
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
ATRP-based polymers with modular ligation points under thermal and thermomechanical stress†
Ozcan
Altintas
abc,
Thomas
Josse
d,
Mahdi
Abbasi
b,
Julien
De Winter
d,
Vanessa
Trouillet
e,
Pascal
Gerbaux
d,
Manfred
Wilhelm
*b and
Christopher
Barner-Kowollik
*ac
aPreparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128 Karlsruhe, Germany. E-mail: christopher.barner-kowollik@kit.edu
bPolymeric Materials, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128 Karlsruhe, Germany. E-mail: manfred.wilhelm@kit.edu
cInstitut für Biologische Grenzflächen, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
dInterdisciplinary Center for Mass Spectrometry (CISMa), Organic Synthesis and Mass Spectrometry Laboratory, University of Mons, 23 Place du Parc, B-7000 Mons, Belgium
eInstitute for Applied Materials (IAM) and Karlsruhe Nano Micro Facility (KNMF), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 12th February 2015
Abstract
Linear polystyrenes carrying a mid-chain triazole, esters as well as terminal secondary bromines functionalities were synthesized via activators regenerated by electron transfer (ARGET) atom transfer radical polymerization (ATRP) using a bifunctional triazole containing initiator (3.8 kDa ≤ Mn,SEC ≤ 125 kDa, 1.08 ≤ Đ ≤ 1.19) with the aim of understanding their behavior under thermal and thermomechanical stress. As reference materials – isolating the influence of individual functional groups – three polystyrene homopolymers carrying an ω-bromine chain-end functionality, α,ω-ester-bromine functionalities as well as α,ω-dibromine/mid-chain ester functionalities (2 kDa ≤ Mn,SEC ≤ 39 kDa, 1.06 ≤ Đ ≤ 1.08) were prepared via ARGET ATRP. Furthermore, a well-defined triazole mid-chain functionalized block homopolymer, i.e. polystyrene-b-polystyrene (PS-b-PS, Mn,SEC = 4.4 kDa, Đ = 1.08), was synthesized via a combination of ARGET ATRP and copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) as a reference material. Reference polymers without bromine and with ester/triazole functionalities were additionally investigated. Thermomechanical stress was applied to the polymers via small scale extrusion as well as a rheological assessment (G′(t), G′′(t)) under processing conditions. The thermally challenged polymers were analyzed by size-exclusion chromatography (SEC), matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-ToF-MS), proton nuclear magnetic resonance (1H NMR) and X-ray photoelectron spectroscopy (XPS) to arrive at a detailed image of the degradation susceptibility of individual functional groups, especially esters, bromines and triazole functions. The findings indicate an enhanced degradation of ATRP polymers via an accelerated ester cleavage due to HBr release at high temperatures accompanied by a concomitant molecular weight increase due to the formation of triazolium salts via the reaction of triazole units with bromine terminal chain ends.
Introduction
The copper-catalyzed azide and alkyne cycloaddition (CuAAC) – a reaction often fulfilling the click criteria – introduced by Sharpless and coworkers in 20011 is a selective and orthogonal reaction that can function under mild conditions with high yields, a simple recovery of the product, compatibility with a wide range of solvents, and often excellent functional group tolerance and no side-products.2,3 The application of the CuAAC reaction in conjunction with living radical polymerization (LRP)4 has contributed to the rapid development of precision materials5 and polymer architectures such as functional polymers,6 block copolymers,7 complex macromolecular structures,8 networks9 and dendrimers.10The most commonly employed LRP techniques include atom transfer radical polymerization (ATRP),11 reversible addition–fragmentation transfer (RAFT) polymerization,12 and nitroxide-mediated radical polymerization (NMP).13 Especially, atom transfer radical polymerization (ATRP) is a highly versatile and efficient LRP technique allowing for the generation of polymers with pre-determined molecular weight, narrow molecular weight distribution, chain-end functionality, adjustable architecture and composition. Activators regenerated by electron transfer (ARGET) ATRP can be utilized to generate block copolymers using a large excess of reducing agent to obtain the activator from the deactivator, allowing transition metal concentrations at low ppm levels without loss of control.14 This advantage could have profound industrial implications for the synthesis of well-defined polymeric materials.
The arguably most prepared polymer architectures are linear, branched and cross-linked structures. A detailed understanding of the molecular changes occurring within precision tailored polymeric materials under the combined effect of thermal and mechanical stress is a key requirement for the processing of thermoplasts via e.g. extrusion and injection molding as well as alternative high-temperature shape-giving processes.
Linear or star block copolymers with various compositions have been synthesized via one of three methodologies, i.e. core-first, arm-first and chain extension.15 These synthetic methods differ from each other based on the sequence employed for the formation of the core and the arms. In the core-first approach, multifunctional initiators are used to grow arms via living polymerization techniques. Significant research effort has been made to achieve block copolymers with complex architectures via the core-first or arm-first approach and combinations of LRP and CuAAC.16 However, detailed knowledge regarding the thermal and thermomechanical stability of the resulting polymeric materials under industrially relevant processing conditions (e.g. melt extrusion) is critically required. We have commenced a research programme into the stability of initially linear and star-shaped RAFT-based polystyrene melts featuring mid-chain trithiocarbonate functions (i.e. a Z-group design) under thermal and thermomechanical stress. Our activities are designed to develop a detailed understanding of the mechanistic changes occurring during thermomechanical and thermal stress of precision polymeric materials while concomitantly understanding changes in their rheological and processing properties under stress.17,18 To the best of our knowledge, there exists no previous study assessing the stability of ATRP-based polymers under thermal stress and thermomechanical stress. In addition, most commercially available ATRP initiators leading to block copolymers and multi-arm star polymers incorporate ester functionalities, which are one of the groups at the center of our current investigation. Thus, any processing of polymers based on ATRP initiators containing ester or triazole functions which connect the polymer arms is covered by the results of our current investigation.
In the present study, we thus investigate the stability of linear polystyrene melts with a diblock homopolymer topology that carry mid-chain triazole and ester as well as bromine chain end functionalities under thermal and thermomechanical stress. The approach taken in the current study entails the detailed analysis of polymer structures of decreasing macromolecular complexity in order to isolate the effect of thermal stress on the individual functional entities. Initially, linear polystyrenes with specific functional groups (refer to Scheme 1 for an overview) were synthesized via ARGET ATRP using a bifunctional triazole and ester containing initiator (3.8 kDa ≤ Mn,SEC ≤ 125 kDa, 1.07 ≤ Đ ≤ 1.19). In addition – enabling the noted isolation of effects – polystyrenes of low molecular weight featuring an ω-bromine chain-end functionality, α,ω-ester-bromine functionalities, α,ω-dibromine/mid-chain ester functionalities as well as α,ω-dibromine/mid-chain ester and triazole functionalities were prepared. Finally, a well-defined triazole mid-chain functionalized polystyrene (with no other functionalities) was additionally synthesized via a combination of ARGET ATRP and CuAAC. To eliminate the effect of the bromine end-group functionalities of the α,ω-dibromine/mid-chain ester functional as well as α,ω-dibromine/mid-chain ester and triazole functional polystyrenes, the bromine termini are readily transformed into hydrogen end groups in the presence of tributyltin hydride.
 | ||
| Scheme 1 Experimental approach for understanding the behavior of modular ATRP polymers carrying various functional groups, specifically ester, triazole and bromine, under mechanical and thermal stress. | ||
The thermal and thermomechanical stability of the functional polystyrenes was studied at elevated temperatures (e.g. 180 or 200 °C) under an inert atmosphere. Low molecular weight variants of the polymers were subjected to identical thermal conditions as those applied during thermal stress as well as extrusion and rheometry of their long chain equivalents prior to MALDI-ToF analysis to explore the mechanism of degradation associated with variable functional groups.
In a subsequent step, the (modular) ATRP polymers were investigated via rheological experiments (G′(t), G′′(t)) to implement a link between the time dependent SEC data and potential processing conditions. Furthermore, rheological assessments are highly sensitive and feature – in contrast to the SEC experiments – a high time resolution (Δt < 1 min) to monitor molecular changes within the polymer structure. Weight-average molecular weight dependency of zero shear viscosity, η0 ∼ Mw3.4, as shown by the reptation theory for linear homopolymers,19–22 provides a fine sensor with a fast time resolution for possible polymer cleavage reactions.
Experimental section
Synthesis
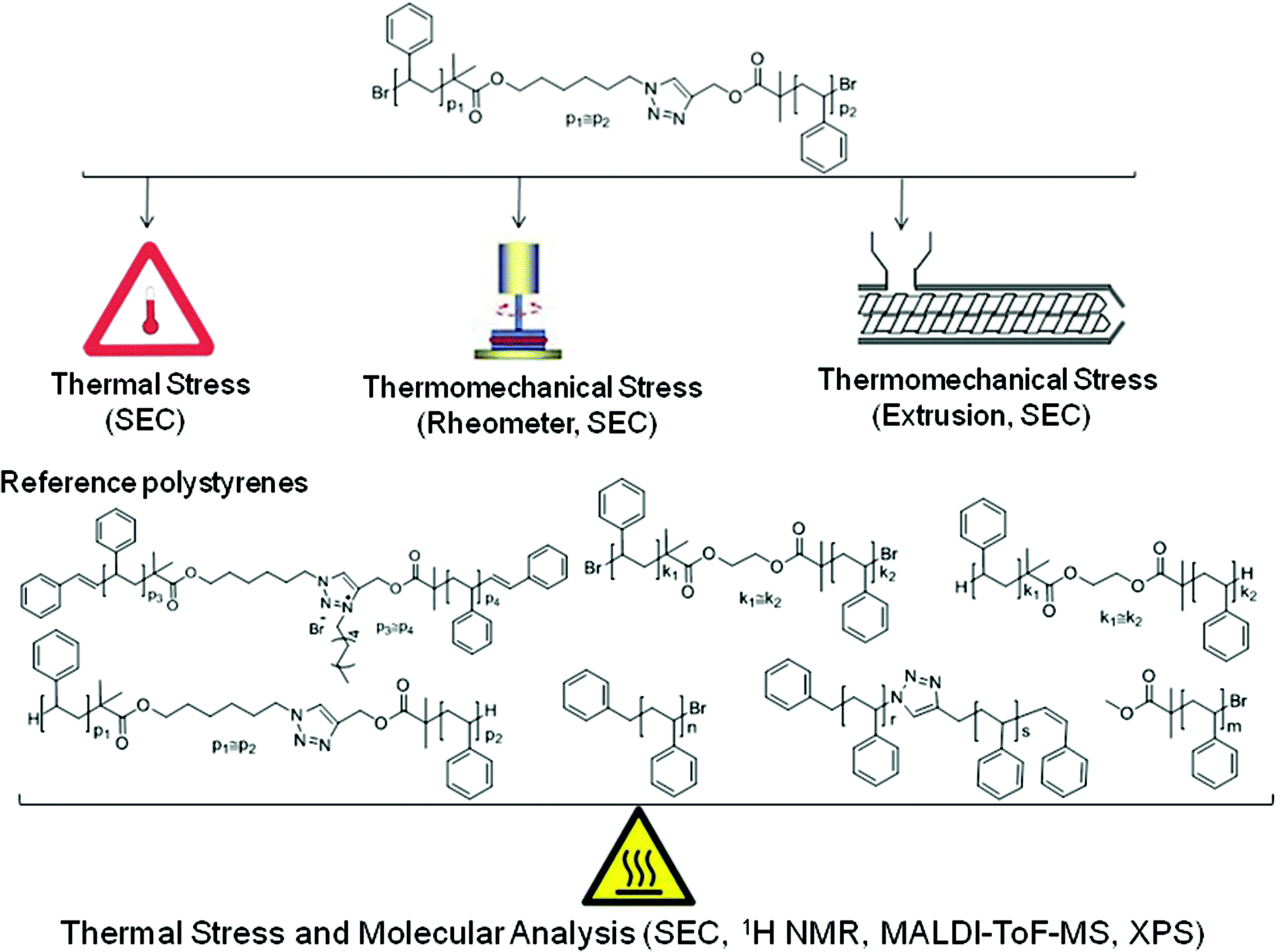
6-Azidohexyl 2-bromo-2-methylpropanoate (4) and prop-2-yn-1-yl 2-bromo-2-methylpropanoate (5) were synthesized according to literature procedures.23,24 The characterization data of all prepared polymers can be found in Fig. 1 and 2 (full molecular weight distribution) as well as in Tables 1 and 2 (numeric data). The ESI† contains further details of the employed materials, synthetic procedures, instrumentation, as well as additional detailed characterization. The interested reader is kindly referred to the ESI† section for in-depth synthetic information as well as further characterization data.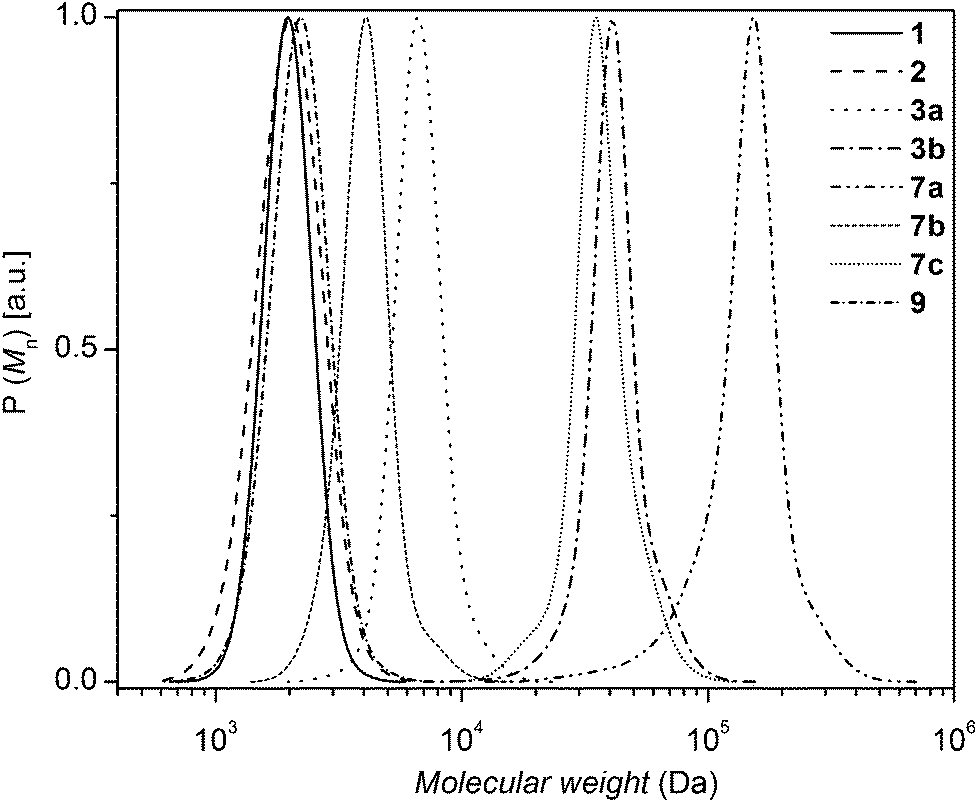 | ||
| Fig. 1 SEC traces of the synthesized functional linear polystyrenes employed in the stability assessments, rheometry and extrusion experiments prior to thermal/mechanical stress: 1 (Mn,SEC = 2 kDa, Đ = 1.06), 2 (Mn,SEC = 2 kDa, Đ = 1.10), 3a (Mn,SEC = 6.5 kDa, Đ = 1.06), 3b (Mn,SEC = 39 kDa, Đ = 1.08), 7a (Mn,SEC = 124.7 kDa, Đ = 1.19), 7b (Mn,SEC = 3.8 kDa, Đ = 1.07), 7c (Mn,SEC = 33.8 kDa, Đ = 1.09), and 9 (Mn,SEC = 2.1 kDa, Đ = 1.08). The molecular weight distributions of the polymers 8, 10 and 11 are depicted in Fig. 2. | ||
 | ||
| Fig. 2 SEC traces of 8 (Mn,SEC = 2 kDa, Đ = 1.06) and 10 (Mn,SEC = 2.1 kDa, Đ = 1.08) building blocks as well as the PS-b-PS diblock homopolymer 11 (Mn,SEC = 4.4 kDa, Đ = 1.08). | ||
| Polymera | Monomer | [M0]/[I0] | Initiator | Reaction time (h) | M n | Đ |
|---|---|---|---|---|---|---|
a [I]0/[Me6TREN]0/[CuBr2]0/[Sn(EH)2]0 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.1:0.1:0.5; at 90 °C.
b Determined via RI detection SEC using linear PS standards. :0.1:0.1:0.5; at 90 °C.
b Determined via RI detection SEC using linear PS standards.
|
||||||
| 1 | Styrene | 50 | BenzBr | 2.5 | 2 kDa | 1.06 |
| 2 | Styrene | 50 | MBriB | 2.5 | 2 kDa | 1.10 |
| 3a | Styrene | 50 | EbBriB | 2.5 | 6.5 kDa | 1.06 |
| 3b | Styrene | 1000 | EbBriB | 48 | 39 kDa | 1.08 |
| 7a | Styrene | 2500 | 6 | 64 | 125 kDa | 1.19 |
| 7b | Styrene | 50 | 6 | 2.5 | 3.8 kDa | 1.07 |
| 7c | Styrene | 1000 | 6 | 48 | 33.7 kDa | 1.09 |
| 9 | Styrene | 50 | TMSPrBr | 2.5 | 2.1 kDa | 1.08 |
| Polymer | M n | Đ |
|---|---|---|
| a Determined via RI detection SEC using linear PS standards. | ||
| 8 | 2 kDa | 1.08 |
| 10 | 2.1 kDa | 1.08 |
| 11 (8-b-10) | 4.3 kDa | 1.08 |
| 12a | 6.1 kDa | 1.06 |
| 12b | 38.5 kDa | 1.08 |
| 13a | 120.1 kDa | 1.19 |
| 13c | 32.5 kDa | 1.09 |
| 14 | 4.1 kDa | 1.06 |
Results and discussion
ATRP has emerged as one of the most powerful synthetic techniques in polymer chemistry, being tolerant towards many solvents, functional groups and impurities often encountered in industrial systems.25 Several variants of the ATRP technique have been developed to overcome the relatively large amounts of copper catalyst present in a classic ATRP system. The new ATRP initiation techniques such as ARGET use very small amounts of catalyst, which could have a beneficial impact on the application of ATRP in industry. In particular, the combination of ATRP and CuAAC allows for the synthesis of well-defined polymers with precise functionality. However, it is mandatory to understand the stability as well as reactivity of ATRP-made polymers, the stability of the functional groups typically present in the initial ATRP initiators (ester linkages) and the triazole unit generated by CuAAC under both thermal and mechanical stress to eventually allow ATRP and CuAAC based polymers to be processed (Scheme 2). | ||
| Scheme 2 Synthetic strategy for the preparation of different linear polystyrenes employed in the present study carrying selected functionalities in a mid-chain and terminal position. (a) Styrene, CuBr2, Me6TREN, Sn(EH)2, anisole, 90 °C; (b) CuSOx5H2O, Na ascorbate, DMF, room temperature; (c) NaN3, DMF, room temperature; (d) TBAF, THF, room temperature; (e) tributyltin hydride (TBTH), toluene, 85 °C, 4.5 h; (f) decyl bromide, DMF, 100 °C, 16 h. | ||
Styrene was polymerized under ARGET ATRP conditions in anisole at 90 °C using the CuBr2/Me6TREN catalytic system and Sn(EH)2 as a reducing agent. Linear mid-chain triazole functional polystyrenes were obtained using 6 as an initiator via the core-first approach to ensure that each chain contains triazole functionality. Commercially available benzyl bromide (BenzBr), methyl 2-bromoisobutyrate (MBriB), ethylene bis(2-bromoisobutyrate) (EbBriB) and 3-(trimethylsilyl)propargyl bromide (TMSPrBr) molecules were employed as initiators for the ARGET ATRP of styrene under the above conditions. First of all, ω-bromine functional polystyrene (1) was synthesized to investigate the effect of the bromine atom during thermal stress. Secondly, the stability of ester and bromine functionalities was jointly studied using α,ω-ester-bromine functional polystyrene (2). Thirdly, α,ω-dibromine functional and mid-chain ester functional linear polystyrene (3a and 3b) were prepared. Finally, the bromine end-group functionalities of the polystyrenes (3a and 3b) and polystyrenes (7a and 7c) were transformed into hydrogen termini in the presence of tributyltin hydride to obtain the polystyrenes (12a and 12b) as well as polystyrenes (13a and 13c), respectively, allowing for the investigation of the stability of the ester group in the absence of bromine end groups. The polystyrenes were subjected to SEC analysis, indicating monomodal molecular weight distributions with low molar mass dispersities. The resulting molecular weight distributions are depicted in Fig. 1 and the associated reaction conditions are collated in Tables 1 and 2. Linear polystyrenes carrying ester and bromine functionalities with low molecular weight were prepared for 1H NMR and MALDI analyses as reference polymers to understand the cleavage processes on the molecular level.
The PS-b-PS diblock homopolymer 11 was characterized by 1H NMR, FT-IR spectroscopy and SEC. As illustrated in Fig. 2, a complete shift of the distribution towards higher molecular weight was observed indicating the formation of the diblock homopolymer.7 The SEC trace of diblock homopolymer (11) indicates the presence of very small amounts of precursor polymer, associated with some unavoidable loss of bromine functionality during the ATRP process or non-stoichiometric conditions.
Assessment of polymer stability and cleavage processes
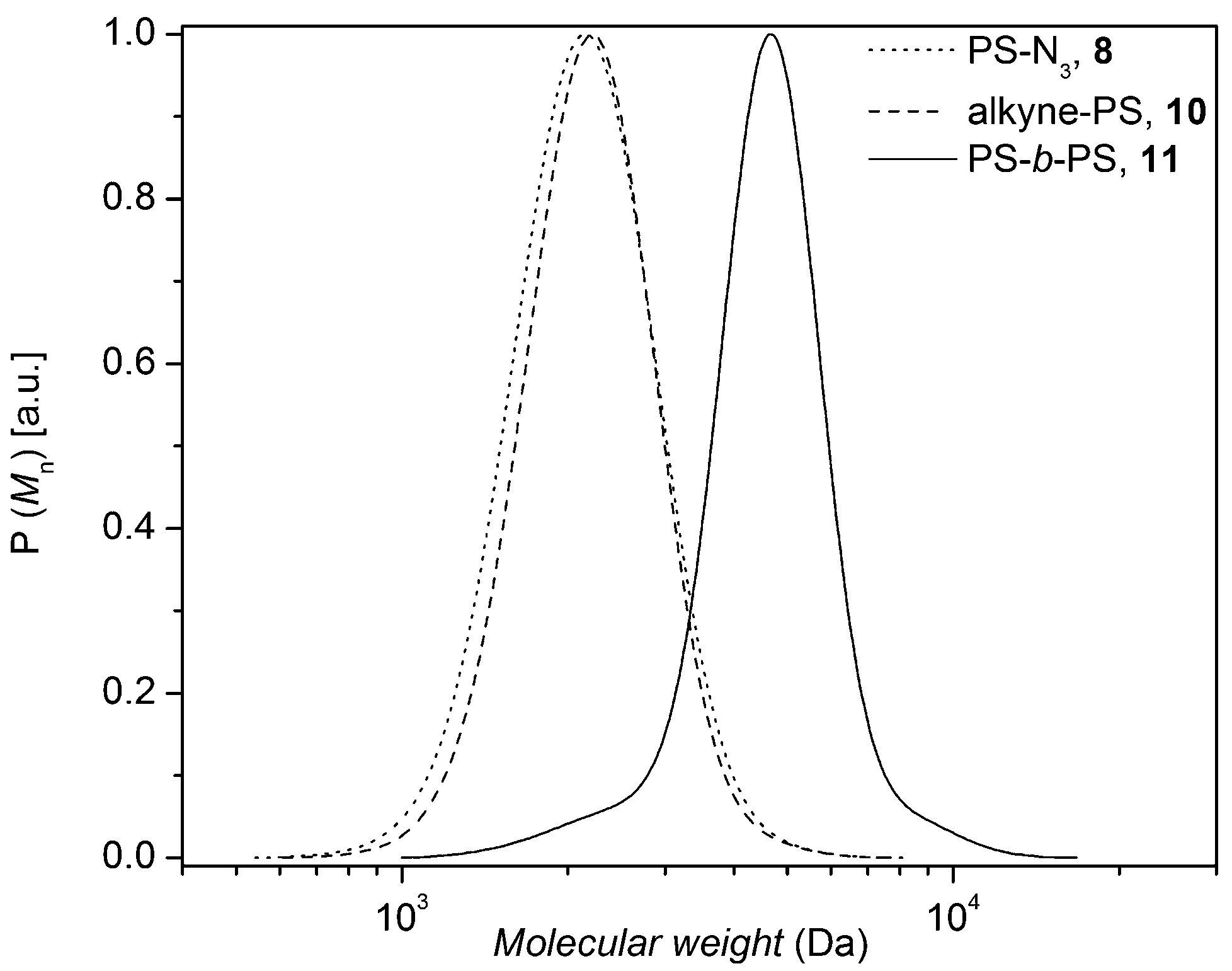
Having a polymer library with well-defined functional groups at hand which allows for a systematic investigation of the effect that different structural elements have on the degradation process and the polymer stability, thermal stress experiments were initially carried out. Understanding the degradation of polymer systems under an oxygen rich atmosphere is rather complex.26–28 Polymer extrusion of thermoplasts is typically conducted in the melt at elevated temperatures (e.g. 200 °C) and pressures (e.g. 300 bar) and thus the oxygen content is relatively low. The thermal stability of the prepared functional linear polymers has been studied at various temperatures under inert atmospheres to mimic the atmospheric conditions under extrusion as closely as possible. A time resolved SEC analysis allows for estimating the thermal stability of linear polystyrenes with several functionalities as a function of time, yet with a limited time resolution.The assessment of the stability of the various macromolecular species is commenced with the most complex system, i.e. polystyrene 7a which features (i) a triazole functionality in mid-chain position, (ii) two ester groups on either side of the triazole ring and next to the styryl chains and (iii) a Br-terminus on either end. The cleavage kinetics of the polystyrene 7a were investigated under isothermal conditions in the melt at different temperatures for pre-set time intervals. For this purpose, the number-average molecular weights, Mn, of the polystyrene 7a were determined via SEC, indicating that the thermal degradation becomes prominent after 2 h and 1 h at 180 °C and 200 °C, respectively. The change in the molecular weight distribution as a function of time is depicted in Fig. 3 and in Fig. S5 (refer to the ESI†) for degradation at 180 °C and 200 °C for polystyrene 7a, respectively, indicating an increasing polydispersity index (Đ) with time, congruent with the formation of higher molecular weight species as well as lower molecular weight species. Subsequently, the degradation products originating from the polymer were systematically investigated via SEC, 1H NMR and MALDI-MS analyses in comparison to the reference polymers (1, 2, 3a, 3b, 11, 12a, 12b, 13a and 13c).
 | ||
| Fig. 3 SEC traces of polystyrene 7a after thermal treatment at 180 °C under an argon atmosphere as a function of time; 0 h (Mn = 119.2 kDa, Đ = 1.19), 2 h (Mn = 112.6 kDa, Đ = 1.25), 4 h (Mn = 109.2 kDa, Đ = 1.32), 8 h (Mn = 109 kDa, Đ = 1.35), 16 h (Mn = 113.5 kDa, Đ = 1.36), 24 h (Mn = 108.4 kDa, Đ = 1.48). All peaks are normalized to the total peak area. | ||
In order to understand the (apparent) complex degradation behaviour of polystyrene 7a under thermal stress and to isolate the contributions of the individual structural elements to the degradation process, the effect of the secondary alkyl halide and ester functionality at the chain-end position was initially investigated. Polystyrenes 1 (ω-bromine functional) and 2 (α,ω-ester-bromine functional) have been kept at 200 °C under an argon atmosphere for 24 h before SEC analysis (refer to Fig. S6 in the ESI†). For both polymers, no reduction in molecular weight was observed. In addition to SEC analysis, the thermally treated polystyrenes 1 and 2 were analyzed by 1H NMR and MALDI-MS. The 1H NMR spectra of polystyrenes 1 and 2 recorded after thermal treatment indicate that the bromine atoms were eliminated from the polystyrene structure generating unsaturated species as well as no effect on the stability of the polystyrene backbone (see Fig. S7 and S8 in the ESI†).
In agreement with a recent report of Ladavière et al.,29 bromine terminated polystyrene is subjected to HBr elimination during the MALDI ionization processes, leading to unsaturated species. In addition, Ladavière et al. have demonstrated the possibility to substitute the halide atom by a hydroxide moiety in the presence of water traces during the sample preparation. Further, it is important to note that this substitution is promoted by the presence of silver ions, usually used as a cationizing agent for the MALDI-MS analysis of polystyrene. Therefore, if only signals characterized by the HBr elimination are observed only in the mass spectrum, bromine elimination occurred both prior to MALDI sample preparation and during analysis. In contrast, the presence of a distribution at 18 mass units higher relative to the unsaturated species confirms the presence of the halide atom at the moment of MALDI sample preparation.
The mass spectra for polystyrenes 1 and 2 before thermal treatment are presented in Fig. S9a† and Fig. 4a, respectively. The main distribution is attributed to the unsaturated species in both cases. The observation of small signals at 18 mass units higher, i.e. m/z 2296 and 2202 (for clarity, the most intense signal (m/z 2206) of this isotopic distribution is also assigned in the MS spectra) respectively, confirms the presence of the halide before MALDI analysis (refer to Schemes 3 and S1† for mechanistic details). For polystyrene 1 and 2 after thermal treatment (Fig. S9b† and Fig. 4b), the recorded mass spectra present also as main distribution the unsaturated species, however the signals at 18 mass unit higher are not present anymore, confirming that the HBr elimination has taken place before the ionization processes, as already attested by 1H NMR (see Fig. S7 and S8†). Moreover, for polystyrene 2, an additional species appears (refer to Fig. 4b) at 14 mass units lower than the unsaturated polystyrene. This ion series is consistent with the hydrolysis of the methylester moiety under thermal stress.
 | ||
| Fig. 4 MALDI-MS spectra of ester and bromine functionalized polystyrene 2 before (a) and after (b) thermal treatment at 200 °C for 24 h under an argon atmosphere. | ||
 | ||
| Scheme 3 Description of the processes involved following the MALDI analysis of polymer 2 (a) before and (b) after thermal treatment (200 °C, 24 h). | ||
To explore the possible mechanism of degradation associated with the ester group in polymer 7a, linear polystyrene with a mid-chain ester and end chain bromine functionality was synthesized via ARGET ATRP utilizing a commercially available bifunctional initiator, affording polystyrene 3a with a molecular weight of 6.1 kDa and a polydispersity index of 1.06. Polystyrene 3a was subjected to identical thermal degradation conditions (i.e. 200 °C for 24 h under an inert atmosphere) as polystyrenes 1 and 2 prior to SEC, 1H NMR and MALDI-MS analyses. Inspection of Fig. 5 clearly indicates a decrease in the peak-average molecular weight from Mp = 6.6 kDa to Mp = 3.3 kDa for polystyrene 3a as a consequence of the appearance of a substantial amount of material within the lower molecular weight region. The 1H NMR spectrum indicates the disappearance of resonances associated with the CH-Br proton and the formation of the unsaturated species as well as a decrease in the intensity of the CH2 proton resonances adjacent to the ester group (see Fig. S10 in the ESI†). While the SEC analysis suggests that the ester functional polymers are cleaved by mid-chain scission at elevated temperature, a more detailed molecular proof is provided by a mass spectrometric investigation. Based on the MALDI mass spectrum depicted in Fig. 6b, the potential mechanism involved during the thermal treatment of polystyrene 3a is presented in Scheme 4b. In addition to the elimination of both bromines, a 1,5-hydrogen shift involving a six membered ring occurred giving rise to the cleavage of the original polystyrene, leading to a carboxylic acid (m/z 2898) end group and a polystyrene with a vinyl function as end-group. However, the latter is not observed by MALDI analysis, yet another ion distribution (m/z 2984) is detected after the thermal treatment.
 | ||
| Fig. 5 SEC traces of polystyrene 3a before and after thermal treatment at 200 °C for 24 h under a nitrogen atmosphere. | ||
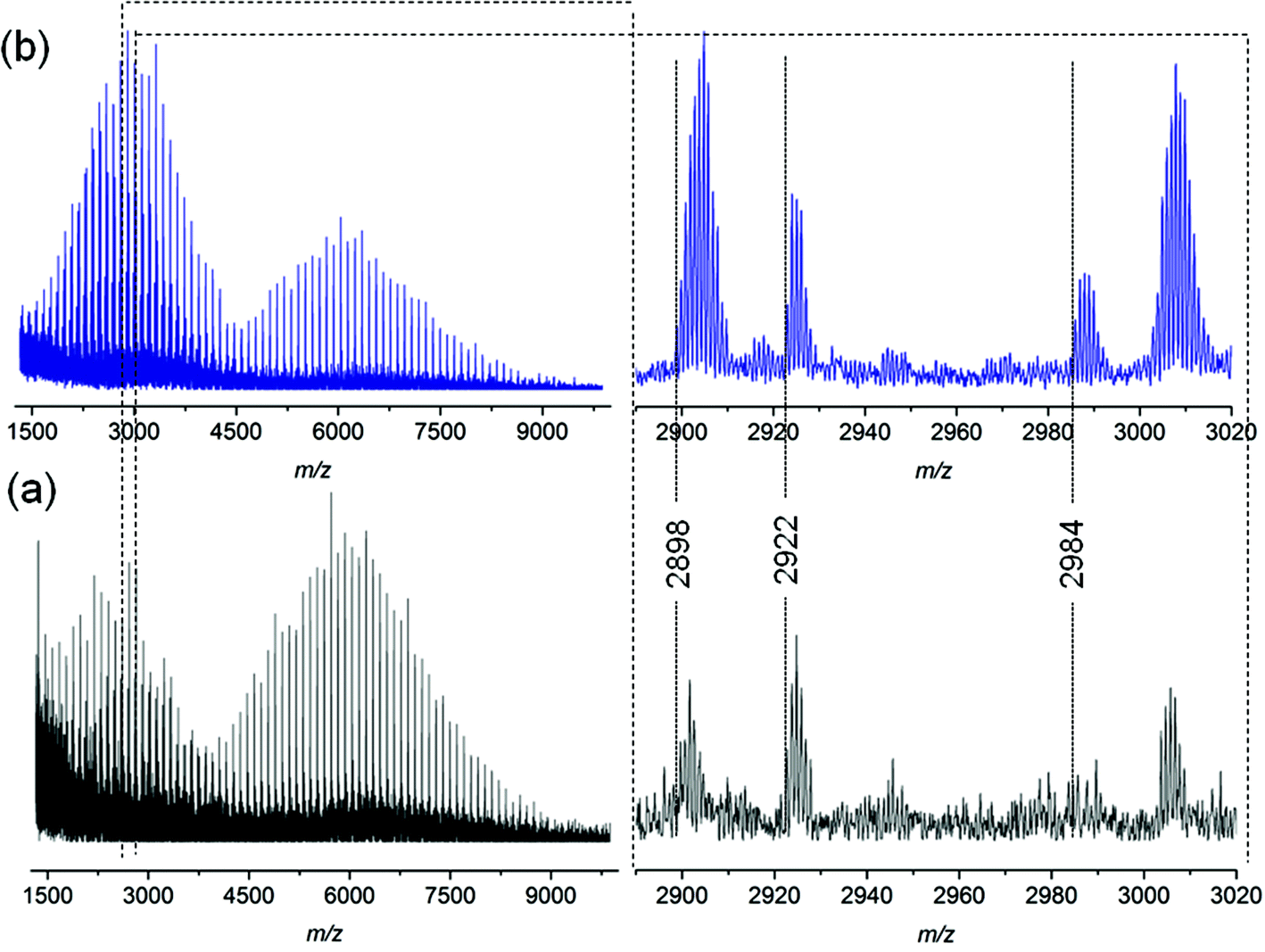 | ||
| Fig. 6 MALDI-MS spectra of polymer 3a before (a) and after (b) thermal treatment at 200 °C for 24 h under an argon atmosphere. | ||
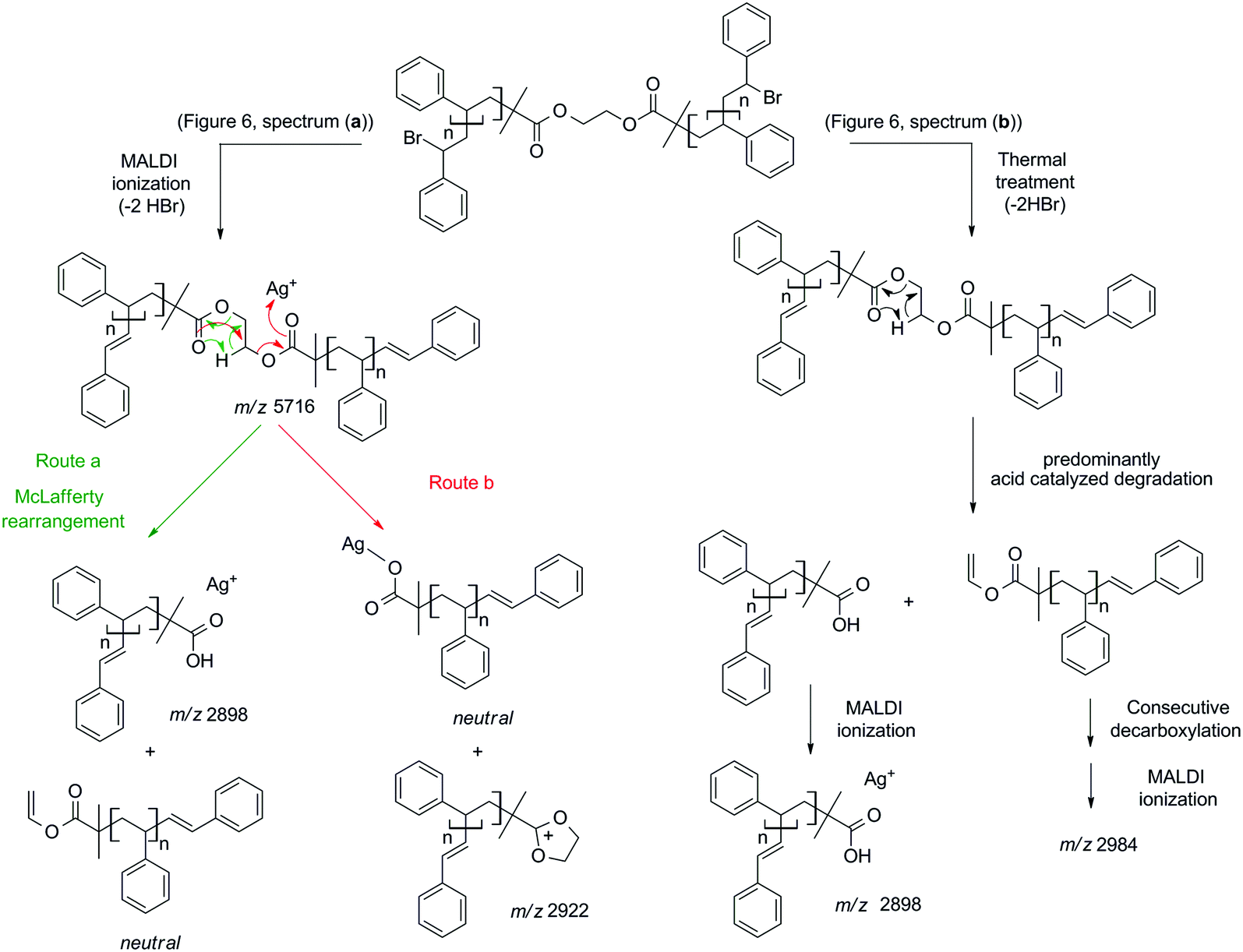 | ||
| Scheme 4 Description of and mechanistic pathway to the ion structures observed by MALDI-MS analysis (Fig. 6) of polystyrene 3a (a) before and (b) after thermal treatment (200 °C, 24 h). | ||
This new distribution is suspected to originate from a consecutive decarboxylation of the vinyl terminal species (see Scheme 4). The pericyclic rearrangement (1,5-hydrogen shift) can also be induced during the MALDI ionization processes.30,31 As a consequence, the MALDI mass spectrum of polystyrene 3a before thermal treatment (Fig. 6a) already shows the presence of a low intensity distribution of polystyrene with carboxylic end-groups (m/z 2898) at a lower mass-to-charge ratio (Scheme 4a, route a). The origin of the second ion distribution (m/z 2922) at lower mass range for the untreated polystyrene is explained by a charge localized fragmentation (Scheme 4a, route b) as reported in the literature.32 Finally, after the thermal treatment of polystyrene 3a, an inversion of the relative intensities of both distributions is observed. Since all mass spectra have been recorded under exactly the same experimental conditions, the modification of the intensities unambiguously confirms the degradation of the polystyrene as attested by SEC. Moreover, the structures of the low mass-to-charge polystyrene ions are evidence for the ester driven degradation. It was initially assumed that the ester group would be stable at elevated temperature as ester pyrolysis only occurs above 300 °C.33 A Chugaev-type elimination reaction of aliphatic esters forming acid and olefin usually prevails when a β-hydrogen atom is available in the alcohol part of the ester,34 yet in the presence of bromine functions that lead to the elimination of HBr, an acid-catalyzed ester degradation prevails. The newly formed end groups decompose by secondary reactions, e.g. decarboxylation, decarbonylation and anhydrate formation.35
Subsequently, the stability of the polystyrene featuring a triazole ring as mid-chain function (11) was investigated by exposing it to 200 °C for 24 h under an inert atmosphere followed by SEC, 1H NMR, MALDI-MS and XPS analysis. After applying thermal treatment as noted above, SEC analysis revealed that the molecular weight distributions of polystyrene 11 remains unchanged (Fig. S11 in the ESI†). The SEC result thus indicates that the triazole mid-chain functional polymer was highly stable under these conditions. The 1H NMR spectrum also indicates that the resonances associated with CH2 next to triazole unit are still present after thermal treatment (see Fig. S12 in the ESI†).
The stability of triazole mid-chain polystyrene is additionally confirmed by MALDI-MS analysis. As depicted in Fig. 7, the mass spectra recorded for polystyrene 11 before and after thermal treatment do not exhibit any changes in both the width of the mass distributions nor the nature of the observed signals, resulting in completely identical spectra confirming a high thermal stability of the triazole group under inert atmosphere conditions.
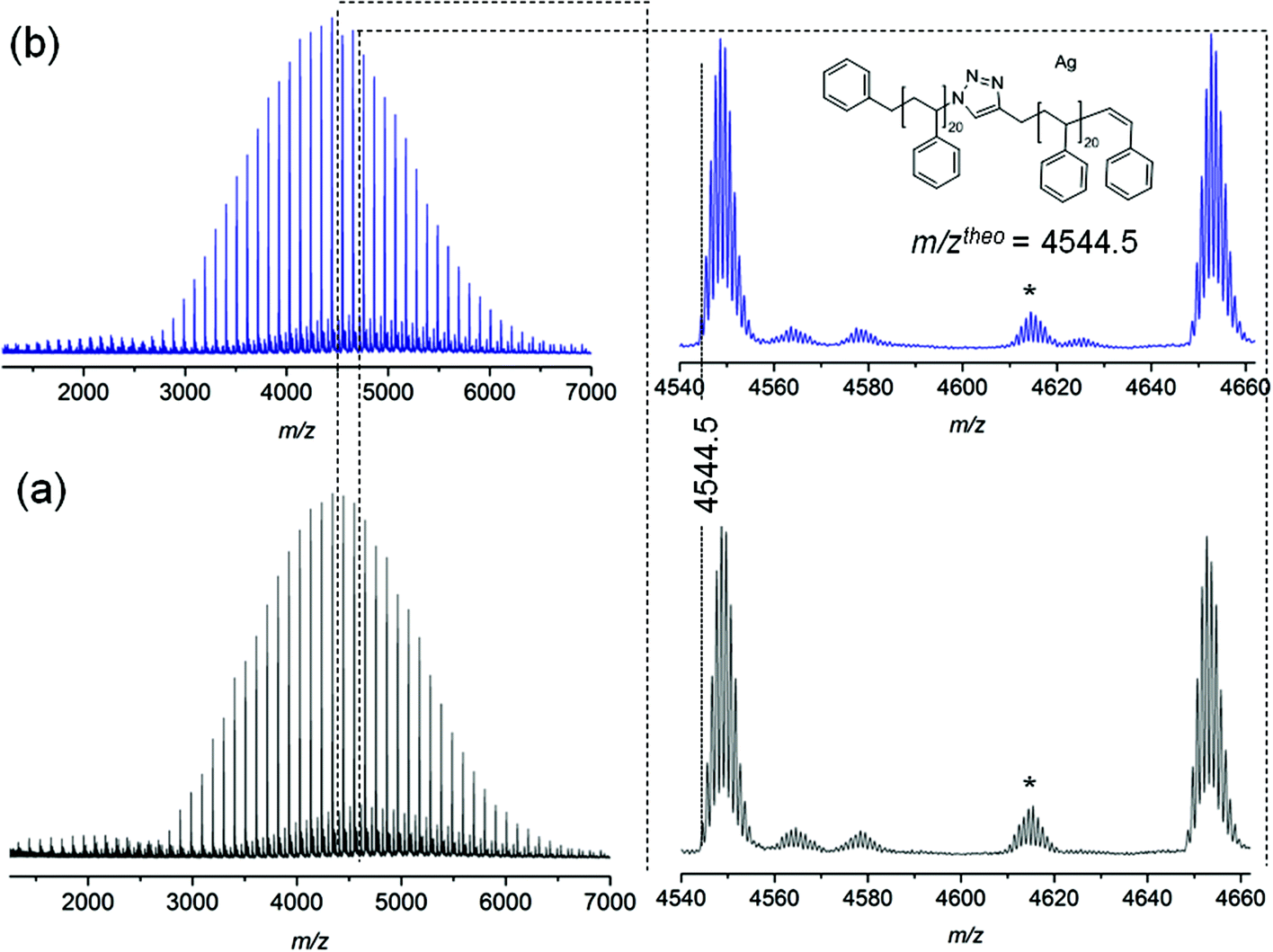 | ||
| Fig. 7 MALDI-MS spectra of polymer 11 before (a) and after (b) thermal treatment at 200 °C for 24 h under an argon atmosphere. *Refers to cationization of PS by Ag2NO3+. | ||
The stability of the triazole unit after thermal treatment was additionally successfully evidenced by XPS. The N 1s spectrum of polystyrene 11 (refer to Fig. S13 in the ESI†) indicates two peaks, one at 400.1 eV (C–N–N–, C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–), which can be attributed to the triazole unit36,37 and a second one at 401.6 eV which is probably due to a protonated species (C–HN+). After thermal treatment of polystyrene 11, the same spectral region can be evaluated (refer to Fig. S13 in the ESI†), indicating that the total amount of nitrogen is stable and exhibits quantities of close to 0.8 (±0.1) at% before and 0.6 (±0.1) at% after thermal treatment, evidencing the stability of the triazole unit. An assessment of the SEC, NMR and XPS data thus demonstrates that the triazole mid-chain functional linear polystyrene 11 does not suffer from degradation, thus indicating that the absence of the mid-chain functional position of the two ester units is critical for the stability of polystyrene 7a.
N–), which can be attributed to the triazole unit36,37 and a second one at 401.6 eV which is probably due to a protonated species (C–HN+). After thermal treatment of polystyrene 11, the same spectral region can be evaluated (refer to Fig. S13 in the ESI†), indicating that the total amount of nitrogen is stable and exhibits quantities of close to 0.8 (±0.1) at% before and 0.6 (±0.1) at% after thermal treatment, evidencing the stability of the triazole unit. An assessment of the SEC, NMR and XPS data thus demonstrates that the triazole mid-chain functional linear polystyrene 11 does not suffer from degradation, thus indicating that the absence of the mid-chain functional position of the two ester units is critical for the stability of polystyrene 7a.
Inspection of the 1H NMR and MALDI-ToF spectra for polystyrenes 1, 2 and 3 clearly indicate that HBr elimination occurs during thermal treatment. As polystyrene 7a indicates the formation of lower and higher molecular species under thermal stress (refer to Fig. 3), it is important to investigate if there is an additional effect on the decomposition of the ester group in polystyrene 7a caused by the elimination of HBr at high temperatures during thermal stress due to the acidic character of HBr. Thus, the bromine end group functionalities of the ATRP-made polymers were transformed to hydrogen end groups in the presence of tributyltin hydride.38 First of all, the bromine functionalities of polystyrenes 3a (Mn,SEC = 6.1 kDa, Đ = 1.06) and 3b (Mn,SEC = 39 kDa, Đ = 1.08) were converted into hydrogen atoms to obtain polymers 12a and 12b in the presence of Bu3SnH, respectively. The low molecular weight polystyrene 12a was prepared for subsequent 1H NMR analysis. The 1H NMR spectrum (refer to Fig. S14 in the ESI†) of polystyrene 12a indicates the disappearance of resonances associated with the CH–Br proton between 4.30 and 4.60 ppm, clearly evidencing the successful conversion of bromine to hydrogen at the chain terminus. Polystyrene 12b (for SEC analysis refer to Fig. S15 in the ESI†) was subjected to identical thermal degradation conditions (i.e. 200 °C for 24 h under an inert atmosphere) as polystyrene 3a and 3b. Inspection of Fig. S15† clearly indicates that thermal degradation of polystyrene 12b in the absence of HBr is substantially less favorable after 24 h at 200 °C compared to polystyrenes 3a and 3b, showing a very small shoulder in the low molecular weight region due to Chugaev-type elimination, while polystyrenes 3a and 3b display a new peak in the low molecular weight region (refer to Fig. 5 for 3a and refer to Fig. S15 for 3b in the ESI†). These observations indicate that the HBr elimination during thermal degradation is the main effect on the ester degradation of polystyrenes 3a and 3b.
It is mandatory to translate the above observation to the assessment of polystyrene 7a. The bromine end group functionalities at the terminus of polystyrene 7a was removed in the presence of the Bu3SnH under identical reaction conditions as for polystyrenes 12a and 12b to obtain polymer 13a, carrying no bromine functionality. Polystyrene 13a was subjected to identical thermal degradation conditions (i.e. 200 °C for 24 h under an inert atmosphere) as polystyrene 7a prior to SEC analysis. The change in the molecular weight distribution as a function of time is depicted in Fig. S16,† indicating a small increase in Đ due to the formation of lower molecular weight species (Chugaev-type elimination of aliphatic esters). However, there is no indication of higher molecular weight species being formed, allowing the conclusion that HBr due to its acidic character has an additional effect on the decomposition of the ester group of polystyrene 7a. Further, the formation of higher molecular weight species may also be related to the bromine end group functionalities, a hypothesis that is explored in detail below.
In addition, polystyrene 7b was synthesized with low weight-average molecular weight for comparison with polystyrene 11 after thermal treatment. Polystyrene 7b was subjected to the same thermal treatment as 11 prior to 1H NMR (refer to Fig. S17†) and SEC (refer to Fig. S18†) analyses. The 1H NMR spectrum of polystyrene 7b indicates the disappearance of resonances associated with the ester protons as well as the protons adjacent to the triazole ring between 3.20 and 4.80 (refer to Fig. S17 in the ESI†), in agreement with ester degradation. Inspection of Fig. S18† clearly indicates a decrease in the number-average molecular weight from Mn = 3.8 kDa to Mn = 1.9 kDa for polystyrene 7b as a consequence of the appearance of a material with lower molecular weight. The mass spectrum for polystyrenes 7b before thermal treatment is depicted in Fig. 8. For polystyrene 7b before thermal treatment (Fig. 8a), the recorded mass spectrum presents as main distribution the unsaturated species. After the thermal treatment of polystyrene 7b, an inversion of the relative intensities of both distributions is observed. Since all mass spectra have been recorded under exactly the same experimental conditions, the change in the intensities shows the presence of a high intensity distribution of polystyrene with carboxylic end-groups (m/z 1962) at lower mass-to-charge ratio (Scheme 5), which unambiguously confirms the ester driven degradation as attested by SEC and 1H NMR. These combined results (refer to Fig. S11 and Fig. S18 in the ESI†) allow the conclusion that the presence of the HBr, which is formed during thermal treatment, enhances ester degradation. However, the ester degradation is faster for low molecular weight polystyrene 7b than the high polymer weight polystyrene 7a (refer to Fig. 3 and S18 in the ESI†). This observation could be associated with the lower glass transition temperature as well as higher diffusion coefficient of the low molecular weight polystyrene 7b compared to polystyrene 7a. Thus, polystyrene 7c (Mn = 33.7 kDa, Đ = 1.09) was synthesized via ARGET ATRP – featuring a higher molecular weight than polystyrene 7b – thus leading to less degradation than polystyrene 7b.
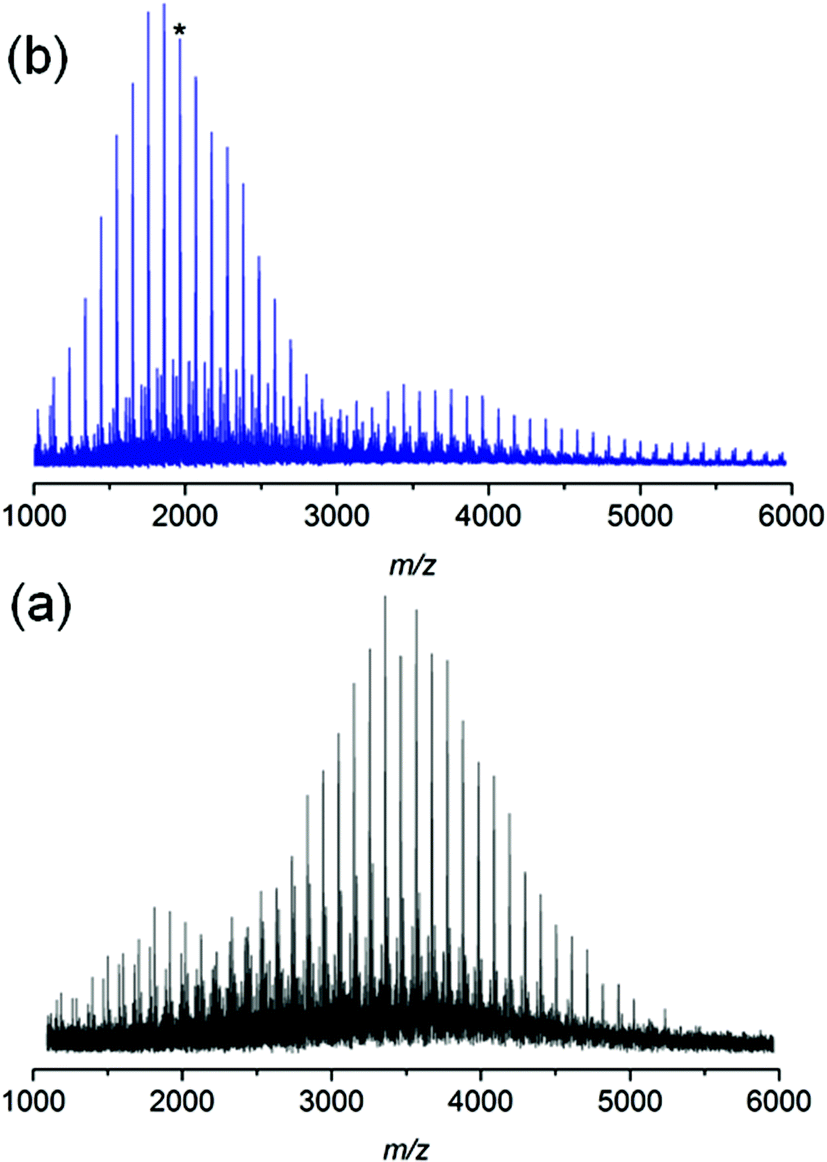 | ||
| Fig. 8 MALDI-MS spectra of polymer 7b before (a) and after (b) thermal treatment at 200 °C for 24 h under an argon atmosphere. After thermal treatment, (b), the presence of polystyrenes with carboxylic end-groups (the peak marked with an asterisk is at m/z 1962) at lower mass-to-charge ratios confirms the ester driven degradation process (Scheme 5). | ||
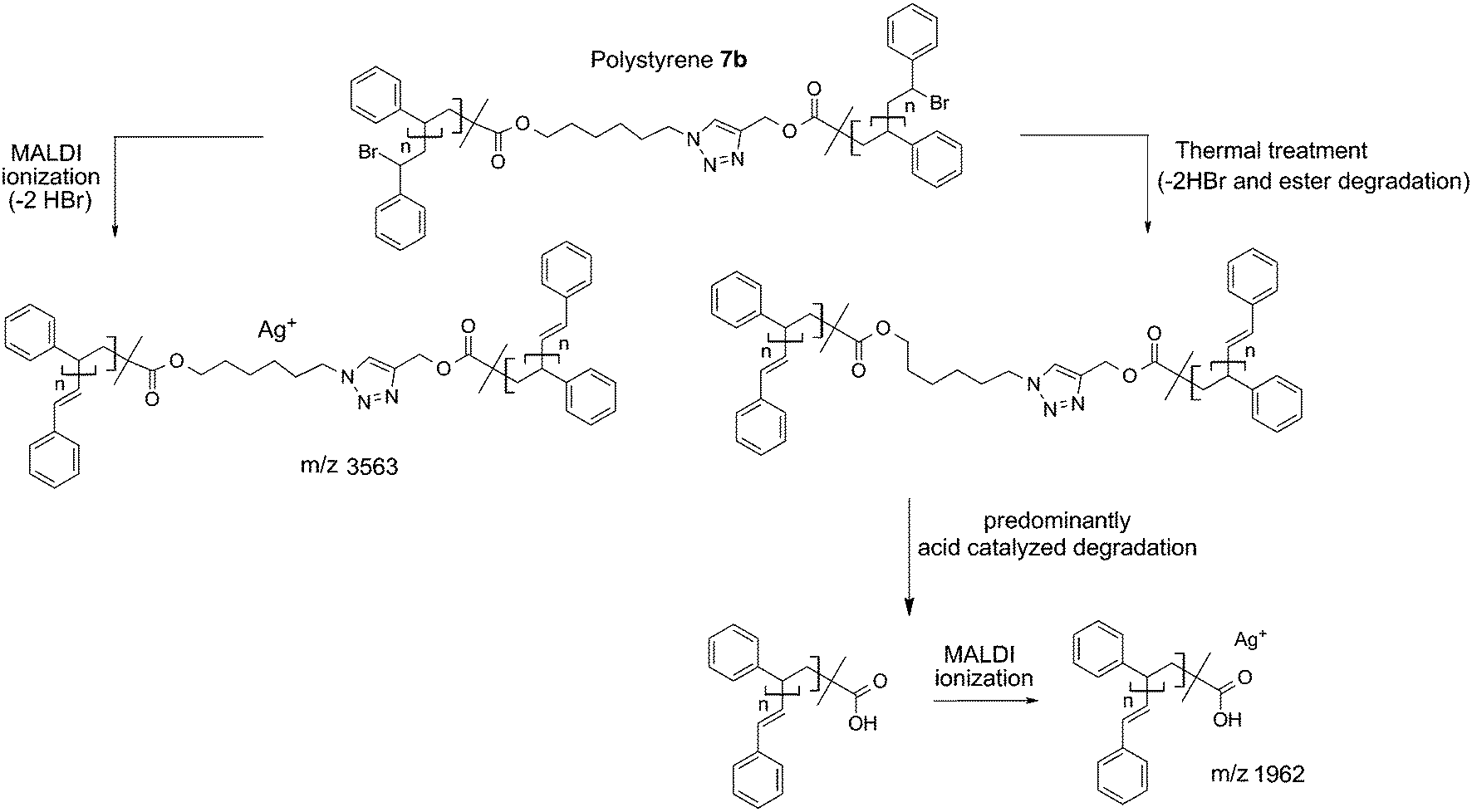 | ||
| Scheme 5 Depiction of the mechanistic pathway to the structures observed by MALDI-MS analysis of polystyrene 7b (a) before and (b) after thermal treatment (200 °C, 24 h). | ||
The same thermal conditions as for polystyrenes 7a and 7b were applied to polystyrene 7c. The change in the molecular weight distribution of polystyrene 7c as a function of time is depicted in Fig. 9, indicating an increasing Đ, congruent with the formation of lower molecular weight species resulting from ester decomposition.36 The bromine functionalities of the polystyrene 7c (Mn,SEC = 33.7 kDa, Đ = 1.09) were subsequently converted to hydrogen atoms to obtain polystyrene 13c, which was subjected to identical thermal treatment conditions as polystyrene 7c. The change in the molecular weight distribution of polystyrene 13c is depicted in Fig. 9, indicating a slight increase in Đ due to the formation of lower molecular weight species with time (Chugaev-type elimination of aliphatic esters). However, there is no indication for the formation of higher molecular weight species and the ester degradation is slow in the absence of HBr. Importantly, in order to investigate the effect of HBr elimination on the ester degradation, polystyrene 7a in the presence of decyl bromide – to increase the HBr content – was subjected to thermal treatment at 200 °C for 24 h under an argon atmosphere. The change in the molecular weight distribution is depicted in Fig. S19 in the ESI,† indicating that ester groups present in polystyrene 7a completely degrade in the presence of excess decyl bromide − to approximately half their initial Mn − at 200 °C within 24 h. This observation underpins the effect of HBr elimination on the decomposition of the ester group in polystyrene 7a.
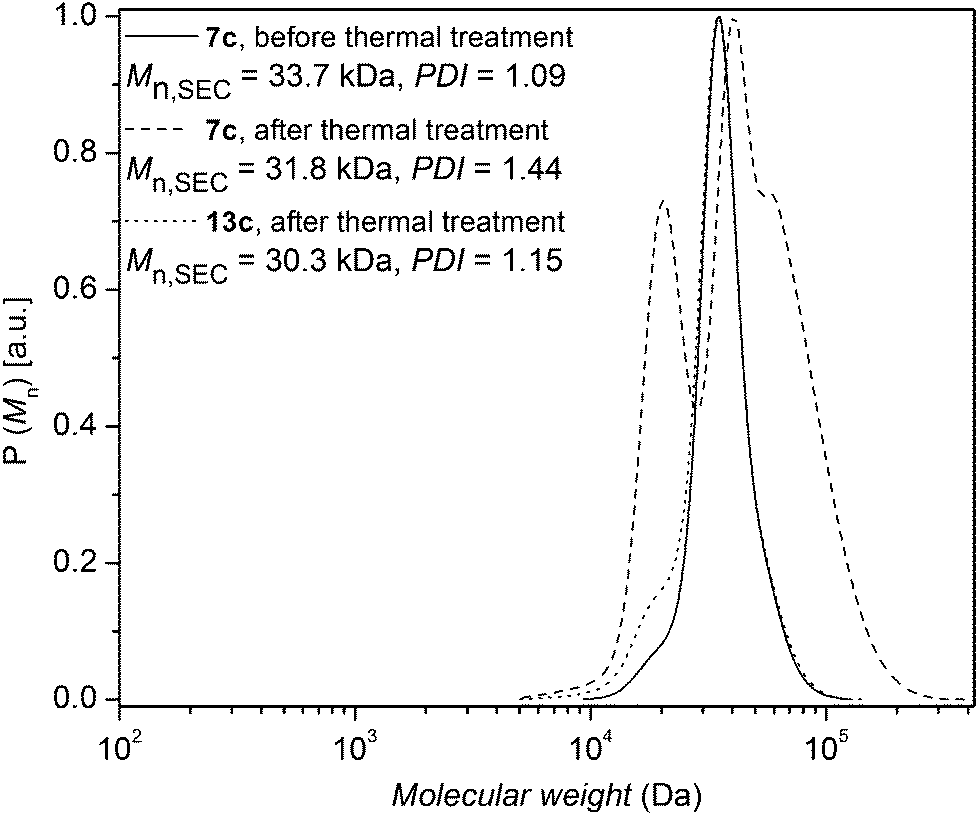 | ||
| Fig. 9 SEC traces of polystyrene 7c before thermal treatment as well as polystyrene 7c and polystyrene 13c after thermal treatment at 200 °C for 24 h under an argon atmosphere. | ||
These observations above demonstrate that (i) polystyrenes 7a and 7c – containing ester functionalities, bromine termini as well as a triazole units – form higher and lower molecular weight species upon thermal stress (refer to Fig. 3 and 9), (ii) there are only lower molecular weight species formed in the presence of ester and bromine end functional polystyrenes 3a and 3b (refer to Fig. 5 and Fig. S15 in the ESI†), (iii) when the bromine termini of polystyrene 3b are removed (12b), very small amounts of the low molecular weight species are observed (refer to Fig. S15 in the ESI†) due to the Chugaev-type elimination of aliphatic esters, (iv) in the absence of the bromine termini and in the presence of ester and triazole units, higher molecular weight species are no longer formed and a very small amount of lower molecular species is formed (polystyrenes 13a and 13c, refer to Fig. 9 and S16 in the ESI†) due to the Chugaev-type elimination of aliphatic esters. These observations suggest that there exists a reaction between the bromine end functionalities and the triazole unit. Interestingly, Drockenmuller and co-workers have proposed a straightforward synthetic route to a new class of ion conducting materials possessing 1,2,3-triazolium charged units via an efficient 1,2,3-triazole quaternization.39,40 The molecular weight of polystyrene 7a is too high for a meaningful end group analysis. Thus, polystyrene 7b was treated with a 20-fold excess of decyl bromide at 100 °C for 16 h in DMF to obtain polystyrene 14. After purification, the 1H NMR spectrum of polystyrene 14 clearly indicates the formation of the triazolium salt in the presence of decyl bromide. Further, the bromine atoms of polystyrene 14 are eliminated from the structure generating unsaturated species in solution (refer to Fig. 10). The 1H NMR spectrum indicates that characteristic resonances associated with the CH2 of the decyl group adjacent to the triazole ring at 4.88 ppm as well as new resonances related to CH in the triazole ring at 10.09 ppm emerged, evidencing the successful ligation between the triazole ring and the alkyl group in excellent agreement with previous observations in the literature40,41 (refer to Fig. 10). The SEC analysis of polystyrene 14 indicates that there is no change in the number-average molecular weight after treatment with decyl bromide (refer to Fig. S20 in the ESI†). The MALDI-ToF spectrum for polystyrene 14 clearly indicates the formation of the triazolium salt (refer to Fig. S21 in the ESI†).
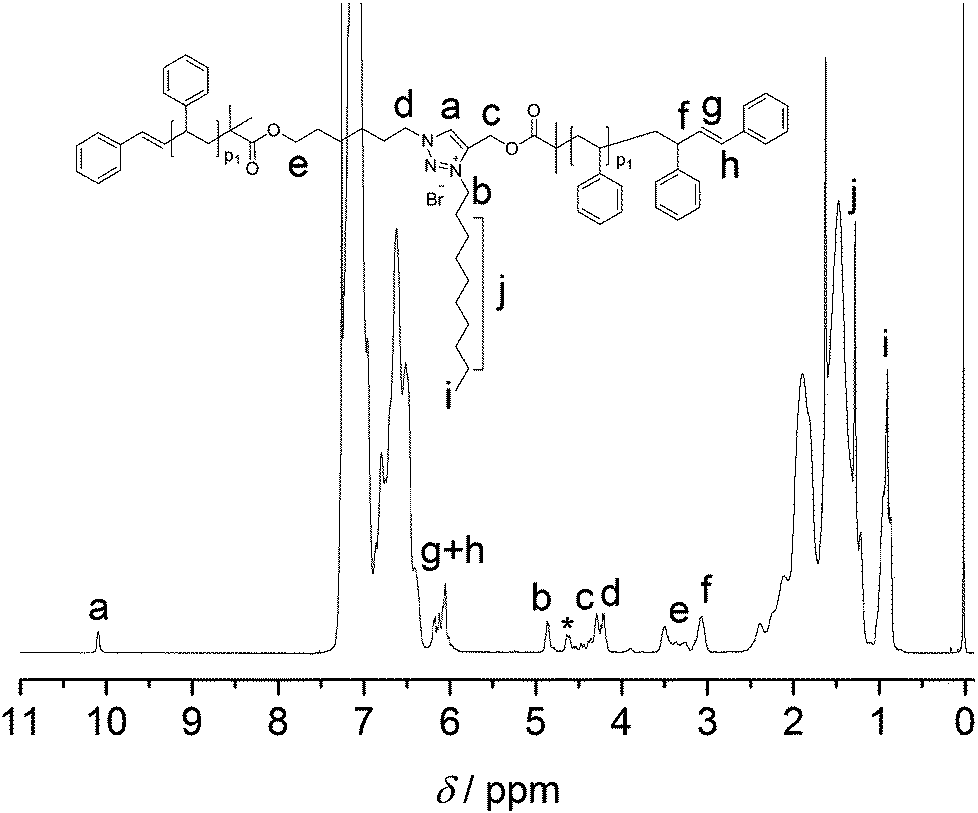 | ||
| Fig. 10 1H NMR spectra of polystyrene (7b) recorded in CDCl3 at ambient temperature after treatment with decyl bromide at 100 °C in DMF for 16 h under an argon atmosphere. The resonance marked with an asterisk is due to the remaining bromine end functional polystyrene 7b. | ||
Polystyrene 14 was subsequently subjected to thermal treatment at 200 °C for 24 h under an argon atmosphere (refer to Fig. S20 in the ESI†). Inspection of Fig. S20† clearly indicates that thermal degradation of the ester group of polystyrene 14 in the absence of HBr is substantially less favorable after 24 h at 200 °C compared to polystyrene 7b in Fig. S18 in the ESI,† showing a very small shoulder in the low molecular weight region due to the Chugaev-type elimination of aliphatic esters. Polystyrene 7b displays a new peak in the low molecular weight region.
These observations (refer to Fig. 3, 8 and 10) support the hypothesis that the formation of higher molecular weight species during thermal treatment of polystyrenes 7a and 7c is due to the formation of triazolium salts.
Rheological assessment and extrusion conditions
In a subsequent step − after establishing the level of stability of the individual functional groups at elevated temperatures − the melt rheological and extrusion behavior of polystyrene 7a featuring all functional groups as well as polystyrene 13a carrying ester and triazole functional groups was investigated.The polymers may undergo degradation caused by temperature under an inert atmosphere as well as additional mechanical stresses during processing. All dynamic measurements were carried out within the linear response domain adjusting the strain amplitude accordingly. The viscoelastic properties of the polymers were measured in small-amplitude oscillatory shear (SAOS) flow as a function of time at certain temperatures and strain amplitudes with a constant frequency.41,42 The rheological properties can be monitored continuously as a function of time (e.g. time resolution of several seconds) and therefore the degradation process of the samples is assessed by rheological properties, especially via the zero shear viscosity,  , throughout the degradation process applying the Cox–Merz rule.20,21 This assumption is reasonable as we investigate linear, monodisperse homopolymers. The following section will detail the results with respect to the zero shear viscosity and in comparison with the weight-average molecular weight obtained by SEC after applying a thermomechanical stress with the weight-average molecular weight (and distribution) of polystyrene 7a. The absolute values of the complex viscosities as a function of time at a temperature of 180 °C at a frequency of ω1/2π = 0.05 Hz and 15% strain amplitude for polystyrene 7a are used to determine the zero shear viscosity,
, throughout the degradation process applying the Cox–Merz rule.20,21 This assumption is reasonable as we investigate linear, monodisperse homopolymers. The following section will detail the results with respect to the zero shear viscosity and in comparison with the weight-average molecular weight obtained by SEC after applying a thermomechanical stress with the weight-average molecular weight (and distribution) of polystyrene 7a. The absolute values of the complex viscosities as a function of time at a temperature of 180 °C at a frequency of ω1/2π = 0.05 Hz and 15% strain amplitude for polystyrene 7a are used to determine the zero shear viscosity, 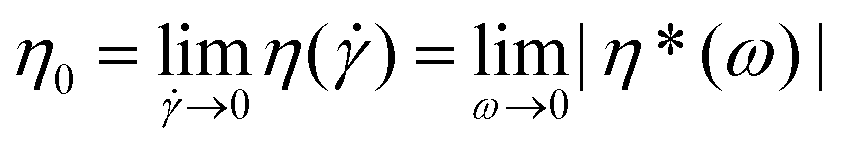 , assuming the validity of the Cox–Merz rule. These data are depicted in Fig. 11. The dynamic frequency sweep test depicted in Fig. S22† shows that the zero shear viscosity,
, assuming the validity of the Cox–Merz rule. These data are depicted in Fig. 11. The dynamic frequency sweep test depicted in Fig. S22† shows that the zero shear viscosity,  , has been reached for shear frequencies below 0.1 Hz for stable polystyrenes (e.g. after 24 hours under a time sweep test at 180 °C where maximum zero shear viscosity was reached, refer to Fig. 11). Inspection of Fig. 11 indicates that the zero shear viscosity of polystyrene 7a increases from the initial value of 10 kPa s up to 17 kPa s during three hours. After this period, the zero shear viscosity was constant during seven hours and subsequently decreases with increasing time at elevated temperatures. It is worth noting that for polystyrene with Mw larger than the critical molecular weight Mc (≈3Me; Me is ca. 18 kDa for polystyrene), reptation theory describes the dependence of the zero shear viscosity on weight-average molecular weight via a cubic power law. The zero shear viscosity of linear polystyrene was studied earlier43 at the reference temperature Tref = 170 °C, η0.Linear PS = 8.8 × 10−14Mw3.4, and the WLF shift factor, logaT = −5.47(T−170)/(119 + (T−170)), where T is given in Celsius. Using this shift factor, the zero shear viscosity for linear polystyrene was re-calculated at T = 180 °C:
, has been reached for shear frequencies below 0.1 Hz for stable polystyrenes (e.g. after 24 hours under a time sweep test at 180 °C where maximum zero shear viscosity was reached, refer to Fig. 11). Inspection of Fig. 11 indicates that the zero shear viscosity of polystyrene 7a increases from the initial value of 10 kPa s up to 17 kPa s during three hours. After this period, the zero shear viscosity was constant during seven hours and subsequently decreases with increasing time at elevated temperatures. It is worth noting that for polystyrene with Mw larger than the critical molecular weight Mc (≈3Me; Me is ca. 18 kDa for polystyrene), reptation theory describes the dependence of the zero shear viscosity on weight-average molecular weight via a cubic power law. The zero shear viscosity of linear polystyrene was studied earlier43 at the reference temperature Tref = 170 °C, η0.Linear PS = 8.8 × 10−14Mw3.4, and the WLF shift factor, logaT = −5.47(T−170)/(119 + (T−170)), where T is given in Celsius. Using this shift factor, the zero shear viscosity for linear polystyrene was re-calculated at T = 180 °C:
| η0.Linear PS = 3.31 × 10−14Mw3.4; T = 180 °C | (1) |
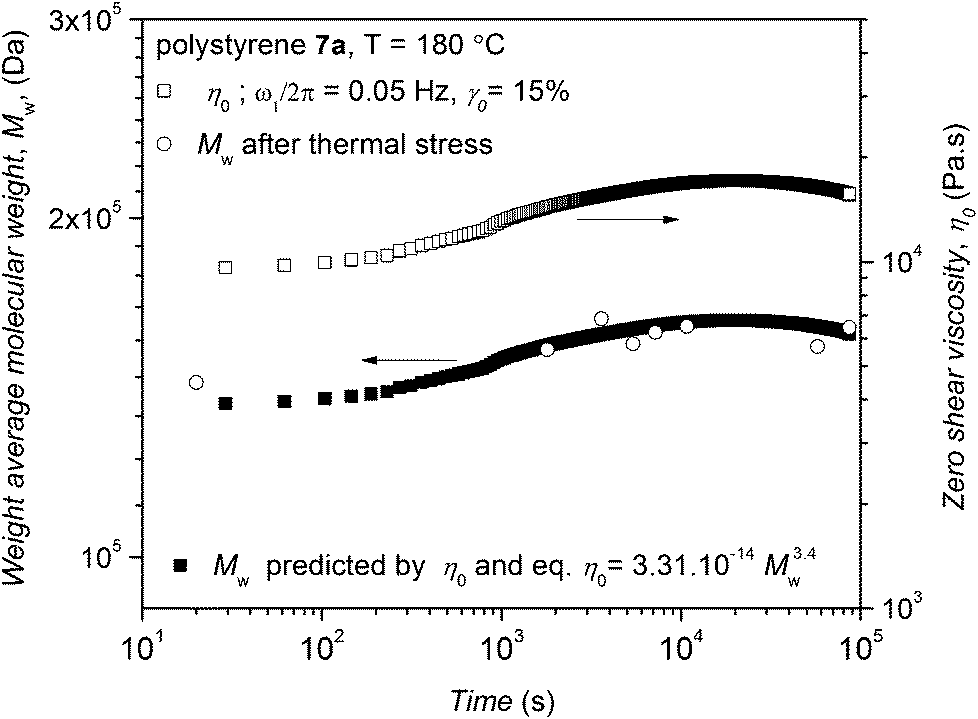 | ||
| Fig. 11 Time dependence of the experimental zero shear viscosity of polystyrene 7a (right y-axis) and weight-average molecular weight of polymer 7a deduced from time dependent SEC (left y-axis) along with the weight-average molecular weight based on the experimental zero shear viscosity and eqn (1). The higher time resolution and lower standard deviation of the rheological assessment can be seen while SEC allows analysis of the full molecular weight distribution. | ||
With respect to the experimental values of zero shear viscosity of polystyrene 7a in Fig. 11 and eqn (1), the weight-average molecular weight of this polymer is predicted and plotted in Fig. 11 on the left y-axis. The weight-average molecular weight evolution of polystyrene 7a (as recorded via time dependent SEC, refer to Fig. 3) is plotted in Fig. 11 (open circles), clearly indicating that the SEC derived weight-average molecular weights (Mw) are in agreement with the values predicted from rheological data (especially the zero shear viscosity) and reptation theory. It should be noted that thermal degradation decreases the number-average molecular weight, Mn, while increasing the polydispersity and subsequently the weight-average molecular weight, Mw, to some extent. The observed increase of the zero shear viscosity during the thermal degradation reflects Mw rather than Mn.
After applying both thermal and mechanical stress (in the linear regime) at a temperature of 180 °C for 24 h at a frequency of ω1/2π = 0.05 Hz and 15% strain amplitude for polystyrene 7a, the molecular weight of polystyrene 7a was determined by SEC (refer to Fig. S23 in the ESI†). The SEC analysis also revealed that the molecular weight of polystyrene 7a has changed. While the weight-average molecular weight of polystyrene 7a under thermomechanical stress decreases from Mw = 148 kDa to Mw = 142 kDa, the number-average molecular weight of the same polystyrene reduces from Mn = 124 kDa to Mn = 87 kDa. The change in number-average molecular weight, Mn, observed for polystyrene 7a is higher than the change observed for the weight-average molecular weight, Mw. On the other hand, the SEC data obtained via the time resolved SEC analysis for polystyrene 7a subjected to only thermal stress for 24 h reflect an increase in weight-average molecular weight from Mw = 148 kDa to Mw = 161 kDa (refer to Fig. S23 in the ESI†) and a reduction in number-average molecular weight from Mn = 124 kDa to Mn = 109 kDa. Thus, combined mechanical and thermal stress has a distinct effect on the cleavage and degradation process of polystyrene 7a compared to thermal stress alone perhaps due to increased convection in the sample. In other words, the degradation process becomes prominent during the rheological characterization of polystyrene 7a.
In order to investigate the effect of HBr elimination on the ester degradation, the zero shear viscosity polystyrene 13a as a function of time for 24 h at a frequency of ω1/2π = 0.05 Hz and 15% strain amplitude was measured (refer to Fig. S24 in the ESI†). Inspection of Fig. S24† indicates that the zero shear viscosity of polymer 13a is constant up to 60 minutes and subsequently slowly decreases with increasing time at elevated temperatures in the linear regime due to degradation and a slight reduction of molecular weight induced by thermomechanical stress. Further inspection of Fig. S24† indicates that there is no increase in the zero shear viscosity with increasing time at elevated temperatures, whereas the same rheological parameter for polystyrene 7a demonstrated an increase in the zero shear viscosity (refer to Fig. 11). The results obtained are in qualitative agreement with the studies invoking thermal stress alone for polystyrene 13a. After applying thermomechanical stress to polystyrene 13a in the above experiment, SEC analysis revealed that the molecular weight distributions of polystyrene 13a had experienced a slight change. The molecular weight reduction observed during thermomechanical stress at elevated temperatures can be compared to the reduction in molecular weight observed in solely pure thermal degradation experiments for polystyrene 13a (refer to Fig. S25 in the ESI†). The relative values for the number-average molecular weight based on SEC before/after thermal stress were found to be close to 118.1 vs. 112.3 kDa for polystyrene 13a. Comparing the number-average molecular weight obtained for polystyrene 13a, it appears that the rheology experiment had no additional effect on the degradation behavior due to thermomechanical stress in the absence of bromine end groups.
Polymer processing allows shaping and manufacturing polymers in powder or pellet form. The extrusion process is a continuous shaping of a polymer in the melt through the orifice of a die and the subsequent solidification into a product. To mimic the extrusion process of polystyrene 7a containing ester, triazole and bromine functionalities, a 10 minute residence time in the extrusion barrel – consistent with the mixing average residence time common in extrusion processes – was selected. However, feeding of the polymers took close to 10 minutes; therefore the total residence time is close to 20 minutes at 200 °C. The change in molar mass after extrusion is a sensor for the combined effects of thermal and mechanical degradation on the polymeric material, due to the orientation and stretch of the polymer chains that can influence their stability. The molecular weight reduction observed during extrusion can be compared to the reduction in molecular weight observed in pure thermal degradation experiments for polystyrene 7a. The change in the number-average molecular weight of polystyrene 7a after extrusion was determined via SEC (refer to Fig. 12). The relative values for the number-average molecular weight based on SEC before/after extrusion were found to be close to 124.6 kDa vs. 108.6 kDa corresponding to a 12% decrease for polystyrene 7a, respectively. In our previous study,17 a 10% reduction in molecular weight after the extrusion at 200 °C after 20 min was observed for anionically prepared non-functional linear polystyrene. Comparing the number-average molecular weight obtained by SEC before and after extrusion, a slight reduction in molecular weight was observed. It appears that the extrusion process for mid-chain triazole functionalized linear polystyrene with mid-chain ester and end chain bromine functionalities had no significant effect on the degradation behavior due to additional mechanical stress at short times.
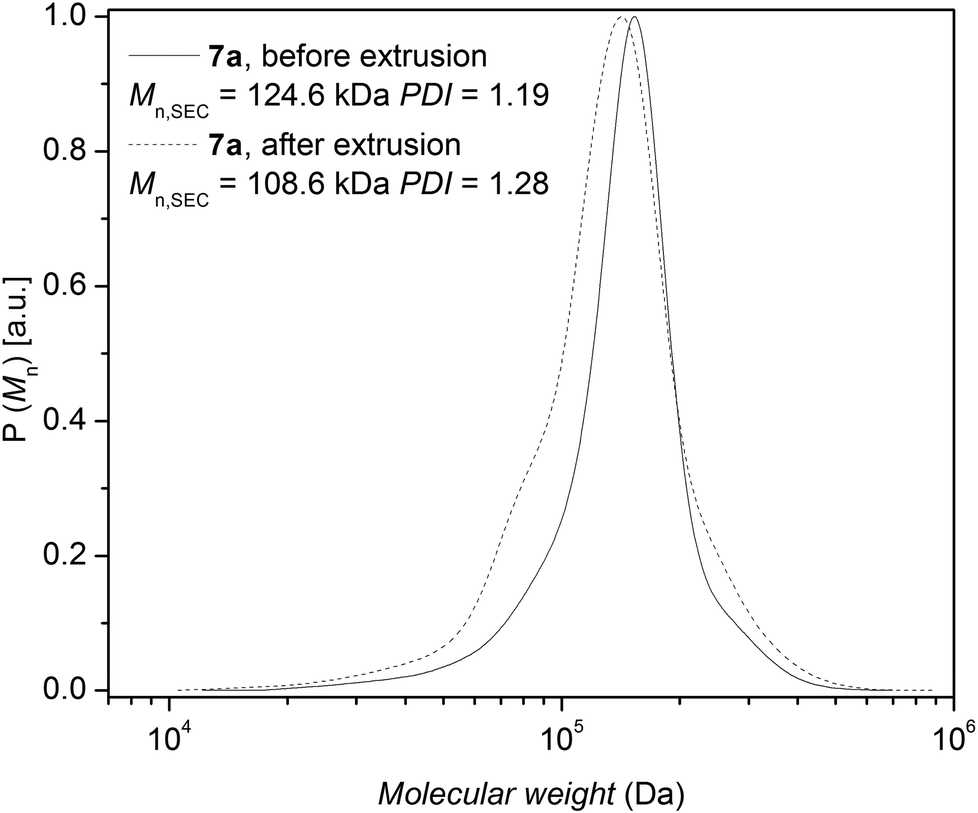 | ||
| Fig. 12 Comparison of the molecular weight distribution of polystyrene 7a before and after extrusion in a twin screw extruder at 200 °C with a total residence time of 20 min. | ||
Conclusions
In summary, the stability of living/controlled polystyrenes generated by activators regenerated by electron transfer (ARGET) atom transfer radical polymerization (ATRP) with ester and triazole mid-chain functionalities and secondary bromine chain-end functionalities was investigated under thermal and thermomechanical stress under an inert atmosphere at elevated temperatures. The thermally treated linear polystyrene containing the above functions (7a) was analyzed by size exclusion chromatography as a function of their exposure time to elevated temperatures (e.g. 200 °C) indicating a degradation of the functional polystyrene. The stability of these three functional groups was systematically investigated under a nitrogen atmosphere at elevated temperatures to identify the cleavage points of polystyrene 7a. Linear polymers having either secondary alkyl halides (1 or 2), ester groups (3a and 3b) or a triazole moiety (11) were prepared as low molecular weight reference polymers and subsequently exposed to elevated temperatures under an inert atmosphere. The thermally treated polystyrenes at elevated temperature were evaluated by SEC, 1H NMR and MALDI-MS, demonstrating the cleavage of the ester group as well as the quaternization of the triazole unit in the presence of the bromine end groups of the ATRP-made polymers. Thus, any high temperature processing or storage of polymers – or at least polystyrenes – prepared via ATRP should avoid the mid-chain presence of any ester groups and triazole unit. When the bromine end groups of the ATRP-made polymers are removed before thermal/thermomechanical treatment, ester degradation almost ceases. Importantly, the chain ends of the ATRP polymers can undergo a quaternization reaction with the triazole units present in the polymer chain, leading to the formation of higher molecular weight material. An in-depth rheological assessment revealed that the number-average molecular weight, Mn, of the mid-chain functional linear polymer decreases under conditions of oscillatory shear at 180 °C with ω1/2π = 0.05 Hz and γ0 = 15% strain amplitude, while the polydispersity and the weight-average molecular weight, Mw, generally increase. These findings are in agreement with the on-line monitoring of the zero shear viscosity under the above oscillatory shear conditions. SEC analyses revealed that the number-average molecular weight of polystyrene 7a was reduced by 30% under thermomechanical stress during rheological characterization of polystyrene 7a, indicating an additional effect on the degradation behavior, while polystyrene 7a shows a 10% reduction in number-average molecular weight with thermal stress alone. Moreover, an assessment of polystyrene 13a carrying no bromine functionalities indicates that there is no increase in the zero shear viscosity with time under the above oscillatory shear conditions. In addition, SEC traces recorded after extrusion for 20 minutes under an inert atmosphere at 200 °C indicate that the number-average molecular weight of the linear polystyrene 7a decreases by 12%, while the chain-end functional or non-functional linear polystyrenes indicate a reduction by close to 10% in number-average molecular weight, suggesting that mechanical stress has a slight effect on the degradation of functional linear polystyrene 7a during the extrusion process.Table 3 collates the identified behavior with regard to the molecular weight averages after thermal insult for all the studied polymer systems.
| Polymer | Ester | Triazole | Bromine | M n , | Đ , | M n , | Đ , |
|---|---|---|---|---|---|---|---|
| a Determined via RI detection SEC using linear PS standards. b Before thermal treatment. c After thermal treatment at 200 °C for 24 h under an inert atmosphere. The molecular weight values are given in kDa. | |||||||
| 1 | − | − | + | 2 | 1.06 | 2 | 1.06 |
| 3a | + | − | + | 6.5 | 1.06 | 4.2 | 1.20 |
| 3b | + | − | + | 39 | 1.08 | 28.1 | 1.18 |
| 7a | + | + | + | 125 | 1.19 | 95.1 | 1.63 |
| 7b | + | + | + | 3.8 | 1.07 | 1.9 | 1.23 |
| 7c | + | + | + | 33.7 | 1.09 | 31.8 | 1.44 |
| 11 | − | + | − | 4.4 | 1.08 | 4.3 | 1.08 |
| 12b | + | − | − | 38.5 | 1.06 | 34.1 | 1.13 |
| 13a | + | + | − | 118.1 | 1.19 | 112.3 | 1.23 |
| 13c | + | + | − | 32.5 | 1.09 | 30.3 | 1.15 |
In summary, it is strongly recommended that ATRP polymers based on initiators with ester linkages in critical positions (as is often the case in those available commercially) and/or triazole units anywhere in the molecular structure are not subjected to any thermal stress or processing, unless the bromine termini are removed.
Acknowledgements
C.B.-K and M.W. gratefully acknowledge financial support from the German Research Council (DFG). The authors acknowledge additional funding for the current project from the Karlsruhe Institute of Technology (KIT). P.G. acknowledges financial support from the Wallonia Region, the European Commission (FSE, FEDER), the National Fund for Scientific Research (F.R.S.-FNRS), and the Belgian Federal Science Policy Office (PAI 6/27). O.A. thanks Dr Eva Blasco (KIT), Andrea Hufendiek (KIT) and Ralf A. A. Bovee (Eindhoven Technical University, TU/e) for helpful discussions.References
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. V. Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS PubMed.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS PubMed.
- P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338–1354 RSC.
- J.-F. Lutz, H. G. Boerner and K. Weichenhan, Macromolecules, 2006, 39, 6376–6383 CrossRef CAS.
- J. A. Opsteen and J. C. M. van Hest, Chem. Commun., 2005, 57–59 RSC.
- O. Altintas, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5699–5707 CrossRef CAS.
- J. A. Johnson, D. R. Lewis, D. D. Diaz, M. G. Finn, J. T. Koberstein and N. J. Turro, J. Am. Chem. Soc., 2006, 128, 6564–6565 CrossRef CAS PubMed.
- M. Malkoch, K. Schleicher, E. Drockenmuller, C. J. Hawker, T. P. Russell, P. Wu and V. V. Fokin, Macromolecules, 2005, 38, 3663–3678 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- R. T. A. Mayadunne, E. Rizzardo, J. Chiefari, Y. K. Chong, G. Moad and S. H. Thang, Macromolecules, 1999, 32, 6977–6980 CrossRef CAS; Handbook of RAFT-Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS PubMed.
- O. Altintas, A. P. Vogt, C. Barner-Kowollik and U. Tunca, Polym. Chem., 2012, 3, 34–45 RSC.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- O. Altintas, K. Riazi, R. Lee, C. Y. Lin, M. L. Coote, M. Wilhelm and C. Barner-Kowollik, Macromolecules, 2013, 46, 8079–8091 CrossRef CAS.
- O. Altintas, M. Abbasi, K. Riazi, A. S. Goldmann, N. Dingenouts, M. Wilhelm and C. Barner-Kowollik, Polym. Chem., 2014, 5, 5009–5019 RSC.
- P. G. de Gennes, Scaling Concepts in Polymer Physics, Cornell University Press, London, UK, 1979 Search PubMed.
- M. T. Shaw, Introduction to Polymer Rheology, John Wiley & Sons, New Jersey, USA, 2012 Search PubMed.
- R. G. Larson, The Structure and Rheology of Complex Fluids, Oxford University Press, New York, USA, 1999 Search PubMed.
- C. W. Macosko, Rheology: Principles, Measurements, and Applications, Wiley-VCH, Weinheim, Germany, 1994 Search PubMed.
- E. H. H. Wong, O. Altintas, M. H. Stenzel, C. Barner-Kowollik and T. Junkers, Chem. Commun., 2011, 47, 5491–5493 RSC.
- W. Lin, Q. Fu, Y. Zhang and J. Huang, Macromolecules, 2008, 41, 4127–4135 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- T. Gruendling, S. Weidner, J. Falkenhagen and C. Barner-Kowollik, Polym. Chem., 2010, 1, 599–617 RSC.
- G. Luo, I. Marginean and A. Vertes, Anal. Chem., 2002, 74, 6185–6190 CrossRef CAS.
- X. Jiang, P. J. Schoenmakers, J. L. J. van Dongen, X. Lou, V. Lima and J. Brokken-Zijp, Anal. Chem., 2004, 75, 5517–5524 CrossRef.
- C. Ladavière, P. Lacroix-Desmazes and F. Delolme, Macromolecules, 2009, 42, 70–84 CrossRef.
- J. De Winter, M. Vachaudez, O. Coulembier, P. Dubois, R. Flammang and P. Gerbaux, e-Polymers, 2010, 103, 1–10 Search PubMed.
- J. De Winter, V. Lemaur, P. Marsal, O. Coulembier, J. Cornil, P. Dubois and P. Gerbaux, J. Am. Soc. Mass Spectrom., 2010, 21, 1159–1168 CrossRef CAS PubMed.
- J. De Winter, O. Coulembier, P. Dubois and P. Gerbaux, Int. J. Mass Spectrom., 2011, 308, 11–17 CrossRef CAS PubMed.
- C. D. Hurda and F. H. Blunck, J. Am. Chem. Soc., 1938, 60, 2419–2425 CrossRef.
- G. L. O'Connor and H. R. Nace, J. Am. Chem. Soc., 1953, 75, 2118–2123 CrossRef.
- H. A. Pohl, J. Am. Chem. Soc., 1951, 73, 5660–5661 CrossRef CAS.
- F. Bentiss, M. Traisnel, L. Gengembre and M. Lagrene, Appl. Surf. Sci., 1999, 152, 237–249 CrossRef CAS.
- E. Blasco, M. Piñol, L. Oriol, B. V. K. J. Schmidt, A. Welle, V. Trouillet, M. Bruns and C. Barner-Kowollik, Adv. Funct. Mater., 2013, 23, 4011–4019 CrossRef CAS.
- V. Coessens and K. Matyjaszewski, Macromol. Rapid Commun., 1999, 20, 66–70 CrossRef CAS.
- P. Dimitrov-Raytchev, S. Beghdadi, A. Serghei and E. Drockenmuller, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 34–38 CrossRef CAS.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, T. N. T. Phan, D. Gigmes and E. Drockenmuller, ACS Macro Lett., 2014, 3, 658–662 CrossRef CAS.
- M. Kempf, D. Ahirwal, M. Cziep and M. Wilhelm, Macromolecules, 2013, 46, 4978–4994 CrossRef CAS.
- K. Hyun and M. Wilhelm, Macromolecules, 2009, 42, 411–422 CrossRef CAS.
- J. Hepperle, H. Muenstedt, P. K. Haug and C. D. Eisenbach, Rheol. Acta, 2005, 45, 151–163 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of compounds 3a, 6, 8, 9, 10, 11 and 12a; 13C NMR spectrum of compound 6; evolution of ATR-IR spectra of polymers 8, 10, and 11; SEC traces of polymer 7a after thermal treatment at 200 °C under an argon atmosphere as a function of time; SEC traces of polystyrenes 3b, 7b, and 12b after thermal treatment at 200 °C under an argon atmosphere for 24 h, 1H NMR spectra of the polymers before and after thermal treatment of 1, 2, 3, 7b, 11; MALDI-MS spectra of polystyrene 1 before and after thermal treatment at 200 °C for 24 h under an argon atmosphere; the storage (G′) and loss modulus (G′′) as a function of frequency at 180 °C for polystyrenes 7a and 13a, supporting SEC data for the rheological experiments; SEC traces and MALDI-MS spectrum of polystyrene 14. See DOI: 10.1039/c5py00036j |
| This journal is © The Royal Society of Chemistry 2015 |