
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA convenient synthesis strategy for microphase-separating functional copolymers: the cyclohydrocarbosilane tool box†
V.
Rittscher
and
M.
Gallei
*
Ernst-Berl-Institut für Technische und Makromolekulare Chemie, Technische Universität Darmstadt, Alarich-Weiss-Str. 4, D-64287 Darmstadt, Germany. E-mail: m.gallei@mc.tu-darmstadt.de
First published on 17th February 2015
Abstract
A novel and feasible strategy for the preparation of functional polyhydrocarbosilane-based homo and block copolymers is described via a combination of anionic ring-opening polymerization of methylsilacyclobutane (MSB) and styrene followed by postmodification applying hydrosilylation protocols. Sterically demanding and functional vinyl compounds such as N-vinylcarbazole (NVC), vinylferrocene (VFc), and 9,10-di(1-naphthalenyl)-2-vinylanthracene (VADN) are block-selectively incorporated into the PMSB backbone with adjustable functional group contents between 26 mol% to 43 mol% with respect to the Si–H functionalities for the PMSB homopolymers and contents ranging from 28 mol% to 63 mol% for the corresponding diblock copolymers as evidenced by 1H NMR spectroscopy. The novel functional diblock copolymers are capable of undergoing microphase separation as shown via TEM.
1 Introduction
Block copolymers which consist of two or more polymer segments covalently connected to each other have attracted enormous attention in the recent past due to their unique capability of undergoing microphase separation in the bulk or self-organization in (selective) solvents.1–6 As a result, various structures such as spheres, cylinders, lamellae and co-continuous structures as well as nanoscale porous structures can be obtained.7,8 Therefore, applications of block copolymers are manifold ranging, e.g. from fields of nanolithography, drug delivery to separation technologies.4 Furthermore, block copolymers and architectures thereof can consist of so-called stimuli-responsive polymers i.e., polymers which are capable of changing their conformation, solubility, or even break or form covalent bonds caused by different external triggers such as the change of temperature, pH value, light, redox reagents or electrical field.9–19 As another important and feasible route towards functional materials, block copolymer self-assembly strategies can be advantageously used for the controlled buildup of ceramic nanostructures obtained by pyrolysis of inorganic/organic block copolymers. There, the inorganic block segment – also referred to as preceramic block segment – is directly converted into the ceramic material after thermal treatment maintaining the block copolymer-templated nanostructure.20,21 Different approaches for the preparation of ceramic materials inclusively nanocomposites are known and excellent reviews within that field are given for example by Orilall and Wiesner22 and other authors.23–29All these above mentioned powerful applications based on block copolymers necessitates the precise synthesis of the underlying block copolymer architectures comprising adjustable block length and exact (block) polymer constitution. For this purpose, especially controlled or living polymerization protocols are applied.30–34 Although there is tremendous effort in terms of functional block copolymer synthesis in the last decades, there are still challenges to overcome in the field of living and controlled polymerization strategies for some functional monomers. Therefore, protocols for postmodification of homopolymers and block copolymers have been applied for many years in order to improve properties and to introduce certain functionalities. Powerful polymer postmodification methods for introducing pendant non-functional side groups or responsive moieties with additional functionalities into silane-, siloxane- and carbosilane-based polymers usually focus on hydrosilylation, hydroboration, and thiol–ene chemistry.35–43 There, especially the group of Frey succeeded in the formation of various functional polycarbosilane-based architectures by using convenient hydrosilylation protocols.44–47 Especially polycarbosilanes gained significant attention due to their outstanding potential to be converted into oxygen-free ceramic materials.48–50 More basic requirements for well-defined microphase separation of block copolymers are e.g., precise control over blocking efficiency and narrowly distributed polymer masses. In order to achieve these demands, especially anionic ring-opening polymerization routes starting from ring-strained cyclic carbosilane monomers has been studied by some groups.51–60 For example, Kim et al. reported the block copolymerization of 1,1-dimethylsilacyclobutane (DMSB) with polystyrene.61 The authors succeeded in preparing microphase-separated block copolymer structures which were converted into highly-ordered silicon-carbide ceramics after thermal treatment. As another example, polycarbosilane-based block copolymer micelles have been synthesized and converted into spherical SiC ceramics by pyrolysis.62
In this contribution polyhydrocarbosilane-containing block copolymers with polystyrene (PS) as second block segment are prepared by using sequential anionic polymerization protocols. For this purpose, the anionic ring-opening polymerization (ROP) of methylsilacyclobutane (MSB) is investigated in order to obtain hydrocarbosilane moieties capable of undergoing efficient subsequent hydrosilylation reaction. Moreover, block copolymers are prepared featuring reactive Si–H moieties in the polycarbosilane segment which are successfully postmodified with three different functional vinyl compounds i.e., N-vinylcarbazole (NVC), vinylferrocene (VFc) and 9,10-di(1-naphthalenyl)-2-vinylanthracene (VADN) (Fig. 1).
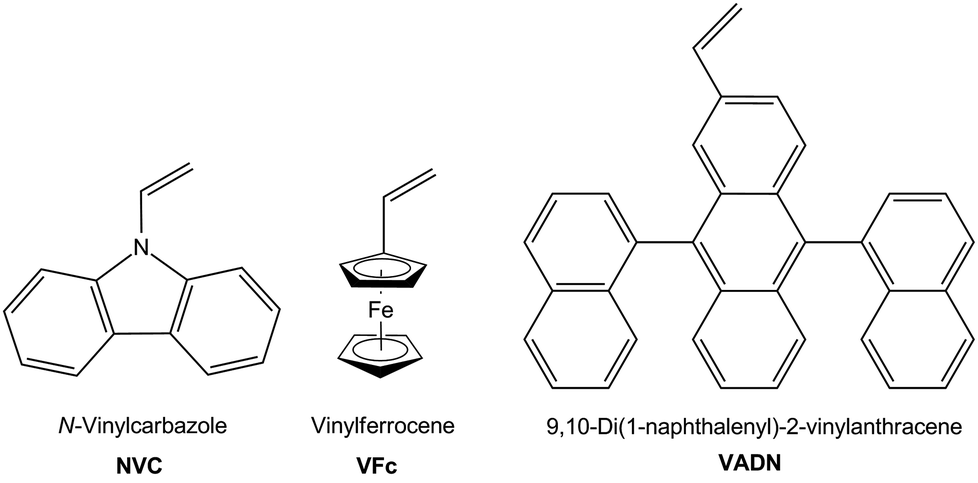 | ||
| Fig. 1 Vinyl compounds for intended postmodification of polyhydrocarbosilane-containing homo- and diblock copolymers. | ||
Although block copolymer formation of PNVC and PVFc were described in literature by applying controlled and living polymerization protocols, preparation of polymers with high molar masses, narrow molecular weight distribution, well-defined constitutions without the presence of side-products and significant yields are still challenging.58,63–66 Within the present work we show how these challenging vinyl compounds i.e., NVC, VFc, and VADN can be successfully incorporated into polyhydrocarbosilanes and corresponding block copolymers with polystyrene (PS). The novel functional polymers and block copolymers are investigated with respect to their molar masses, constitution, and thermal behaviour. Finally, polyhydrocarbosilane-containing block copolymers with functional moieties are shown to undergo microphase-separation. In the case of PMSB with pendant VFc groups it is shown that pyrolysis yields oxygen-free SiC/Fe-containing ceramic materials. We expect this postmodification route of polyhydrocarbosilane-based block copolymers by using hydrosilylation protocols as universal methodology for the controlled buildup of functional block copolymers which will be useful for a manifold of applications in the field of nanotechnology.
2 Experimental
2.1 Materials and methods
All solvents and reagents were purchased from Alfa Aesar, Sigma Aldrich, Fisher Scientific, ABCR and Merck and used as received unless otherwise stated. Platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex in xylene (2.1–2.4% Pt) and N-vinylcarbazole (NVC) were used as received. 1-Methylsilacyclobutane (MSB) and styrene were stirred over calcium hydride (CaH2), distilled and stored in a glove-box at −16 °C. MSB58 and VADN67 were synthesized as described elsewhere. Tetrahydrofuran (THF) and toluene were distilled from sodium/benzophenone under reduced pressure (cryo-transfer) prior to the addition of 1,1-diphenylethylene and n-butyllithium (n-BuLi) followed by a second cryo-transfer. Methylene chloride, diethylether, and ethanol were stirred over molecular sieves overnight, and degassed before use. Vinylferrocene (VFc) was synthesized and purified described elsewhere.66 Deuterated solvents were purchased from Deutero GmbH, Kastellaun, Germany. All syntheses were carried out under an atmosphere of nitrogen using Schlenk techniques or a glovebox equipped with a Coldwell apparatus.2.2 Preparation of PMSB
1H-NMR (CDCl3, 300 MHz, 300 K): δ = 3.827–3.716 (m, Si–H), 1.514–1.360 (m, 3-H2), 0.775–0.556 (m, 2-H4), 0.041 (d, 1-H3) ppm. J1-H, Si–H = 3.57 Hz.
13C-NMR (CDCl3, 75 MHz, 300 K): δ = 19.923 (3-C), 17.067 (2-C), −6.058 (1-C) ppm.
2.3 Preparation of polystyrene-block-polymethylsilacyclobutane
1H-NMR (CD2Cl2, 300 MHz, 300 K): δ = 7.382–6.886 (m, 5-H2, 6-H1), 6.880–6.295 (m, 4-H2), 3.931–3.628 (m, Si–H), 2.378–1.168 (m, 1-H2, 2-H1, 9-H2), 0.894–0.461 (m, 8-H4), 0.049 (d, 7-H3) ppm. J7-H, Si–H = 3.68 Hz.
13C-NMR (CD2Cl2, 75 MHz, 300 K): δ = 145.809 (3-C); 128.425, 128.035, 126.073 (4-C, 5-C, 6-C); 41.015 (2-C); 40.766 (1-C); 20.160 (9-C), 17.274 (8-C), −6.091 (7-C) ppm.
2.4 Hydrosilylation of PMSB with functional vinyl compounds
NVC@PMSB: 1H-NMR (CDCl3, 300 MHz, 300 K): δ = 8.22–7.89 (m, 15-H2), 7.54–7.05 (m, 12-H2, 13-H2, 14-H2), 4.43–4.10 (m, 10-H2), 3.91–3.70 (m, Si–H), 1.63–1.20 (m, 3-H2, 7-H2), 1.20–0.99 (m, 9-H2), 0.79–0.42 (m, 2-H2, 4-H2, 6-H2, 8-H2), 0.19–(−0.10) (m, 1-H3, 5-H3) ppm.
VFc@PMSB: 1H-NMR (CDCl3, 300 MHz, 300 K): δ = 4.28–4.00 (m, 14-H5, 13-H2, 12-H2), 2.47–2.27 (m, 10-H2), 3.90–3.76 (m, Si–H), 1.68–1.19 (m, 3-H2, 7-H2), 0.96–0.46 (m, 2-H2, 4-H2, 6-H2, 8-H2, 9-H2), 0.22–(−0.14) (m, 1-H3, 5-H3) ppm.
VADN@PMSB: 1H-NMR (CD2Cl2, 300 MHz, 300 K): δ = 8.21–6.78 (m, 12-H1, 13-H1, 17-H1, 18-H1, 19-H1, 20-H1, 24-H1, 26-H2, 27-H2, 28-H2, 30-H2, 31-H2, 32-H2, 33-H2), 3.81–3.46 (m, Si–H, 10-H2), 2.56–(−0.62) (m, 1-H3, 2-H2, 3-H2, 4-H2, 5-H3, 6-H2, 7-H2, 8-H2, 9-H2) ppm.
2.5 Hydrosilylation of PS-b-PMSB with NVC
1H-NMR (CD2Cl2, 500 MHz, 300 K): δ = 8.18–7.95 (m, 21-H2), 7.54–6.29 (m, 4-H2, 5-H2, 6-H1, 18-H2, 19-H2, 20-H2,), 4.46–4.14 (m, 16-H2), 3.92–3.66 (m, Si–H), 2.33–1.25 (m, 1-H2, 2-H1, 9-H2, 13-H2), 1.22–0.99 (15-H2), 0.83–0.45 (m, 8-H2, 10-H2, 12-H2, 14-H2), 0.23–(−0.13) (m, 7-H3, 11-H3) ppm.
2.6 Hydrosilylation of PS-b-PMSB with VFc
1H-NMR (CD2Cl2, 500 MHz, 300 K): δ = 7.39–6.30 (m, 4-H2, 5-H2, 6-H1), 4.28–3.87 (m, 18-H2, 19-H2, 20-H5), 3.87–3.69 (m, Si–H), 2.48–1.45 (m, 1-H2, 2-H1, 9-H2, 13-H1, 16-H2), 1.00–0.37 (m, 8-H2, 10-H2, 12-H2, 14-H2, 15-H2), 0.22–(−0.22) (m, 7-H3, 11-H3) ppm.
2.7 Hydrosilylation of PS-b-PMSB with VADN
1H-NMR (CD2Cl2, 500 MHz, 300 K): δ = 8.21–7.78 (m, 36-H2, 39-H2), 7.79–6.31 (m, 4-H2, 5-H2, 6-H1, 18-H1, 19-H1, 23-H1, 24-H1, 25-H1, 25-H1, 30-H1, 32-H2, 33-H2, 34-H2, 37-H2, 38-H2), 3.83–3.50 (m, Si–H, 16-H2), 2.64–(−0.70) (m, 1-H2, 2-H1, 7-H3, 8-H2, 9-H2, 10-H2, 11-H3, 12-H2, 13-H2, 14-H2, 15-H2) ppm.
2.8 Block copolymer film preparation
Block copolymer film samples for investigation of the bulk morphologies are obtained by slowly evaporating methylene chloride solvent from a polymer solution (at room temperature over a period of four weeks). After that time, polymer films are heated to 150 °C with a heating rate of 50 K min−1, keeping the temperature at 150 °C for 1 h and again cooled to room temperature with a cooling rate of 50 K min−1.2.9 Characterization
NMR spectra were recorded on a Bruker DRX 500 NMR or on a Bruker DRX 300 spectrometer working at 500 MHz or 300 MHz. NMR chemical shifts are referenced relative to tetramethylsilane. Standard SEC was performed with THF as the mobile phase (flow rate 1 mL min−1) on a SDV column set from PSS (SDV 1000, SDV 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, SDV 1000000) at 30 °C. Calibration was carried out using PS standards (from Polymer Standard Service, Mainz). For the SEC-MALLS experiments, a system composed of a Waters 515 pump (Waters, Milford, CT), a TSP AS100 autosampler, a Waters column oven, a Waters 486 UV detector operating at 254 nm, a Waters 410 RI-detector, and a DAWN DSP lightscattering detector (Wyatt Technology, Santa Barbara, CA) was used. For data acquisition and evaluation of the light-scattering experiments, Astra version 4.73 (Wyatt Technology, Santa Barbara, CA) was used. The light-scattering instrument was calibrated using pure toluene, assuming a Rayleigh ratio of 9.7810−6 cm−1 at 690 nm. An injection volume of 118 μL, a sample concentration of 1–2 g L−1, a column temperature of 35 °C, and a THF flow rate of 1 mL min−1 have been applied. SEC analysis was performed on a high resolution column set from PSS (SDV 5 μm 106 Å, SDV 5 μm 105 Å, SDV 5 μm 1000 Å). TEM experiments were carried out on a Zeiss EM 10 electron microscope operating at 60 kV. All shown images were recorded with a slow-scan CCD camera obtained from TRS (Tröndle) in bright field mode. Camera control was computer-aided using the ImageSP software from TRS. CV measurements were carried out on a multi-potentiostat VMP2 (Princeton Applied Research) with a custom-made cell in dry methylene chloride with tetrabutylammoniumhexafluorophosphate (TBAHFP) as electrolyte (0.1 M) and ferrocene as standard under nitrogen atmosphere. Ag/AgCl reference electrode and Pt counter electrodes were chosen and a scan rate of 100 mV s−1 in a range of −0.2 V to 0.8 V was applied. TGA was measured using a STA429 (Netzsch Gerätebau GmbH, Selb/Bavaria). The experiments were carried out in TG/DTA mode with an argon flow of 75 mL min−1 using a heating rate of 10 K min−1. WAXS measurements were conducted using a Stoe Stadi P with Ge111 as monochromator and a wavelength of MoK,α = 0.7093 Å. A PSD Klein detector was used. For a qualitative analysis the program Match! from Crystal Impact was used. The corresponding structures were taken from Crystallography Open Database. A quantitative analysis was partially performed by the program Full-Prof using the Rietveld refinement algorithm. Thermal properties of the polymers were characterized using a differential scanning calorimeter (DSC) (Mettler Toledo DSC-1) in the temperature range from −110 °C to 180 °C with a heating rate of 10 K min−1.
000, SDV 1000000) at 30 °C. Calibration was carried out using PS standards (from Polymer Standard Service, Mainz). For the SEC-MALLS experiments, a system composed of a Waters 515 pump (Waters, Milford, CT), a TSP AS100 autosampler, a Waters column oven, a Waters 486 UV detector operating at 254 nm, a Waters 410 RI-detector, and a DAWN DSP lightscattering detector (Wyatt Technology, Santa Barbara, CA) was used. For data acquisition and evaluation of the light-scattering experiments, Astra version 4.73 (Wyatt Technology, Santa Barbara, CA) was used. The light-scattering instrument was calibrated using pure toluene, assuming a Rayleigh ratio of 9.7810−6 cm−1 at 690 nm. An injection volume of 118 μL, a sample concentration of 1–2 g L−1, a column temperature of 35 °C, and a THF flow rate of 1 mL min−1 have been applied. SEC analysis was performed on a high resolution column set from PSS (SDV 5 μm 106 Å, SDV 5 μm 105 Å, SDV 5 μm 1000 Å). TEM experiments were carried out on a Zeiss EM 10 electron microscope operating at 60 kV. All shown images were recorded with a slow-scan CCD camera obtained from TRS (Tröndle) in bright field mode. Camera control was computer-aided using the ImageSP software from TRS. CV measurements were carried out on a multi-potentiostat VMP2 (Princeton Applied Research) with a custom-made cell in dry methylene chloride with tetrabutylammoniumhexafluorophosphate (TBAHFP) as electrolyte (0.1 M) and ferrocene as standard under nitrogen atmosphere. Ag/AgCl reference electrode and Pt counter electrodes were chosen and a scan rate of 100 mV s−1 in a range of −0.2 V to 0.8 V was applied. TGA was measured using a STA429 (Netzsch Gerätebau GmbH, Selb/Bavaria). The experiments were carried out in TG/DTA mode with an argon flow of 75 mL min−1 using a heating rate of 10 K min−1. WAXS measurements were conducted using a Stoe Stadi P with Ge111 as monochromator and a wavelength of MoK,α = 0.7093 Å. A PSD Klein detector was used. For a qualitative analysis the program Match! from Crystal Impact was used. The corresponding structures were taken from Crystallography Open Database. A quantitative analysis was partially performed by the program Full-Prof using the Rietveld refinement algorithm. Thermal properties of the polymers were characterized using a differential scanning calorimeter (DSC) (Mettler Toledo DSC-1) in the temperature range from −110 °C to 180 °C with a heating rate of 10 K min−1.
3 Results and discussion
3.1 Anionic ring-opening polymerization of MSB and sequential polymerization of styrene with MSB
Liao et al. were the first to report the anionic ring-opening polymerization (ROP) of methylsilacyclobutane (MSB).52 Although anionic polymerization using alkyllithium initiator took place, it proved impossible to obtain PMSB homopolymers with narrow molar mass distributions and polydispersity values Đ were typically close to 2.0.35,58 However, within these studies no detailed information about the character of “livingness” or the kinetic for this anionic ROP was described. Before we turn to the copolymerization of styrene with MSB, we performed experiments to clarify some of these aspects. Polymerization of MSB was carried out as described in the Experimental section and as given in Fig. 2 using n-butyllithium as initiator and THF as solvent at −78 °C.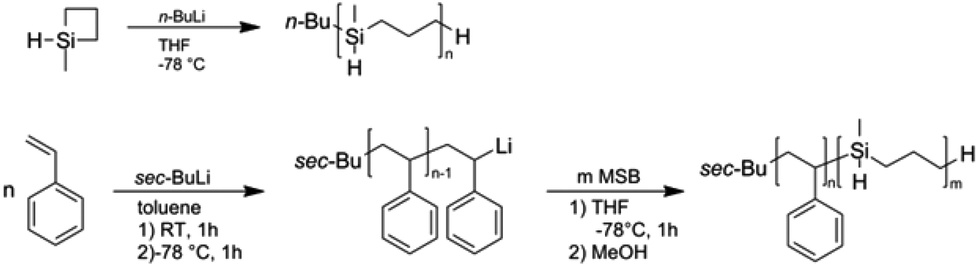 | ||
| Fig. 2 Anionic ring-opening polymerization of MSB (top) and block copolymerization by sequential monomer addition of styrene and MSB (bottom). | ||
Results of four different exemplary polymerizations are compiled in Table S1 of the ESI.† From the obtained yields of the experiments and from results of SEC measurements (Fig. S1†) it can be concluded that it is challenging to control the anionic ROP of MSB. The molar masses obtained strongly differ from expected values. Remarkably, under these conditions almost quantitative yields of PMSB could be obtained even after 5 minutes of reaction time independent of monomer concentrations or varying reaction temperatures between −78 °C and −90 °C. We additionally performed this reaction in n-hexane instead of THF as solvent applying the same reaction protocols. This led to slightly higher molar masses (Mw = 41 kDa) accompanied with a broadening of the molecular weight distribution (Đ = 1.84). Attempts to subsequently add MSB monomer in order to prove a “living” character of the anionic polymerization also failed as the PMSB chains were not capable of further reaction with MSB monomer. Also the addition of 1,1-diphenylethylene (DPE) immediately after initiator addition to a precooled monomer solution did not result in a typical deep red colour. Hence it can be concluded that it is not possible to control the MSB polymerization by applying herein investigated synthesis protocols due to the remarkably high reactivity of MSB monomer. Nevertheless, it would be feasible to (i) form block copolymers with a structure-directing polymer such as polystyrene (PS) which can be polymerized in a well-defined manner and to (ii) further functionalize PMSB and PS-block-PMSB in a subsequent step by using e.g., hydrosilylation strategies. For this purpose, styrene was polymerized by anionic polymerization and active polymer chains were used as macro-initiator for MSB monomer (Fig. 2). The results are compiled in Table 1 while an exemplary SEC measurement for PS-b-PMSB3 is given as Fig. S2.† It can be concluded that active PS polymer chains were capable of initiating MSB monomer under these conditions (see Experimental section). Due to the excellent control of styrene polymerization, low values for Đ could be obtained. In more detail, values of Đ for formed PS-b-PMSB diblock copolymers were still low and range from 1.03 to 1.08. These results were surprising as homopolymerization could not be performed in a controlled way as discussed above. A possible explanation is that the apparent molar mass of the corresponding block copolymers obtained by using SEC vs. PS calibration is undervalued leading to lower molar masses for PMSB. For comparison, SEC-MALLS measurements of a PMSB featuring an apparent molar mass (Mw) of 41400 g mol−1vs. PS calibration revealed a value of 56300 g mol−1 (Mw) for the absolute molar mass (refractive index increment was determined to be 0.106 mL g−1). Therefore, values obtained by using SEC vs. PS calibrations for PMSB-containing block copolymers should be treated with caution.
| s-BuLi (μmol) |
M
wa PS (g mol−1) |
M
wa (block) (g mol−1) |
Đ (block) | Composition wt% PS/PMSBb | |
|---|---|---|---|---|---|
| a Molar masses determined by using SEC measurements with PS calibration. b Composition determined by using 1H NMR spectroscopy. | |||||
| PS-b-PMSB1 | 50 | 24200 |
26400 |
1.04 | 83/17 |
| PS-b-PMSB2 | 94 | 16900 |
19200 |
1.07 | 79/21 |
| PS-b-PMSB3 | 50 | 18100 |
21900 |
1.08 | 60/40 |
| PS-b-PMSB4 | 25 | 27100 |
28800 |
1.03 | 58/42 |
Additionally, 1H NMR spectroscopy measurements revealed that the composition of corresponding diblock copolymers can be adjusted to a maximum content of 42% per weight for the PMSB block (Table 2). An exemplary 1H NMR spectrum of PS-b-PMSB3 is given as Fig. S3† while selective areas of stacked 1H NMR spectra showing the aromatic PS region and the Si–H signal at 3.77 ppm for all block copolymers is given in Fig. 3.
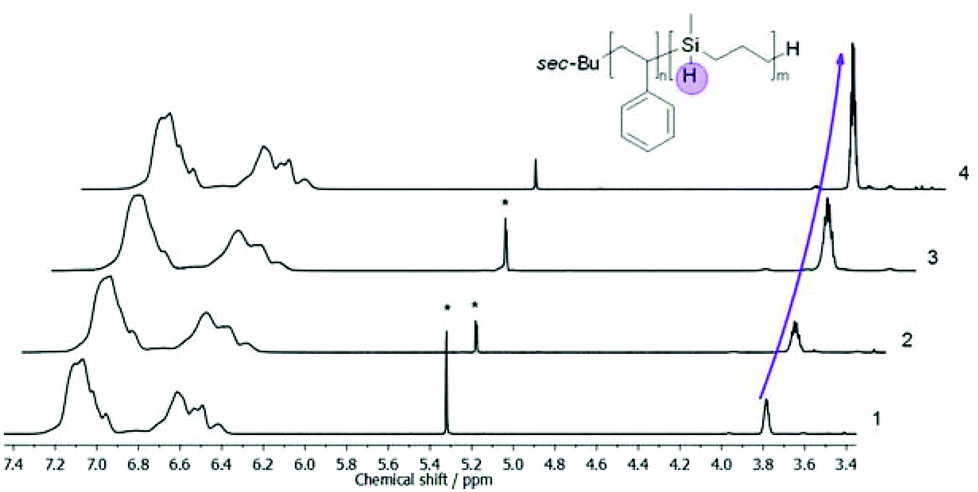 | ||
| Fig. 3 Stacked 1H NMR spectra of PMSB and all PS-b-PMSB diblock copolymers (1–4) as given in Table 1. The increasing amount of PMSB is determined by integration of the Si–H signal at 3.77 ppm compared to the aromatic region. | ||
|
M
na (g mol−1) |
M
wa (g mol−1) |
Đ | DSb (mol%) | T g (°C) | |
|---|---|---|---|---|---|
| a Molar masses determined by using SEC measurements with PS calibration. b DS: Degree of substitution of Si–H moieties for all functional groups (NVC, VFc, VADN) were determined by using 1H NMR spectroscopy. | |||||
| PMSB | 21300 |
34200 |
1.60 | — | −82 |
| NVC@PMSB | 14000 |
39000 |
2.80 | 31 | 22 |
| VFc@PMSB | 19500 |
42000 |
2.16 | 43 | 0 |
| VADN@PMSB | 22100 |
47700 |
2.16 | 26 | 124 |
In order to investigate the thermal properties of the new PMSB-containing copolymers, DSC measurements were performed for all investigated block copolymers. The glass transition temperatures of PS and PMSB were observable in the expected regions i.e., 100 °C for PS68 and −91 °C for PMSB.35 A DSC thermogram of PS-b-PMSB3 is given as Fig. S4 in the ESI.† As diblock copolymers consisting of PS as structure-directing block segment and PMSB with adjustable length ranging from 17 to 42% per weight could be obtained, we turn to the investigation of applying hydrosilylation protocols for both PMSB homopolymers as well as the corresponding diblock copolymers with functional monomers in the next section.
3.2 Hydrosilylation of polycarbosilane homopolymers and block copolymers with pendant functional groups
Postmodification both of PMSB and PS-b-PMSB diblock copolymers was carried out by hydrosilylation using Karstedt's catalyst (Fig. 4). In order to prove the universality of this synthetic approach, functional moieties based on the functional vinyl compounds N-vinylcarbazole (NVC), vinylferrocene (VFc), and 9,10-di(1-naphthalenyl)-2-vinylanthracene (VADN) were investigated.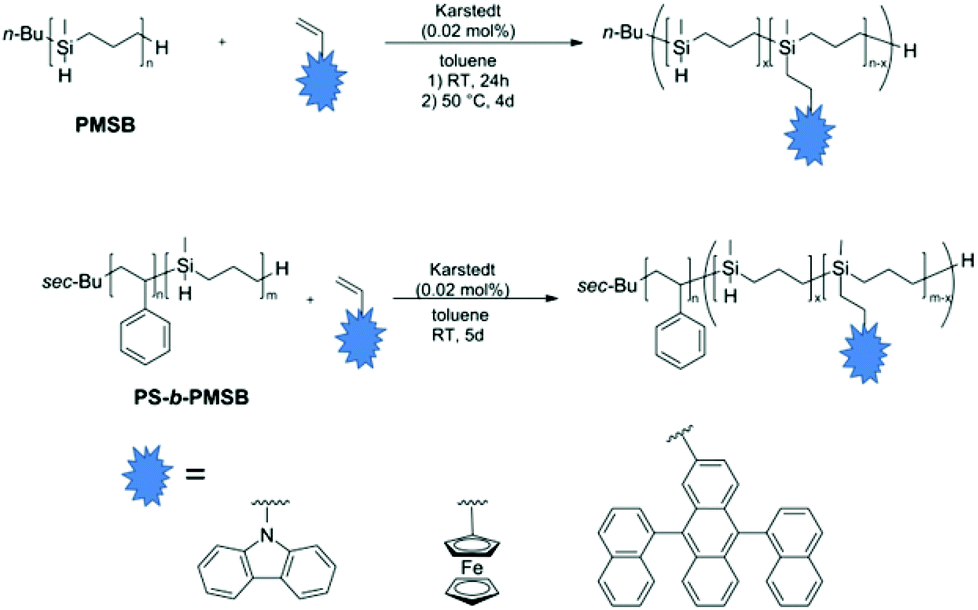 | ||
| Fig. 4 Hydrosilylation reaction of PMSB with functional vinyl compounds using Karstedt's catalyst (top) and hydrosilylation reaction of diblock copolymers consisting of polystyrene and PMSB (PS-b-PMSB). | ||
The content of newly introduced functional groups was first determined by using 1H NMR spectroscopy. For calculation of the polymer composition, integration of the aromatic region for the functional moieties NVC, VADN and the cyclopentadienyl-region of ferrocene was compared to the signals stemming from Si–CH3 signal at −0.02–0.02 ppm (Fig. S5–7†). SEC measurements of all hydrosilylated PMSB homopolymers featuring NVC, VFc or VADN revealed an increase in molar mass for all prepared polymers again proving the feasibility of used postmodification strategy. All results obtained by using SEC and 1H NMR spectroscopy are compiled in Table 2.
DSC measurements of corresponding substituted PMSB polymers revealed an increase in glass transition temperatures, Tg, compared to non-functionalized PMSBs. Shifted Tg values are additionally compiled in Table 2 while the corresponding DSC thermograms for NVC@PMSB, VFc@PMSB, and VADN@PMSB are given in the ESI as Fig. S8–S10.† As expected, increase of Tg values nicely fits to the degree of substitution (DS) with sterically demanding moieties which results in a hindered polymer chain segment mobility. It is worthy to mention that the residual Si–H moieties after postmodification with investigated bulky vinyl compounds were still capable of undergoing further functionalization with less bulky vinyl compounds. Exemplarily in the case of VFc@PMSB polymers, further hydrosilylation with e.g. styrene could be carried out up to a degree of substitution of 97% with respect to the Si–H moieties. As functionalization of PMSB homopolymers with all three functional monomers could be carried out without problems, we turned to postmodification of PS-b-PMSB diblock copolymers again by hydrosilylation in the next step. By applying exactly the same hydrosilylation protocols as for PMSB homopolymers (Fig. 4, middle), various novel functional block copolymers could be obtained. Exemplarily, postmodification of PS-b-PMSB having molar masses of 26400 g mol−1 (PS-b-PMSB1) and 21900 g mol−1 (PS-b-PMSB3) with NVC, VFc, and VADN was carried out (Fig. 4). Corresponding SEC measurements (Fig. S11–13†) and DSC measurements (stacked Fig. S14†) proved the successful postmodification of the polyhydrosilane-segment with functional moieties also for the diblock copolymers. As can be concluded from DSC thermograms, there was no shift of Tg values for the PS segment, while Tg for the PMSB segment shifted according to the degree of substitution with different functional sterically demanding vinyl compounds. Results obtained by SEC, DSC and 1H NMR measurements for the modified block copolymers are compiled in Table 3. Moreover, in order to vary the content of functional groups which are covalently attached to the PMSB backbone, the degree of substitution was modified simply by the addition of certain amounts of vinyl compounds in the hydrosilylation step. Exemplarily, in the case of VFc the content was varied in the range of 28 mol% to remarkable 63 mol% with respect to the reactive Si–H units.
| Polymer |
M
na (g mol−1) |
M
wa (g mol−1) |
Đ | DSb (mol%) | T g (PMSB/PS) (°C) |
|---|---|---|---|---|---|
| a Molar masses determined by using SEC measurements with PS calibration. b DS: Degree of substitution for all functional groups were determined by using 1H NMR spectroscopy. | |||||
| NVC@PS-PMSB1 | 35600 |
43200 |
1.21 | 31 | 22/98 |
| NVC@PS-PMSB3 | 27000 |
30500 |
1.13 | 36 | 16/99 |
| VFc@PS-PMSB1 | 27000 |
29300 |
1.08 | 28 | −23/100 |
| VFc@PS-PMSB3 | 29900 |
39800 |
1.33 | 63 | 6/95 |
| VADN@PS-PMSB1 | 34200 |
37700 |
1.10 | 51 | 109 |
| VADN@PS-PMSB3 | 25600 |
27200 |
1.06 | 48 | 100 |
However, in some cases an additional signal appeared at 3.40 ppm in the corresponding 1H NMR spectra after hydrosilylation reaction of PMSB or corresponding PS-b-PMSB using Karstedt's catalyst. Our first assumption was that this signal corresponded to a methyl group of the Markovnikov product (Fig. 5A), which is maybe a by-product during hydrosilylation reaction.
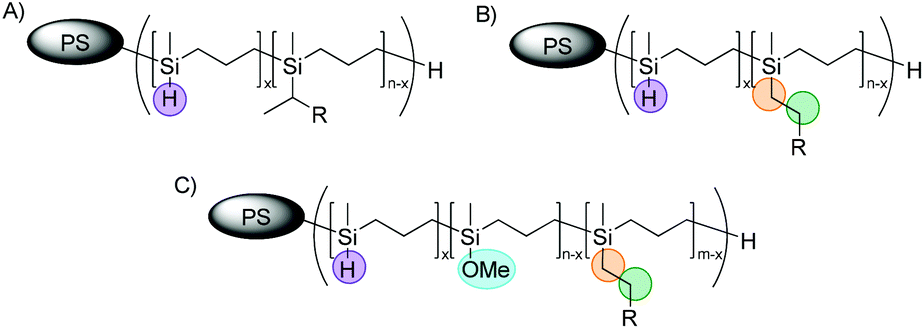 | ||
| Fig. 5 Possible Markovnikov (A) and expected anti-Markovnikov (B) products formed after hydrosilylation reaction of PS-b-PMSB with vinyl compounds and the methoxy-substituted by-product after polymer work-up (C). | ||
In general, following hydrosilylation strategies, predominantly formation of the anti-Markovnikov product (Fig. 5B) is expected. In order to confirm this first assumption – the formation of a Markovnikov product (A) – we studied NVC@PS-b-PMSB1 in more detail via 2D NMR spectroscopy. In detail, in order to assign this signal the scalar proton coupling was analyzed through 2D homonuclear correlation measurement (COSY). A section of the aliphatic region of NVC@PS-b-PMSB as well as the corresponding 1H NMR spectrum is shown in Fig. 6. For the anti-Markovnikov product the cross-peak appeared due to the spin coupling between the methylene groups. In the case of a Markovnikov product, a correlation between a methyl group and a methine proton would be expected. In fact, however, for the signal at 3.40 ppm no cross-peak was observed leading to the conclusion that possible product A was not formed.
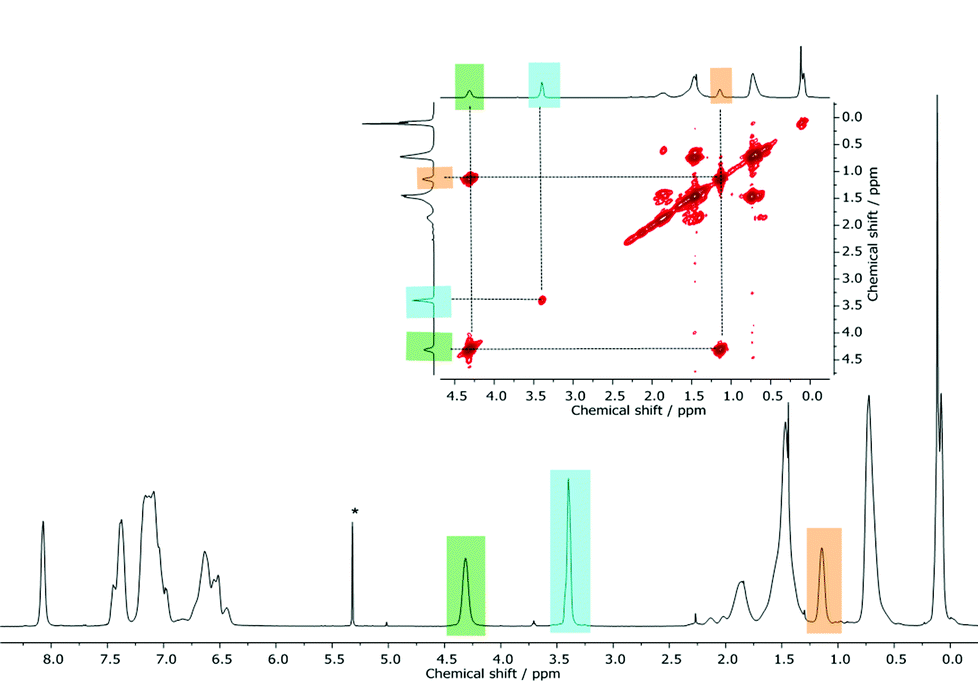 | ||
| Fig. 6 2D homonuclear correlation measurement (COSY) of NVC@PS-b-PMSB1 for exclusion of by-product A shown in Fig. 5 (see text). | ||
As by-product A could be excluded using 2D NMR techniques, we followed the working hypothesis that the by-product was formed during polymer work-up protocols after hydrosilylation. Mirza-Aghayan et al. reported the transformation of silanes to corresponding silyl ethers in the presence of transition metal catalysts.69 Thus, if the presence of alcohol during the polymer precipitation step can be avoided, no formation of the supposed product C (Fig. 5) will take place. We note that the corresponding 1H NMR signal at 3.40 ppm significantly increases, if the polyhydrocarbosilane is treated with methanol in the presence of Karstedt's catalyst for a prolonged time. This is exemplarily shown for NVC@PS-b-PMSB1 which was stirred for 30 min in the methanol precipitation step. An even more intensive peak appeared at 3.40 ppm of the corresponding 1H NMR spectrum (Fig. S15†).
In order to prove the assumption of silylether formation with methanol, NVC@PMSB1 was precipitated either in methanol or pentane followed by studying the corresponding polymers by using 1H NMR spectroscopy. In Fig. 7, corresponding stacked 1H NMR spectra are given.
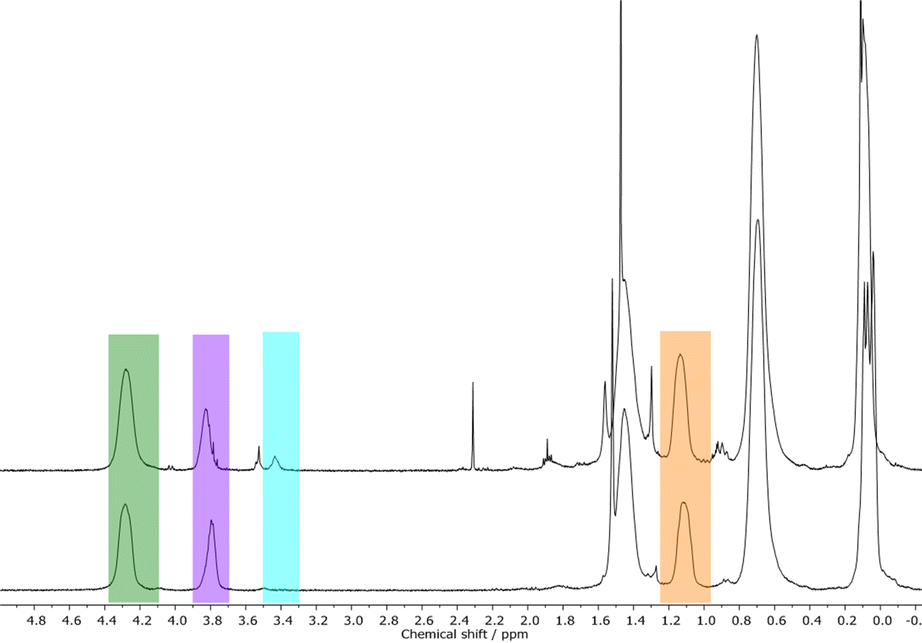 | ||
| Fig. 7 Stacked 1H NMR spectra of NVC@PMSB1 precipitated in methanol to form the methoxy-containing by-product C (top) and precipitated in pentane to obtain the anti-Markovnikov product B (bottom). | ||
It can be concluded from both spectra that pentane is suitable to precipitate (functionalized) hydrosilylated polycarbosilanes without the formation of the methoxy-containing by-product C (corresponding signals are highlighted in blue in Fig. 7). As these results comprise the successful formation of functional polyhydrocarbosilane block copolymers featuring different contents of pendant functional groups, first attempts for microphase-separation and potential applications for these novel materials will be provided in the ensuing sections.
3.3 Microphase-separation of BC with pendant functional groups
The block copolymer film preparation is described in detail in the Experimental section. Unfortunately, the pristine PS-b-PMSB samples were too soft to prepare thin sections by ultramicrotoming due to the glass transition temperature of −91 °C to −86 °C for the PMSB segment. However, as the content of Si–H group functionalization with sterically demanding NVC, VFc, and VADN monomers increased, also Tg values significantly increased (Table 3). Ultramicrotoming became possible and corresponding thin sections of all samples of Table 3 could be investigated by using TEM. Exemplary images are given in Fig. 8. | ||
| Fig. 8 TEM images of VADN@PS-b-PMSB1 (top), NVC@PS-b-PMSB1 (middle) and VFc@PS-b-PMSB1 (bottom). Scale bars correspond to 500 nm (left column) and 250 nm (right column). | ||
All samples were investigated without any staining as functionalized polyhydrocarbosilanes revealed sufficient contrast due to the presence of silicon or iron. Especially ferrocene-containing samples were rich in contrast (Fig. 8C) as it is observed for other ferrocene-containing polymers before.58,66,70,71 It can be concluded from all investigated block copolymer samples that a microphase separation with domain sizes in the range of 20–50 nm were obtained. Due to the fact that no typical nanostructures could be observed the system seems not to be in the equilibrium state. Nevertheless, the domain sizes are of high interest especially for preceramic block copolymers, e.g. for the preparation of porous structures featuring high surface areas after thermal treatment.
3.4 Stimuli-responsiveness and ceramization of VFc@PMSB
In order to prove the potential of these novel functional materials, VFc@PMSB polymers were exemplarily investigated in terms of redox-responsiveness and ceramization.In general, applications based on ferrocene-containing polymers is an emerging field of recent research comprising e.g., redox-mediated release out of the interior of nanocapsules,19,72 for switching the surface wettability and membrane gating,73,74 optical applications,75,76 or for reversible activity modulation of immobilized catalysts.77 All these feasible applications necessitate the chemical or electrical response of the ferrocene moiety. Therefore, cyclic voltammetry (CV) studies for VFc@PMSB were performed (Fig. S16†). As can be concluded from the corresponding cyclic voltammogram, also functionalized ferrocene-containing polymers prepared and investigated within this contribution clearly revealed a redox-responsive behaviour for at least 24 cycles. The slight shift in the peak potentials is due to the worse solubility of polyvinylferrocenium compared to PVFc in acetonitrile forming a film at the electrodes as described in earlier studies.76 We expect our synthetic strategy and easily adjustable protocol for polymer postmodification as excellent model in order to study the influence of the amount of functional groups over a wide range. A deeper understanding of stimuli-responsive polymers is thereof expected which is part of ongoing work.
As already stated in the introduction, metallopolymers and silicon-containing polymers are highly attractive as preceramic polymers. For widening the potential of herein studied functional polymers, VFc@PMSB1 was investigated by using thermogravimetric analysis (TGA). The corresponding diagram of an exemplary TGA of this polymer up to a temperature of 1500 °C is given as Fig. S17.† Two steps accompanied with a slight weight loss at 210 °C and a more significant weight loss at 470 °C could be observed. The final material after ceramization under inert conditions (argon) was analyzed by using XRD. The corresponding XRD pattern (Fig. S18†) consists of different series of peaks. As a first result, thermal treatment of VFc@PMSB1 up to a temperature of 1500 °C under argon enabled the formation of an interesting composite material consisting of Fe3Si, Fe3Si5, SiC, and graphite. Investigations of properties and potential applications for such type of materials which can be tailored and modified further by using herein established protocols are under way.
4 Conclusions
In the current work we present a feasible strategy for the preparation of polyhydrocarbosilane-based block copolymers with polystyrene. Methylsilacyclobutane (MSB) monomer is polymerized by anionic ring-opening polymerization (ROP) followed by postmodification of PMSB homopolymers with three functional vinyl compounds i.e., N-vinylcarbazole (NVC), vinylferrocene (VFc), and 9,10-di(1-naphthalenyl)-2-vinylanthracene (VADN) by applying hydrosilylation protocols. Degree of substitution (DS) range between 26 mol% to 43 mol% with respect to the residual Si–H moieties of the PMSB polymer backbone. In the case of polystyrene-block-polymethylsilacyclobutane (PS-b-PMSB) corresponding DS values between 28 mol% to 63 mol% are obtained. All polymers are investigated by using SEC, NMR, and DSC proving the suitability of herein established synthesis protocols for convenient modification of the PMSB segment. As precipitation of homo- and block copolymers in methanol leads to undesired silylether by-products, polymer work-up procedures are presented in order to obtain well-defined oxygen-free polyhydrocarbosilanes featuring functional moieties. Microphase separation for all functional block copolymer samples are evidenced by TEM measurements after film formation although obtained structures are obviously not in the equilibrium state. Due to the presence of a PMSB segment consisting of polymer chains with significantly broader molecular weight distribution compared to the PS block, worm-like morphologies with domain sizes of approximately 50 nm are observed. The herein described block-selective hydrosilylation strategy for PMSB and the capability for convenient postmodification with adjustable contents of functional groups are expected to give insight into the mechanism of stimuli-responsive polymers. Moreover, such type of preceramic block copolymers will be advantageously used for the preparation of innovative tailor-made ceramics with adjustable domain sizes.Acknowledgements
The authors thank Claudia Fasel (group of Prof. R. Riedel) and Jean-Christoph Jaud (group of Prof. W. Donner) from the Materials Science Department Darmstadt for performing the TGA and XRD measurement, Sabrina Mehlhase, Marion Trautmann, Johannes Elbert, and Daniel Scheid for help with DSC, TEM, and SEC measurements. Jochen Wulff is acknowledged for providing VADN monomer. Additionally, Haiko Didzoleit and Prof. Bernd Stühn (department of physics, TU Darmstadt) are acknowledged for helpful discussions concerning the XRD measurements.Notes and references
- I. W. Hamley, The Physics of Block Copolymers, Oxford University Press, Oxford, 1998 Search PubMed.
- M. Lazzari, G. Liu and S. Lecommandoux, Block Copolymers in Nanoscience, Wiley-VCH, Weinheim, 2006 Search PubMed.
- N. A. Lynd, A. J. Meuler and M. A. Hillmyer, Prog. Polym. Sci., 2008, 33, 875–893 CrossRef CAS PubMed.
- H.-C. Kim, S.-M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146–177 CrossRef CAS PubMed.
- D. Klinger, M. J. Robb, J. M. Spruell, N. A. Lynd, C. J. Hawker and L. A. Connal, Polym. Chem., 2013, 4, 5038–5042 RSC.
- R. C. Hayward and D. J. Pochan, Macromolecules, 2010, 43, 3577–3584 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32 CrossRef CAS PubMed.
- M. A. Hillmyer, Adv. Polym. Sci., 2005, 190, 137–181 CrossRef CAS.
- J. K. Kim, S. Y. Yang, Y. Lee and Y. Kim, Prog. Polym. Sci., 2010, 35, 1325–1349 CrossRef CAS PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. luzinov and S. Minko, Nature, 2010, 9, 101–113 CrossRef PubMed.
- A. E. Smith, X. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS PubMed.
- D. Han, X. Tong and Y. Zhao, Macromolecules, 2011, 44, 437–439 CrossRef CAS.
- J. Du, L. Fan and Q. Liu, Macromolecules, 2012, 45, 8275–8283 CrossRef CAS.
- J. Hu, H. Meng, G. Li and S. I. Ibekwe, Smart Mater. Struct., 2012, 21, 053001 CrossRef.
- F. H. Schacher, P. A. Rupar and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 7898–7921 CrossRef CAS PubMed.
- J. I. Clodt, V. Filiz, S. Rangou, K. Buhr, C. Abetz, D. Höche, J. Hahn, A. Jung and V. Abetz, Adv. Funct. Mater., 2013, 23, 731–738 CrossRef CAS.
- M. Gallei, S. Rangou, V. Filiz, K. Buhr, S. Bolmer, C. Abetz and V. Abetz, Macromol. Chem. Phys., 2013, 214, 1037–1046 CrossRef CAS.
- B. V. K. J. Schmidt, J. Elbert, C. Barner-Kowollik and M. Gallei, Macromol. Rapid Commun., 2014, 35, 708–714 CrossRef CAS PubMed.
- R. H. Staff, M. Gallei, K. Landfester and D. Crespy, Macromolecules, 2014, 47, 4876–4883 CrossRef CAS.
- P. R. Malenfant, J. Wan, S. T. Taylor and M. Manoharan, Nat. Nanotechnol., 2007, 2, 43–46 CrossRef CAS PubMed.
- S. Kaur, M. Gallei and E. Ionescu, Adv. Polym. Sci., 2015, 267, 143–185 CrossRef.
- M. C. Orilall and U. Wiesner, Chem. Soc. Rev., 2011, 40, 520–535 RSC.
- M.-S. She, T.-Y. Lo, H.-Y. Hsueh and R.-M. Ho, NPG Asia Mater., 2013, 5, e42 CrossRef CAS.
- Y. Ren, Z. Ma and P. G. Bruce, Chem. Soc. Rev., 2012, 41, 4909–4927 RSC.
- P. Innocenzi and L. Malfatti, Chem. Soc. Rev., 2013, 42, 4198–4216 RSC.
- N. D. Petkovich and A. Stein, Chem. Soc. Rev., 2013, 42, 3721–3739 RSC.
- M. Rawolle, M. A. Niedermeier, G. Kaune, J. Perlich, P. Lellig, M. Memesa, Y. J. Cheng, J. S. Gutmann and P. Müller-Buschbaum, Chem. Soc. Rev., 2012, 41, 5131–5142 RSC.
- I. E. Rauda, R. Buonsanti, L. C. Saldarriaga-Lopez, K. Benjauthrit, L. T. Schelhas, M. Stefik, V. Augustyn, J. Ko, B. Dunn, U. Wiesner, D. J. Milliron and S. H. Tolbert, ACS Nano, 2012, 6, 6386–6399 CrossRef CAS PubMed.
- J. Zhou, G. R. Whittell and I. Manners, Macromolecules, 2014, 47, 3529–3543 CrossRef CAS.
- A. Gregory and M. H. Stenzel, Prog. Polym. Sci., 2012, 37, 38–105 CrossRef CAS PubMed.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068–1132 CrossRef CAS PubMed.
- S. Ito, R. Goseki, T. Ishizone and A. Hirao, Polym. Chem., 2014, 5, 5523–5534 RSC.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- C. Herfurth, D. Voll, J. Buller, J. Weiss, C. Barner-Kowollik and A. Laschewsky, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 108 CrossRef CAS.
- C. X. Liao and W. P. Weber, Macromolecules, 1993, 26, 563–566 CrossRef CAS.
- T. Ganicz, W. Staficzyk, E. Biatecka-Florjaficzyk and I. Sledzifiska, Polymer, 1996, 37, 4167–4174 CrossRef CAS.
- R. Shankar, A. Saxena and A. S. Brar, J. Organomet. Chem., 2001, 628, 262–270 CrossRef CAS.
- F. Wurm, H. Schüle and H. Frey, Macromolecules, 2008, 41, 9602–9611 CrossRef CAS.
- F. Wurm, S. Hilf and H. Frey, Chem. – Eur. J., 2009, 15, 9068–9077 CrossRef CAS PubMed.
- H. Kudo, Y. Fujiwara, M. Miyasaka and T. Nishikubo, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5746–5751 CrossRef CAS.
- L. Sacarescu, M. Simionescu, G. Sacarescu and V. Hamciuc, J. Appl. Polym. Sci., 2012, 125, 1284–1291 CrossRef CAS.
- S. Hilf, P. W. Cyr, D. A. Rider, I. Manners, T. Ishida and Y. Chujo, Macromol. Rapid Commun., 2005, 26, 950–954 CrossRef CAS.
- I. Martínez-Montero, S. Bruña, A. M. González-Vadillo and I. Cuadrado, Macromolecules, 2014, 47, 1301–1315 CrossRef.
- G. de la Cruz, H. Schüle, J. Losada, M. P. García-Armada, H. Frey, B. Alonso and C. M. Casado, Eur. J. Inorg. Chem., 2013, 2013, 44–53 CrossRef CAS.
- P. Boehm, M. Mondeshki and H. Frey, Macromol. Rapid Commun., 2012, 33, 1861–1867 CrossRef CAS PubMed.
- J. Kong, T. Schmalz, G. Motz and A. H. E. Müller, Macromolecules, 2011, 44, 1280–1291 CrossRef CAS.
- A. G. Marcos, T. M. Puse, R. Thomann, T. Pakula, L. Okrasa, S. Geppert, W. Gronski and H. Frey, Macromolecules, 2006, 39, 971–977 CrossRef CAS.
- D. Schawaller, B. Clauß and M. R. Buchmeiser, Macromol. Mater. Eng., 2012, 297, 502–522 CrossRef CAS.
- M. Birot, J.-P. Pillot and J. Dunogues, Chem. Rev., 1995, 95, 1443–1477 CrossRef CAS.
- R. Riedel, G. Mera, R. Hauser and A. Klonczynski, J. Ceram. Soc. Jpn., 2006, 114, 425–444 CrossRef CAS.
- S. Q. Zhou, Y. T. Park, G. Manuel and W. P. Weber, Polym. Bull., 1990, 23, 491–496 CrossRef CAS.
- C. X. Liao and W. P. Weber, Polym. Bull., 1992, 28, 281–286 CrossRef CAS.
- R. Rulkens, A. J. Lough and I. Manners, J. Am. Chem. Soc., 1994, 116, 797–798 CrossRef CAS.
- M. Suzuki, J. Kotani, S. Gyobu, T. Kaneko and T. Saegusa, Macromolecules, 1994, 27, 2360–2363 CrossRef CAS.
- V. Bellas and M. Rehahn, Angew. Chem., Int. Ed., 2007, 46, 5082–5104 CrossRef CAS PubMed.
- R. Knischka, H. Frey, U. Rapp and F. J. Mayer-Posner, Macromol. Rapid Commun., 1998, 19, 455–459 CrossRef CAS.
- K. Matsumoto and H. Yamaoka, Macromolecules, 1995, 28, 7029–7031 CrossRef CAS.
- M. Gallei, S. Tockner, R. Klein and M. Rehahn, Macromol. Rapid Commun., 2010, 31, 889–896 CrossRef CAS PubMed.
- M. Gallei, J. Li, J. Elbert, M. Mazurowski, A. Schönberger, C. Schmidt, B. Stühn and M. Rehahn, Polymers, 2013, 5, 284–302 CrossRef PubMed.
- J. Li, B. Kuttich, M. Gallei, J. Elbert, M. Rehahn and B. Stühn, Polymer, 2013, 54, 5703–5711 CrossRef CAS PubMed.
- Q. D. Nghiem and D.-P. Kim, Chem. Mater., 2008, 20, 3735–3739 CrossRef CAS.
- K. Matsumoto and H. Matsuoka, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3778–3787 CrossRef CAS.
- I. Natori, Macromolecules, 2006, 39, 6017–6024 CrossRef CAS.
- N.-G. Kang, Y.-H. Hur, M. Changez, B.-G. Kang, Y.-G. Yu and J.-S. Lee, Polymer, 2013, 54, 5615–5625 CrossRef CAS PubMed.
- H. Mori, H. Ookuma and T. Endo, Macromolecules, 2008, 41, 6925–6934 CrossRef CAS.
- M. Gallei, R. Klein and M. Rehahn, Macromolecules, 2010, 43, 1844–1854 CrossRef CAS.
- J. Wang and L. M. Leung, Eur. Polym. J., 2013, 49, 3722–3733 CrossRef CAS PubMed.
- D. S. Fryer, P. F. Nealey and J. de Pablo, Macromolecules, 2000, 703, 6439–6447 CrossRef.
- M. Mirza-Aghayan, R. Boukherroub and M. Bolourtchian, J. Organomet. Chem., 2005, 690, 2372–2375 CrossRef CAS PubMed.
- M. Gallei, B. V. K. J. Schmidt, R. Klein and M. Rehahn, Macromol. Rapid Commun., 2009, 30, 1463–1469 CrossRef CAS PubMed.
- R. G. H. Lammertink, M. A. Hempenius, E. L. Thomas and G. J. Vancso, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 1009–1021 CrossRef CAS.
- R. H. Staff, M. Gallei, M. Mazurowski, M. Rehahn, R. Berger, K. Landfester and D. Crespy, ACS Nano, 2012, 6, 9042–9049 CrossRef CAS PubMed.
- J. Elbert, M. Gallei, C. Rüttiger, A. Brunsen, H. Didzoleit, B. Stühn and M. Rehahn, Organometallics, 2013, 32, 5873–5878 CrossRef CAS.
- J. Elbert, F. Krohm, C. Rüttiger, S. Kienle, H. Didzoleit, B. N. Balzer, T. Hugel, B. Stühn, M. Gallei and A. Brunsen, Adv. Funct. Mater., 2014, 24, 1591–1601 CrossRef CAS.
- D. Scheid, C. Lederle, S. Vowinkel, C. G. Schäfer, B. Stühn and M. Gallei, J. Mater. Chem. C, 2014, 2, 2583–2590 RSC.
- D. P. Puzzo, A. C. Arsenault, I. Manners and G. A. Ozin, Angew. Chem., Int. Ed., 2009, 48, 943–947 CrossRef CAS PubMed.
- J. Elbert, J. Mersini, N. Vilbrandt, C. Lederle, M. Kraska, M. Gallei, B. Stühn, H. Plenio and M. Rehahn, Macromolecules, 2013, 46, 4255–4267 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data on PMSB homopolymers, 1H NMR spectra, SEC, and DSC of all functional hydrosilylated PMSB homo- and block copolymers, 1H NMR spectrum of NVC@PS-b-PMSB1 precipitated in methanol, cyclic voltammogram of VFc@PMSB1, TGA of VFc@PMSB1, and XRD pattern of ceramized VFc@PMSB. See DOI: 10.1039/c5py00065c |
| This journal is © The Royal Society of Chemistry 2015 |