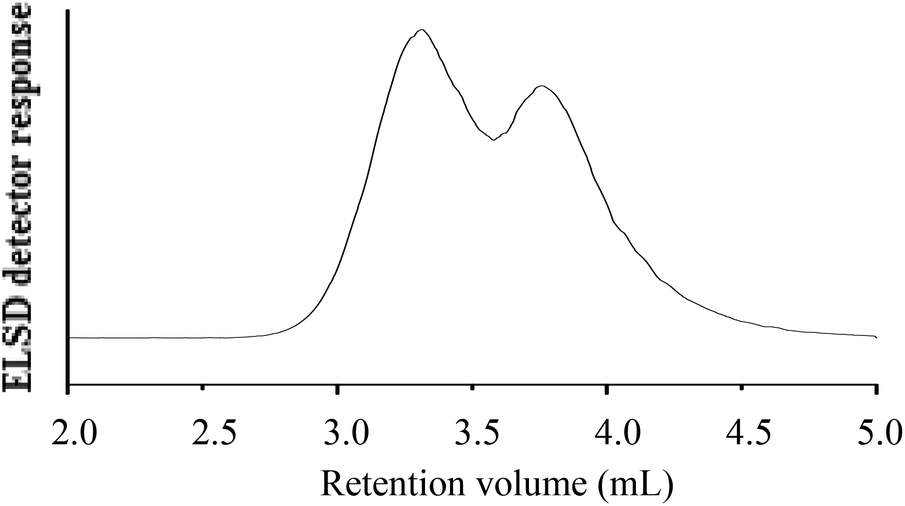 | ||
| Fig. 10 HPLC chromatogram of the hPS-b-dPS block copolymer (run 3, Table 3) at 46 °C and THF–ACN (48:52) (v/v) as the mobile phase; stationary phase: Phenomenex C18 300 Å–5 μm; (A) hPS-b-dPS (Mn, SEC = 42700 g mol−1), (B) h-PS precursor (Mn, SEC = 35200 g mol−1). | ||
No baseline separation for the block copolymer could be achieved, as seen in Fig. 10. This was somehow expected, since the polarities of the two components are so similar. In the present case, the block copolymer had a molar mass of 77900 g mol−1, while the h-PS precursor had a molar mass of 35200 g mol−1. It is expected that a significantly better separation is obtained when the d-PS block of the block copolymer is significantly longer than the h-PS block.
Two-dimensional liquid chromatography of hPS-b-dPS
The chromatographic conditions used to separate the two components of the hPS-b-dPS block copolymer (run 3) were used as the first dimension in two-dimensional liquid chromatography (2D-LC).Fig. 11 shows the contour plot of the two-dimensional separation of the hPS-b-dPS block copolymer sample. The separation in the first dimension (in ordinate direction) was based on a separation due to isotopic effect, while the second dimension (in vertical direction) separated according to molar mass. The 2D contour plot shows a separation of three components in the first dimension. Based on the assignment in Fig. 10, the first eluting component can be assigned to the block copolymer while the last eluting component is the h-PS precursor block. The component eluting between those is tentatively assigned to d-PS homopolymer, however, this assignment needs further confirmation. The separation according to molar mass is rather poor. No distinct differences in the molar masses of the three components can be seen. This poor molar mass resolution is due to the fact that only one high-throughput SEC column was used and the separation was conducted at very high flow rate. Therefore, the separation in the second dimension needs further optimization.
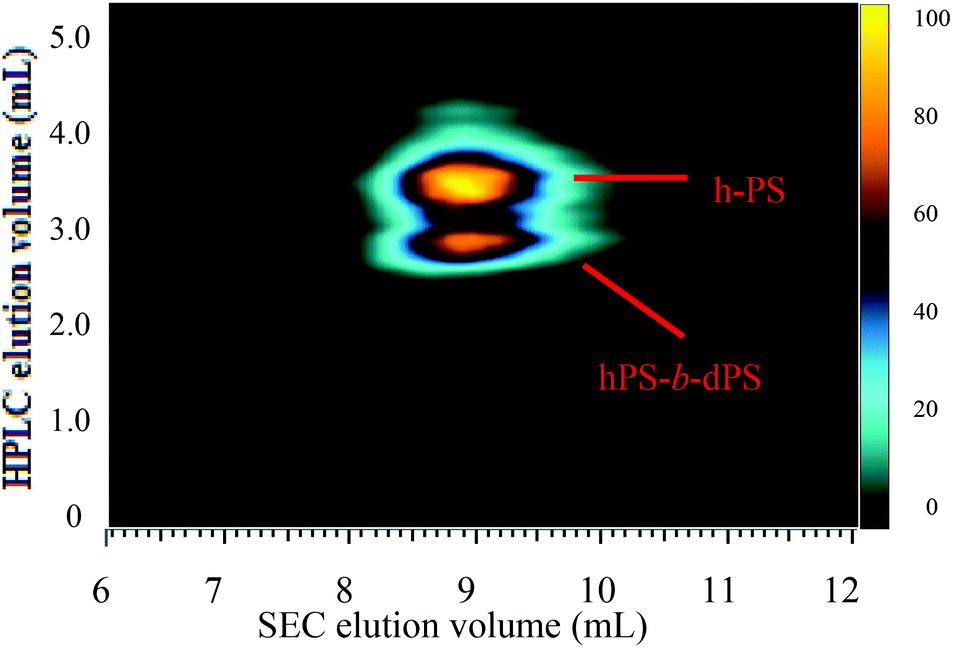 | ||
| Fig. 11 Contour plot of the two-dimensional separation of the hPS-b-dPS block copolymer (run 3) measured at a THF–ACN composition of 48:52 (v/v) and column oven temperature of 46 °C. | ||
Conclusions
RITP was used to prepare hydrogenous polystyrene samples. These h-PS samples were used as macro-initiators in the block copolymerisation of hydrogenous-polystyrene-block-deuterated-polystyrene (hPS-b-dPS) via RITP. Deuterated polystyrene was successfully prepared via RITP for the first time, and the results were comparatively similar to those obtained for the h-PS results. The livingness of the h-PS precursors was demonstrated by the successful synthesis, for the first time, of hPS-b-dPS block copolymers via RITP. The kinetics of the polymerisation was investigated by 1H NMR and it was found that RITP of h-styrene and d-styrene behave in a similar manner. Liquid chromatography at critical conditions was used to analyse the hPS-b-dPS block copolymers. At the critical conditions established for the h-PS homopolymers, the hPS-b-dPS block copolymers eluted in SEC mode due to the directing effect of the d-PS block. An improved separation was observed at chromatographic conditions where one component (d-PS) eluted in SEC mode, while the other component (h-PS) eluted in LAC mode. Two-dimensional liquid chromatography (2D-LC) showed that this separation was due to isotopic effects. Lastly, 2D-LC showed a third component that was not observed in the one-dimensional analysis.Notes and references
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- R. B. Grubbs, Polym. Rev., 2011, 51, 104–137 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Chem. – Asian J., 2013, 8, 1634–1644 CrossRef CAS PubMed.
- G. David, C. Boyer, J. Tonnar, B. Ameduri, P. Lacroix-Desmazes and B. Boutevin, Chem. Rev., 2006, 106, 3936–3962 CrossRef CAS PubMed.
- P. Lacroix-Desmazes, R. Severac and B. Boutevin, Macromolecules, 2005, 38, 6299–6309 CrossRef CAS.
- C. Boyer, P. Lacroix-Desmazes, J.-J. Robin and B. Boutevin, Macromolecules, 2006, 39, 4044–4053 CrossRef CAS.
- J. Tonnar, R. Severac, P. Lacroix-Desmazes and B. Boutevin, Polym. Prepr., 2008, 49, 68–69 CAS.
- P. Lacroix-Desmazes and J. Tonnar, Degenerative Transfer with Alkyl Iodide in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier BV, Amsterdam, 2012, vol. 3, pp. 159–180 Search PubMed.
- K. Matyjaszewski and T. P. Davis, Handbook of Radical Polymerization, Wiley-Interscience, Canada, 2002, pp. 361–406 Search PubMed.
- P. Lacroix-Desmazes, R. Severac, B. Otazaghine and B. Boutevin, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2003, 44, 683–684 CAS.
- C. Fringant, Y. Vanderveken, P. Lacroix-Desmazes and J. Tonnar, Free-radical polymerization process in aqueous dispersion for the preparation of a block copolymer comprising at least one halogenated polymer block, (Solvay (Societe Anonyme), Belg.), PCT Int. Appl, WO 2008003728 A1 20080110, 2008. Application: WO 2007-EP56778 20070704. Priority: FR 2006-6079 20060704; US 2006-818276 20060705. AN 2008:43752 Search PubMed.
- P. Lacroix-Desmazes, R. Severac, B. Boutevin, V. Bodart and V. Kurowski, Iodinated organic oligomer and its preparation and application in free-radical polymerization, (Solvay Societe Anonyme, Belg.), PCT Int. Appl, 28 pp. WO 2004094356 A1 20041104, 2004. Application: WO 2004-EP50591 20040422. Priority: BE 2003-255 20030422; US 2003-487258 20030716; FR 2003-9561 20030801. CAN 141:380281; AN 2004:927153 Search PubMed.
- P. Lacroix-Desmazes, R. Severac, B. Boutevin, V. Bodart and V. Kurowsky, Radical polymerization method for preparing halogenated polymers, and the halogenated polymers, (Solvay SA, Belg.). Fr. Demande, 23 pp., FR 2839724 A1 20031121, 2003. Application: FR 2002-6246 20020517. Extension PCT, WO 2003097704 A1 20031127, Application: WO 2003-EP5314. Priority: FR 2002-6246. CAN 139:381915; AN 2003:914240 Search PubMed.
- P. Lacroix-Desmazes and J. Tonnar, Process of free-radical polymerization in aqueous dispersion for the preparation of polymers, (Ecole Nationale Superieure de Chimie de Montpellier, Fr.), PCT Int. Appl, WO 2008003729 A2 20080110, 2008. Application: WO 2007-EP56779 20070704. Priority: FR 2006-6079 20060704; US 2006-818276 20060705. AN 2008:42934 Search PubMed.
- F. J. Enríquez-Medrano, R. Guerrero-Santos, M. Hernandez-Valdez and P. Lacroix-Desmazes, J. Appl. Polym. Sci., 2011, 119, 2476–2484 CrossRef.
- N. Greesh, R. Sanderson and P. Hartmann, J. Appl. Polym. Sci., 2012, 126, 1773–1783 CrossRef CAS.
- B. N. Patra, D. Rayeroux and P. Lacroix-Desmazes, React. Funct. Polym., 2010, 70, 408–413 CrossRef CAS PubMed.
- D. Rayeroux, V. Lapinte and P. Lacroix-Desmazes, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4589–4593 CrossRef CAS.
- D. Rayeroux, B. N. Patra and P. Lacroix-Desmazes, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4389–4398 CrossRef CAS.
- D. G. H. Ballard, M. G. Rayner and J. Schelten, Polymer, 1976, 17, 640–641 CrossRef CAS.
- F. S. Bates, J. Appl. Crystallogr., 1988, 21, 681–691 CrossRef CAS.
- F. S. Bates, L. J. Fetters and G. D. Wignall, Macromolecules, 1988, 21, 1086–1094 CrossRef CAS.
- F. S. Bates and G. D. Wignall, Phys. Rev. Lett., 1986, 57, 1429–1432 CrossRef CAS.
- R. G. Kirste and W. A. Kruse, Polymer, 1975, 16, 120–124 CrossRef CAS.
- A. Maconnachie, R. P. Kambour and R. C. Bopp, Polymer, 1984, 25, 357–364 CrossRef CAS.
- M. Warner, J. S. Higgins and A. J. Carter, Macromolecules, 1983, 16, 1931–1935 CrossRef CAS.
- M. T. Baker, W. C. Ronnenberg, J. A. Ruzicka, C. K. Chiang and J. H. Tinker, Drug Metab. Dispos., 1993, 21, 1170–1171 CAS.
- L. D. Costanzo, M. Moulin, M. Haertlein, F. Meilleur and D. W. Christianson, Arch. Biochem. Biophys., 2007, 465, 82–89 CrossRef PubMed.
- N. Modutlwa, T. Maegawa, Y. Monguchi and H. Sajiki, J. Labelled Compd. Radiopharm., 2010, 53, 686–692 CrossRef CAS.
- S. D. Nelson and W. F. Trager, Drug Metab. Dispos., 2003, 31, 1481–1497 CrossRef CAS PubMed.
- G. Pons and E. Rey, Pediatrics, 1999, 104, 633–639 CAS.
- R. Sharma, T. J. Strelevitz, H. Gao, A. J. Clark, K. Schildknegt, R. S. Obach, S. L. Ripp, D. K. Spracklin, L. M. Tremaine and A. D. N. Vaz, Drug Metab. Dispos., 2012, 40, 625–634 CrossRef CAS PubMed.
- J. Thierrin, G. B. Davis and C. Barber, v, 1995, 33, 469–475 CAS.
- D. J. Gillich, A. Kovanen and Y. Danon, J. Nucl. Mater., 2010, 405, 181–185 CrossRef CAS PubMed.
- Z. Lin, T. Yongjian, Z. Chifeng, L. Xuan and Z. Houqiong, Nucl. Instrum. Methods Phys. Res., Sect. A, 2002, 480, 242–245 CrossRef CAS.
- K. Nemoto, A. Maksimchuk, S. Banerjee, K. Flippo, G. Mourou, D. Umstadter and V. Y. Bychenkov, Appl. Phys. Lett., 2001, 78, 595–597 CrossRef CAS PubMed.
- P. Sinha, G. W. Harding, K. Maiko, W. Hiller and H. Pasch, J. Chromatogr., A, 2012, 1265, 95–104 CrossRef CAS PubMed.
- X. Wang, Z. Xu, Y. Wan, T. Huang, S. Pispas, J. W. Mays and C. Wu, Macromolecules, 1997, 30, 7202–7205 CrossRef CAS.
- S. Ahn, K. Im, T. Chang, P. Chambon and C. M. Fernyhough, Anal. Chem., 2011, 83, 4237–4242 CrossRef CAS PubMed.
- D. Boschmann, M. Mänz, A.-C. Pöppler, N. Sörensen and P. Vana, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7280–7286 CrossRef CAS.
- D. I. Shiman, S. V. Kostyuk, L. V. Gaponik and F. N. Kaputskii, Russ. J. Appl. Chem., 2010, 83, 2028–2034 CrossRef CAS.
- J. Tonnar, R. Severac, P. Lacroix-Desmazes and B. Boutevin, Polym. Prepr., 2008, 49, 187–188 CAS.
- T. Wright, H. Chirowodza and H. Pasch, Macromolecules, 2012, 45, 2995–3003 CrossRef CAS.
- P. Lacroix-Desmazes, A.-M. Villa-Hernandez and D. Rayeroux, Progress in Controlled Radical Polymerization: Mechanisms and Techniques, ACS Symposium Series, 2012, vol. 1100, pp. 317–331 Search PubMed.
- S. Lee, H. Lee, L. Thieu, Y. Jeong and T. Chang, Macromolecules, 2013, 46, 9114–9121 CrossRef CAS.
- S. Kayillo, M. J. Gray, R. A. Shalliker and G. R. Dennis, J. Chromatogr., A, 2005, 1073, 83–86 CrossRef CAS PubMed.
- Y. Kim, S. Ahn and T. Chang, Anal. Chem., 2010, 82, 1509–1514 CrossRef CAS PubMed.
- S. Perny, J. Allgaier, D. Cho, W. Lee and T. Chang, Macromolecules, 2001, 34, 5408–5415 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |