
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nucleobase-functionalized acrylic ABA triblock copolymers and supramolecular blends†
Keren
Zhang
a,
Motohiro
Aiba
b,
Gregory B.
Fahs
a,
Amanda G.
Hudson
a,
William D.
Chiang
a,
Robert B.
Moore
a,
Mitsuru
Ueda
b and
Timothy E.
Long
*a
aDepartment of Chemistry, Macromolecules and Interfaces Institute, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: Telong@vt.edu
bDepartment of Organic and Polymeric Materials, Graduate School of Science and Engineering, Tokyo Institute of Technology, 2-12-1, O-okayama, Meguro-ku, Tokyo 152-8552, Japan
First published on 30th January 2015
Abstract
Reversible addition-fragmentation chain transfer (RAFT) polymerization afforded the unprecedented synthesis of well-defined acrylic ABA triblock copolymers with nucleobase-functionalized external blocks and a central poly(n-butyl acrylate) (PnBA) block. Size exclusion chromatography (SEC) confirmed the molecular weight and molecular weight distribution of the central block. 1H NMR spectroscopy revealed the successful chain extension of the PnBA macro-chain transfer agent (CTA) using adenine or thymine-functionalized acrylic monomers. The acrylic monomer with a flexible spacer to the pendant nucleobases promoted intermolecular recognition of nucleobases and long range segmental motion of polymer main chains. The external block glass transition temperatures (Tg's) of thymine (T) and adenine (A) functionalized blocks were 52 °C and 76 °C, respectively. Thermomechanical and morphological analysis revealed the effect of processing conditions on self-assembly and microphase-separated morphology of nucleobase-functionalized ABA copolymers. Thymine and adenine-functionalized ABA triblocks formed a thermodynamically stable, hydrogen-bonded complex upon blending. The supramolecular blend exhibited a cylindrical microphase-separated morphology with an extended plateau window compared to the individual block copolymers. The complementary hydrogen bonding between adenine and thymine formed a thermally labile, physically crosslinked, network that exhibited enhanced mechanical performance with melt processability. Thus, these ABA nucleobase-functionalized block copolymers demonstrate potential as thermoplastic elastomers for hot melt adhesives and coatings.
Introduction
Specific noncovalent interactions are a critical design feature for tailoring polymeric material properties.1 Noncovalent interactions, including hydrogen bonding and pi–pi stacking, create supramolecular physically crosslinked networks that significantly influence polymer properties. The intermediate bond strength between van der Waals forces and covalent bonds leads to reversible bonds that respond to external stimuli such as temperature, moisture, solvent, and pH.1,2 Noncovalent interactions also improve cohesive strength for adhesives,3,4 enhance temperature dependence of melt viscosity,5 and promote self-assembly.4–9 Block copolymers containing physical crosslinking attract both academia and industry due to the potential synergy between microphase-separation and noncovalent interactions. Localized hydrogen bonding within the hard phase induces or reinforces microphase separation of block copolymers.1,10–12 Long et al. observed a phase-separated morphology of triblock copolymers with short nucleobase external blocks of molecular weights between 1–4 kDa.12 Long et al. also recently showed the capability of hydrogen bonding to facilitate self-assembly of ABC block copolymers.13 Nowick et al. and McHale et al. both noted enhanced molecular recognition through segregation of block copolymers in solution.14,15 The commercial impact of block copolymer thermoplastic elastomers (TPEs) continues to increase, including hot melt adhesives, synthetic elastomers, automobile parts, and membranes. The past two decades have also witnessed various fundamental studies of block copolymer TPEs with structure–property relationships and emerging applications.16–21 The microphase-separated morphology of block copolymers is crucial for their thermoplastic properties and elasticity. The hard phase (high Tg or semicrystalline) provides mechanical integrity, while the soft phase (low Tg) affords flexibility. ABA triblock copolymers, such as styrene–butadiene–styrene (SBS) and styrene–isoprene–styrene (SIS), represent TPEs with wide industrial appeal due to their unique synergy of elastomeric properties and melt processibility. Noncovalent interactions have shown great potential for inducing thermal responsiveness and enhancing mechanical strength of polymers.1,5,12,22,23 It is hypothesized that reversible physical crosslinking in block copolymers allows supramolecular thermoplastic elastomers with a self-assembled morphology, enhanced mechanical integrity, and tailored thermal responsiveness.Nucleobases in deoxyribonucleic acid (DNA) exemplify biology-inspired candidates for introducing noncovalent interactions to synthetic polymers due to their thermal stability and synthetic versatility.24 The unique molecular recognition between purine and pyrimidine rings is of particular interest for their potential in self-assembly, template polymerization, thermal responsiveness, and information storage.25,26 Nucleobase-functionalized polymers also offer unique biological properties including selective protein adsorption, suppressed bacterial adherence, and biocompatibility.27 Furthermore, nucleobase-containing polymers suggest potential as biosensors, molecular probes, biomedicine, biomimetic information storage, and materials with enhanced mechanical properties.25,28–30 However, introducing nucleobases to synthetic polymers is challenging due to their limited solubility and susceptibility to multiple substitutions during monomer synthesis.22
Most nucleobase-containing polymers in the literature are synthesized through the polymerization of nucleobase-functionalized monomers, and styrenic and methacrylic adenine/thymine monomers are extensively described. Various controlled polymerization strategies are amendable to synthesize nucleobase-functionalized polymers, including ring-opening metathesis polymerization,31,32 nitroxide mediated polymerization,12,14,33 and atom transfer radical polymerization (ATRP).34–36 Lutz et al. synthesized nucleobase-functionalized styrenic polymers and observed the association and dissociation of hydrogen bonding in solution.36,37 McHale et al. combined segregation in solution with templating to obtain well-controlled, high molecular weight styrenic nucleobase polymers.14 Our research group reported styrenic triblock copolymers with adenine and thymine external blocks and showed their potential as drug delivery vehicles and biological probes.12,33 Inaki et al. synthesized methacrylamide adenine and uracil polymers with an ethylene spacer for template polymerization.38–40 Others probed the effect of molecular recognition on copolymer composition with nonpolar and hydrogen-bonding disrupting solvent using methacrylic adenine and thymine monomers with ethylene diester spacers.30 Haddleton et al. synthesized methacrylic and acrylic nucleoside monomers for subsequent template polymerization.29,41 Other researchers synthesized and observed assembly of PEGylated methacrylic nucleobase polymer in water, and also reported nucleobase-functionalized monomers for step-growth polymerization including norbornene derivatives.31,32,42–44
Despite the intensive research interest in nucleobase-containing polymers, most nucleobase-containing polymers mentioned above either presented sterically constrained side groups or rigid polymer backbones with Tg's above the hydrogen bonding dissociation temperature range. Literature values for styrenic and methacrylic adenine-functionalized polymer Tg's range from 105 °C to 204 °C; thymine-functionalized polymers show a Tg from 87 °C to 191 °C, depending on the monomer structure.12,29,30,41,45 The only previously reported acrylic nucleoside homopolymers also showed Tg's around 140–170 °C.29 Steric effects and polymer chain rigidity significantly restricted molecular association and dissociation in the bulk.46 This also accounts for predominate research focus in literature on supramolecular polymer self-assembly and template polymerization in solution, where solvation and mobility promote molecular recognition. Previous investigations of bulk and mechanical properties of supramolecular polymers only include telechelic polymers with physical crosslinking sites located only at the chain ends.5,47–49 As a result, nucleobase-functionalized block copolymers with sufficiently low Tg backbone and less steric hindrance are of particular interest for ascertaining the influence of nucleobase pendant groups on solid-state properties.
Acrylic polymers exhibit 20–100 °C lower Tg values than their methacrylic analogs due to the absence of a pendent methyl on the backbone.50 However, controlled polymerization of acrylate monomers is more challenging due to the presence of an α-proton, which facilitates branching due to chain transfer to polymer.51 Long et al. previously synthesized acrylic adenine and thymine polymers with Tg of 65 °C and 43 °C, respectively, with flexible spacers to promote molecular recognition. Both rheological and adhesive analyses demonstrated the effect of A–T complementary hydrogen bonding on random copolymer blends due to supramolecular association in the bulk.4
This manuscript focuses on the synthesis of novel, bio-inspired, supramolecular block copolymers with well-defined compositions and nanoscale phase-separated morphologies. An optimized, two-step, RAFT polymerization afforded ABA triblock copolymers with either adenine acrylic (AdA) or thymine acrylic (ThA) external blocks and a PnBA central block. The structural design of an acrylic backbone with a flexible spacer to the nucleobase pendant groups ensured sufficiently low Tg's and flexibility for molecular recognition.4 Dynamic mechanical analysis (DMA), small angle X-ray scattering (SAXS), and atomic force microscopy (AFM) revealed the effect of noncovalent interactions on block copolymer morphologies and thermomechanical properties. We further discuss the effect of processing conditions on film performance. The complementary hydrogen bonding between adenine and thymine contributed to a microphase-separated supramolecular blend with an elongated plateau region and similar melt processibility compared to the precursors. The reversible supramolecular network provided enhanced mechanical properties and thermal responsiveness.
Experimental section
Materials
n-Butyl acrylate (nBA, 99+%) was purchased from Aldrich and passed through a neutral alumina column before use. α,α′-Azobis(isobutyronitrile) (AIBN, Fluka, 99%) was recrystallized from methanol. N,N′-dicyclohexylcarbodiimide (DCC, 99%), 4-(dimethylamino) pyridine (DMAP, ≥99%), and 1,4-butanediol diacrylate (Alfa Aesar, 99%) were used without further purification. Adenine (A, 99%), thymine (T, 99%), triethylamine (TEA, 99%), potassium carbonate (99%), 1,6-hexanediamine (98%), 4-cyano-4-(dodecylsulfanylthiocarbonylsulfanyl)pentanoic acid (CDP, 97%), and 2,6-di-tert-butyl-4-methylphenol (BHT, 99%) were purchased from Aldrich and used without further purification. Hexane (HPLC grade), chloroform (CHCl3, HPLC), tetrahydrofuran (THF, HPLC grade), N,N-dimethylsulfoxide (DMSO, HPLC grade) and N,N-dimethylformamide (DMF, HPLC grade, anhydrous) were purchased from Fisher Scientific and used as received.Analytical methods
1H NMR and 13C NMR spectra were collected in CDCl3 or DMSO-d6 on a Varian INOVA spectrometer operating at 400 MHz at 23 °C. Size exclusion chromatography (SEC) was performed using a Waters size exclusion chromatograph. The instrument was equipped with an auto sampler, three 5 μm PLgel Mixed-C columns, a Waters 2410 refractive index (RI) detector operating at 880 nm, and a Wyatt Technologies miniDAWN multi-angle laser light scattering (MALLS) detector operating at 690 nm with a flow rate of 1 mL min−1 at 30 °C in THF. Reported molecular weights are absolute values from the light scattering detector with dn/dc value of 0.07. Differential scanning calorimetry (DSC) was performed under a nitrogen flush of 50 mL min−1 at a heating rate of 10 °C min−1 on a TA instruments Q1000 DSC, which was calibrated using indium (mp = 156.60 °C) and zinc (mp = 419.47 °C) standards. Glass transition temperatures were measured as the midpoint of the transition in the second heating scan. DMA was conducted on a TA Instruments Q800 in tension mode at a frequency of 1 Hz, an oscillatory amplitude of 8 μm, and a static force of 0.01 N. The temperature ramp was 3 °C min−1. The glass transition temperature (Tg) was determined at the peak maximum of the tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ curve. All FTIR experiments were performed using a Varian 670-IR spectrometer (DTGS detector) with Pike Technologies variable temperature GladiATR™ attachment (Diamond crystal). The spectra were collected at 4 cm−1 resolution and as an average of 32 scans. The samples were subjected to a temperature ramp of 1 °C min−1, starting from 30 °C to 180 °C and FTIR spectra were collected every 10 °C beginning from 30 °C.
δ curve. All FTIR experiments were performed using a Varian 670-IR spectrometer (DTGS detector) with Pike Technologies variable temperature GladiATR™ attachment (Diamond crystal). The spectra were collected at 4 cm−1 resolution and as an average of 32 scans. The samples were subjected to a temperature ramp of 1 °C min−1, starting from 30 °C to 180 °C and FTIR spectra were collected every 10 °C beginning from 30 °C.
A Veeco MultiMode scanning probe microscope was used for tapping-mode AFM imaging. Samples were imaged at a set-point ratio of 0.60 with a magnification of 1 μm × 1 μm. Veeco nanosensor silicon tips with a spring constant of 42 N m−1 were utilized for imaging. SAXS experiments were performed using a Rigaku S-Max 3000 3 pinhole SAXS system, equipped with a rotating anode emitting X-ray with a wavelength of 0.154 nm (Cu Kα). The sample-to-detector distance was 1600 mm, and q-range was calibrated using a silver behenate standard. Two-dimensional SAXS patterns were obtained using a fully integrated 2D multiwire, proportional counting, gas-filled detector, with an exposure time of 2 h. All SAXS data were analyzed using the SAXSGUI software package to obtain radically integrated SAXS intensity versus scattering vector q, where q = (4π/λ)sin(θ), θ is one half of the scattering angle and λ is the wavelength of X-ray.
Polymerization of difunctional PnBA macro-CTA
1,6-Bis(4-cyano-4-(dodecylsulfanyl-thiocarbonylsulfanyl)pentanoic acid)-hexane diamide (dCDP–NH2) was synthesized according to previous literature.52 DMF (13.0 g, 20 wt%), dCDP–NH2 (35.6 mg, 0.4 mmol), nBA (3.2 g, 25.1 mmol), and AIBN (0.7 mg, 0.04 mmol) were charged into a single-necked Schlenk flask. The monomer:initiator:CTA ratio was 630:1:10. The flask was subjected to four freeze–pump–thaw cycles with subsequent refilling with argon. The flask was then sealed and thermostated at 65 °C for 6 h. 1H NMR determined a monomer conversion of 55%. After the polymerization, residual monomer and solvent were removed by distillation. SEC analysis in THF revealed molecular weight data Mn = 44.8 kDa. Mw/Mn = 1.13. Yield was approximately 50%.
Polymerization of adenine-functionalized ABA triblock copolymers
AdA (613.8 mg, 1.8 mmol), AIBN (0.2 mg, 1.2 μmol), PnBA macro-CTA (549.0 mg, 12.3 μmol), and DMF (4.6 g, 20 wt%) were charged into a single-necked Schlenk flask and subjected to four cycles of freeze–pump–thaw and subsequent refilling with argon. The flask was then sealed and maintained at 65 °C for 6 h. The copolymer was isolated from precipitation into methanol and dried in vacuo at room temperature for 24 h. The monomer:initiator:CTA ratio was 1500:1:10. 1H NMR revealed the number-average molecular weight for each external block of poly(AdA-b-nBA-b-AdA) Mn = 13.8 kDa, monomer conversion 56%, assuming an equal chain growth rate from both macro-CTA ends.
Polymerization of thymine-functionalized ABA triblock copolymers
ThA (613.0 mg, 1.9 mmol), AIBN (0.2 mg, 1.2 μmol), PnBA macro-CTA (549.0 mg, 12.3 μmol), and DMF (4.7 g, 20 wt%) were charged into a single-necked Schlenk flask and subjected to four cycles of freeze–pump–thaw and subsequent refilling with argon. The flask was then sealed and maintained at 65 °C for 6 h. The copolymer was isolated from precipitation into methanol and dried under vacuum at room temperature for 24 h. The monomer:initiator:CTA ratio was 1600:1:10. 1H NMR revealed number-average molecular weight for each external block of poly(ThA-b-nBA-b-ThA) Mn = 15.0 kDa for each block, monomer conversion 56%, assuming an equal chain growth rate from both macro-CTA ends.
Triblock copolymer films and supramolecular blend preparation
Poly(AdA-b-nBA-b-AdA) and poly(ThA-b-nBA-b-ThA) were melt pressed at 120 °C for 1 h and annealed at 120 °C in vacuo for 24 h. poly(AdA-b-nBA-b-AdA)/poly(ThA-b-nBA-b-ThA) were also dissolved in DMSO (2 wt% solids) at 50 °C for 18 h and casted to a Teflon® mold. A Teflon® mold was used to minimize deformation of the polymeric films during removal. The mold was placed at 80 °C for 48 h to slowly evaporate DMSO. The dried copolymer film was then annealed under vacuum at 120 °C for 24 h. To obtain the supramolecular blend, poly(AdA-b-nBA-b-AdA) (50 mg) and poly(ThA-b-nBA-b-ThA) (47 mg) (A:T = 1:1) were dissolved in DMSO (5 mL) at 50 °C for 18 h and casted to a Teflon® mold, using an identical drying and annealing procedure.
Results and discussion
Synthesis of acrylic ABA triblock copolymers with nucleobase-functionalized external blocks
Controlled radical polymerization allowed the synthesis of nucleobase-functionalized ABA triblock copolymers with an acrylic backbone. Michael addition of a butanediol diacrylate with adenine or thymine afforded a convenient synthesis of the acrylic nucleobase monomers.4 The acrylic backbone and flexible spacer linking the backbone to pendent nucleobases facilitated noncovalent interactions and tuned the Tg's of the external blocks (Scheme 1). The butanediol spacer also promoted the solubility of nucleobase monomers in organic solvents, which allowed homogeneous copolymerization in DMF at typical concentrations.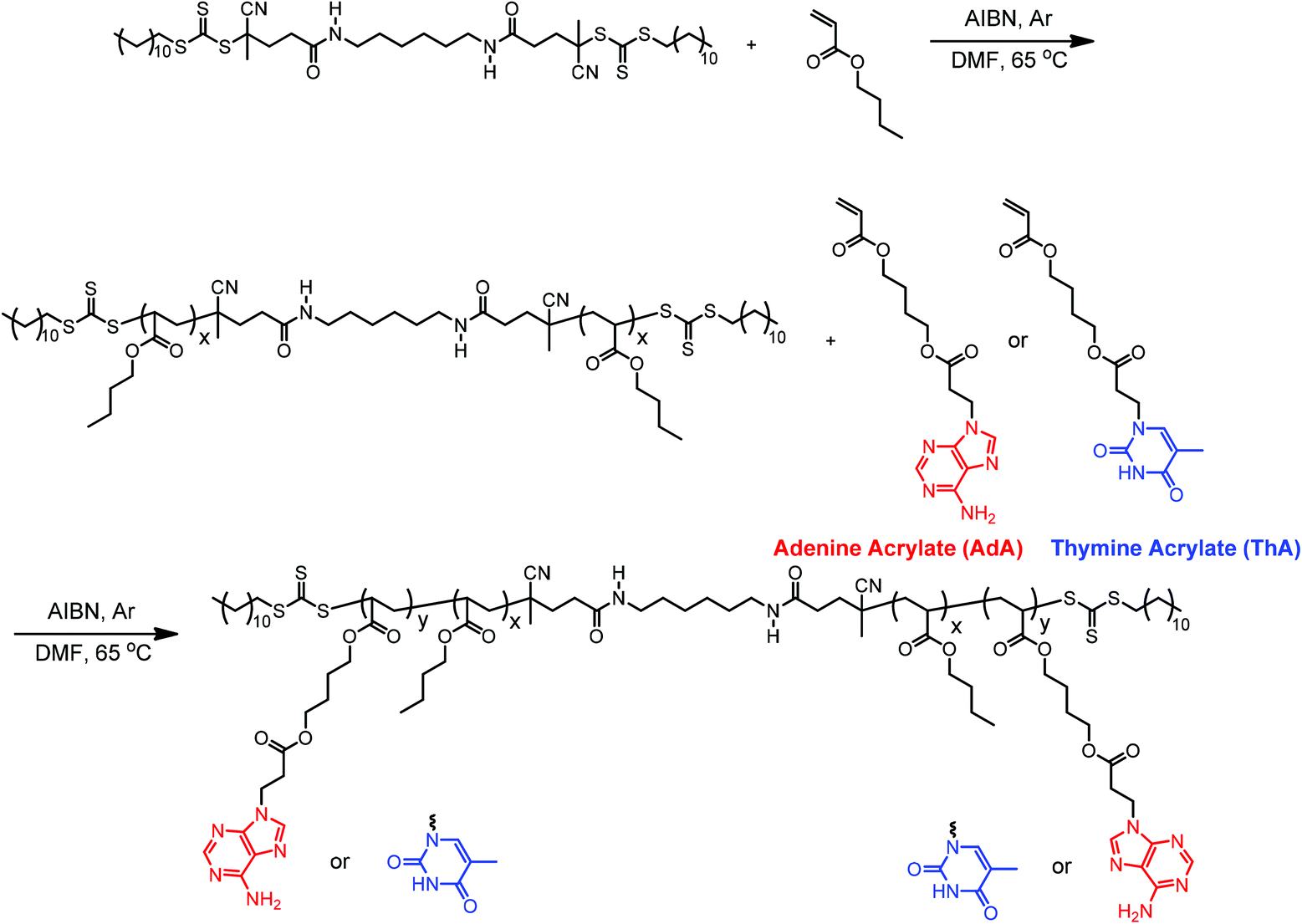 | ||
| Scheme 1 Synthesis of adenine- and thymine-functionalized acrylic ABA triblock copolymers using RAFT polymerization. *For visual guidance, all adenine copolymers are labeled red; thymine copolymers are labeled blue. | ||
Reversible addition-fragmentation chain transfer (RAFT) polymerization is an efficient route for synthesizing block copolymers of various compositions due to excellent functional group tolerance.53 Despite the many advantages of RAFT polymerization, only a few previous reports used RAFT to synthesize nucleobase-functionalized polymers.13,14,30,54 Long et al. previously designed and synthesized two difunctional chain transfer agents (CTA) for RAFT polymerization of ABA diblock copolymers through divergent chain growth.52 A diamide-linked difunctional CTA was used to polymerize acrylic nucleobase monomers. The difunctional CTA afforded a divergent polymerization of ABA triblock in two steps (Scheme 1). In the first step, dCDP–NH2 controlled the polymerization of nBA and yielded PnBA macro-initiator with absolute Mn of 44.8 kDa and PDI of 1.13 according to SEC (Fig. S1†). Chain transfer to polymer for acrylics was suppressed due to preference for chain transfer to the CTA. Several parameters proved necessary for obtaining good control of RAFT polymerization of acrylates, including strict freeze–pump–thaw cycles, proper CTA-to-initiator ratio (10:1 for dCDP–NH2), low monomer conversion (<70%), and constant polymerization temperature. A practical threshold molecular weight for trithiocarbonate was observed near 80–100 kDa in the polymerization of acrylic monomers, where controlled polymerization diminished.
In the second step, PnBA macro-initiator enabled the polymerization of nucleobase-functionalized external blocks in a divergent fashion. This divergent chain growth method eliminates possible diblock formation if cleavage of the trithiocarbonate occurs.52 The diamide linker also contributed to thermal and hydrolytic stability. 1H NMR spectroscopy was the primary tool for collecting structural and molecular weight information due to limited solubility of the triblock copolymers for molecular weight determination using SEC. 1H NMR spectroscopic analysis of the reaction mixture immediately after polymerization provided number-average molecular weights of the nucleobase block through a comparison of monomer conversion and macroCTA molecular weight. For example, the ratio of integration of acrylic peaks (3H) at 5.8–6.4 ppm to the methylene peak (2H) adjacent to adenine at 4.2–4.4 ppm yielded a 56% conversion of the polymerization (Fig. S2†). 1H NMR spectroscopic analysis of the purified copolymers determined number-average molecular weights for the nucleobase blocks through the ratio of nucleobase to PnBA (Fig. S4, 5†). The molecular weight results of purified block copolymer products (DP2 in Table S1†) agreed with results from experimental predictions (DP1 in Table S1†). Number-average molecular weights of poly(AdA-b-nBA-b-AdA) and poly(ThA-b-nBA-b-ThA) were 27.6–44.8–27.6 kDa and 30.1–44.8–30.1 kDa, respectively. The degree of polymerization (DP) of the nucleobase-functionalized external block was calculated from the average of two calculation methods. DP of AdA external block for poly(AdA-b-nBA-b-AdA) was 83; DP of ThA external block for poly(ThA-b-nBA-b-ThA) was 93. Total number-average molecular weights of poly(AdA-b-nBA-b-AdA) and poly(ThA-b-nBA-b-ThA) were 72.4 kDa and 74.9 kDa, respectively. Similarity of the acrylic nucleobase monomers to nBA also eliminated possible crossover problems associated with reactivity ratio difference of monomers in controlled radical polymerization.4
Hydrogen bonding between monomers using NMR titration
NMR titration is a common tool to monitor reversibility of hydrogen bonding and supramolecular assembly.1,55–58 NMR titration experiments at 22 °C for the AdA and ThA monomers quantified the association constant for their complementary hydrogen bonding. The concentration of ThA monomer (guest) in CDCl3 remained constant while the concentration of AdA monomer (host) varied. Actual AdA concentration was calculated from the ratio of AdA to ThA in CDCl3. The hydrogen bonded proton resonance for ThA shifted from high field to low field with increasing AdA concentration (Fig. S6†). The Conners’ method provided a more accurate calculation using a non-linear fitting compared to the commonly used Benesi-Hildebrand model.58,59Fig. 1 depicts a plot of the concentration of AdA versus the chemical shift changes (δi) of ThA hydrogen bonded proton, and non-linear fitting determined the binding constant (K) to be 128 M−1 using eqn (1). [H]0 and [G]0 represent the concentration of AdA (host) and ThA (guest), respectively. δi is the difference between the chemical shift of observed ThA and the free ThA; δc is the difference between the chemical shift of completely bonded ThA and free ThA. The binding constant of AdA and ThA agreed well with literature values.56,60 The acrylic substitution on the nucleobase did not interfere with complementary hydrogen bonding between the AdA and ThA monomers.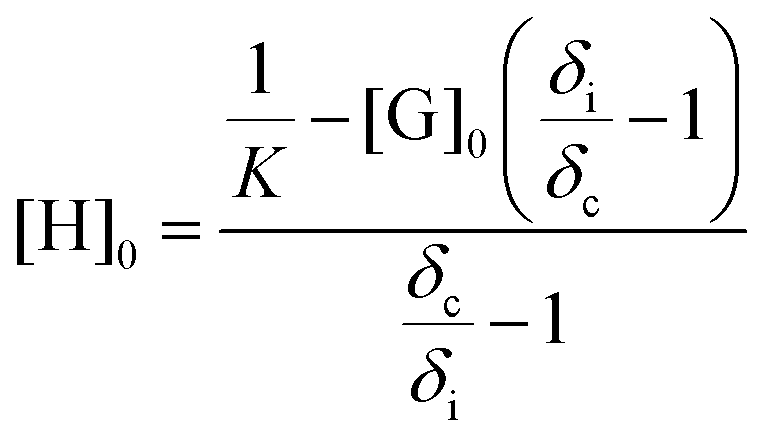 | (1) |
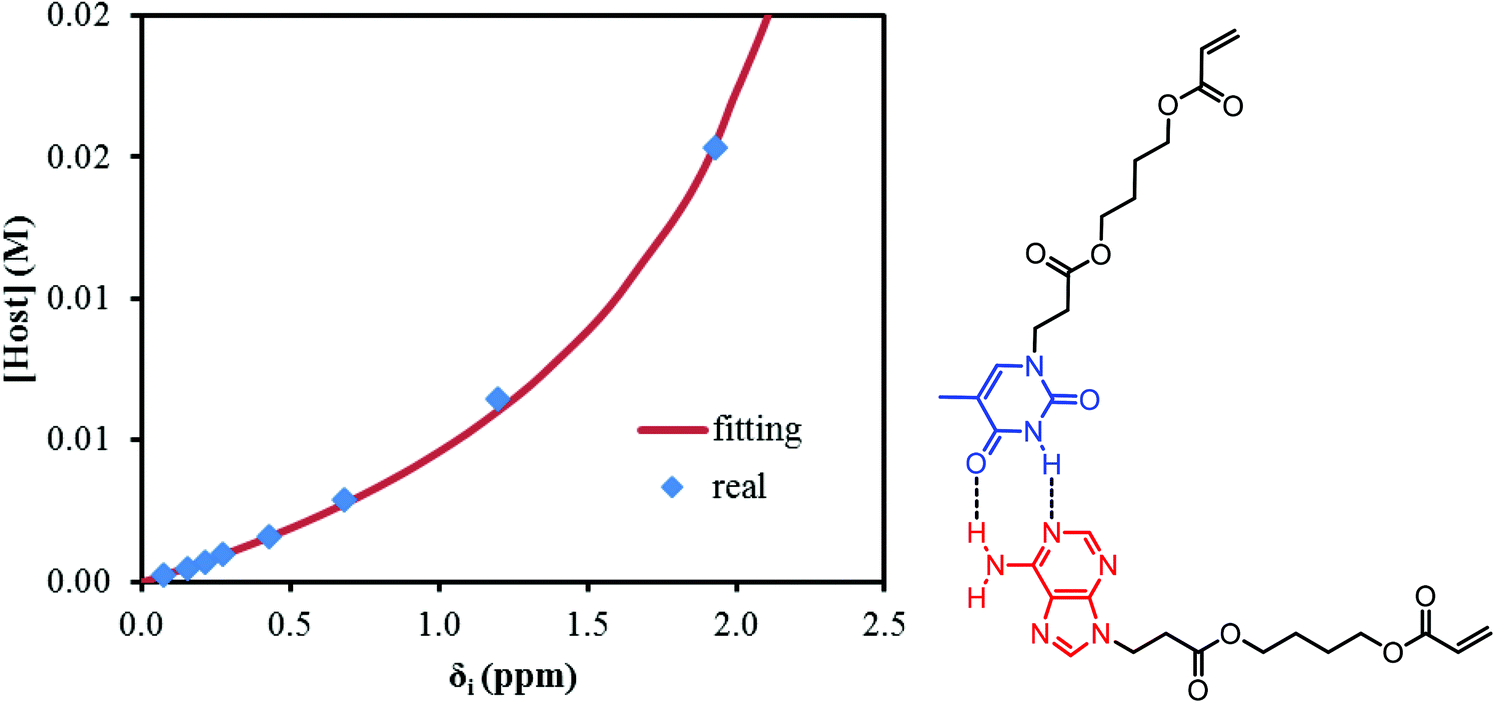 | ||
| Fig. 1 Non-linear fitting of NMR titration results to determine the binding constant of AdA and ThA in CDCl3 at 22 °C. | ||
Thermal transitions
In the design of amorphous TPEs, the hard-soft-hard triblock copolymer structure is essential for mechanical integrity and elasticity. The application temperature window for TPEs lies between the Tg of the hard external blocks, which afford mechanical integrity, and the soft central block, which provides flexibility.20 The nucleobase-functionalized ABA triblock copolymers contained acrylic adenine or thymine external blocks designed as the hard blocks and PnBA central block for the soft block. DSC determined Tg's of poly(ThA-b-nBA-b-ThA) to be −40 °C and 52 °C, and Tg's of poly(AdA-b-nBA-b-AdA) to be −39 °C and 76 °C (Fig. S7†). The presence of two Tg's confirmed a block structure of the nucleobase-functionalized copolymers. The similar lower Tg's for both ABA triblock copolymers corresponded to PnBA block, while the higher Tg's correlated well with the previously reported homopolymers Tg's.4 The soft blocks Tg's were slightly higher than polynBA homopolymer Tg (−47 °C) due to phase mixing of the soft and hard phases, which was confirmed in mechanical analysis. The hard blocks Tg's were slightly higher than the reported homopolymers Tg's, which was presumably attributed to molecular weight differences of the nucleobase blocks from previously reported homopolymers.4 The Tg difference between adenine and thymine blocks resulted from the bulkier pyrimidine structure and additional pi–pi stacking of adenine. Self-association of adenine is stronger than thymine despite their similar self-hydrogen bonding constants.60 This structural difference also affected self-assembly and morphology, which is discussed below. TGA showed 5 wt% loss temperatures of poly(AdA-b-nBA-b-AdA) and poly(ThA-b-nBA-b-ThA) to be 282 °C and 285 °C, respectively, which demonstrated their thermal stability for high temperature applications such as hot melt adhesives and injection molding (Fig. S8†). To the best of our knowledge, the acrylic nucleobase block copolymers showed the lowest Tg's among all previously reported nucleobase-functionalized polymers.Thermomechanical analysis
All triblock copolymer films showed superior mechanical integrity compared to diblock copolymers with similar nucleobase content and molecular weight. Poly(nBA-b-AdA) and poly(nBA-b-ThA) synthesized using a monofunctional CTA did not form free-standing films. As expected, the hard-soft-hard block copolymer structure proved crucial for TPE behavior. Higher Tg external blocks anchored both ends of the polymer chain, while the soft central block provided flexibility. Two different processing methods prepared block copolymer films with an identical annealing procedure. Both melt-pressed and solution-cast copolymer films exhibited microphase-separation in their thermomechanical analysis and surface morphology (Fig. 2, 3a–d). In Fig. 2, the first transition in the storage modulus-temperature curve and the first tan delta peak from the lowest temperature corresponded to the glass transition of the soft phase, followed by a plateau region where modulus remained constant with temperature. The bright and dark regions related to hard and soft phases, respectively, in AFM phase images (Fig. 3). Both processing conditions resulted in self-assembled, microphase-separated ABA triblock copolymer films. However, DMA and AFM revealed significant morphological and mechanical differences, presumably resulting from differing processing conditions. In Fig. 2, the solution-cast films showed higher plateau moduli on storage modulus curves compared to the melt-pressed analogs for both copolymers. The tan delta curves and AFM images (Fig. 3a–d) also indicated more defined microphase-separation with slow evaporating solvent. Soft phase Tg (tan delta peak) of melt-pressed poly(ThA-b-nBA-b-ThA) and poly(AdA-b-nBA-b-AdA) films were −19 °C and −17 °C, which were higher than solution-cast samples. Restricted mobility and shorter self-assembly time in the melt impeded phase separation compared to solution. A mixed soft phase that contained both nucleobase and PnBA blocks resulted in a higher soft block Tg and a lower plateau modulus, forming a kinetically trapped morphology. Further annealing at 120 °C for 5 d did not have a significant impact on the partially mixed phase separation for the melt-pressed samples. Chain mobility and time are keys to the self-assembly kinetics of block copolymers in the presence of physical crosslinks. Intermolecular hydrogen bonding also presumably further restricted chain mobility even at the annealing temperature. As a polar solvent and a hydrogen bonding acceptor, DMSO dilutes hydrogen bonding groups and screens intermolecular hydrogen bonding. Proper solvent was necessary to disrupt physical crosslinking and facilitate self-assembly. The self-assembled morphology significantly affected mechanical properties as temperature increased.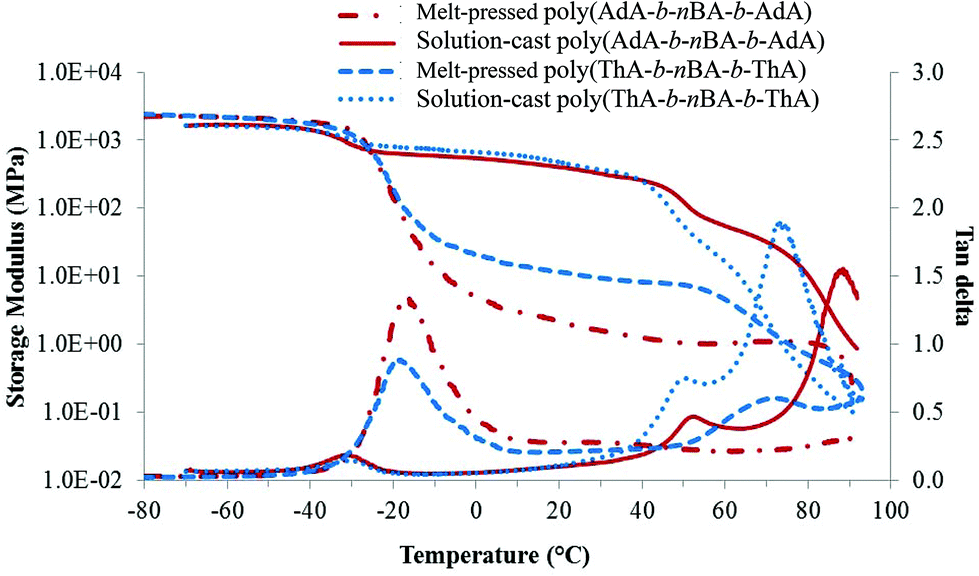 | ||
| Fig. 2 Effect of processing conditions on the thermomechanical properties of nucleobase-functionalized triblock copolymers. | ||
 | ||
| Fig. 3 Tapping mode AFM phase image of (a) solution-cast poly(AdA-b-nBA-b-AdA), (b) solution-cast poly(ThA-b-nBA-b-ThA), (c) melt-pressed poly(AdA-b-nBA-b-AdA), (d) melt-pressed poly(ThA-b-nBA-b-ThA), and (e) solution-cast supramolecular blend. | ||
Plateau moduli of melt-pressed ABA triblock copolymers were in the range of typical TPEs, while plateau moduli of solution-cast copolymers were too high for elastomers.21 38 wt% of adenine-functionalized blocks and 40 wt% of thymine-functionalized blocks self-assembled into hard phases, which restricted the flexibility of copolymers in the plateau temperature range. Decreasing the nucleobase content and block length will potentially afford nucleobase-functionalized triblock copolymer TPEs with improved elasticity.
DMA also elucidated the effect of noncovalent interactions on block copolymer thermomechanical properties. Above the Tg of the soft phase, the second drop of modulus and intermediate tan delta peak related to the Tg of the hard block for both solution-cast samples. A small second plateau followed the hard block Tg before the terminal flow. These secondary plateaus were attributed to self-association of the adenine-adenine and thymine-thymine hydrogen bonding and pi–pi stacking. Variable temperature FTIR of poly(ThA-b-nBA-b-ThA) and poly(AdA-b-nBA-b-AdA) verified the presence of weak self-hydrogen bonding (Fig. S9, 10†). Poly(AdA-b-nBA-b-AdA) showed a higher modulus above the second Tg compared to the poly(ThA-b-nBA-b-ThA), which correlated to stronger pi–pi stacking of the purine rings.4 The noncovalent-interaction dominated region was not obvious for melt-pressed poly(ThA-b-nBA-b-ThA) and absent for melt-pressed poly(AdA-b-nBA-b-AdA) due to the poor assembly of the hard phase in the melt. The lower plateau modulus for the melt-pressed poly(AdA-b-nBA-b-AdA) resulted from a slower self-assembly compared to poly(ThA-b-nBA-b-ThA). The additional pi–pi stacking and bulkier pendant group further inhibited chain mobility of the adenine block. Melt-pressed poly(AdA-b-nBA-b-AdA) exhibited more phase mixing than melt-pressed poly(ThA-b-nBA-b-ThA) under the same annealing time. Certain features of block copolymers were necessary to reveal the noncovalent interaction-dominated plateau region. In particular, the Tg of the physically crosslinked block needs to be lower than the dissociation temperature of physical crosslinks. In addition, sufficient physical crosslinking strength is also needed. Strong crosslinks will restrict segmental motion and potentially lead to an order–disorder transition temperature above the hard block Tg as shown for supramolecular blend in Fig. 4. Sufficient degrees of polymerization for the nucleobase blocks also ensured sufficient noncovalent interactions to influence the mechanical properties.
 | ||
| Fig. 4 Thermomechanical properties of solution-cast nucleobase-functionalized triblock copolymers and their blend. | ||
Supramolecular blend
Solution blend of poly(AdA-b-nBA-b-AdA) and poly(ThA-b-nBA-b-ThA) were prepared to examine the influence of complementary hydrogen bonding of A–T on mechanical performance. The supramolecular blend of poly(AdA-b-nBA-b-AdA) and poly(ThA-b-nBA-b-ThA) at 1:1 adenine:thymine molar ratio showed microphase-separation rather than macrophase-separation due to collectively strong intermolecular interactions.61 AFM and DMA revealed a well-organized microphase-separated morphology for the supramolecular blend (Fig. 3e, 4). Complementary hydrogen bonding between adenine and thymine is approximately 50× stronger than A–A/T–T self-association.4,60 The physically crosslinked thymine and adenine-functionalized hard blocks self-assembled into a single high Tg phase, while the soft PnBA block formed a soft phase. The plateau region of the supramolecular blend in Fig. 4 extended beyond the Tg's of either hard blocks. Table 1 summarizes observed Tg's from DSC and DMA and the major plateau range of solution-cast poly(AdA-b-nBA-b-AdA), poly(ThA-b-nBA-b-ThA), and their blend. The blend did not exhibit an external block Tg, and an approximately 40 °C wider plateau window was observed compared to the individual components. The molecular recognition between adenine and thymine restricted the mobility of the hard blocks above Tg's of either hard blocks. In addition, the blend showed an onset of terminal flow at a similar temperature as poly(AdA-b-nBA-b-AdA) and poly(ThA-b-nBA-b-ThA) near 90 °C, corresponding to the temperature where a majority of the hydrogen bonding dissociated. These results illustrated that noncovalent interactions extend the temperature independent modulus region, while maintaining melt processibility due to thermoreversibility of the physical crosslinks.
| Temperature (°C) | T A1g (°C) | T A2g (°C) | T B2g (°C) | T B2g (°C) | Plateau (°C) |
|---|---|---|---|---|---|
| Poly(AdA-b-nBA-b-AdA) | −31 | 52 | −39 | 76 | 76 |
| Poly(ThA-b-nBA-b-ThA) | −29 | 49 | −40 | 52 | 71 |
| A:T 1:1 Blend |
−30 | NA | −47 | 74 | 109 |
Dynamic mechanical analysis revealed potential benefits of the nucleobase-functionalized supramolecular blend for TPE applications. Inter-chain recognition contributed to well-defined microphase-separation, extended plateau range, and maintained melt processibility for the supramolecular blend. However, a higher plateau modulus, was observed compared to a typical TPE modulus range, limiting the elasticity of the solution-cast supramolecular blend. Further studies are underway to tune the triblock copolymer structure for a supramolecular blend with improved TPE performance.
SAXS and bulk morphology
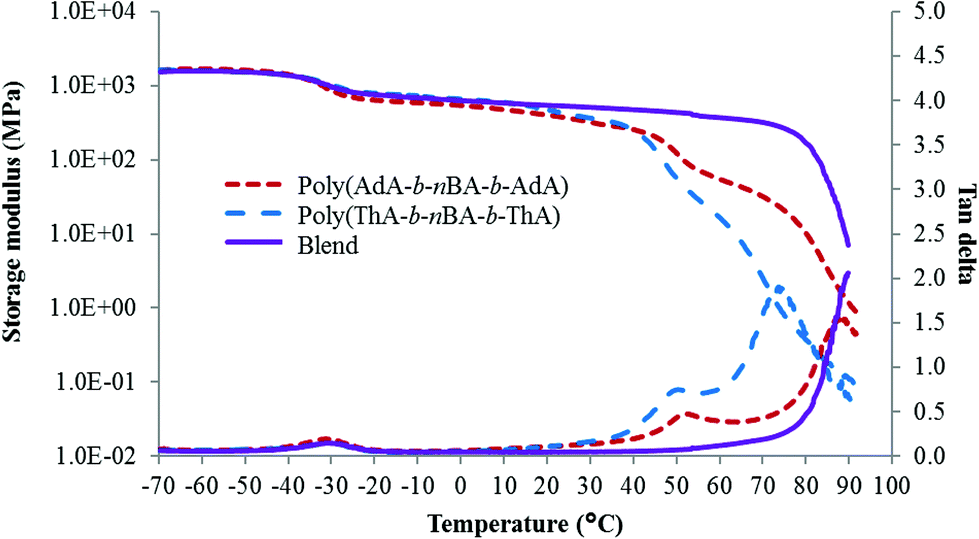
SAXS results were used to probe the bulk morphology of solution-cast poly(AdA-b-nBA-b-AdA), poly(ThA-b-nBA-b-ThA), and blends (Fig. 5). The SAXS profile for each of the samples contains relatively broad scattering maxima with periodic intensity oscillations, indicative of a morphology with a distribution of characteristic dimensions (in agreement with the AFM data in Fig. 3a, b, and e). Poly(AdA-b-nBA-b-AdA) and the blend show periodic scattering maxima that roughly match the expected q,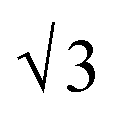 q,
q, 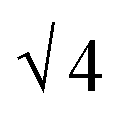 q,
q,  q, and
q, and 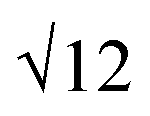 q peak positions that are characteristic of hexagonally packed cylinders, where q is the position of the first maximum. In contrast, for poly(ThA-b-nBA-b-ThA), both Fig. 3b and 5 indicate a much less ordered morphology. The well-assembled cylindrical phase separation morphology of poly(AdA-b-nBA-b-AdA) and the blend was attributed to enhanced ordering originating from pi–pi stacking and complementary hydrogen bonding, respectively. Purine rings of poly(AdA-b-nBA-b-AdA) were shown to afford stronger pi–pi interaction than pyrimidine rings in poly(ThA-b-nBA-b-ThA).4 However, the WAXD profile (Fig. S11†) only shows a broad diffraction peak characteristic of inter-chain correlations (amorphous halo) of the PnBA block.4 The absence of a signature diffraction peak for pi–pi stacking is presumably due to the small, nanometer-scale size of the packed AdA domains, as compared to much longer range order of packed AdA units in the homopolymers.4,5 The d spacings were 60.4 nm, 63.4 nm, and 58.7 nm for poly(AdA-b-nBA-b-AdA), poly(ThA-b-nBA-b-ThA), and the blend, respectively. These d spacings were attributed to the inter-particle distance between the phase separated domains, which also corresponded well with inter-particle distances observed in AFM. All average spacings were close to 60 nm due to the similar volume fractions of the external and internal blocks for all three samples. Overall, noncovalent interactions within the hard phase facilitated the self-assembly of block copolymers at the equilibrium self-assembled morphology. The bulk morphology and surface morphology agreed well, and both demonstrated the positive effect of noncovalent interactions on block copolymer self-assembly.
q peak positions that are characteristic of hexagonally packed cylinders, where q is the position of the first maximum. In contrast, for poly(ThA-b-nBA-b-ThA), both Fig. 3b and 5 indicate a much less ordered morphology. The well-assembled cylindrical phase separation morphology of poly(AdA-b-nBA-b-AdA) and the blend was attributed to enhanced ordering originating from pi–pi stacking and complementary hydrogen bonding, respectively. Purine rings of poly(AdA-b-nBA-b-AdA) were shown to afford stronger pi–pi interaction than pyrimidine rings in poly(ThA-b-nBA-b-ThA).4 However, the WAXD profile (Fig. S11†) only shows a broad diffraction peak characteristic of inter-chain correlations (amorphous halo) of the PnBA block.4 The absence of a signature diffraction peak for pi–pi stacking is presumably due to the small, nanometer-scale size of the packed AdA domains, as compared to much longer range order of packed AdA units in the homopolymers.4,5 The d spacings were 60.4 nm, 63.4 nm, and 58.7 nm for poly(AdA-b-nBA-b-AdA), poly(ThA-b-nBA-b-ThA), and the blend, respectively. These d spacings were attributed to the inter-particle distance between the phase separated domains, which also corresponded well with inter-particle distances observed in AFM. All average spacings were close to 60 nm due to the similar volume fractions of the external and internal blocks for all three samples. Overall, noncovalent interactions within the hard phase facilitated the self-assembly of block copolymers at the equilibrium self-assembled morphology. The bulk morphology and surface morphology agreed well, and both demonstrated the positive effect of noncovalent interactions on block copolymer self-assembly.
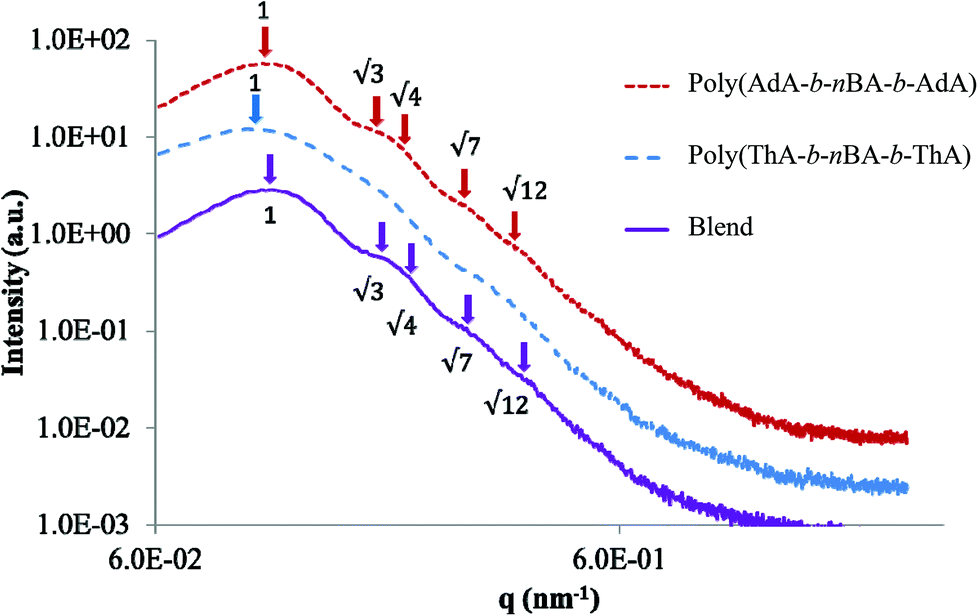 | ||
| Fig. 5 SAXS of solution-cast nucleobase-functionalized triblock copolymers and their blend. For clarity, data were vertically shifted by arbitrary factors. | ||
SAXS results also agreed with the trend in dynamic mechanical analysis of the solution-cast poly(AdA-b-nBA-b-AdA), poly(ThA-b-nBA-b-ThA), and the blend. Poly(ThA-b-nBA-b-ThA) exhibited the lowest order–disorder transition temperature due to the least ordered microphase-separation. Fig. 6 depicts the self-assembled morphology of the supramolecular blend. The complementary hydrogen bonding facilitated self-assembly of supramolecular block copolymer blends and enhanced the mechanical performance as a function of temperature. The thermal dissociation of hydrogen bonding led to a disruption of the physically crosslinked network in the hard phase, which resulted in a modulus drop. Overall, a synergy between the noncovalent interactions within the hard phase and the equilibrium, self-assembled morphology contributed to enhanced supramolecular polymer properties. Better phase separation led to more physical crosslinking within the hard phase and higher modulus when comparing solution-cast to melt-pressed samples. Stronger physical crosslinking within the hard phase facilitated self-assembly and reinforced the microphase-separated morphology when comparing solution-cast supramolecular blend with poly(AdA-b-nBA-b-AdA) and poly(ThA-b-nBA-b-ThA). However, noncovalent interactions decreased the rate of self-assembly in the melt.
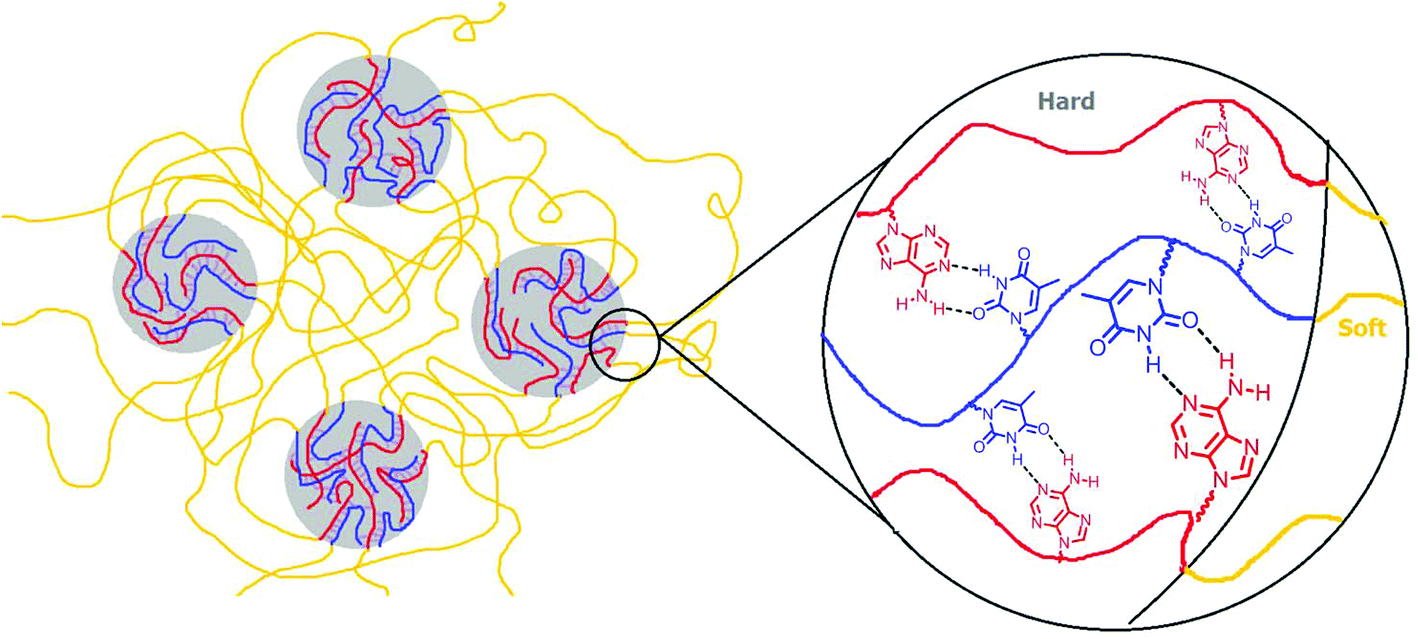 | ||
| Fig. 6 Pictorial representation of supramolecular blend of adenine and thymine-functionalized triblock copolymers. | ||
Variable temperature FTIR
Variable temperature FTIR is suitable to monitor the thermal reversibility of hydrogen bonding in the bulk.4,55 The low solubility of nucleobase-functionalized block copolymers in nonpolar solvents limited the feasibility of NMR titration despite monomer solubility. Variable temperature FTIR on poly(AdA-b-nBA-b-AdA), poly(ThA-b-nBA-b-ThA), and the blend verified the presence of hydrogen bonding and thermal reversibility. Fig. 7 shows the scale-expanded, ATR-corrected, spectral overlay of the supramolecular blend upon a temperature ramp. Hoogsteen base pairing is shown in Fig. 7 to label signature peaks in the spectrum. However, the association of adenine and thymine units most likely combined both Hoogsteen and Watson-Crick base pairing.22 The primary absorbance at 1730 cm−1 corresponded to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the carbonyl peak from the PnBA block and the non-conjugated, carbonyl peak from nucleobase blocks. The broad absorbance centered at 1670 cm−1 related to the hydrogen bonded CO on thymine, which shifted to higher wavenumber upon heating (red arrow). Two N-H bending vibration peaks at 1600 cm−1 and 1645 cm−1 both shifted toward lower wavenumbers with increasing temperature (blue arrows). Spectra collected after cooling to 30 °C shows that all hydrogen bonded absorbances shifted to their original wavenumbers. Upon cooling, hydrogen bonding formation led to a red-shift of CO stretching vibration and a blue-shift of N–H bending vibration.4,62,63 Red-shift of CO stretching vibration was due to CO bonding lengthening when hydrogen-bonded, where frequency decreases as force constant decreases in equation
O stretching of the carbonyl peak from the PnBA block and the non-conjugated, carbonyl peak from nucleobase blocks. The broad absorbance centered at 1670 cm−1 related to the hydrogen bonded CO on thymine, which shifted to higher wavenumber upon heating (red arrow). Two N-H bending vibration peaks at 1600 cm−1 and 1645 cm−1 both shifted toward lower wavenumbers with increasing temperature (blue arrows). Spectra collected after cooling to 30 °C shows that all hydrogen bonded absorbances shifted to their original wavenumbers. Upon cooling, hydrogen bonding formation led to a red-shift of CO stretching vibration and a blue-shift of N–H bending vibration.4,62,63 Red-shift of CO stretching vibration was due to CO bonding lengthening when hydrogen-bonded, where frequency decreases as force constant decreases in equation 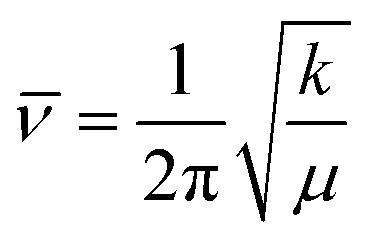 (k: force constant; μ: reduced mass).64 Blue-shift of N–H bending vibration was attributed to restriction of the bending motion from hydrogen bonding. The spectral reversibility over a heat-cool cycle demonstrated the thermal reversibility of the supramolecular network. The gray peaks corresponded to free CO and N–H that were not hydrogen bonded. The same FTIR experiment on poly(AdA-b-nBA-b-AdA) and poly(ThA-b-nBA-b-ThA) (Fig. S9, 10†) indicated the presence of weaker hydrogen bonding from the self-association of A–A and T–T, respectively. The FTIR results confirmed our hypothesis for the dynamic mechanical performance of the nucleobase-functionalized copolymers and the blend. The FTIR results also provided support for the morphology depicted in Fig. 6. Thermoreversible hydrogen bonding within the hard phase contributed to a supramolecular network with a more defined, self-assembled phase-separated morphology, mechanical performance, and thermal responsiveness.
(k: force constant; μ: reduced mass).64 Blue-shift of N–H bending vibration was attributed to restriction of the bending motion from hydrogen bonding. The spectral reversibility over a heat-cool cycle demonstrated the thermal reversibility of the supramolecular network. The gray peaks corresponded to free CO and N–H that were not hydrogen bonded. The same FTIR experiment on poly(AdA-b-nBA-b-AdA) and poly(ThA-b-nBA-b-ThA) (Fig. S9, 10†) indicated the presence of weaker hydrogen bonding from the self-association of A–A and T–T, respectively. The FTIR results confirmed our hypothesis for the dynamic mechanical performance of the nucleobase-functionalized copolymers and the blend. The FTIR results also provided support for the morphology depicted in Fig. 6. Thermoreversible hydrogen bonding within the hard phase contributed to a supramolecular network with a more defined, self-assembled phase-separated morphology, mechanical performance, and thermal responsiveness.
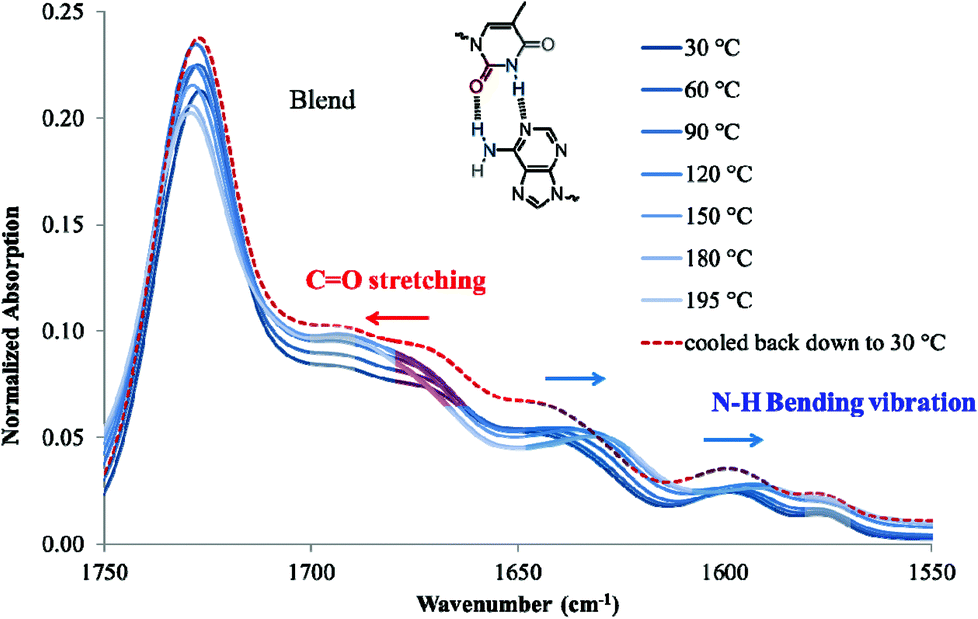 | ||
| Fig. 7 Variable temperature FTIR spectra in the 1500–1700 cm−1 region for the supramolecular blend. | ||
Conclusions
Divergent RAFT polymerization afforded synthesis of nucleobase-functionalized acrylic ABA triblock copolymers using a difunctional CTA. The acrylic backbone and flexible linker were crucial to lower the Tg's for probing the effect of noncovalent interactions. Nucleobase-functionalized triblock copolymers self-assembled into microphase-separated morphologies. Hydrogen bonding and pi-stacking created physically crosslinked networks with enhanced thermomechanical properties. The performance of block copolymer films was largely dependent on their self-assembly behavior, which was affected by processing conditions. The complementary hydrogen bonding of A–T in the hard phase of the supramolecular blend contributed to an elongated plateau, while maintaining melt processibility. We observed the synergy between physical crosslinking within the hard phase and self-assembled microphase-separation. Complementary noncovalent interactions showed potential in promoting self-assembly, enhancing mechanical strength, and introducing thermal responsiveness to improve melt processibility for block copolymers. Nucleobase-functionalized ABA copolymers offered a potential platform for future thermoplastic elastomer fabrication.Acknowledgements
This research was supported in part by the U.S. Army Research Laboratory and the U.S. Army Research Office under Contract/grant W911NF-07-1-0452, Ionic Liquids in Electro-Active Devices Multidisciplinary University Research Initiative (ILEAD MURI). Additionally, this work was supported in part by Henkel Corporation, and we thank Charles Paul, Cristina DeJesus, and Eric Silverberg for insightful discussions. This material is also partially based upon work supported by the National Science Foundation under grant no. DMR-0923107. We also thank Professor Mitsuru Ueda and the Academy for Co-creative Education of Environment and Energy Science at Tokyo Institute of Technology for their support of summer research for Mr Motohiro Aiba at Virginia Tech.References
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS PubMed.
- Analytical Methods in Supramolecular Chemistry, ed. C. Schalley, Wiley-VCH Verlag GmbH & Co. KGaA, 2007 Search PubMed.
- K. Yamauchi, J. R. Lizotte and T. E. Long, Macromolecules, 2003, 36, 1083–1088 CrossRef CAS.
- S. Cheng, M. Zhang, N. Dixit, R. B. Moore and T. E. Long, Macromolecules, 2012, 45, 805–812 CrossRef CAS.
- S. Sivakova, D. A. Bohnsack, M. E. Mackay, P. Suwanmala and S. J. Rowan, J. Am. Chem. Soc., 2005, 127, 18202–18211 CrossRef CAS PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- D. C. Sherrington and K. A. Taskinen, Chem. Soc. Rev., 2001, 30, 83–93 RSC.
- J. M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH, 1995 Search PubMed.
- C. Fouquey, J. M. Lehn and A. M. Levelut, Adv. Mater., 1990, 2, 254–257 CrossRef CAS.
- J. Pan, M. Chen, W. Warner, M. He, L. Dalton and T. E. Hogen-Esch, Macromolecules, 2000, 33, 7835–7841 CrossRef CAS.
- J. Kriz, J. Dybal and J. Brus, J. Phys. Chem. B, 2006, 110, 18338–18346 CrossRef CAS PubMed.
- B. D. Mather, M. B. Baker, F. L. Beyer, M. A. G. Berg, M. D. Green and T. E. Long, Macromolecules, 2007, 40, 6834–6845 CrossRef CAS.
- K. Zhang, G. B. Fahs, M. Aiba, R. B. Moore and T. E. Long, Chem. Commun., 2014, 50, 9145–9148 RSC.
- R. McHale, J. P. Patterson, P. B. Zetterlund and R. K. O'Reilly, Nat. Chem., 2012, 4, 491–497 CrossRef CAS PubMed.
- J. S. Nowick, J. S. Chen and G. Noronha, J. Am. Chem. Soc., 1993, 115, 7636–7644 CrossRef CAS.
- R. J. Spontak and N. P. Patel, Curr. Opin. Colloid Interface Sci., 2000, 5, 334–341 CrossRef CAS.
- S. Amin and M. Amin, Rev. Adv. Mater. Sci., 2011, 29, 15–30 CAS.
- P. Antony and S. K. De, J. Macromol. Sci., Polym. Rev., 2001, C41, 41–77 CrossRef CAS PubMed.
- R. P. Quirk and J. Kim, Rubber Chem. Technol., 1991, 64, 450–468 CrossRef CAS.
- V. Duchacek, J. Macromol. Sci., Phys., 1998, B37, 275–282 CrossRef CAS.
- J. G. Drobny, Handbook of Thermoplastic Elastomers, William Andrew Publishing/Plastics Design Library, 2007 Search PubMed.
- S. Sivakova and S. J. Rowan, Chem. Soc. Rev., 2005, 34, 9–21 RSC.
- R. P. Sijbesma and E. W. Meijer, Chem. Commun., 2003, 5–16 RSC.
- S. T. Hemp and T. E. Long, Macromol. Biosci., 2012, 12, 29–39 CrossRef CAS PubMed.
- R. McHale and R. K. O'Reilly, Macromolecules, 2012, 45, 7665–7675 CrossRef CAS.
- P. Yakovchuk, E. Protozanova and M. D. Frank-Kamenetskii, Nucleic Acids Res., 2006, 34, 564–574 CrossRef CAS PubMed.
- J. C. Kim, J. Jung, Y. Rho, M. Kim, W. Kwon, H. Kim, I. J. Kim, J. R. Kim and M. Ree, Biomacromolecules, 2011, 12, 2822–2833 CrossRef CAS PubMed.
- M. Egholm, O. Buchardt, P. E. Nielsen and R. H. Berg, J. Am. Chem. Soc., 1992, 114, 1895–1897 CrossRef CAS.
- A. Khan, D. M. Haddleton, M. J. Hannon, D. Kukulj and A. Marsh, Macromolecules, 1999, 32, 6560–6564 CrossRef CAS.
- Y. Kang, A. Lu, A. Ellington, M. C. Jewett and R. K. O'Reilly, ACS Macro Lett., 2013, 581–586 CrossRef CAS.
- H. S. Bazzi and H. F. Sleiman, Macromolecules, 2002, 35, 9617–9620 CrossRef CAS.
- P. K. Lo and H. F. Sleiman, J. Am. Chem. Soc., 2009, 131, 4182–4183 CrossRef CAS PubMed.
- B. D. Mather, M. B. Baker, F. L. Beyer, M. D. Green, M. A. G. Berg and T. E. Long, Macromolecules, 2007, 40, 4396–4398 CrossRef CAS.
- H. J. Spijker, A. J. Dirks and H. J. C. M. Van, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4242–4250 CrossRef CAS.
- H. J. Spijker, D. F. L. van and H. J. C. M. van, Macromolecules, 2007, 40, 12–18 CrossRef CAS.
- J.-F. Lutz, A. F. Thuenemann and R. Nehring, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4805–4818 CrossRef CAS.
- J.-F. Lutz, A. F. Thuenemann and K. Rurack, Macromolecules, 2005, 38, 8124–8126 CrossRef CAS.
- Y. Inaki, K. Ebisutani and K. Takemoto, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 3249–3262 CrossRef CAS.
- Y. Inaki, Prog. Polym. Sci., 1992, 17, 515–570 CrossRef CAS.
- K. Takemoto and Y. Inaki, in Speciality Polymers, Springer, Berlin Heidelberg, 1981, vol. 41, ch. 1, pp. 1–51 Search PubMed.
- A. Marsh, A. Khan, D. M. Haddleton and M. J. Hannon, Macromolecules, 1999, 32, 8725–8731 CrossRef CAS.
- K. P. Nair, J. M. Pollino and M. Weck, Macromolecules, 2006, 39, 931–940 CrossRef CAS.
- H. S. Bazzi, J. Bouffard and H. F. Sleiman, Macromolecules, 2003, 36, 7899–7902 CrossRef CAS.
- P. K. Lo and H. F. Sleiman, Macromolecules, 2008, 41, 5590–5603 CrossRef CAS.
- A. Marsh, A. Khan, D. M. Haddleton and M. J. Hannon, Macromolecules, 1999, 32, 8725–8731 CrossRef CAS.
- G. T. F. A. De, M. J. Kade, K. E. Feldman, E. J. Kramer, C. J. Hawker and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4253–4260 CrossRef.
- W. H. Binder, M. J. Kunz, C. Kluger, G. Hayn and R. Saf, Macromolecules, 2004, 37, 1749–1759 CrossRef CAS.
- A. S. Karikari, W. F. Edwards, J. B. Mecham and T. E. Long, Biomacromolecules, 2005, 6, 2866–2874 CrossRef CAS PubMed.
- K. Yamauchi, J. R. Lizotte and T. E. Long, Macromolecules, 2002, 35, 8745–8750 CrossRef CAS.
- Thermal Transitions of Homopolymers, https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Aldrich/General_Information/thermal_transitions_of_homopolymers.pdf, Sigma Aldrich Search PubMed.
- N. M. Ahmad, B. Charleux, C. Farcet, C. J. Ferguson, S. G. Gaynor, B. S. Hawkett, F. Heatley, B. Klumperman, D. Konkolewicz, P. A. Lovell, K. Matyjaszewski and R. Venkatesh, Macromol. Rapid Commun., 2009, 30, 2002–2021 CrossRef CAS PubMed.
- M. H. Allen, Jr., S. T. Hemp, M. Zhang, M. Zhang, A. E. Smith, R. B. Moore and T. E. Long, Polym. Chem., 2013, 4, 2333–2341 RSC.
- A. E. Smith, X. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS PubMed.
- Y. Tao, K. Satoh and M. Kamigaito, Macromol. Rapid Commun., 2011, 32, 226–232 CrossRef CAS PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS.
- M. Tamami, S. T. Hemp, K. Zhang, M. Zhang, R. B. Moore and T. E. Long, Polymer, 2013, 54, 1588–1595 CrossRef CAS PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- L. Fielding, Tetrahedron, 2000, 56, 6151–6170 CrossRef CAS.
- K. A. Connors, Binding constants: the measurement of molecular complex stability, Wiley, New York, 1987 Search PubMed.
- J. Sartorius and H.-J. Schhneider, Chem. – Eur. J., 1996, 2, 1446–1452 CrossRef CAS.
- Z. Yang and C. D. Han, Macromolecules, 2008, 41, 2104–2118 CrossRef CAS.
- M. Tamami, K. Zhang, N. Dixit, R. B. Moore and T. E. Long, Macromol. Chem. Phys., 2014, 215, 2337–2344 CrossRef CAS.
- M. Falk and T. A. Ford, Can. J. Chem., 1966, 44, 1699–1707 CrossRef CAS.
- E. Arunan, R. Desiraju Gautam, A. Klein Roger, J. Sadlej, S. Scheiner, I. Alkorta, C. Clary David, H. Crabtree Robert, J. Dannenberg Joseph, P. Hobza, G. Kjaergaard Henrik, C. Legon Anthony, B. Mennucci and J. Nesbitt David, Pure Appl. Chem., 2011, 83, 1637 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4py01798f |
| This journal is © The Royal Society of Chemistry 2015 |