
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Low volume shrinkage of polymers by photopolymerization of 1,1-bis(ethoxycarbonyl)-2-vinylcyclopropanes†
Paul Pineda
Contreras‡
,
Payal
Tyagi‡
and
Seema
Agarwal
*
University of Bayreuth, Macromolecular Chemistry II and Bayreuth Center for Colloids and Interfaces, Universitätsstraße 30, 95440, Bayreuth, Germany. E-mail: agarwal@uni-bayreuth.de; Fax: +49-921-553393; Tel: +49-921-553397
First published on 19th January 2015
Abstract
Low volume shrinkage of polymers by photoring-opening polymerization of 1,1-bis(ethoxycarbonyl)-2-vinylcyclopropane (VCP) and its methyl substituted derivate 1,1-bis(ethoxycarbonyl)-2-(prop-1-en-2-yl)-cyclopropane (Me-VCP) is presented. The aim of this study was to evaluate the photopolymerization behavior and structure of the corresponding polymers from VCP and its methyl substituted derivates 1,1-bis(ethoxycarbonyl)-2-(prop-1-en-2-yl)-cyclopropane (Me-VCP) and 1,1-bis(ethoxycarbonyl)-2-methyl-2-(prop-1-en-2-yl)-cyclopropane (DiMeVCP). VCP monomers were polymerized using camphorquinone (CQ) as a photo-initiator either in a binary or ternary photo-initiator system. The binary systems were formulated with ethyl 4-dimethylaminobenzoate (EDMAB) in relation to the monomer and 1 mol% of CQ. The ternary system was a mixture of 1 mol% of CQ, 2 mol% of EDMAB and 2 mol% of DPIHFP. Ternary photo-initiator systems showed high polymerization rates leading to high conversion in a short photo-activation time. One of the important findings is the formation of higher amounts of 1,5-type ring-opened units and lower amounts of cyclobutane-containing units in photopolymerization, which can make VCP an attractive component of dental composites, as the volume shrinkage is proportional to the 1,5-ring opening. Earlier it was observed that thermal free radical polymerization of VCPs gives higher amounts of cyclobutane structures compared to 1,5-type ring-opened units in the corresponding polymers, causing VCP to be excluded from the list of promising dental composites, as the volume shrinkage was higher than expected.
1. Introduction
2-Vinylcyclopropane-1,1-dicarboxylates (VCPs) are attractive monomers as they display less polymerization shrinkage than other vinyl monomers, such as methacrylates.1–3 This characteristic makes such cyclic monomers attractive as low-shrinking components in dental adhesives or filling composites, curing resins, molding and filling materials. As a consequence, VCPs have attracted considerable interest among researchers in publications and patents.4–6 Many studies have been carried out to investigate the influence of the type and position of substituents on the rate, molecular weight, volume shrinkage and polymer structure of VCPs by thermal radical polymerization.7 It has been reported that thermal radical polymerization provides a polymer consisting of a 1,5-ring opened unit along with a new unit, which is believed to have a cyclobutane structure (Scheme 1).3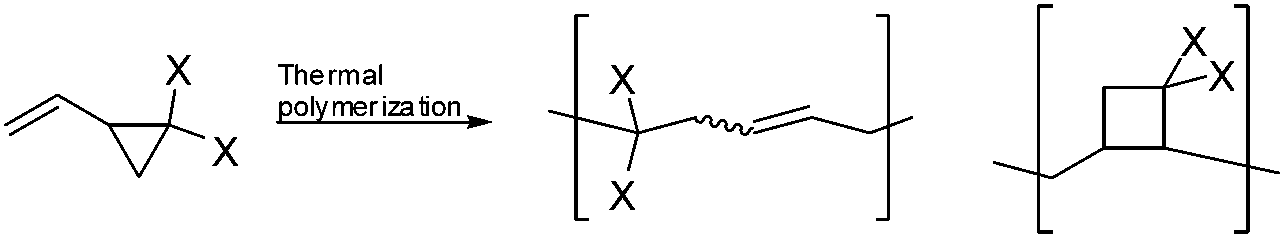 | ||
| Scheme 1 Reported polymer structures of the radical ring-opening polymerization of VCPs. | ||
By comparing the dependence between the shrinkage on polymerization and the reciprocal of the molecular weight of several VCP monomers, a higher shrinkage was observed than expected for ring-opening monomers. Furthermore the obtained volume shrinkage was in the range of vinyl monomers. This additional shrinkage was attributed to the formation of the cyclobutane containing unit during polymerization.3 But since VCPs are possible ring-opening monomers, they are expected to be useful materials with lower shrinkage in volume, by reducing the formation of cyclobutane containing units during radical polymerization.
It is interesting to note that not many detailed studies in the open literature are provided for ring-opening photopolymerization of VCPs. Camphorquinone (CQ) and ethyl-4-dimethylamino-benzoate (EDMAB) and 2,4,6-trimethylbenzoyldiphenyl-phosphinoxide (Lucirin), a three component initiator system was used for photocopolymerization of bifunctional methacrylate monomers like bis-GMA and UDMA with a vinylcyclopropane derivate (1,3-bis[(1-ethoxycarbonyl-2-vinylcyclopropane-1-yl)carboxy)]benzene to form crosslinked networks.8
Till now, the practical application of VCPs has been restricted, as in comparison with standard methacrylate monomers they are less reactive.3,9 However, the research group of N. Moszner et al. in particular, has synthesized and investigated with great effort a number of different 1,1-di-substituted VCPs to increase the reactivity during thermal radical polymerization; unfortunately the many different approaches resulted only in moderate improvements.10 In order to enhance the polymerization reactivity of VCPs regardless, not by synthetic methods, but rather by a variation of the photopolymerization conditions, diaryliodonium salts were introduced to the photo-initiator system. Diaryliodonium salts are efficient photo-initiators for both radical and ionic (mostly cationic) polymerization.11,12 When light is irradiated on the excited iodonium it decomposes to an aryliodo radical cation, a reactive aryl radical and an anion.13 However, these salts do not absorb significantly above 300 nm, therefore UV light sources were used, which restrict their biological applications, where UV light is not recommended. However, it was reported that using dyes that absorb in the visible light region as sensitizers, would allow a reaction with the onium salts, promoting the decomposition.14 In a similar way, onium salts can also act in radical polymerization.15,16 Several kinds of visible light sensitizer dyes have been investigated, such as camphorquinone,17 safranine,18 acridine derivatives16 and acetophenone dyes.14 Camphorquinone, a dye widely used as a photo-initiator for radical polymerization, can act as a photo-sensitizer to diphenyliodonium hexafluorophosphate, which does not absorb light in the visible region, improving the reactivity of the photo-initiator system.17
The aim of the present work was to study the effect of the photoinitiated ring-opening polymerization of 2-vinylcyclopropane-1,1-dicarboxylate and the corresponding monomethyl substituted VCP on the polymer structure, kinetics and volume shrinkage. Camphorquinone was used as a photosensitizer with ethyl-4-dimethylaminobenzoate for binary, and together with diphenyliodonium hexafluorophosphate, for ternary initiation processes to evaluate the influence of an onium salt on the radical photopolymerization kinetics and polymer structure of VCPs.
2. Experimental section
2.1 Measurements
1H- (300 MHz) and 13C- (75 MHz) NMR spectra were recorded on a Bruker Ultrashield-300 spectrometer in CDCl3, using tetramethylsilane (TMS) as an internal standard. GC measurements have been performed by a GC-FID system of the type QP-5050 from Shimadzu Company with nitrogen as carrier gas. GC-MS measurements have been performed with a system Agilent 5977A MSD with helium as carrier gas. Molecular weights and polydispersity index (PDI) of the polymers were determined by GPC with a Knauer system equipped with two columns, PSS-SDV (linear, 10 μl, 60 cm × 0.8 cm), and a differential refractive index (RI) detector. THF was used as an eluent at a flow rate of 0.8 ml min−1. A polystyrene calibration was employed as a reference. A Mettler thermal analyzer of the type 821c DSC was used for the thermal characterization of polymers. DSC scans were recorded under a nitrogen atmosphere at a flow rate of 70 ml min−1 at heating rates of 10 K min−1. Each sample weighed about 10 ± 2 mg for DSC analysis. The polymerization shrinkage was calculated from the difference in density of the monomer and the formed polymers (Archimedes principle). The densities of the monomers were measured by a 1 mL pycnometer, and the densities of the polymers, by water displacement of 400 mg cured samples. The polymer samples were prepared by inserting the monomer–initiator mixture into the bottom of a 10 mL standard test tube at light exclusion, flushing the tubes with an argon gas for 30 s and irradiating each sample for 48 h at room temperature. Afterwards the samples were cooled and bounced out of the glass tubes to a glass plate of similar density and weight, as the VCP polymers occurred as not totally solid but rather as viscous polymer melts. The polymerization shrinkage was determined separately for the three samples.2.2 Materials
1,4-Dibromo-2-butene, diethyl malonate, sodium, isoprene, camphorquinone (CQ) and tert-butyl peroxide (DTBP) were supplied by Aldrich and used without further purification. Azobisisobutyronitrile (AIBN) was obtained from Bayer AG and recrystallized from methanol before use. Ethyl 4-dimethylaminobenzoate (EDMAB) (Fluka) and diphenyliodonium hexafluorophosphate (DPIHFP) (ABCR GmbH & Co. KG) were used without further purification. Tetrahydrofuran (BASF), ethanol (BASF), chloroform and pentane were distilled before use.2.3 Monomer synthesis
1,1-Disubstituted 2-vinylcyclopropanes were obtained from diethyl malonate and the corresponding dibromo butane as per the published procedure.3,19 1,4-Dibromo-2-methyl-2-butene was prepared previously in a reaction between isoprene and bromine in chloroform (Scheme 2).20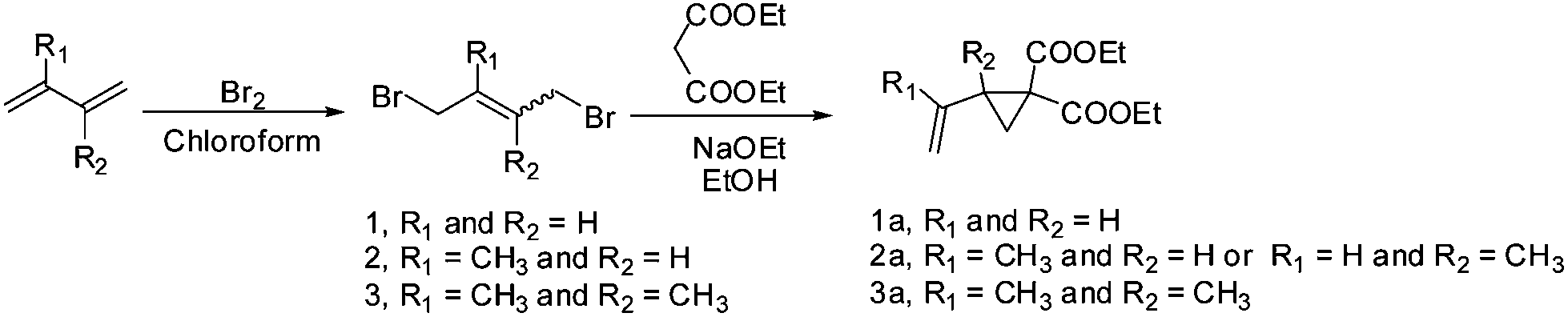 | ||
| Scheme 2 Schematic pathway to synthesize monomers 1a–3a. | ||
The detailed reaction procedures, including purification and characterization, are given in the relevant ESI.† The structures were characterized by NMR and GC. In contrast to the reported literature, we observed for the synthesis of 2a a constitution isomer of around 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 of diethyl 2-(prop-1-en-2-yl)cyclopropane-1,1-dicarboxylate to diethyl 2-methyl-2-vinylcyclopropane-1,1-dicarboxylate, that could not be separated by distillation or column chromatography as shown in the NMR (Fig. 1). Therefore this mixture was used for polymerizations.
:20 of diethyl 2-(prop-1-en-2-yl)cyclopropane-1,1-dicarboxylate to diethyl 2-methyl-2-vinylcyclopropane-1,1-dicarboxylate, that could not be separated by distillation or column chromatography as shown in the NMR (Fig. 1). Therefore this mixture was used for polymerizations.
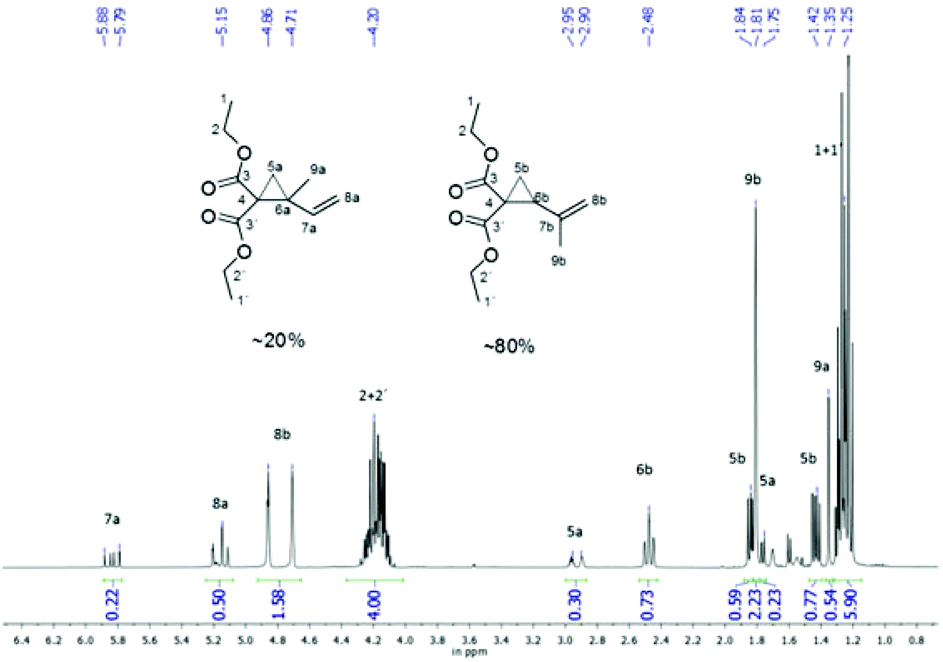 | ||
| Fig. 1 1H-NMR of the monomer (2a), which consists of diethyl 2-(prop-1-en-2-yl)cyclopropane-1,1-dicarboxylate (∼20%) and diethyl 2-methyl-2-vinylcyclopropane-1,1-dicarboxylate (∼80%) (CDCl3, 300 MHz). | ||
2.4 Polymerization
3. Results and discussion
Thermal radical polymerization of VCPs such as 2-vinylcyclopropane-1,1-dicarboxylates and derivatives is well-studied in the literature.1,3,7 The radical from the thermal initiator adds at the vinyl double bond of VCP followed by isomerization with ring-opening leading to the formation of a stable t-radical for further propagation. In the present work, photopolymerization of 2-vinylcyclopropane-1,1-dicarboxylate (1a) and the corresponding mono and di-α-methyl substituted VCPs (2a and 3a) was studied using different photo-initiator systems in different ratios (camphorquinone (CQ) and ethyl 4-dimethylaminobenzoate (EDMAB) (1:2 mol%), and CQ and EDMAB (1:2 mol%) with different ratios of DPIHFP as the ternary initiator (1, 2 and 4 mol%)17 (Scheme 3).
 | ||
| Scheme 3 Schematic polymerization equation of photosynthesized polymers. | ||
The reaction kinetics of the photo-polymerization with the binary initiator system (1 mol% CQ and 2 mol% EDMAB) is depicted in Fig. 2. White to light yellow colored polymers, which were soluble in common solvents were obtained from 1a and 2a. However, 3a the di-methyl substituted monomer did polymerize only in negligible yields, which confirms the reported literature for thermal polymerization of methyl substituted 1-ethoxycarbonyl-2-vinylcyclopropanes21 and methyl-substituted 1,1-diethoxycarbonyl-2-vinylcyclopropanes.19 This trend was explained with the assumption of the higher chain transfer reaction of the di-methyl substituted VCP.19 Nevertheless it should be considered that the presence of two methyl substituents, one in the α-position and the other on the cyclopropane ring might provide the cause of hyperconjugation, a relative stable radical, whereby the rate of ring-opening is reduced to a range within the radical termination reaction, and becomes a byword of the propagation.
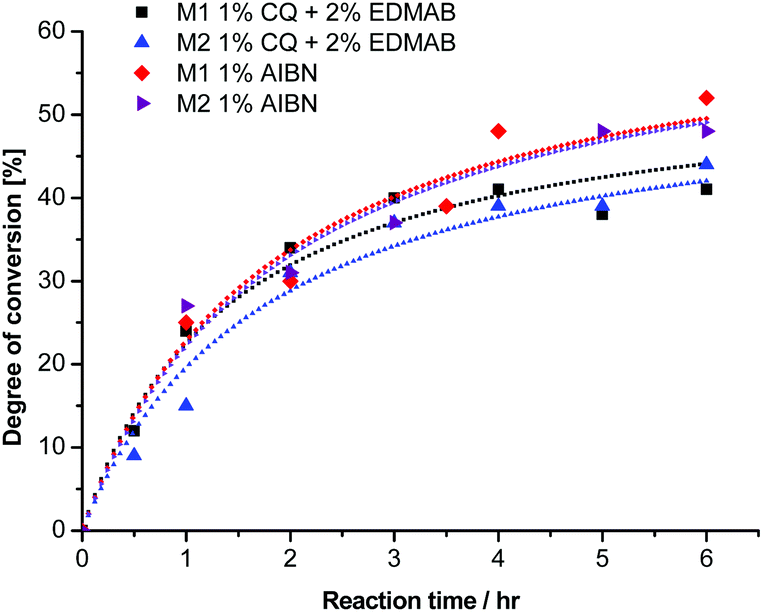 | ||
| Fig. 2 Degree of conversion for the polymerization of monomers 1a (M1) and 2a (M2) with different initiators. Concentrations of 1 mol% CQ and 2 mol% EDMAB as well as 1 mol% AIBN were used. | ||
The rates of polymerization (Rp) of the photo-polymerization with the binary initiator system were 0.30 and 0.25% per min for 1a and 2a, respectively. As a matter of fact it seemed that by conventional radical polymerization, the radical ring opening process, independent of the radical initiator and α-substituent, was very slow. Therefore for providing a comparison we added in Fig. 2 the thermal polymerization kinetics of 1a and 2a in the presence of 1 mol% AIBN, a thermal initiator, where we observed almost identical Rp values of 0.29 and 0.28% per min. These Rp values were still slow, which is in line with the literature.3,19
However the ternary photoinitiator system with additional DPIHFP could provide faster vinylcyclopropane polymerizations. It is worth mentioning that the ternary photoinitiator systems are already known to provide enhanced rates of polymerizations to the conventional vinyl monomers owing to an increased concentration of initiating radicals or because of the concurrent cationic or radical initiating species.16,17,22 The observed Rp values for ring-opening polymerization of VCPs were much higher with higher overall conversions (around 80%) compared to the conventional CQ-EDMAB system. Therefore for a detailed investigation we added DPIHFP as an initiator at different molar concentrations.
We observed for the degree of conversion as a function of the photo-activation time for the ternary initiator system with 2 mol% DPIHFP Rp values of 11.37 and 2.60% per min for 1a and 2a, respectively (Fig. 3). Thereby the Rp values, compared to the conventional photopolymerization system CQ-EDMAB and to the thermal polymerization AIBN (Fig. 2), can be significantly improved by using a ternary system to the factor ∼35 and ∼10 for poly(1a) and by factor ∼38 and ∼9 for poly(2a). In general, an increase in the molar mass of polymers was also seen on using the ternary initiator system (Table 1).
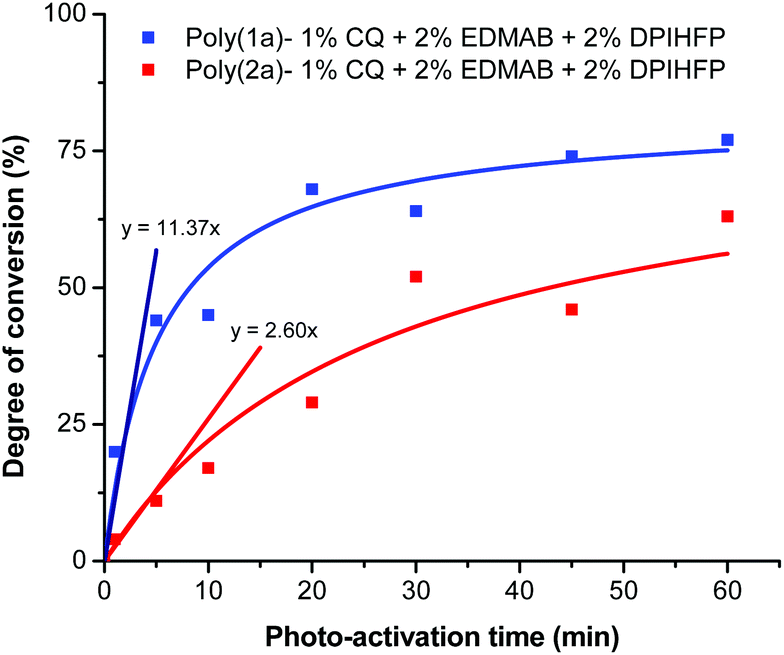 | ||
| Fig. 3 Degree of conversion for the photo-polymerization of monomers 1a and 2a with 1 mol% CQ + 2 mol% EDMAB + 2 mol% DPIHFP as the ternary initiator. | ||
| Entry | Initiatorb | Temp./°C | Ring opened polymer structurec/% | Cyclo-butane polymer structurec/% |
M
nd/Da × 10−4 |
D | Yield/% | |
|---|---|---|---|---|---|---|---|---|
| a Monomer: 0.3 g. b 1 mol% CQ; 2 mol% EDMAB, 2 mol% DPIHFP. c Estimated by 1H NMR in CDCl3, polymer structure illustrated in Schemes 7 and 9 and Fig. 5 and S13–S17.† d Estimated by GPC; bimodal distribution of poly(2a). e Yield in 12 h. f Yield in 8 h. g Yield in 1 h. | ||||||||
| 1 | Poly(1a) | CQ/EDMAB | 25 | 78 | 22 | 0.8 | 1.5 | 62e |
| 2 | Poly(2a) | CQ/EDMAB | 25 | 93 | 7 | 1.3 | 1.9 | 64e |
| 3 | Poly(1a) | AIBN | 70 | 59 | 41 | 8.9 | 1.8 | 57f |
| 4 | DTBP | 120 | 50 | 50 | 4.4 | 1.9 | 51f | |
| 5 | Poly(2a) | AIBN | 70 | 86 | 14 | 1.3 | 2.5 | 56f |
| 6 | DTBP | 120 | 75 | 25 | 1.1 | 2.9 | 58f | |
| 7 | Poly(1a) | CQ/EDMAB | 25 | 86 | 14 | 5.5 | 2.9 | 24g |
| 8 | CQ/EDMAB/DPIHFP | 25 | 80 | 20 | 2.0 | 2.1 | 77g | |
| 9 | Poly(2a) | CQ/EDMAB | 25 | 95 | 5 | 1.0 | 2.7 | 15g |
| 10 | CQ/EDMAB/DPIHFP | 25 | 86 | 14 | 1.7 | 2.2 | 63g | |
Further the concentration level of DPIHFP within the ternary photo-initiator system showed a significant effect both on the rate of polymerization and the degree of monomer conversion (Fig. 4). Polymerization with a lower concentration of DPIHFP (below 2%) showed higher monomer conversions at low reaction times. 10 min of light radiation and just 0.5 mol% DPIHFP in the ternary initiator system was sufficient to polymerize almost 25% of the monomers. However, for further photo-activation, there was almost no further increment in the yield of polymerization. One of the possible reasons could be that the onium salt decomposes after a certain period of time. The highest conversion was obtained with 4% onium salt concentration (about 80% conversion in 15 minutes), although there were solubility problems with onium salts having a concentration of 2% or higher in 1,1-disubstituted 2-vinylcyclopropanes.
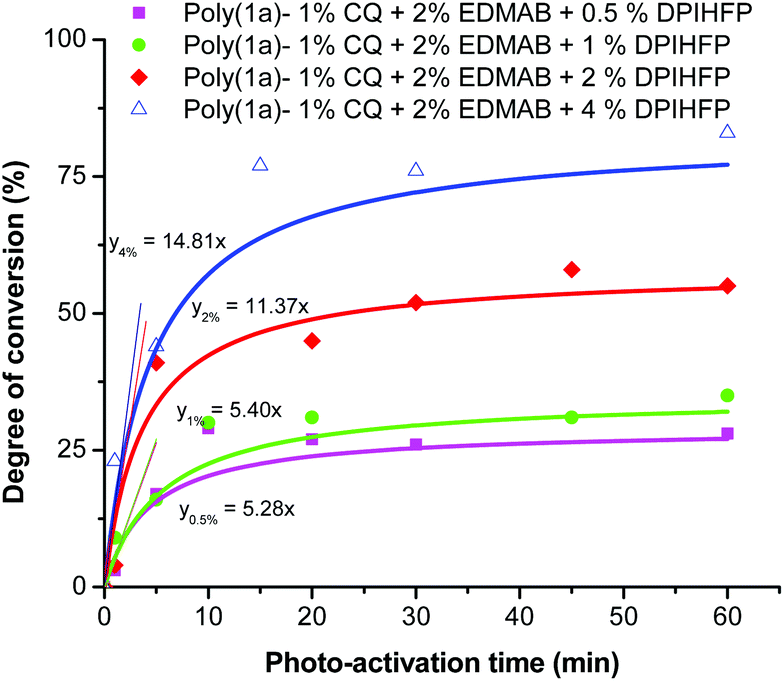 | ||
| Fig. 4 Degree of conversion for the photo-polymerization of monomer 1a with different concentrations of DPIHFP as the ternary initiator. | ||
The photo-initiator system CQ/EDMAB is frequently used for dentistry applications as the absorption maximum of CQ is around 470 nm, beyond the UV wavelength. Camphorquinone as a 1,2-diketone compound forms a reactive excited species (CQ*) during light absorption (eqn (1)).23 These excited species interact with the EDMAB to form an exciplex, and within CQ* abstracts, with a hydrogen atom from the tertiary amine to give a ketyl radical and the α-amino-alkyl radicals (eqn (2)), which initiate the polymerization at the carbon double bond of the monomer (eqn (6)). Meanwhile the ketyl radical mainly dimerizes or disproportionates.23 The simplified reaction mechanism of CQ/EDMAB is shown in Scheme 4.23,24
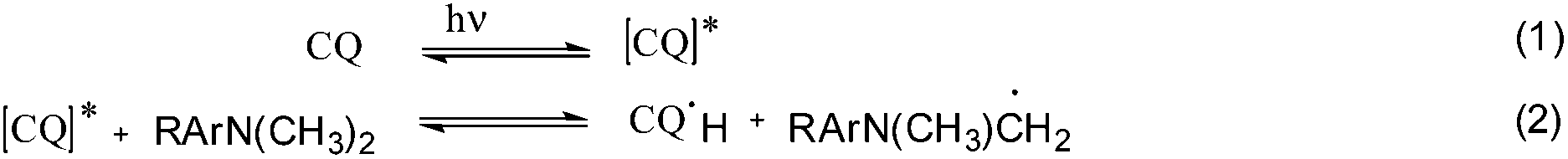 | ||
| Scheme 4 Schematic simplified reaction scheme of CQ/EDMAB initiation. | ||
The reaction mechanism using a ternary photo-initiator system is more complex to understand, as Ogliari et al. and other authors have already described.17,24 For consideration of our mechanism only the radical pathway has been described, as we observed that polymerization propagation was based only on radical species and not the cationic one (description further below). The amine radical generated from the exciplex state between CQ* and EDMAB (eqn (2)) could react with DPIHFP, irreversibly cleaving the C–I bond to generate a reactive phenyl radical (eqn (3)). This phenyl radical can either react with the remaining amine, abstracting a proton and generating a new amine free radical or simply start propagation (eqn (7)). It is also reported elsewhere that an exciplex between CQ* and DPIFHP is initially formed, which reacts further with the EDMAB, giving a radical amine species that initiates the polymerization and regenerates the DPIFHP (eqn (5)).18 Beyond that the DPIFHP can be reduced by an electron transfer within the exciplex with CQ* to a diphenyliodine free radical, which is unstable and decomposes into a phenyliodine and again to a phenyl free radical (eqn (4)).17,24 Photopolymerization experiments with monomer 1a were performed in the presence of the radical inhibitor, hydroquinone. It was observed that the polymerization of 1a was completely inhibited in ternary systems (CQ/EDMAB/DPIHFP). This result indicated that the polymerization mechanism of the ternary initiation system (CQ/EDMAB/DPIHFP) was the only free radical mechanism for VCP polymerization, and not concurrently radical and cationic, as known for ternary initiator systems with onium salts (Schemes 5 and 6).12
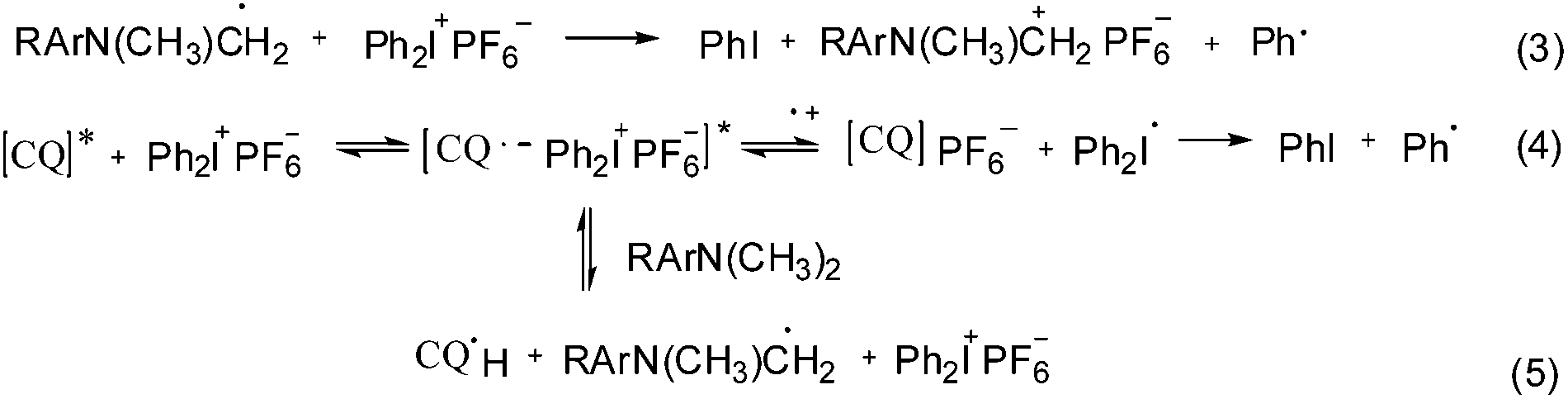 | ||
| Scheme 5 Simplified scheme of the radical initiation reaction of the ternary photo-initiator system using DPIHFP as the onium salt. | ||
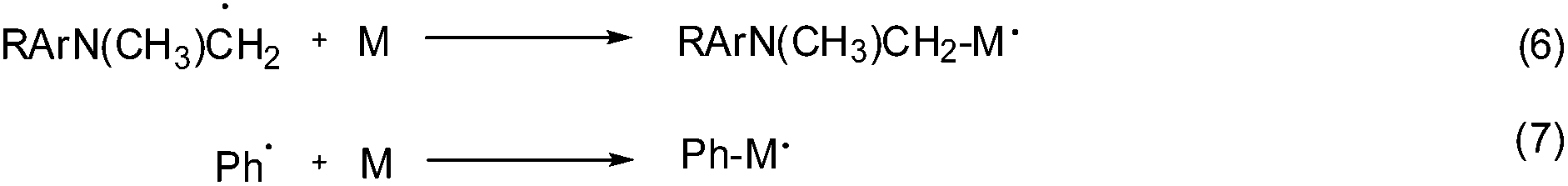 | ||
| Scheme 6 Schematic propagation initiations during photo polymerization. | ||
Altogether, it can be concluded that the addition of the iodonium salt DPIHFP opens further pathways capable of generating radicals, whereby compared to the binary initiator system CQ/EDMAB, on the one hand the relative efficiency of the radical initiation is increased, while on the other hand an increased concentration of initiating radicals results simply by using DPIHFP as the ternary initiator.
3.1 Structure of the polymer
It has been described by Sanda et al. that a 1,5-type ring-opened polymer unit is primarily formed in the thermal polymerization of 1,1-disubstituted 2-vinylcyclopropanes with minor amounts of cyclobutane containing units (Scheme 7).3,19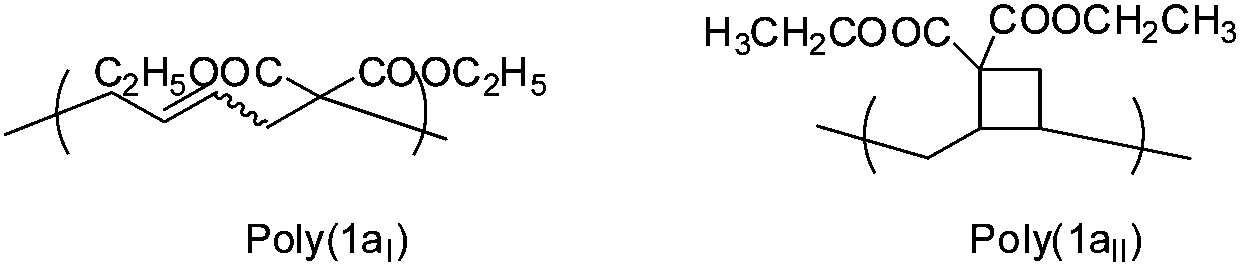 | ||
| Scheme 7 Expected polymer structures for monomer (1a). | ||
The polymer structure of poly(1a) made by photopolymerization was examined in detail by 1H-NMR (Fig. 5), and compared with the structures formed using thermal radical polymerization. According to the reported literature a broad signal between 1.5 and 2.5 ppm in 1H NMR is caused by the formation of a cyclobutane containing unit (1aII) for thermally polymerized samples.3 This broad signal was not as clearly observed in poly(1a) obtained from photopolymerization, as in the polymers obtained during thermal polymerization with AIBN or DTBP (Fig. 5).
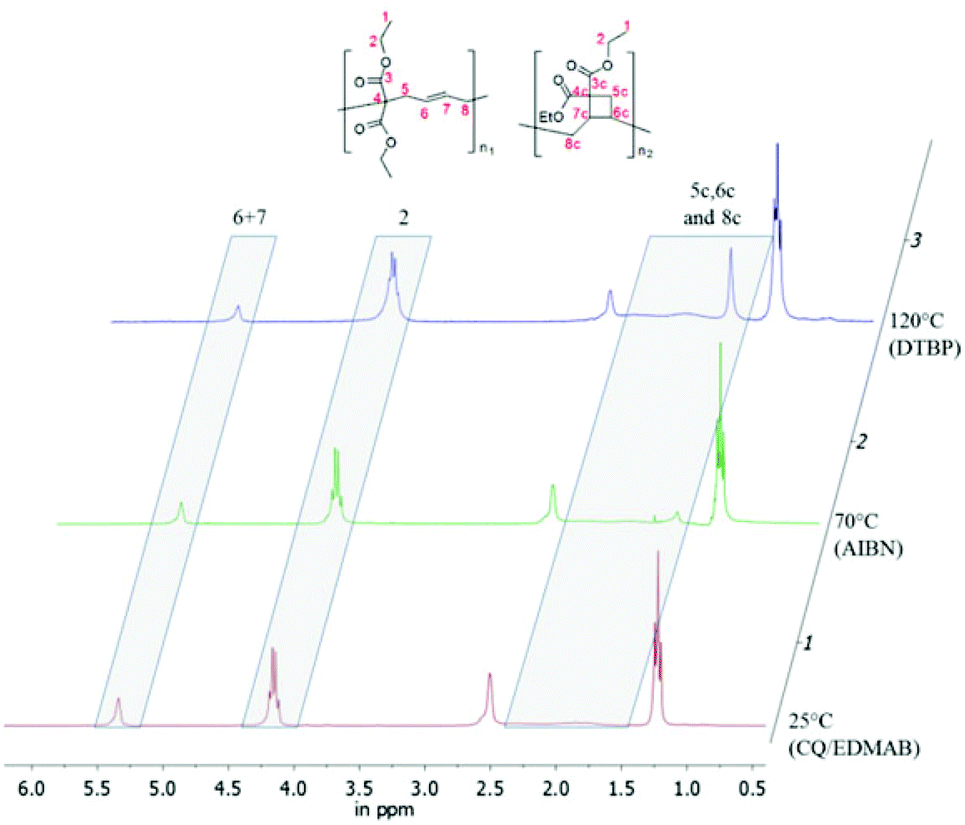 | ||
| Fig. 5 Overlay of three 1H-NMRs of poly(1a) (solvent CDCl3, 300 MHz), polymerized by (1) 1 mol% CQ and 2 mol% EDMAB at 25 °C; (2) 1 mol% AIBN at 70 °C; (3) 1 mol% DTBP at 120 °C. | ||
Nevertheless we could not observe a 92% percent ring opened structure (1aI) for poly(1a), polymerized thermally with AIBN at 70 °C,3 rather we calculated only 59% of the ring opened structure (Table 1). To determine the amount of the ring opened structure for poly(1a) and poly(2a) equally, the 1H-NMR integral values of the ethyl ester protons (proton signal 2 within Fig. 4) have been normalized to a value of four, as these exhibit an analogous chemical shift for the ring opened and the cyclobutane units. Thereby the total proton values were congruent to the expected one (16 protons for poly(1a) and 18 for poly(2a)). The ratio of the olefinic protons (signal 6 + 7 in Fig. 5) to the expected protons (for 100% ring opening) distinguished directly the amount of the ring opened structure.
A reduced amount of cyclobutane-containing units 1aII (Scheme 7) for poly(1a) and 2aIII and 2aIV (Scheme 9) for poly(2a) could be observed, as the temperature for the polymerization was reduced to room temperature for photopolymerizations (Table 1). This is in accordance with the literature3,4 in which the cyclobutane units increased as the polymerization temperature of the thermal polymerization increased.
From 1H-NMR data, it was also evident that the presence of the onium salt DPIHFP and the reaction time did influence the ring opened structure of the polymers 1a and 2a (Table 1). Naturally with rising reaction time, viscosity as well as conversion increased leading to increased amounts of cyclobutane-containing units (entries 1 and 7; Table 1). The increase in cyclobutane units with increasing conversion, as a matter of increasing melt viscosity was also evident on addition of the onium salt DPIHFP (i.e. use of the ternary initiator system). Using a binary initiator system, the conversions of 1a and 2a were very low and unlike ternary initiator systems with DPIHFP, no glass-like state was observed by equivalent exposure times (please compare entries 7 with 8 and 9 with 10 in Table 1) giving less cyclobutane units. The cyclobutane units were formed by the addition of a newly formed radical during propagation to the double bond in the polymer main chain. It is a competitive reaction with the reaction of the ring-opened propagating radical with a new VCP monomer. With an increase in reaction viscosity and prolonged reaction time the addition of the ring-opened radical at the double bond of the same molecule giving cyclobutane units became more probable.
The effect of temperature on volume shrinkage has been mentioned in various publications,25–27 usually as a matter of thermal curing effects and changes in the rates of polymerization. Nevertheless, by comparison of the volume shrinkages of poly(1a) and (2a) polymerized thermally by AIBN at 70 °C3 with the volume shrinkages of those polymerized by photopolymerization, the effect of volume shrinkage could not be only explained by curing effects or changed rates in polymerization. Rather the polymerization behavior, and much more, the percentage of the ring opened polymer structure (Table 1) are relevant to the volume shrinkage, as the volume shrinkage of poly(1a) and poly(2a) is disproportionately reduced, compared to the effect of curing and changed rates of polymerizations for methylmethacrylate (MMA) and styrene (St) (Fig. 6).
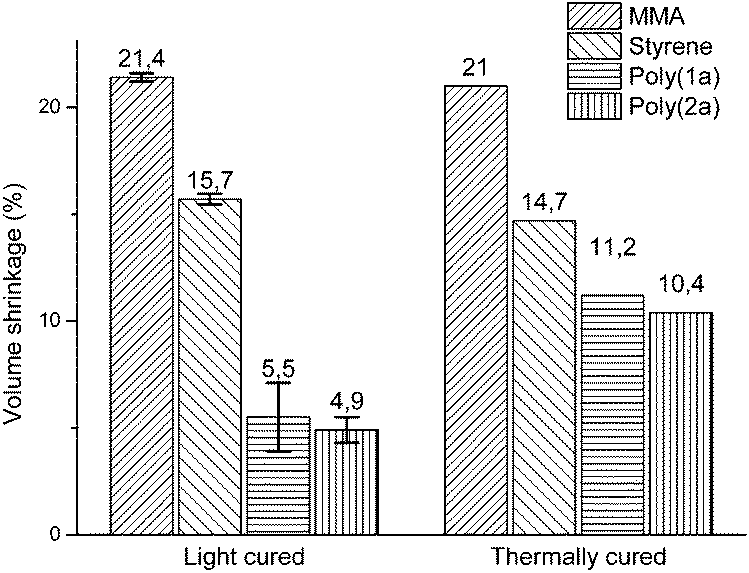 | ||
| Fig. 6 Comparison of volume shrinkages of MMA, St, poly(1a) and poly(2a) dependent on the polymerization method (light curing at 25 °C with CQ and EDMAB as initiators; thermal curing with AIBN at 70 °C.3,19 The volume shrinkage of the light cured samples was adjusted to complete monomer conversion, by determination of the polymerization yield by 1H-NMR (Fig. S18†). | ||
As mentioned in the Experimental part the synthesized monomer 2a, other than mentioned by Sanda et al.,19 consists of a constitution isomer of around 80:20 (Fig. 1). Nevertheless, it was exhibited that the existence of the constitution isomer did not influence the polymer structure overall, as head and tail conjunctions results in nearly the same polymer structure (Scheme 8).
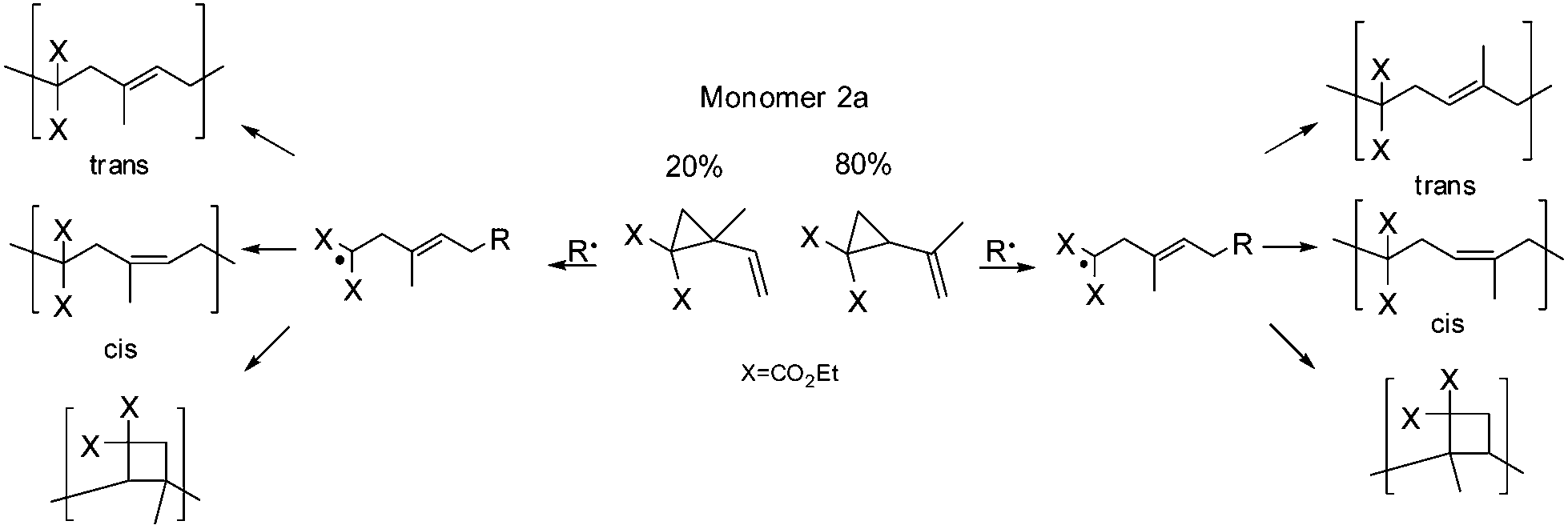 | ||
| Scheme 8 Schematic description of the expected polymer structures for monomer (2a). | ||
In addition the main structure of poly(2a) was concluded to be chiefly composed of 1,5-ring-opened units, with lower ratios of cyclobutane units compared to polymer 1a (Table 1). Sanda et al.19 examined intensely through 13C-NMR spectroscopy, that within the polymer structure of poly(2a) a cis and a trans unit were assigned, which could be confirmed, as we observed within two-dimensional NOESY measurements only cross peaks for 9a-5 (trans isomer), whereas the cis configuration was proved by the absence of an NOE correlation (Fig. S17†).
Furthermore the constitution isomer of the methyl substituted monomer (2a) seemed to be promising for the verification of the cyclobutane unit, as this unit could not be explicitly proved currently, because of the difficult characterization, as only a broad signal for poly(1a) was observed, that could not be assigned to any protons. Therefore previously the cyclobutane unit was assumed with the missing of the olefinic protons and the respectively higher volume shrinkage.3,19 In contrast to the polymer structure of poly(1a) for poly(2a) further cross correlations for the methyl group (9a–c′) of the substructure 2aIII and 2aIV (Scheme 9) should be verifiable during a two-dimensional HMBC measurement (Fig. 7). Thus a unique interpretation of the polymer signals has been iterated by 1H-, 13C-, HSQC-, NOESY (all shown in the ESI†), HMBC-NMR (Fig. 7) measurements.
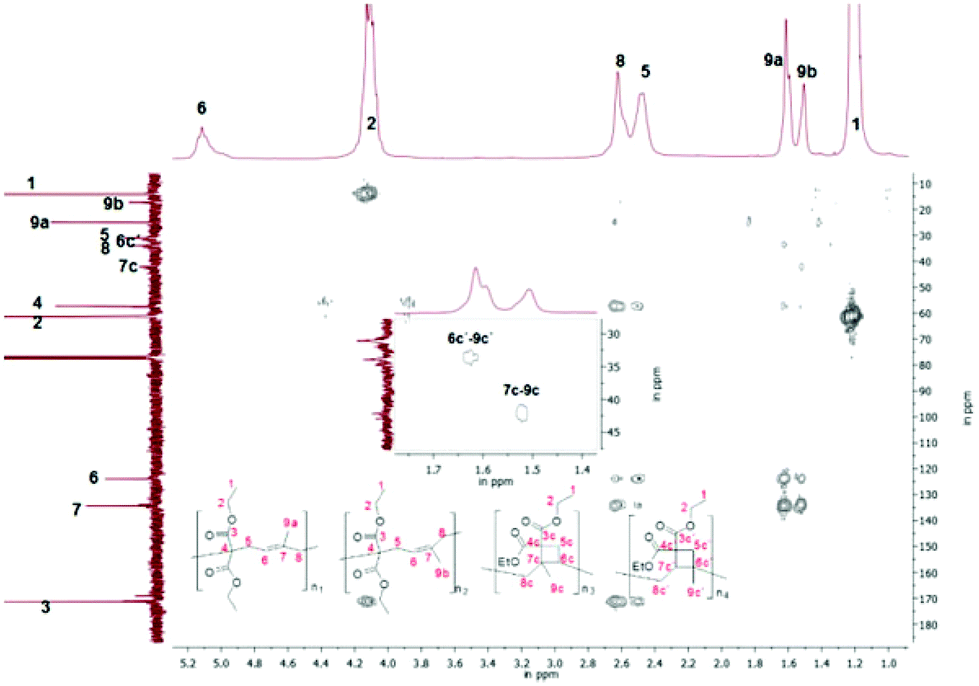 | ||
| Fig. 7 2D-HMBC NMR of poly(2a) (solvent CDCl3, 300 MHz), (polymerization conditions: 1 mol% CQ; 2 mol% EDMAB, 2 mol% DPIHFP), inclusive an expansion to verify the correlation between 6c′–9c′ and 7c-9c. | ||
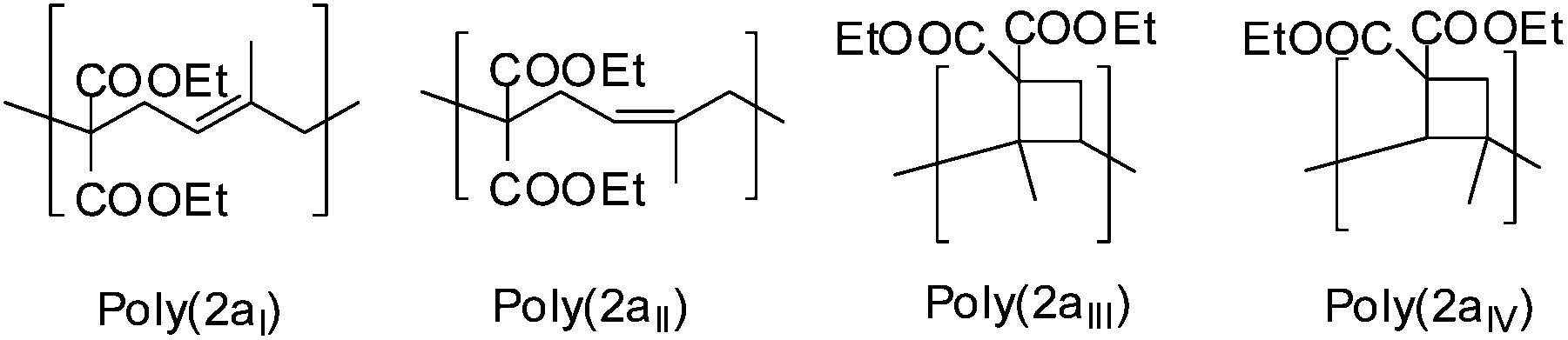 | ||
| Scheme 9 Proved polymer structures for monomer (2a). | ||
Indeed from the HMBC spectra for poly(2a) further correlation signals of the methyl groups were present that are assumed to belong to the correlation signals of 6c′–9c′ (2aIII) and 7c–9c (2aIV) of the cyclobutane unit, whereby to our knowledge, this cyclobutane unit has been detected directly for 1,1-bis(ethoxycarbonyl)-2-vinylcyclopropane for the first time.
4. Conclusions
In summary, 1,1-disubstituted 2-vinylcyclopropanes undergo radical photopolymerization with a reduced formation of cyclobutane units and increased 1,5-type ring-opening compared to thermally polymerized specimens. The reduced amount of cyclobutane units was proportional to the volume shrinkage. In addition, we varied the photopolymerization conditions to observe the impact on reaction kinetics. The addition of the onium salt DPIHFP as a ternary initiator to the radical initiator system drastically reduced the curing time for bulk polymerizations. This makes VCPs attractive as a monomer component in low shrinking dental adhesives or composites.Acknowledgements
We thank the German Federal Ministry for Economic Affairs and Energy (BMWi) for financial support. Dr. A. Bublewitz and Dr. A. Theis are thanked for useful discussions during the work.Notes and references
- N. Moszner, T. Völkel and V. Rheinberger, Macromol. Chem. Phys., 1996, 197, 621–631 CrossRef CAS.
- N. Moszner, F. Zeuner, U. K. Fischer, V. Rheinberger, A. d. Meijere and V. Bagutski, Macromol. Rapid Commun., 2003, 24, 269–273 CrossRef CAS.
- F. Sanda, T. Takata and T. Endo, Macromolecules, 1993, 26, 1818–1824 CrossRef CAS.
- N. Moszner and U. Salz, Prog. Polym. Sci., 2001, 26, 535–576 CrossRef CAS.
- T. E. F. Sanda, J. Polym. Sci., Part A: Polym. Chem, 2001, 39, 265–276 CrossRef.
- R. G. Fayter, US, 4321406, 1980 Search PubMed.
- N. Moszner, T. Völkel, U. K. Fischer and V. Rheinberger, Macromol. Chem. Phys., 1999, 200, 2173–2187 CrossRef CAS.
- J. Pavlinec and N. Moszner, J. Appl. Polym. Sci., 2003, 89, 579–588 CrossRef CAS.
- F. Sanda, T. Takata and T. Endo, Macromolecules, 1994, 27, 3982–3985 CrossRef CAS.
- N. Moszner and U. Salz, Macromol. Mater. Eng., 2007, 292, 245–271 CrossRef CAS.
- J. H. W. L. J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4241 CrossRef CAS.
- H.-J. Timpe, Pure Appl. Chem., 1988, 60, 1033 CrossRef CAS.
- J. V. Crivello and J. H. W. Lam, Macromolecules, 1977, 10, 1307–1315 CrossRef CAS.
- M. S. J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem, 2001, 39, 343 CrossRef.
- J. G. Kloosterboer and G. F. C. M. Lijten, Polymer, 1990, 31, 95–101 CrossRef CAS.
- H. J. Timpe, S. Ulrich, C. Decker and J. P. Fouassier, Macromolecules, 1993, 26, 4560–4566 CrossRef CAS.
- F. A. Ogliari, C. Ely, C. L. Petzhold, F. F. Demarco and E. Piva, J. Dent., 2007, 35, 583–587 CrossRef CAS PubMed.
- M. L. Gómez, V. Avila, H. A. Montejano and C. M. Previtali, Polymer, 2003, 44, 2875–2881 CrossRef.
- F. Sanda, T. Takata and T. Endo, Macromolecules, 1995, 28, 1346–1355 CrossRef CAS.
- O. J. Sweeting and J. R. Johnson, J. Am. Chem. Soc., 1946, 68, 1057–1061 CrossRef CAS.
- T. Endo, M. Watanabe, K. Suga and T. Yokozawa, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 3039–3048 CrossRef CAS.
- K. Sirovatka Padon and A. B. Scranton, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2057–2066 CrossRef.
- W. D. Cook, Polymer, 1992, 33, 600–609 CrossRef CAS.
- J. Pączkowski and D. C. Neckers, Photoinduced Electron Transfer Initiating Systems for Free-Radical Polymerization, Electron Transfer in Chemistry ed. V. Balzani, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2001 Search PubMed.
- K. S. Anseth, C. N. Bowman and N. A. Peppas, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 139–147 CrossRef CAS.
- B. Lu, P. Xiao, M. Sun and J. Nie, J. Appl. Polym. Sci., 2007, 104, 1126–1130 CrossRef CAS.
- C.-M. Chung, J.-G. Kim, M.-S. Kim, K.-M. Kim and K.-N. Kim, Dent. Mater., 2002, 18, 174–178 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR, GC, GC-MS and experimental notices of the synthesis and evaluation. See DOI: 10.1039/c4py01705f |
| ‡ Paul Pineda Contreras and Payal Tyagi have contributed equally to the experimental part. |
| This journal is © The Royal Society of Chemistry 2015 |