
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoinduced topological transformation of cyclized polylactides for switching the properties of homocrystals and stereocomplexes†
Naoto
Sugai
,
Shigeo
Asai
*,
Yasuyuki
Tezuka
* and
Takuya
Yamamoto
*
Department of Organic and Polymeric Materials, Tokyo Institute of Technology, O-okayama, Meguro-ku, Tokyo 152-8552, Japan. E-mail: asai.s.aa@m.titech.ac.jp; ytezuka@o.cc.titech.ac.jp; yamamoto.t.ay@m.titech.ac.jp; Fax: +81 3 5734 2876
First published on 4th March 2015
Abstract
Cyclized poly(L-lactide) and poly(D-lactide) (Mn ∼ 3 kDa) incorporating an o-nitrobenzyl group as a photocleavable linker were synthesized and photoirradiated for topological transformation to form photocleaved linear polylactides. By DSC, Tm of the cyclized stereocomplex (167 °C) decreased by more than 40 °C from that of the linear prepolymers (209 °C) despite their essentially identical molecular weights. Upon the photocleavage, the resulting linear stereocomplex showed almost the same Tm (211 °C) as that before the cyclization. The enthalpy of melting of crystals having an infinite thickness, i.e. ΔHm(100%), and the surface free energy (σe) were determined by the combination of WAXD, SAXS, and DSC. Both ΔHm(100%) and σe were considerably smaller for the cyclized polylactide homocrystals and stereocomplexes than those of the linear prepolymers and photocleaved products. These suggest that the absolute enthalpy of the melt state is lower, and the crystalline–amorphous interface is more stable for the cyclized polylactides arising from the topology.
Introduction
Stimuli-responsive polymers have attracted great interest and been developed for numerous applications.1 For example, poly(N-isopropylacrylamide) is known for a thermal responsive polymer that shows a lower critical solution temperature at around 32 °C by dehydration.2 pH-responsive polymers are often employed for lithography3 and drug delivery systems (DDS)4 by switching their solubility through deprotection and (de)protonation. Furthermore, DDS5 and self-healing materials6 using redox-responsive polymers were reported. However, these responses require a stimulus on essentially every monomer unit, and thus a considerable amount of heat, acid/base, or oxidant/reductant is indispensible. In order to establish a notably more efficient stimuli-responsive polymer system, we expected cyclic polymers to have substantial potential because only one cleaving reaction per polymer molecule leads to cyclic-to-linear topological transformation to trigger changes in the properties. Furthermore, the molecular weight and chemical structure of the main chain are unaffected through the topological transformation. In this regard, cyclic polymers were reported to show significantly enhanced material properties compared to relevant linear polymers, including the stability of micelles.7The reversible linear–cyclic topological transformation of polystyrene and poly(ethylene oxide) was reported by making use of thiol–disulfide conversion,8 hydrogen bonding,9 and dimerization of porphyrin.10 In addition, photocleavage of cyclic morpholino11,12 and oligonucleotide13 was sometimes employed to control gene expression. Nevertheless, topological transformation has never been applied to stimuli-responsive polymeric materials.
We previously synthesized cyclic poly(L-lactide), PLLA (2L), and poly(D-lactide), PDLA (2D) (number average molecular weight (Mn) ∼3 kDa, Fig. 1a), as well as cyclic stereoblock polylactides, and reported the melting points (Tm) of their homocrystals and stereocomplexes14,15 using differential scanning calorimetry (DSC).16 The linear and cyclic stereocomplexes showed distinguishable Tm, and we expected that this stereocomplex system can be applied for a topology-dependent stimuli-responsive polymeric material. Meanwhile, Waymouth and co-workers reported DSC, wide-angle X-ray diffraction (WAXD), and small-angle X-ray scattering (SAXS) of cyclic PLLA and PDLA having a relatively large molecular weight (≥26 kDa) synthesized by zwitterionic ring-opening polymerization.17 These cyclic polylactides exhibited similar thermal and crystallographic properties to their linear counterparts. In regard to this, polylactide stereocomplexes are known to form folded chain crystals of molecular weights larger than several thousands.18 The folded chain crystals likely resulted in diminished and/or complicated influences by the polymer topology. In addition, the optical purity of these cyclic polylactides was modest due to partial racemization during the ring-expansion polymerization.
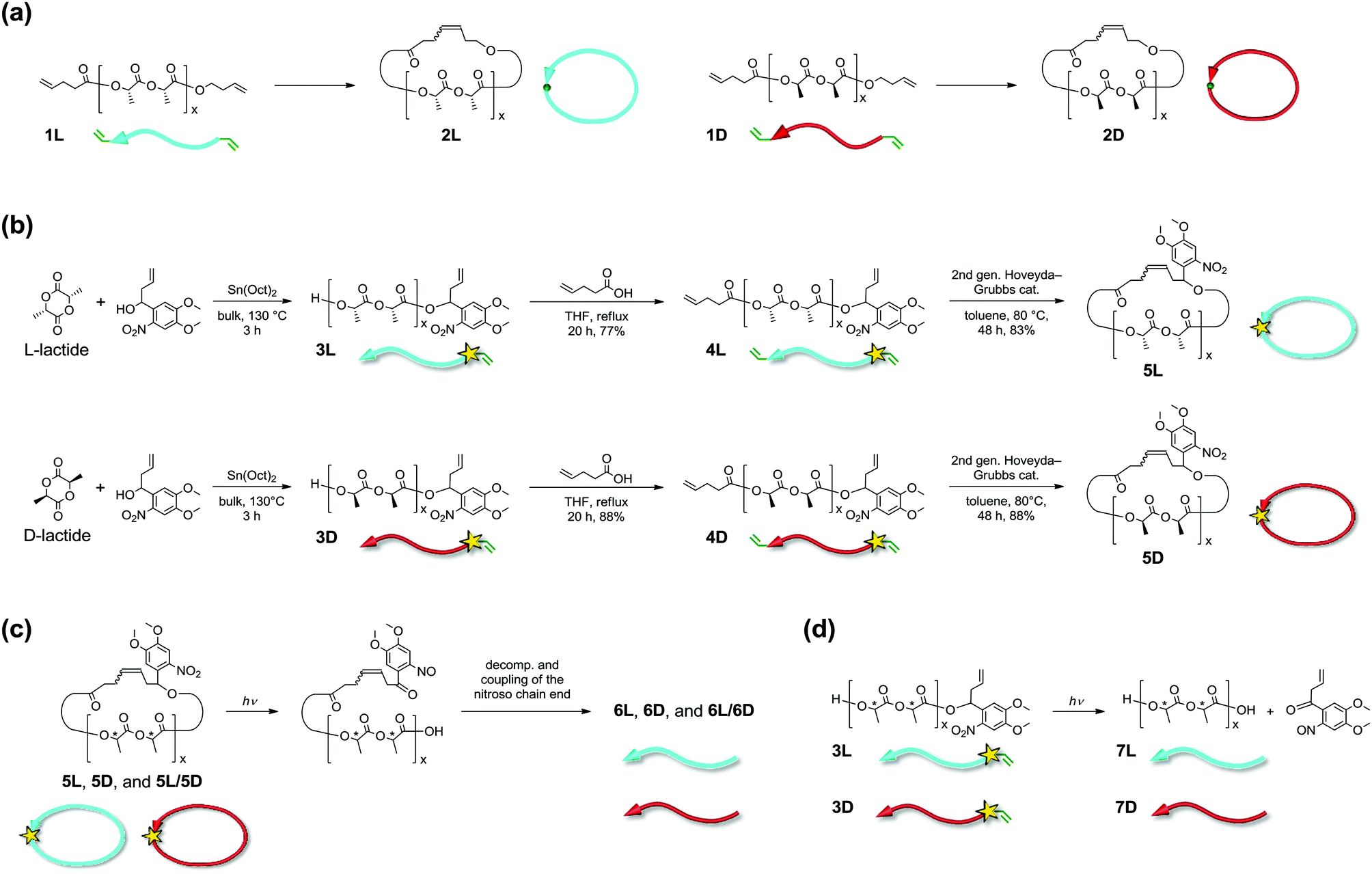 | ||
| Fig. 1 Chemical structures and schematic representation of linear and cyclic PLLA and PDLA used in the present study. (a) Non-photocleavable 1L, 2L, 1D, and 2D. (b) Synthetic scheme for photocleavable 3L, 4L, 5L, 3D, 4D, and 5D. (c) Photocleavage of 5L, 5D, and their blend (5L/5D) to form 6L, 6D, and 6L/6D, respectively. (d) Photocleavage of 3L and 3D to form 7L and 7D, respectively. For convenience, the direction of the polylactide main chains is indicated by an arrow; the head of the arrows indicates the hydroxyl end, and the tail of the arrows indicates the carboxylic acid end. PLLA and PDLA are shown in light blue and red, respectively. The yellow stars indicate a photocleavable NB linker. | ||
In the present work, cyclic PLLA and PDLA with an Mn of approximately 3 and 1.5 kDa incorporating an o-nitrobenzyl group (NB) as a photocleavable linker19 were synthesized via highly optically pure polymerization (Fig. 1b). These homopolymers and their stereocomplexes were subjected to WAXD, SAXS, and DSC measurements to study the effects of the polymer topology on the extended chain crystals. Furthermore, cyclic-to-linear topological transformation by photoirradiation was performed. Tm of the stereocomplex, which decreased upon cyclization by 42 °C (Tm before the cyclization, 209 °C; Tm after the cyclization, 167 °C), was restored to 211 °C by photocleavage. The difference in Tm arose from the crystal thickness (lc) in the lamellar structure,20 directly reflecting the topology of the polymers. Furthermore, the enthalpy of melting of crystals having an infinite thickness, i.e. ΔHm(100%), and the surface free energy (σe) of the crystalline layers were significantly smaller for the homocrystals and the stereocomplex of the cyclic polylactides. Coupled with the recently attracted bio-based, bio-degradable, and carbon-neutral features of polylactides, the switch of the properties by topological transformation should find various applications.
Results and discussion
Synthesis of photocleavable cyclic PLLA (5L) and PDLA (5D) with a NB group
A NB group, which is one of the most widely used photolabile protecting groups due to its prompt removal, was chosen as the photocleavable linker.19 The cyclic-to-linear topological transformation was expected to be enabled by the molecular design that incorporates the photocleavable NB linker in the main chain of the cyclic polymers (Fig. 1c). Cyclic PLLA (5L) and PDLA (5D) with a photocleavable linker were prepared using a similar method to that of 2L and 2D, which were reported previously (Fig. 1a and b).16 In this regard, the employed polymerization is known to form highly optically pure polylactides without degradation in the stereochemistry.21 The intermediate and final products were fully characterized using 1H NMR, SEC, and MALDI-TOF MS (Fig. S1–S3†).Photocleavage
To achieve selective photoinduced cyclic-to-linear topological transformation, the conditions for the photocleavage reaction were investigated. The photoirradiation experiments were first conducted with the most available linear polylactides with a photocleavable linker at the chain end (3L and 3D) under various conditions to form 7L and 7D, respectively (Fig. 1d). The experiments were performed using a wavelength of 365 nm and an intensity of 700 mW cm−2.19 A polylactide has a very weak absorption at 365 nm; the main chain should not be directly decomposed by photoirradiation.22 Good solvents for polylactide homopolymers in the absence of absorption around 365 nm such as CH2Cl2, CHCl3, THF, toluene, CH3CN, 1,2-dichloroethane, and EtOAc were used.22 At a polymer concentration of 0.5 mg mL−1, 30 min of irradiation caused decomposition and crosslinking of the main chains, as observed by SEC for all of these solvents (Fig. S4†). The photocleavage of the NB group generated unstable nitroso compounds, which spontaneously collapsed to form radicals, leading to the radical-mediated degradation of the polylactide main chain.19 Therefore, a radical scavenger (butylated hydroxytoluene, BHT) was added, and the decomposition and crosslinking were found to be effectively suppressed (Fig. S4b,† bottom).On the basis of these results, the photocleavage of cyclic 5L and 5D was investigated. The SEC traces after photoirradiation (6L and 6D) in the presence of BHT exhibited suppression of the main chain degradation (Fig. S5†). The photocleavage was observed after 2 h by noticeable peak top shifts toward the smaller elution volume. The peak molecular weight of 6L (Mp = 5.0 kDa), which represented the hydrodynamic volume, was nearly equal to that of linear prepolymer 4L (Mp = 5.2 kDa), in agreement with previously reported linearization reactions.8,23 Moreover, the SEC trace of 6L also has a shoulder peak at Mp = 11 kDa, which is comparable to that of a dimer of 6L (2 × 5.0 kDa = 10 kDa). A similar shoulder also appeared in 6D (Fig. S5†). The dimers presumably formed by the intermolecular coupling of the nitroso groups, which were resulted by the photocleavage (Fig. S6†).24 During the photocleavage process, the nitroso group remained at the terminus of the polylactide chains and likely formed the dimer by the nitroso–nitroso association.25 Furthermore, trimer or higher multimer formation was not detected, suggesting that the dimerization occurred through this mechanism. This hypothesis was also supported by the photocleavage reaction of linear PLLA possessing a NB group (3L) in Fig. S4.† The dimerization of photocleaved 3D in the presence of BHT was not observed because 3D had a molecular structure to detach the nitroso group from the main chain upon the photocleavage (Fig. 1d). By comparing the 1H NMR spectra of 5L and 6L, the signals for the NB group (“f” at 6.38–6.46 ppm, “d” at 6.91 ppm, and “e” at 7.63 ppm) clearly decreased after photoirradiation (Fig. S7†). The small peaks remaining in the aromatic region of 6L could be due to the nitroso–nitroso association products.25 A decrease of signal “o” in 6L (Fig. S7†) suggests that this photocleavage process is accompanied by some other side reactions on the alkene due to the reactivity of the intermediate radicals and/or the nitroso group. Based on these results, it was concluded that the cyclic-to-linear topological transformation was successfully complete. The photocleavage in the solid state was also studied. Despite many attempts under various conditions, the decomposition of the polylactides occurred before the completion of the intended photocleavage reaction. Thus, the solid-state topological conversion is currently investigated.
X-ray structures
The X-ray structural analysis of the solvent-cast samples of the previously reported non-photocleavable linear and cyclic polylactides without a NB group (1L, 1D, 2L, 2D, 1L/1D, 1L/2D, and 2L/2D)16 was first conducted. The WAXD profiles of individual 1L, 1D, 2L, and 2D showed diffraction peaks at 2θ = 15, 17, and 19° that are ascribed to the α- or α′-forms of the polylactide homocrystals (Fig. S8,† left).26 The degree of crystallinity (χc) of the homocrystals was estimated by the profiles (1L, 63%; 1D, 58%; 2L, 64%; 2D, 50%). Fig. S8† (right) shows the WAXD profiles of the enantiomeric blends of 1L/1D, 1L/2D, and 2L/2D. The diffraction peaks at 2θ = 12, 21, and 24° are attributed to the stereocomplex.26 The χc values of the stereocomplexes were also estimated (1L/1D, 79%; 1L/2D, 62%; 2L/2D, 69%).The SAXS profiles of the individual polymers (1L, 1D, 2L, and 2D) and the blends (1L/1D, 1L/2D, and 2L/2D) are shown in Fig. S9.† The values of the long spacing (L), i.e. the distance between the centers of the adjacent crystallites, the crystal thickness (lc), and the thickness of the amorphous intercrystalline layers (la) of 1L (L = 12 nm, lc = 8.2 nm, la = 2.1 nm), 1D (L = 12 nm, lc = 9.4 nm, la = 2.5 nm), 2L (L = 6.3 nm, lc = 3.9 nm, la = 2.4 nm), and 2D (L = 6.1 nm, lc = 3.6 nm, la = 2.5 nm) were determined (Table S1†).27 The direction of the thickness of the polylactide homocrystals is the same as the c axis of the unit cell, which is 2.886 nm consisting of ten lactic acid units.28 Thus, based on the above-mentioned lc, the number of lactic acid units and its molecular weight in the crystal layers were 1L, 28 units for Mn = 2.0 kDa; 1D, 33 units for Mn = 2.3 kDa, 2L, 14 units for Mn = 1.0 kDa, and 2D, 12 units for Mn = 0.9 kDa (Table S1†). On the basis of Mn determined by NMR (∼3 kDa), these results suggest that linear 1L and 1D form extended chain crystals with the residual lengths of the polymer chains existing in the amorphous layers (Fig. 2a).29 In the case of 2L and 2D, the polymer chain supposedly formed the flattened conformation shown in Fig. 2b, and thus lc became nearly half of that of 1L and 1D.30 Furthermore, the SAXS peak became dull for cyclized 2L and 2D (Fig. S9†). This indicates that the crystal lattice was distorted likely due to the difficulty in the alignment of the polymer chains restricted by the cyclic topology.
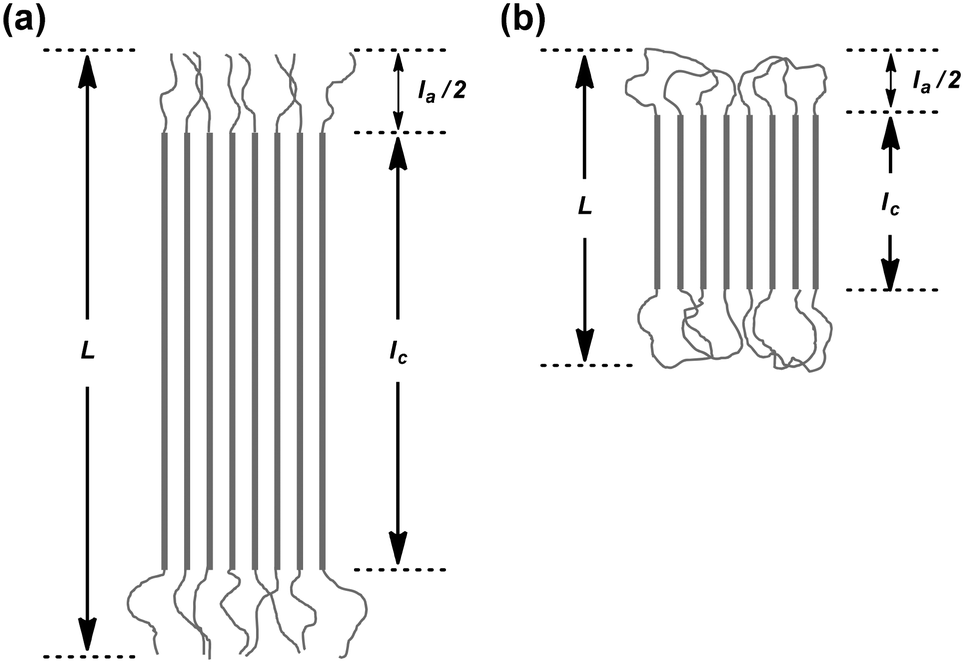 | ||
| Fig. 2 Schematic representation of the lamellar structures of (a) linear and (b) cyclic homocrystals and stereocomplexes of the polylactides determined by SAXS. L, long period; la, amorphous thickness; lc, crystal thickness. | ||
The SAXS profiles of the enantiomeric blends are also shown in Fig. S9,† and the values of L, lc and la were estimated (1L/1D, L = 12 nm, lc = 10 nm, la = 2.1 nm; 1L/2D, L = 8.8 nm, lc = 7.2 nm, la = 1.6 nm; 2L/2D, L = 7.0 nm, lc = 5.4 nm, la = 1.6 nm) as shown in Table S1.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 The direction of the thickness of a polylactide stereocomplex is the same as the c axis of the unit cell, which is 0.87 nm consisting of three lactic acid units.18,31,32 Thus, based on the above-mentioned lc, the number of lactic acid units and the molecular weights in the crystal layers were 1L/1D, 34 units for Mn = 2.5 kDa; 1L/2D, 25 units for Mn = 1.8 kDa, and 2L/2D, 17 units for Mn = 1.3 kDa (Table S1†). Given that Mn ∼ 3 kDa, these results suggest that, similar to the homocrystals, 1L/1D forms extended chain crystals with the residual lengths of the polymer chains existing in the amorphous layers (Fig. 2a). In the case of 2L/2D (lc = 5.4 nm), the polymer chains form the flattened conformation shown in Fig. 2b, and thus lc became nearly half of that of 1L/1D (lc = 10 nm).30 In the case of the linear/cyclic blend (1L/2D), the L and lc values were in between the linear/linear and cyclic/cyclic blends.
27 The direction of the thickness of a polylactide stereocomplex is the same as the c axis of the unit cell, which is 0.87 nm consisting of three lactic acid units.18,31,32 Thus, based on the above-mentioned lc, the number of lactic acid units and the molecular weights in the crystal layers were 1L/1D, 34 units for Mn = 2.5 kDa; 1L/2D, 25 units for Mn = 1.8 kDa, and 2L/2D, 17 units for Mn = 1.3 kDa (Table S1†). Given that Mn ∼ 3 kDa, these results suggest that, similar to the homocrystals, 1L/1D forms extended chain crystals with the residual lengths of the polymer chains existing in the amorphous layers (Fig. 2a). In the case of 2L/2D (lc = 5.4 nm), the polymer chains form the flattened conformation shown in Fig. 2b, and thus lc became nearly half of that of 1L/1D (lc = 10 nm).30 In the case of the linear/cyclic blend (1L/2D), the L and lc values were in between the linear/linear and cyclic/cyclic blends.
The newly synthesized photocleavable linear and cyclic PLLA and PDLA (4L, 4D, 5L, and 5D) were subsequently subjected to the WAXD and SAXS measurements. Although 4L, 4D, 5L, and 5D possess the photocleavable linker, the crystallographic properties of these polylactides were relevant to those without the photocleavable linker (1L, 1D, 2L, and 2D). The WAXD showed diffraction peaks at 2θ = 15, 17, and 19° that are ascribed to the α- or α′-forms of polylactide homocrystals (Fig. 3).26 The χc values of these samples were estimated from the profiles (4L, 62%; 4D, 60%; 5L, 58%; 5D, 56%) as in Table 1. The SAXS profiles are shown in Fig. 4, and L, lc, and la were determined: 4L (L = 13 nm, lc = 9.3 nm, la = 3.6 nm), 4D (L = 13 nm, lc = 8.6 nm, la = 4.0 nm), 5L (L = 7.5 nm, lc = 4.8 nm, la = 2.7 nm), and 5D (L = 8.1 nm, lc = 5.3 nm, la = 2.8 nm). Therefore, L and lc of cyclic 5L and 5D were considerably smaller than linear 4L and 4D (Fig. 2),27 similar to the polylactides without the photocleavable linker.30 The molecular weights of the polylactide units in the crystalline layers were also calculated (Table 1).
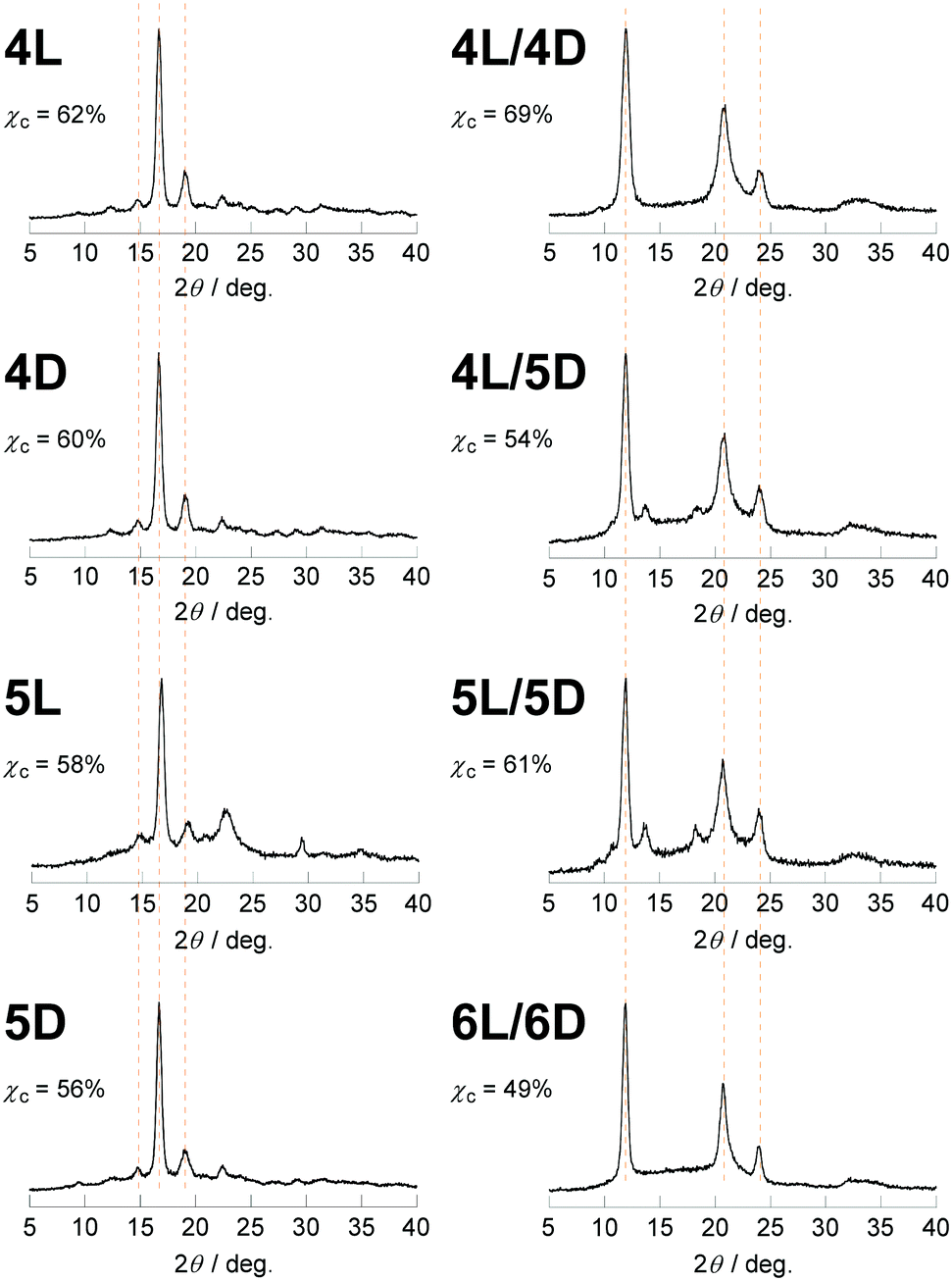 | ||
| Fig. 3 WAXD profiles of 4L, 4D, 5L, 5D, 4L/4D, 4L/5D, 5L/5D, and 6L/6D. | ||
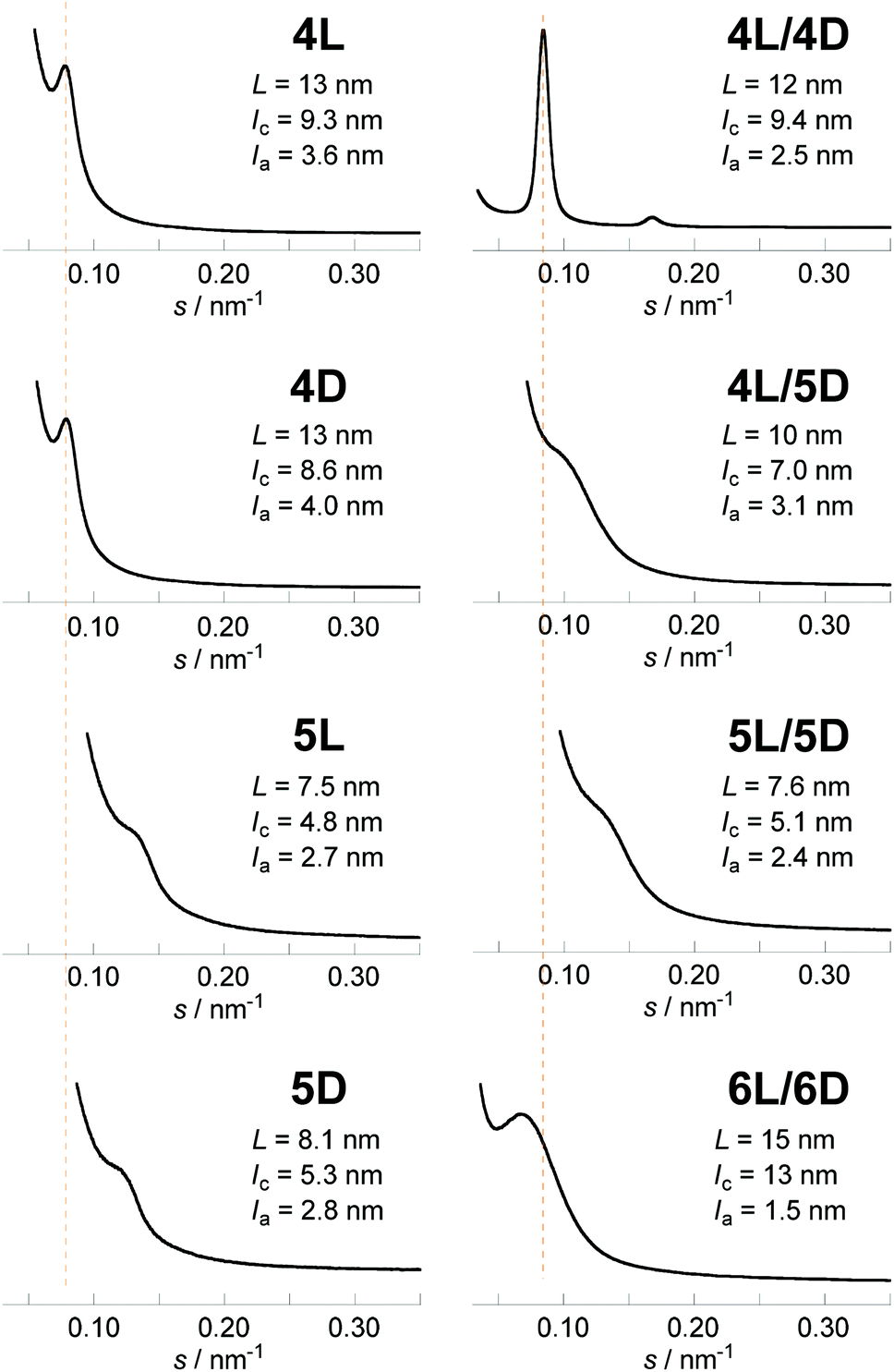 | ||
| Fig. 4 SAXS profiles of 4L, 4D, 5L, 5D, 4L/4D, 4L/5D, 5L/5D and 6L/6D. | ||
| Description | L (nm) | l c (nm) | M n for lc (kDa) | l a (nm) | χ c | T m (°C) | ΔHm (J g−1) | ΔHm(100%) (J g−1) | σ e (mJ m−2) | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Values reported in H. Tsuji, K. Ikarashi, N. Fukuda, Polym. Degrad. Stab., 2004, 84, 515–523.36 b Values calculated based on those reported in H. Tsuji, K. Ikarashi, N. Fukuda, Polym. Degrad. Stab., 2004, 84, 515–523.36 c Values reported in H. Tsuji, F. Horii, M. Nakagawa, Y. Ikada, H. Odani, R. Kitamaru, Macromolecules, 1992, 25, 4114–4118.34 d Values calculated based on those reported in H. Tsuji, F. Horii, M. Nakagawa, Y. Ikada, H. Odani, R. Kitamaru, Macromolecules, 1992, 25, 4114–4118.34 | ||||||||||
| 4L | Photocleavable linear PLLA (Mn ∼ 3 kDa) | 13 | 9.3 | 2.3 | 3.6 | 62% | 142 | 37 | 60 | 54 |
| 4D | Photocleavable linear PDLA (Mn ∼ 3 kDa) | 13 | 8.6 | 2.1 | 4.0 | 60% | 142 | 41 | 68 | 55 |
| 5L | Photocleavable cyclic PLLA (Mn ∼ 3 kDa) | 7.5 | 4.8 | 1.2 | 2.7 | 58% | 128 | 26 | 44 | 24 |
| 5D | Photocleavable cyclic PDLA (Mn ∼ 3 kDa) | 8.1 | 5.3 | 1.3 | 2.8 | 56% | 125 | 25 | 44 | 28 |
| ref. | Linear PLLA (Mn(PS) = 360–450 kDaa) | 22.2–35.1a | 7.6–19.6a | 1.9–4.9b | 11.9–20.6a | 34.2%–55.8%a | 176.5–190.1a | 46.2–55.8a | 100,a 135a | 52–64b |
| 4L/4D | Blend of 4L and 4D | 12 | 9.4 | 2.3 | 2.5 | 69% | 209 | 60 | 87 | 66 |
| 4L/5D | Blend of 4L and 5D | 10 | 7.0 | 1.7 | 3.1 | 54% | 173 | 22 | 41 | 35 |
| 5L/5D | Blend of 5L and 5D | 7.6 | 5.1 | 1.3 | 2.4 | 61% | 167 | 26 | 43 | 28 |
| 6L/6D | Photoirradiated 5L/5D | 15 | 13 | 3.2 | 1.5 | 49% | 211 | 44 | 89 | 91 |
| ref. | Blend of linear PLLA (Mv = 27 kDa) and PDLA (Mv = 25 kDa)c | 12.0c | 8.2c | 2.0d | 3.8c | 70%c | 231c | 102c | 146d | 66d |
The WAXD peaks at 2θ = 12, 21, and 24°, attributing to a stereocomplex, were observed for 4L/4D, 4L/5D, and 5L/5D (Fig. 3).26 Additional diffractions at 2θ = 13 and 18° were detected for the stereocomplexes involving the cyclic polylactides (4L/5D and 5L/5D). We assume that the photocleavable linker in the cyclic polymer that is incorporated in the crystalline layer caused these diffractions (Fig. 2b). In contrast, the photocleavable linker in the linear polymers, which was attached at the chain end, was presumably in the amorphous layer (Fig. 2a). Based on the WAXD profiles, the χc values of the stereocomplexes were estimated as shown in Table 1 (4L/4D, 69%; 4L/5D, 54%; 5L/5D, 61%). In this regard, the linear/linear blends showed several percent higher χc values than the cyclic/cyclic blends for both photocleavable (4L/4D, 69%; 5L/5D, 61%) and non-photocleavable (1L/1D, 79%; 2L/2D, 69%) polylactides. The χc value for the linear/cyclic blends was the lowest (4L/5D, 54%; 1L/2D, 62%) likely due to the difficulty of crystallization arising from the unmatched topologies of the enantiomeric pairs. By comparing the lamellar structures estimated from the SAXS profiles of 4L/4D (L = 12 nm, lc = 9.4 nm, la = 2.5 nm) and 5L/5D (L = 7.6 nm, lc = 5.1 nm, la = 2.4 nm) (Table 1), a significant decrease of L and lc due to the cyclization was revealed. Therefore, the polylactides possessing a photocleavable linker (4L, 4D, 5L, and 5D) and their blends have similar effects on the crystallographic properties via cyclization30 as the polylactides without a photocleavable linker (1L, 1D, 2L, and 2D) and their blends.
Lastly, the photoirradiated PLLA/PDLA blend (6L/6D) was also subjected to the WAXD and SAXS measurements. The crystal form was unchanged; the diffraction peaks at 2θ = 12, 21, and 24° in the WAXD profile (Fig. 3) were ascribed to the stereocomplex. The χc value was estimated to be 49% (Table 1). The additional peaks at 2θ = 13 and 18°, which were found in cyclic/cyclic 5L/5D before the photocleavage as well as in linear/cyclic 4L/5D, disappeared indicating that these diffractions were indeed from the photocleavable units in the crystalline layers. Despite the same crystal form determined by WAXD, SAXS revealed an increase of L and lc upon photoirradiation. The estimated values for 6L/6D (L = 15 nm, lc = 13 nm) were more than double in comparison with those of 5L/5D (L = 7.6 nm, lc = 5.1 nm) (Table 1), indicating that the cyclic polymers were photocleaved to likely form extended chain crystals (Fig. 2). When compared with 4L/4D (L = 12 nm, lc = 9.4 nm), those for 6L/6D were found to be significantly larger. This is probably because some degree of dimerization through the nitroso–nitroso association occurred (Fig. S6†).25 The number of lactic acid units and their molecular weight in the crystal layers were 45 units and Mn = 3.2 kDa, respectively. Furthermore, la for both homocrystals and stereocomplexes was relatively large for the polylactides containing the photocleavable linker (4L, 4D, 5L, 5D, 4L/4D, 4L/5D, and 5L/5D) compared with those without the photocleavable linker including the one after the photocleavage (1L, 1D, 2L, 2D, 1L/1D, 1L/2D, 2L/2D and 6L/6D), as shown in Tables 1 and S1.† These results were likely due to the volume of the photocleavable linkers in the amorphous layer. Moreover, the SAXS peak of photocleaved 6L/6D was somewhat sharpened in comparison with that of cyclic 5L/5D (Fig. 4), suggesting that the order of the crystal lattice is restored to some extent after the photocleavage.
Melting points (Tm)
The Tm values of the non-photocleavable linear and cyclic polylactides were previously reported.16 In the present study, those of the linear and cyclic polylactides with a photocleavable linker and the photocleaved stereocomplex were measured by DSC to investigate the effects of the topological conversion. The homocrystals of linear 4L and 4D had a Tm of 142 °C, whereas cyclic 5L and 5D showed a lowered Tm of 128 and 125 °C, respectively (Table 1 and Fig. 5).15 The blend of linear 4L/4D showed a Tm of 209 °C, which was higher than the respective homopolymers due to typical stereocomplexation.15,33 In contrast, the blends involving a cyclic polylactide showed relatively low Tm values: 4L/5D, 173 °C; 5L/5D, 167 °C. Notably, the effect of cyclization was more prominent for the stereocomplexes (Tm(5L/5D) − Tm(4L/4D) = −42 °C) than for the homocrystals (Tm(5L) − Tm(4L) = −14 °C; Tm(5D) − Tm(4D) = −17 °C). After the photocleavage, Tm of 6L/6D increased to 211 °C, which was more than 40 °C higher than that of the cyclized 5L/5D (167 °C) and comparable to that before the cyclization (4L/4D, 209 °C). These results suggest that Tm of the stereocomplexes of the present polylactides is switchable by approximately 40 °C through controlling their polymer topology.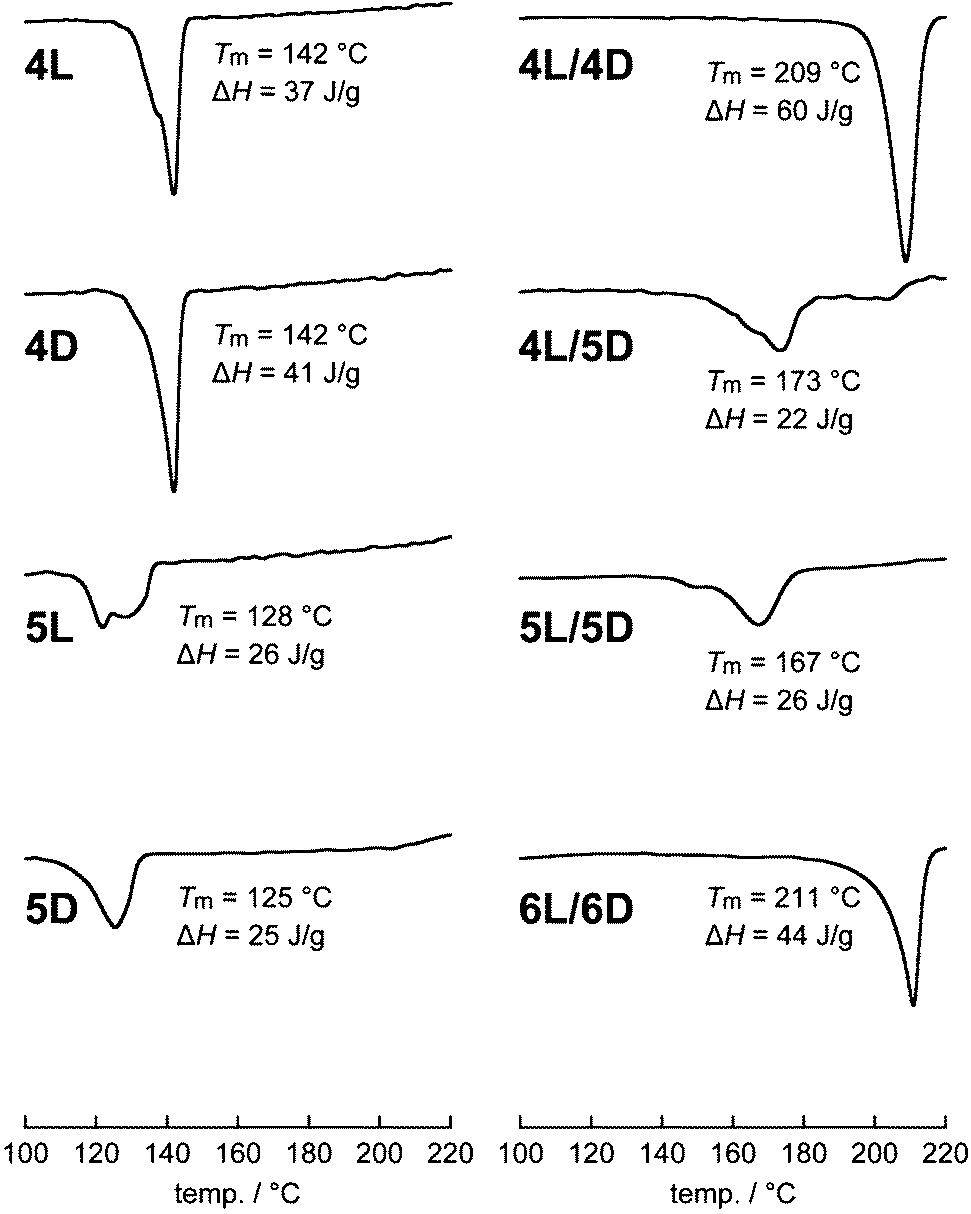 | ||
| Fig. 5 DSC thermograms of 4L, 4D, 5L, 5D, 4L/4D, 4L/5D, 5L/5D, and 6L/6D. | ||
Enthalpies of melting (ΔHm, ΔHm(100%))
The ΔHm values were calculated from the DSC thermograms (Fig. 5) and converted to those of crystals having an infinite thickness, i.e., ΔHm(100%), based on the crystallinity determined by WAXD (Fig. 3).34 Interestingly, we found that the homocrystals consisting of a cyclic polymer (5L and 5D) have significantly smaller ΔHm(100%) (44 J g−1) than those of the linear counterparts (4L, 60 J g−1; 4D, 68 J g−1) as shown in Table 1. This difference presumably arose from the absolute enthalpy of the respective melt states rather than those of the crystalline states because the line widths of the WAXD peaks, thus crystallite size and lattice distortion, were comparable (Fig. 3). In other words, the absolute enthalpy of cyclic 5L and 5D in the melt state is expected to be lower by roughly 20 J g−1 than that of linear 5L and 5D due to the topology. In this regard, linear polymers are relatively easy to entangle in the melt state, possibly leading to a higher absolute enthalpy, while cyclic polymers are less likely.35 Moreover, the ΔHm(100%) values of the present polylactides (4L, 60 J g−1; 4D, 68 J g−1; 5L, 44 J g−1; 5D, 44 J g−1) were much smaller than those used for PLLA with high molecular weights (100 and 135 J g−1 for linear PLLA (Mn(PS) = 360–450 kDa), where Mn(PS) indicates that Mn determined by SEC calibrated using standard PS) as shown in Table 1.36 Presumably, this was also caused by the high degree of entanglement in the melt state due to the length of the polymers. The trend for the ΔHm(100%) values of the stereocomplexes is parallel to that of the homocrystals; the cyclic/cyclic blend (5L/5D, 43 J g−1) as well as the linear/cyclic blend (4L/5D, 41 J g−1) has notably smaller ΔHm(100%) than the linear/linear blend (4L/4D, 87 J g−1) and that after the photocleavage (6L/6D, 89 J g−1). In addition, the ΔHm(100%) value for a stereocomplex calculated based on the previous report using longer PLLA (Mv = 27 kDa) and PDLA (Mv = 25 kDa)34 was also considerably larger (146 J g−1).Furthermore, photocleavable linear PLLA (4L′), linear PDLA (4D′), cyclic PLLA (5L′), and cyclic PDLA (5D′) with a molecular weight of approximately 1.5 kDa were also synthesized and subjected to DSC measurements in order to study the molecular weight dependence. No Tm was found for any of these individual polylactides likely due to the insufficient length for crystallization. However, the blends of linear/linear 4L′/4D′, cyclic/cyclic 5L′/5D′, and photocleaved 6L′/6D′ had Tm of 138, 127, and 155 °C with ΔHm of 29, 6, and 16 J g−1, respectively. This trend concerning the polymer topology is the same for the polylactides with a molecular weight of approximately 3 kDa, but a lower Tm and smaller ΔHm were likely due to the smaller molecular weight.
Surface free energy (σe)
σ e of the interface between the crystal and amorphous was calculated by the Gibbs–Thomson equation (1) using the reported values of the densities (homocrystal, 1.285 g cm−3;37 stereocomplex, 1.27 g cm−3)32,38 and equilibrium melting points, T°m (homocrystal, 215 °C;39 stereocomplex, 279 °C).40| Tm(lc) = T°m × [1 − 2σe/(ΔHm(100%) × lc)] | (1) |
The σe value for linear 4L (54 mJ m−2) and 4D (55 mJ m−2) was close to that of high-molecular-weight linear PLLA (Mn(PS) = 360–450 kDa36 (52–64 mJ m−2)), which has the chain-folding surfaces of the crystalline layers. On the other hand, σe for cyclic 5L (24 mJ m−2) and 5D (28 mJ m−2) was significantly smaller. This suggests that cyclic 5L and 5D form a well-ordered looped structure at the crystal surfaces (Fig. 2b) to help stabilize the crystalline–amorphous interface compared to linear 4L and 4D having chain ends (Fig. 2a). On the other hand, the folding segments of high-molecular-weight PLLA were probably not as organized as those of cyclic 5L and 5D, resulting in high σe values (52–64 mJ m−2). The less ordered folding segments of high-molecular-weight PLLA are also suggested by the thicker amorphous layers (la = 11.9–20.6 nm in Table 1). The higher stability of the interface was also determined for the stereocomplex of cyclic 5L/5D (28 mJ m−2) compared to linear 4L/4D (66 mJ m−2) and the reported blend of linear PLLA (Mv = 27 kDa) and PDLA (Mv = 25 kDa) (66 mJ m−2).34 The linear/cyclic blend of 4L/5D (35 mJ m−2) was comparable to cyclic/cyclic 5L/5D (28 mJ m−2). On the other hand, considerably higher σe was found for photocleaved 6L/6D (91 mJ m−2), probably caused by the non-uniform chemical structure of the photoreacted chain ends and/or the partial intermolecular coupling through the nitroso–nitroso association.
Interestingly, Müller and coworkers studied the crystallization of linear and cyclic poly(ε-caprolactone) and found higher Tm and larger σe for the cyclic polymers,41 in contrast to the present results of the polylactides. The different crystal systems were expected to cause the opposite topology effects from the respective polymers. For reference, they also reported higher spherulitic growth rates for cyclic poly(ε-caprolactone) than linear analogues.41,42 Conversely, cyclic poly(tetrahydrofuran)43 and cyclic polyethylene44 showed a slower rate than their linear counterparts. Presumably, the higher growth rate for cyclic poly(ε-caprolactone) was due to faster diffusion.45 On the other hand, the unfavorable entropic process governed the crystallization of cyclic poly(tetrahydrofuran) and cyclic polyethylene, resulting in slower rates. The elucidation of the relationship between the polymer topology and Tm and σe of these crystal systems is strongly desired and currently in progress.
Conclusions
Photocleavable cyclic PLLA and PDLA possessing a NB group were synthesized and subjected to a photocleavage reaction to transform the topology. The WAXD and SAXS measurements of the present polylactides, along with previously reported non-photocleavable polylactides, were performed. Despite WAXD indicating that the essentially crystallographically identical homocrystals and stereocomplexes are formed by the linear and cyclic polylactides, SAXS of the cyclic polylactides showed that L and lc are roughly half of the linear counterparts due to the flattened ring conformation. Moreover, the photocleaved PLLA/PDLA blend showed L and lc values comparable to those before the cyclization. DSC showed that Tm of the stereocomplex reflects lc; more than 40 °C of Tm was controlled through the linear–cyclic topological transformation. The combination of WAXD, SAXS, and DSC revealed that ΔHm(100%) is smaller for the homocrystals and stereocomplex of the cyclic polylactides than the respective linear counterparts. Furthermore, the interface between the crystalline and amorphous phases was significantly more stable for the cyclic polylactides, shown by smaller σe.The present results provide new insights into the structure and properties of crystalline polymeric materials. Novel designs based on the cyclic polymer topology and simple photoirradiation would lead to versatile control on the properties such as Tm, ΔHm(100%), and σe, which cannot be achieved by traditional means. Moreover, a photocleavage reaction to increase Tm by approximately 40 °C is appropriate for the efficient processing of polylactide materials. Altogether, the present methodology is expected to lead to the development of novel topology-based stimuli-responsive materials, which require only one reaction per polymer molecule and allow for switching the properties without altering the chemical structure and molecular weight of the polymer.
Experimental section
Materials
Unless otherwise noted, all commercial reagents were used as received. 3-Buten-1-ol (>98.0%, Tokyo Chemical Industry Co., Ltd) was distilled prior to use. L-Lactide (>99%, Musashino Chemical Laboratory, Ltd) and D-lactide (>99%, Musashino Chemical Laboratory, Ltd) were recrystallized from dry toluene twice prior to use. According to the previously reported procedure,11 1-(4,5-dimethoxy-2-nitro-phenyl)-but-3-ene-1-ol was prepared. THF (>99.0%, Kanto Chemical Co., Inc.) was distilled over a Na wire. Toluene (>99%, Godo Co., Inc.) was distilled over CaH2. For column chromatography, Wakosil C-300 (Wako Pure Chemical Industries, Ltd) was used. SiliaMetS® DMT (SiliCycle Inc.) was used to remove the residue of the second-generation Hoveyda–Grubbs catalyst. Synthesis was repeated several times for some polymers, resulting in different (although slight) Mn, Mp, and PDI values for such polymers.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 3.98 (t, 6H, J = 7.7, Ar–OCH3), 4.35 (q, 1H, J = 6.8 Hz, –CH(CH3)OH), 5.06–5.36 (m, –CO2CH(CH3)–, –CHCH2), 5.76–5.92 (m, 1H, –CHCH2), 6.45–6.51 (m, 1H, Ar–CH–), 6.94 (d, 1H, J = 24.6 Hz, Ar–H ortho to N), 7.64 (d, 1H, J = 15.0 Hz, Ar–H meta to N).
CHCH2CH–, CH2CHCH2CH2CO2–), 3.98 (t, 6H, J = 7.7, Ar–OCH3), 5.06–5.37 (m, –CO2CH(CH3)–, –CHCH2), 5.76–5.90 (m, 1H, –CHCH2), 6.45–6.51 (m, 2H, Ar–CH–), 6.94 (d, 1H, J = 24.9 Hz, Ar–H ortho to N), 7.64 (d, 1H, J = 14.4 Hz, Ar–H meta to N).
CHCH2–), 3.97 (t, 6H, J = 4.2, Ar–OCH3), 4.98–5.33 (m, –CO2CH(CH3)–), 5.48–5.56 (s, 2H, –CHCH–), 6.38–6.46 (m, 1H, Ar–CH–), 6.91 (d, 1H, J = 23.1 Hz, Ar–H ortho to N), 7.63 (d, 1H, J = 14.4 Hz, Ar–H meta to N).
CH2), 3.98 (t, 6H, J = 7.7, Ar–OCH3), 4.35 (q, 1H, J = 6.8 Hz, –CH(CH3)OH), 5.06–5.36 (m, –CO2CH(CH3)–, –CHCH2), 5.76–5.92 (m, 1H, –CHCH2), 6.45–6.51 (m, 1H, Ar–CH–), 6.94 (d, 1H, J = 24.6 Hz, Ar–H ortho to N), 7.64 (d, 1H, J = 15.0 Hz, Ar–H meta to N).
CHCH2CH–, CH2CHCH2CH2CO2–), 3.98 (t, 6H, J = 7.7, Ar–OCH3), 5.06–5.37 (m, –CO2CH(CH3)–, –CHCH2), 5.76–5.90 (m, 1H, –CHCH2), 6.45–6.51 (m, 2H, Ar–CH–), 6.94 (d, 1H, J = 24.9 Hz, Ar–H ortho to N), 7.64 (d, 1H, J = 14.4 Hz, Ar–H meta to N).
CHCH2–), 3.97 (t, 6H, J = 4.2, Ar–OCH3), 4.98–5.33 (m, –CO2CH(CH3)–), 5.48–5.56 (s, 2H, –CHCH–), 6.38–6.46 (m, 1H, Ar–CH–), 6.91 (d, 1H, J = 23.1 Hz, Ar–H ortho to N), 7.63 (d, 1H, J = 14.4 Hz, Ar–H meta to N).
Photocleavage reaction
Photocleavable polylactides (3L, 3D, 5L, 5D, or 5L/5D) were dissolved in CH2Cl2, CHCl3, THF, toluene, CH3CN, 1,2-dichloroethane, or EtOAc. To the solution was added butylated hydroxy toluene (BHT, 0.05%) where applicable. The solution was photoirradiated at 365 nm using an Asahi Spectra POT-365 light source to form 7L, 7D, 6L, 6D, or 6L/6D, respectively.Measurements
1H NMR spectra were recorded at ca. 20 °C on a JEOL JNM-AL300 spectrometer operating at 300 MHz. CDCl3 was used as a solvent, and the proton chemical shifts were reported relative to the signal of tetramethylsilane. SEC measurements were performed at 40 °C on a Tosoh model CCPS equipped with a TSK G3000HXL column and a refractive index detector model RI 8020. CHCl3 was used as an eluent at a flow rate of 1.0 mL min−1. Linear polystyrene standards were used for calibration, and Mp of the sample was reported as a linear polystyrene equivalent. MALDI-TOF mass spectra were recorded on a Shimadzu AXIMA Performance spectrometer equipped with a nitrogen laser (λ = 337 nm). The spectrometer was operated at an accelerating potential of 20 kV in linear positive ion mode with pulsed ion extraction. A CH2Cl2 solution (3.0 μL) of a polymer sample (1.0 mg mL−1), an acetone solution (3.0 μL) of trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propylidene]malonitrile (DCTB) (10 mg mL−1), and an acetone solution (3.0 μL) of sodium trifluoroacetate (10 mg mL−1) were deposited onto a sample target plate in sequence. Mass values were calibrated using the four-point method using peaks from SpheriCal®, monodisperse dendritic calibration standards that had m/z = 1715.68, 2255.88, 2796.08, and 3422.35 for polymers in the molecular weight range of 1200–3800 and m/z = 3636.44, 4816.89, 5997.34, and 7263.87 for polymers in the molecular weight range of 3400–8000. SEC fractionation was performed on a JAI LC-908 equipped with JAIGEL-2H and JAIGEL-3H columns, and CHCl3 was used as an eluent at a flow rate of 3.5 mL min−1. WAXD measurements were performed on a Rigaku RINT-2100 spectrometer with CuKα radiation (λ = 0.15418 nm) operating at 40 kV and 40 mA. The optical system with a pinhole geometry consisted of a graphite monochromator and scintillation counter. The intensity data were collected in the 2θ range between 3 and 60°. SAXS measurements were conducted on a Rigaku NANO-Viewer system with CuKα radiation (λ = 0.15418 nm) operating at 40 kV and 20 mA. The SAXS intensity measured by pin-hole collimation was corrected for parasitic scattering. The intensity data were collected in the 2θ range between 0.1 and 2.5°. A powder sample was filled in the center space of a 0.8 mm-thick stainless folder and sealed both faces with amorphous polyethylene thin films of less than 10 μm thickness. DSC measurements were performed on a Seiko DSC 6200, and all samples were cast from CH2Cl2 solutions. Thermograms were recorded during the first heating cycle at a rate of 5 or 10 °C min−1 under a nitrogen gas flow of 50 mL min−1.Acknowledgements
The authors are grateful to Prof. M. Kakimoto and Prof. T. Hayakawa for access to measurement facilities. This work was supported by Grant-in-Aid for JSPS Fellows (239581, N.S.), Grants for Excellent Graduate Schools (N.S.), and KAKENHI (26288099, T.Y.).References
- (a) M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed; (b) J. F. Mano, Adv. Eng. Mater., 2008, 10, 515–527 CrossRef CAS; (c) X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- Y. N. Xia and G. M. Whitesides, Annu. Rev. Mater. Sci., 1998, 28, 153–184 CrossRef CAS.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- M. Huo, J. Yuan, L. Tao and Y. Wei, Polym. Chem., 2014, 5, 1519–1528 RSC.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- (a) S. Honda, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2010, 132, 10251–10253 CrossRef CAS PubMed; (b) S. Honda, T. Yamamoto and Y. Tezuka, Nat. Commun., 2013, 4, 1574 CrossRef PubMed.
- M. R. Whittaker, Y.-K. Goh, H. Gemici, T. M. Legge, S. Perrier and M. J. Monteiro, Macromolecules, 2006, 39, 9028–9034 CrossRef CAS.
- (a) O. Altintas, P. Gerstel, N. Dingenouts and C. Barner-Kowollik, Chem. Commun., 2010, 46, 6291–6293 RSC; (b) O. Altintas, E. Lejeune, P. Gerstel and C. Barner-Kowollik, Polym. Chem., 2012, 3, 640–651 RSC.
- (a) M. Schappacher and A. Deffieux, J. Am. Chem. Soc., 2011, 133, 1630–1633 CrossRef CAS PubMed; (b) M. Schappacher and A. Deffieux, Macromolecules, 2011, 44, 4503–4510 CrossRef CAS.
- S. Yamazoe, I. A. Shestopalov, E. Provost, S. D. Leach and J. K. Chen, Angew. Chem., Int. Ed., 2012, 51, 6908–6911 CrossRef CAS PubMed.
- Y. Wang, L. Wu, P. Wang, C. Lv, Z. Yang and X. Tang, Nucleic Acids Res., 2012, 40, 11155–11162 CrossRef CAS PubMed.
- (a) L. Wu, Y. Wang, J. Wu, C. Lv, J. Wang and X. Tang, Nucleic Acids Res., 2013, 41, 677–686 CrossRef CAS PubMed; (b) J. C. Griepenburg, B. K. Ruble and I. J. Dmochowski, Bioorg. Med. Chem., 2013, 21, 6198–6204 CrossRef CAS PubMed.
- (a) K. Fukushima and Y. Kimura, Polym. Int., 2006, 55, 626–642 CrossRef CAS; (b) M. Kakuta, M. Hirata and Y. Kimura, Polym. Rev., 2009, 49, 107–140 CrossRef CAS; (c) L. Yu, K. Dean and L. Li, Prog. Polym. Sci., 2006, 31, 576–602 CrossRef CAS PubMed.
- H. Tsuji, Macromol. Biosci., 2005, 5, 569–597 CrossRef CAS PubMed.
- N. Sugai, T. Yamamoto and Y. Tezuka, ACS Macro Lett., 2012, 1, 902–906 CrossRef CAS.
- E. J. Shin, A. E. Jones and R. M. Waymouth, Macromolecules, 2012, 45, 595–598 CrossRef CAS.
- H. Marubayashi, T. Nobuoka, S. Iwamoto, A. Takemura and T. Iwata, ACS Macro Lett., 2013, 2, 355–360 CrossRef CAS.
- (a) C. G. Bochet, J. Chem. Soc., Perkin Trans. 1, 2002, 125–142 CAS; (b) H. Zhao, E. S. Sterner, E. B. Coughlin and P. Theato, Macromolecules, 2012, 45, 1723–1736 CrossRef CAS.
- H. D. Keith, in Physics and Chemistry of the Organic Solid State, ed. D. Fox, M. M. Labes and A. Weissberger, Interscience Publishers, New York, 1963, vol. 1, pp. 462–542 Search PubMed.
- S. J. Huang and J. M. Onyari, J. Macromol. Sci., Part A: Pure Appl. Chem., 1996, 33, 571–584 CrossRef.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed.
- M. M. Stamenović, P. Espeel, E. Baba, T. Yamamoto, Y. Tezuka and F. E. Du Prez, Polym. Chem., 2013, 4, 184–193 RSC.
- K. G. Orrell, V. Šik and D. Stephenson, Magn. Reson. Chem., 1987, 25, 1007–1011 CrossRef CAS.
- (a) R. R. Holmes, R. P. Bayer, L. A. Errede, H. R. Davis, A. W. Wiesenfeld, P. M. Bergman and D. L. Nicholas, J. Org. Chem., 1965, 30, 3837–3840 CrossRef CAS; (b) J. Li and Q. Wang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 810–815 CrossRef CAS.
- (a) L. Bouapao, H. Tsuji, K. Tashiro, J. Zhang and M. Hanesaka, Polymer, 2009, 50, 4007–4017 CrossRef CAS PubMed; (b) H. Tsuji, T. Wada, Y. Sakamoto and Y. Sugiura, Polymer, 2010, 51, 4937–4947 CrossRef CAS PubMed.
- (a) L. B. Li, Z. Y. Zhong, W. H. de Jeu, P. J. Dijkstra and J. Feijen, Macromolecules, 2004, 37, 8641–8646 CrossRef CAS; (b) M. Fujita, T. Sawayanagi, H. Abe, T. Tanaka, T. Iwata, K. Ito, T. Fujisawa and M. Maeda, Macromolecules, 2008, 41, 2852–2858 CrossRef CAS; (c) L. Chang and E. M. Woo, Macromol. Chem. Phys., 2011, 212, 125–133 CrossRef CAS.
- (a) W. Hoogsteen, A. R. Postema, A. J. Pennings, G. ten Brinke and P. Zugenmaier, Macromolecules, 1990, 23, 634–642 CrossRef CAS; (b) K. Wasanasuk and K. Tashiro, Polymer, 2011, 52, 6097–6109 CrossRef CAS PubMed.
- H. Takeshita, M. Poovarodom, T. Kiya, F. Arai, K. Takenaka, M. Miya and T. Shiomi, Polymer, 2012, 53, 5375–5384 CrossRef CAS PubMed.
- G.-E. Yu, T. Sun, Z.-G. Yan, C. Price, C. Booth, J. Cook, A. J. Ryan and K. Viras, Polymer, 1997, 38, 35–42 CrossRef CAS.
- L. Cartier, T. Okihara and B. Lotz, Macromolecules, 1997, 30, 6313–6322 CrossRef CAS.
- D. Sawai, Y. Tsugane, M. Tamada, T. Kanamoto, M. Sungil and S. H. Hyon, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 2632–2639 CrossRef CAS.
- Y. Ikada, K. Jamshidi, H. Tsuji and S. H. Hyon, Macromolecules, 1987, 20, 904–906 CrossRef CAS.
- H. Tsuji, F. Horii, M. Nakagawa, Y. Ikada, H. Odani and R. Kitamaru, Macromolecules, 1992, 25, 4114–4118 CrossRef CAS.
- M. Kapnistos, M. Lang, D. Vlassopoulos, W. Pyckhout-Hintzen, D. Richter, D. Cho, T. Chang and M. Rubinstein, Nat. Mater., 2008, 7, 997–1002 CrossRef CAS.
- H. Tsuji, K. Ikarashi and N. Fukuda, Polym. Degrad. Stab., 2004, 84, 515–523 CrossRef CAS PubMed.
- T. Miyata and T. Masuko, Polymer, 1997, 38, 4003–4009 CrossRef CAS.
- (a) J. Kobayashi, T. Asahi, M. Ichiki, A. Oikawa, H. Suzuki, T. Watanabe, E. Fukada and Y. Shikinami, J. Appl. Phys., 1995, 77, 2957–2973 CrossRef CAS PubMed; (b) J. Puiggali, Y. Ikada, H. Tsuji, L. Cartier, T. Okihara and B. Lotz, Polymer, 2000, 41, 8921–8930 CrossRef CAS.
- B. Kalb and A. J. Pennings, Polymer, 1980, 21, 607–612 CrossRef CAS.
- H. Tsuji and Y. Ikada, Macromol. Chem. Phys., 1996, 197, 3483–3499 CrossRef CAS.
- H.-H. Su, H.-L. Chen, A. Díaz, M. T. Casas, J. Puiggalí, J. N. Hoskins, S. M. Grayson, R. A. Pérez and A. J. Müller, Polymer, 2013, 54, 846–859 CrossRef CAS PubMed.
- (a) R. A. Pérez, M. E. Córdova, J. V. López, J. N. Hoskins, B. Zhang, S. M. Grayson and A. J. Müller, React. Funct. Polym., 2014, 80, 71–82 CrossRef PubMed; (b) M. E. Córdova, A. T. Lorenzo, A. J. Müller, J. N. Hoskins and S. M. Grayson, Macromolecules, 2011, 44, 1742–1746 CrossRef.
- Y. Tezuka, T. Ohtsuka, K. Adachi, R. Komiya, N. Ohno and N. Okui, Macromol. Rapid Commun., 2008, 29, 1237–1241 CrossRef CAS.
- T. Kitahara, S. Yamazaki and K. Kimura, Kobunshi Ronbunshu, 2011, 68, 694–701 CrossRef CAS.
- H. Takeshita and T. Shiomi, in Topological Polymer Chemistry: Progress of Cyclic Polymers in Syntheses, Properties and Functions, ed. Y. Tezuka, World Scientific, Singapore, 2013, pp. 317–328 Search PubMed.
- (a) J. Watanabe, T. Eriguchi and K. Ishihara, Biomacromolecules, 2002, 3, 1109–1114 CrossRef CAS PubMed; (b) H. Shinoda and K. Matyjaszewski, Macromolecules, 2001, 34, 6243–6248 CrossRef CAS.
- T. Yamamoto, T. Fukushima, Y. Yamamoto, A. Kosaka, W. Jin, N. Ishii and T. Aida, J. Am. Chem. Soc., 2006, 128, 14337–14340 CrossRef CAS PubMed.
- Y. Tezuka and R. Komiya, Macromolecules, 2002, 35, 8667–8669 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S9 and Table S1. See DOI: 10.1039/c5py00158g |
| This journal is © The Royal Society of Chemistry 2015 |