
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of molecular weight on the properties and organic solar cell device performance of a donor–acceptor conjugated polymer†
Zeyun
Xiao
a,
Kuan
Sun
a,
Jegadesan
Subbiah
a,
Tianshi
Qin
b,
Shirong
Lu
 a,
Balaji
Purushothaman
a,
David J.
Jones
a,
Andrew B.
Holmes
ab and
Wallace W. H.
Wong
*a
a,
Balaji
Purushothaman
a,
David J.
Jones
a,
Andrew B.
Holmes
ab and
Wallace W. H.
Wong
*a
aSchool of Chemistry, Bio21 Institute, the University of Melbourne, 30 Flemington Road, Parkville, Victoria 3010, Australia. E-mail: wwhwong@unimelb.edu.au; Fax: +61 3 83442384; Tel: +61 3 83442371
bCSIRO Materials Science and Engineering, Private Bag 10, Clayton South, Victoria 3169, Australia
First published on 4th February 2015
Abstract
Donor–acceptor conjugated polymers with 2-(2-ethylhexyl)-3-hexyl thienyl substituted benzo[1,2-b:4,5-b′]dithiophene (BDT) as donor building block and 5,6-difluorobenzo[c][1,2,5]thiadiazole as acceptor building block have been synthesized by Stille coupling polymerization. The polymerization conditions were optimized to achieve high molecular weight polymers (number-average molecular weight, Mn, up to 139 kg mol−1). The molecular weight dependent polymer properties were studied and compared. Photovoltaic applications of the polymers in bulk heterojunction (BHJ) solar cells revealed that the power conversion efficiency increased significantly (from 0.9% to 4.1%) as the Mn increased from 10 kg mol−1 to 73 kg mol−1 while further increase of the molecular weight had less influence on the solar cell performance.
Introduction
Stille coupling polymerization is one of the most widely used polymerization methods for the synthesis of conjugated polymers which are the basis of modern organic electronics.1–7 The advantage of using Stille coupling polymerization lies in its compatibility with various functional groups, which have made the syntheses of numerous functional polymers possible.4 However, it is often difficult to achieve high molecular weight polymers as a result of a combination of factors including monomer purity and stability, degree of conversion and rate of reaction.4 On the other hand, studies on the influence of molecular weight on polymer electronic properties repeatedly showed that bulk heterojunction (BHJ) solar cell performance improved significantly as the molecular weight of the conjugated polymers increased.8–11 For example, BHJ solar cells based on poly[(4,4-didodecyldithieno[3,2-b:2′,3′-d]silole)-2,6-diyl-alt-(2,1,3-benzothiadiazole)-4,7-diyl] active layer showed increased short-circuit current density (Jsc) from 4.2 to 17.3 mA cm−2 and increased fill factor (FF) from 0.35 to 0.61 as the Mn increased from 7 to 34 kg mol−1.9 These changes resulted in nearly a five-fold enhancement in the power conversion efficiency (PCE) from 1.2 to 5.9%. BHJ solar cells using polymer fractions with number average molecular weight, Mn, of 10, 18, and 31 kg mol−1 exhibited PCE of 3.0%, 5.2%, and 7.5%, respectively, displaying a nearly linear increase in solar cell performance with increasing molecular weight.10 It was thought that polymers with high molecular weight exhibited more isotropic films and enhanced interconnectivity, potentially leading to larger values for charge-carrier mobility.12 The enhanced performance in BHJ solar cells with increasing polymer molecular weight was also attributed to the more favoured active layer morphology and decreased series resistance.8,10 However, in the aforementioned studies, the molecular weight ranges were quite limited (with the highest Mn around only 30 kg mol−1). Questions may be raised: “Can the solar cell performance increase linearly with the molecular weight, or there is a limit beyond which the performance saturates?” To address these questions, conjugated polymers of extremely high molecular weight need to be synthesized. In this work, we optimized the Stille coupling polymerization conditions to get extremely high molecular weight donor–acceptor conjugated polymers and studied their molecular-weight-dependent BHJ solar cell performances.Benzo[1,2-b:4,5-b′]dithiophene (BDT) based conjugated polymers are frequently used in BHJ solar cells as donor materials.13–25 By replacing the substituent of 2-ethylhexyloxy group with 2-(2-ethylhexyl)thienyl group, the PCE increased from 6.43% to 7.59%.26 The introduction of two alkyl chains on the same thiophene ring were found to enhance the solubility and processability of the resulting polymers.15,27
In this study, we use 4,8-di(2-(2-ethylhexyl)-3-hexyl-thiophen-5-yl)-benzo[1,2-b:4,5-b′]dithiophene (DTBDT) as the donor building block and 5,6-difluoro-2,1,3-benzothiadiazole (DFBT) as the acceptor building block for the polymer (Fig. 1) with the aim of getting deep-lying highest occupied molecular orbital (HOMO) energy level and thus high open circuit voltage (Voc) in the BHJ solar cells.28–33 The bis(trimethylstannyl)-DTBDT monomer (compound 1) was coupled with diiodo-DFBT monomer (compound 2) using a variety of Stille polycondensation conditions to give poly[(4,8-di(2-(2-ethylhexyl)-3-hexylthiophen-5-yl)-benzo[1,2-b:4,5-b′]dithio-phene)-2,6-diyl-alt-(5,6-difluoro-2,1,3-benzothiadiazole)-4,7-diyl] (P1) samples with different molecular weight ranges.
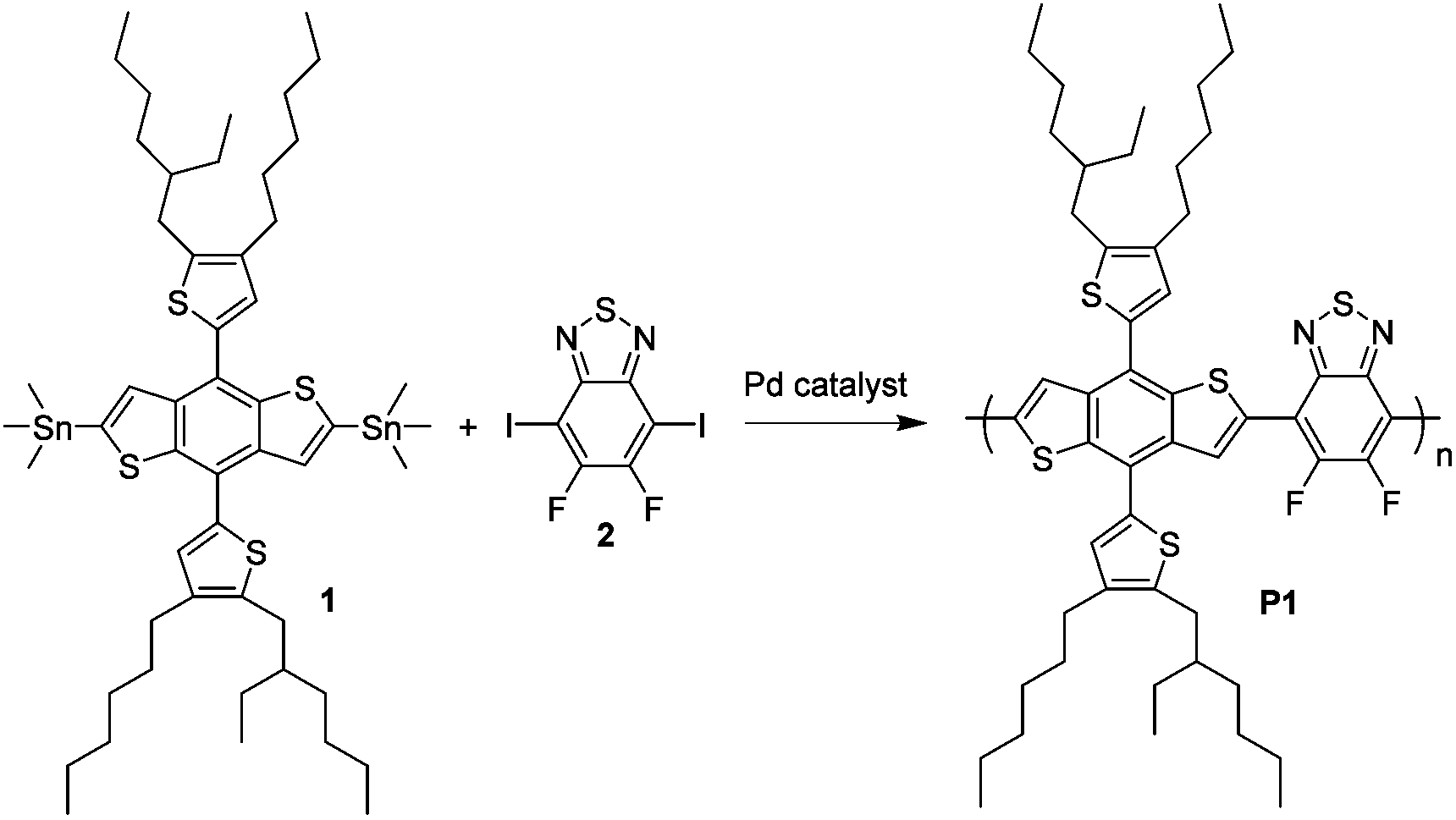 | ||
| Fig. 1 Chemical structures of compound 1 and 2 and the Stille polymerization for conjugated polymer P1. | ||
Synthesis and characterization
Synthesis of the polymer by Stille coupling polymerization
To ensure monomer purity for the Stille polycondensation, compound 1 was recrystallized from dichloromethane and isopropanol at 5 °C and compound 2 was recrystallized from dichloromethane. The Stille coupling polymerization was first conducted with a widely-used catalyst, Pd(PPh3)4, and toluene/DMF as the solvent. Under this condition, most of the polymer material was isolated by Soxhlet extraction with dichloromethane. The yield of the chloroform extract was low (13%, see Table 1) and the Mn was 32 kg mol−1. Microwave heating was used to try and increase the molecular weight and the yield of the chloroform fraction but there was little improvement. A more active catalyst system Pd2(dba)3/P(o-tolyl)3 was then used along with chlorobenzene as solvent to better solvate the sparingly soluble conjugated polymers.34,35 This condition was found to be particularly efficient in increasing the molecular weight and the chloroform fraction of the polymer. Polymer with Mn of 72.9 kg mol−1 was obtained in 77% yield. Increasing the monomer concentration from 0.1 M to 0.25 M further increased the Mn to 138.9 kg mol−1 (see ESI† for GPC traces). Other phosphine ligands were also tested, including P(o-anisolyl)3, but none worked as well as the P(o-tolyl)3 ligand.4,36,37| Entry | Catalystb | Temperaturec | Monomer concentration | Solvent | Yield | Chloroform fraction Mn (kg mol−1) | PDI |
|---|---|---|---|---|---|---|---|
| a All the reactions were run on 0.15 mmol scale and the polymers were extracted successively with methanol, acetone, petroleum spirits, dichloromethane and chloroform. b The catalyst loadings were 5% mol Pd(PPh3)4 for entry 1 and 2, 1.5 mol% Pd2(dba)3 and 9 mol% P(o-tolyl)3 for entries 3 and 4, 1.5 mol% Pd2(dba)3 and 9 mol% P(o-anisolyl)3 for entry 5. c Entry 2 was heated with microwave. d Molecular weight obtained by using high-temperature GPC at 120 °C with 1,2,4-trichlorobenzene as eluent. | |||||||
| 1 | Pd(PPh3)4 | 120 °C | 0.1 M | Toluene–DMF 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
13% | 32.0 | 1.70 |
| 2 | Pd(PPh3)4 | 150–170 °C | 0.1 M | Xylene | 15% | 31.5 | 1.21 |
| 3 | Pd2(dba)3, P(o-tolyl)3 | 130 °C | 0.1 M | Chlorobenzene | 77% | 72.9d | 3.51d |
| 4 | Pd2(dba)3, P(o-tolyl)3 | 130 °C | 0.25 M | Chlorobenzene | 64% | 138.9d | 5.33d |
| 5 | Pd2(dba)3, P(o-anisolyl)3 | 130 °C | 0.1 M | Chlorobenzene | Trace | ||
Five samples of polymer P1, whose chemical structure was shown in Fig. 1, were collected and their optoelectronic and photovoltaic properties were studied. The molecular weight range of the polymer samples is shown in Table 2. Sample P1-9.6k (Mn 9.6 kg mol−1, PDI 1.57) was obtained from dichloromethane extract of the reaction described in entry 2 of Table 1 while P1-17.1k (Mn 17.1 kg mol−1, PDI 1.50) was obtained from dichloromethane extract and P1-32.0k (Mn 32.0 kg mol−1, PDI 1.70) was obtained from chloroform extract of the reaction described in entry 1 of Table 1. Samples P1-72.9k (Mn 72.9 kg mol−1, PDI 3.51) and P1-138.9k (Mn 138.9 kg mol−1, PDI 5.33) were extracted with chloroform from the reactions described in entries 3 and 4 of Table 1 respectively. The five samples of polymer P1 showed similar FT-IR spectra indicating the same chemical composition (Fig. S6†). However, as the molecular weight increased, the 1H NMR spectra lost some fine structures and the peaks became broadened owing to the increased number of repeating units (Fig. S7†). Differential scanning calorimetry (DSC) of the polymers showed no thermal transitions for any of the polymers between 25 °C and 300 °C (Fig. S8†).
| Polymer |
M
na (kg mol−1) |
PDIa | UV-vis λmaxb (nm) |
UV-vis λonsetc (nm) |
Optical gapd (eV) |
E
HOMOe (eV) |
E
LUMOf (eV) |
|---|---|---|---|---|---|---|---|
| a HT-GPC data in brackets. b UV-vis absorption maxima in chloroform solution and as thin films in bracket. c UV-vis onset absorption in chloroform solution and as thin films in bracket. d Calculated from thin film absorption onset. e Measured using CV. f Calculated from the HOMO energy level and the optical HOMO–LUMO gap. | |||||||
| P1-9.6k | 9.6 (13.4) | 1.57 (1.53) | 651 (656) | 705 (723) | 1.72 | −5.53 | −3.81 |
| P1-17.1k | 17.1 (15.8) | 1.50 (3.79) | 653 (664) | 705 (734) | 1.69 | −5.52 | −3.83 |
| P1-32.0k | 32.0 (19.1) | 1.70 (4.85) | 656 (665) | 707 (735) | 1.69 | −5.52 | −3.83 |
| P1-72.9k | (72.9) | (3.51) | 658 (670) | 710 (741) | 1.67 | −5.52 | −3.85 |
| P1-138.9k | (138.9) | (5.33) | 658 (670) | 710 (741) | 1.67 | −5.49 | −3.82 |
Characterization of optoelectronic properties
The UV-vis absorption spectra of the five fractions of P1 in chloroform solutions and in films are shown in Fig. 2. In solution, as the Mn increased from 9.6 kg mol−1 to 72.9 kg mol−1, the absorption maximum (λmax) increased from 651 nm to 658 nm and the onset absorption (λonset) increased from 705 nm to 710 nm due to the increased conjugation length. However, the λmax and the λonset of polymer samples with Mn 72.9 kg mol−1 and 138.9 kg mol−1 were the same, implying that an effective conjugated length for the HOMO–LUMO energy gap was already reached at the Mn of 72.9 kg mol−1.9 The relative intensity of the main absorption to the shoulder peak at 594 nm also increased possibly due to the increased percentage of large molecular weight polymers. This phenomenon was also observed in other polymer systems.9,38 The absorption of different fractions of P1 in solid films showed similar trends as in the chloroform solution. The solid film λmax increased from 656 nm to 670 nm and the λonset increased from 723 nm to 741 nm, resulting in a reduction of optical energy gap from 1.72 eV to 1.67 eV as the molecular weight increased. The polymers showed solution emission maxima around 700 nm and slightly red-shifted emission maxima of 714 nm in the solid films (Fig. S9†).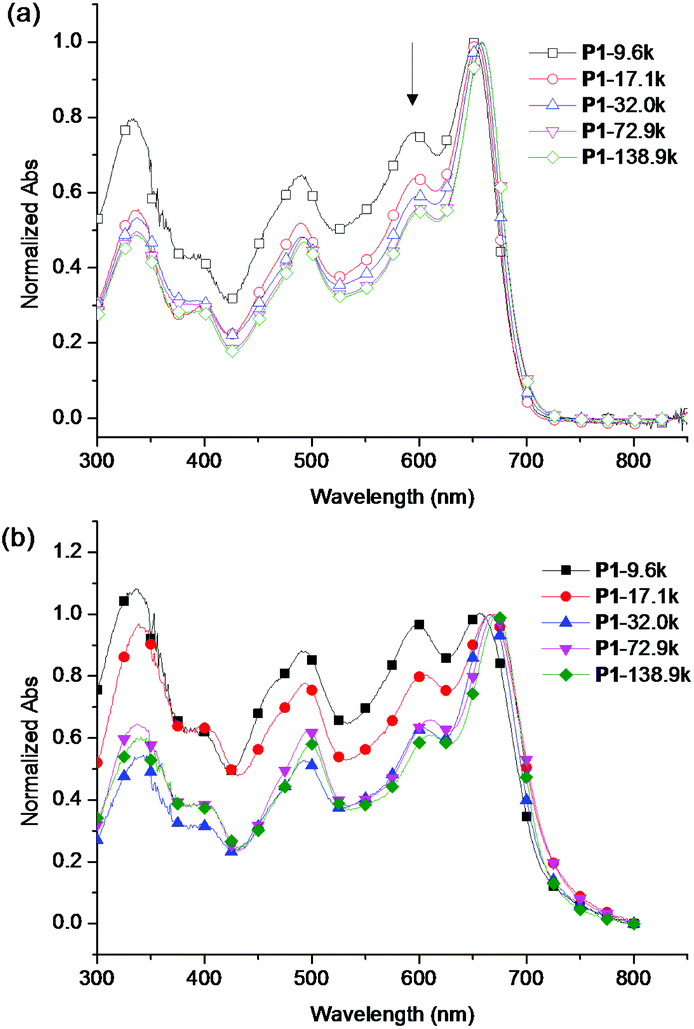 | ||
| Fig. 2 UV-Vis absorption spectra of the five polymer fractions in (a) chloroform and (b) solid film. The spectra were normalized at the absorption maximum. | ||
The HOMO energy levels of the five polymer fractions were estimated by cyclic voltammetry measurements (see ESI for experimental details, Fig. S10†). Owing to the incorporated 5,6-difluoro-2,1,3-benzothiadiazole acceptor moiety, all the polymers possessed deep-lying HOMO energy levels (around −5.5 eV), which is promising for achieving high Voc in organic photovoltaic cells.39–41 It was also observed that the polymer fractions showed a slight HOMO level increase as the molecular weight increased (Table 2). The lowest molecular weight polymer P1-9.6k exhibited a HOMO level of −5.53 eV while the highest molecular weight polymer P1-138.9k displayed a HOMO level of −5.49 eV.
BHJ solar cell fabrication and characterization
The photovoltaic properties of the five polymer fractions were investigated in BHJ solar cell devices. A schematic diagram of the solar cells with inverted device architecture [ITO/ZnO/active layer/MoO3/Ag] is shown in the ESI.† 1,2-Dichlorobenzene (oDCB) was chosen as the processing solvent owing to its good solvent properties and low evaporation rates. An optimized blend ratio of 1:2 between polymer and PC71BM acceptor was used for spin coating the active layer. The blend film UV-vis absorption spectra showed similar profiles for samples containing different molecular weight polymers (Fig. S11†). The films containing higher molecular weight material showed slightly enhanced absorption at the longer wavelength absorption band (670 nm). This is consistent with the absorption spectrum of the neat polymer films (Fig. 2b).
The devices were studied under the illumination of AM 1.5 G, 100 mW cm−2. The current density–voltage (J–V) curves of the polymer/PC71BM devices are displayed in Fig. 3a and the photovoltaic performance (average of 10 devices) is listed in Table 3. The open circuit voltage (Voc) and the fill factor (FF) of the BHJ solar cells were essentially the same for all devices containing the five different molecular weight polymer samples (Fig. 3c and d). In great contrast, the short circuit current density (Jsc) increased significantly from 2.0 mA cm−2 to 9.9 mA cm−2 as the Mn increased from 9.6 kg mol−1 to 72.9 kg mol−1 (Fig. 3c). It was found that further increase of the molecular weight did not increase the Jsc further, instead the P1-138.9k even showed a slightly decreased Jsc of 8.6 mA cm−2. As a result, the overall power conversion efficiency (PCE) increased from 0.85% to 4.10% as the Mn increased from 9.6 kg mol−1 to 72.9 kg mol−1 and the device containing P1-138.9k showed lower PCE than the devices containing P1-72.9k. Literature reports have shown a similar increase of PCE in the low molecular weight range (Mn up to about 30 kg mol−1).9,10,42 However the higher molecular weight range has been seldom discussed with only a few examples for donor–acceptor polymers in recent literature.43,44 Our results showed that the PCE reached a peak value of 4.10% as the Mn increased to 72.9 kg mol−1. These results indicated that a maximum PCE could be reached at a certain molecular weight for a specific polymer and further increase of the molecular weight might not result in further improvements in device performance. It is important to note that device fabrication optimisation, including adjusting substrate temperature and spin coating speed, was attempted on the high molecular weight material but failed to give improvements in device performance.45 It is also interesting to note that an analogous polymer system18 with Mn of 22.6 kg mol−1 gave very similar device performance to that of P1-17.1k at around 1% PCE.
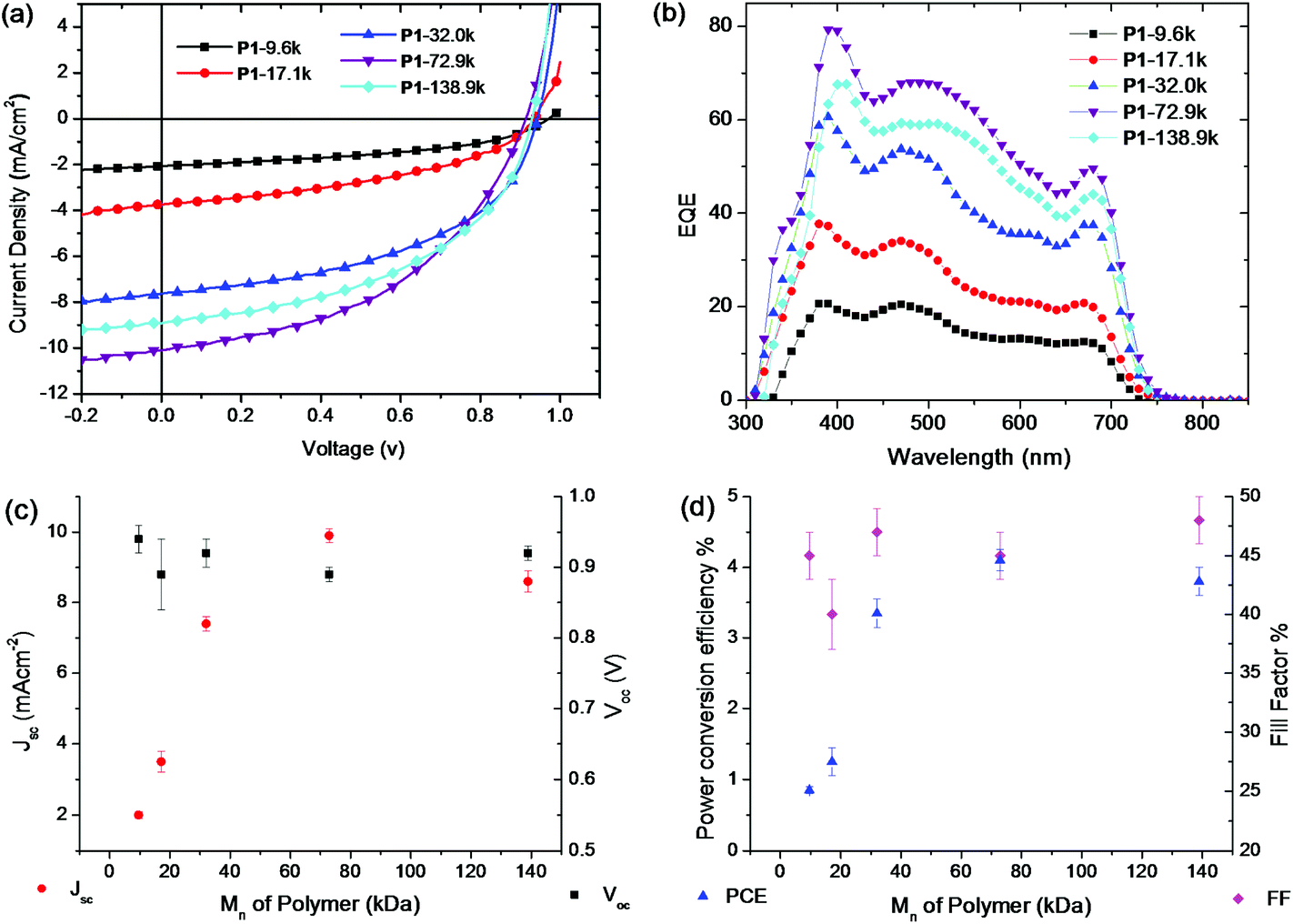 | ||
| Fig. 3 (a) J–V characteristics of photovoltaic devices fabricated with different molecular weight P1/PC71BM under AM 1.5G irradiation (100 mW cm−2). (b) EQE curves of the BHJ solar cells fabricated under the same optimized conditions as used for the J–V measurements. (c) Mn of polymer plotted against Jsc and Voc and (d) Mn of polymer plotted against FF and PCE. | ||
| Polymera | V oc (V) | J sc (mA cm−2) | FF (%) | PCE (%) |
μ
hb (cm2 V−1 s−1) |
|---|---|---|---|---|---|
|
a The weight ratio of P1/PC71BM was 1:2 and the average values (10 devices) are shown with standard deviation.
b Measured by using the space-charge-limited current (SCLC) method (see ESI†).
|
|||||
| P1-9.6k | 0.94 ± 0.02 | 2.0 ± 0.1 | 45 ± 2 | 0.85 ± 0.05 | 2.5 × 10−6 |
| P1-17.1k | 0.89 ± 0.05 | 3.5 ± 0.3 | 40 ± 3 | 1.25 ± 0.20 | 5.4 × 10−6 |
| P1-32.0k | 0.92 ± 0.02 | 7.4 ± 0.2 | 47 ± 2 | 3.35 ± 0.20 | 2.6 × 10−5 |
| P1-72.9k | 0.89 ± 0.01 | 9.9 ± 0.2 | 45 ± 2 | 4.10 ± 0.15 | 3.7 × 10−5 |
| P1-138.9k | 0.92 ± 0.01 | 8.6 ± 0.3 | 48 ± 2 | 3.80 ± 0.20 | 3.8 × 10−5 |
The external quantum efficiencies (EQEs) of the solar cell devices were also studied. Fig. 3b shows the EQE curves of the solar cells fabricated under the same optimized conditions as those used for the J–V measurements. Clearly, the EQE values for devices containing the high molecular weight polymers are higher than those of low molecular weight polymer based solar cells, which agree with the J–V measurements. The EQE curves of the BHJ solar cells made from the polymers of higher molecular weights showed significant improvement across the whole spectral response region, indicating that the enhancement in Jsc was mainly due to more efficient exciton dissociation and charge collection from both the polymer and the PC71BM domains.10
To gain further insight into the PCE change as a result of the different polymer molecular weights, the hole mobility was derived using the space-charge-limited current (SCLC) method, with a hole-only device configuration of ITO/PEDOT:PSS/polymer:PC71BM/Au. The mobility data in Table 3 clearly showed that the hole mobility was enhanced when the molecular weight increased, which is consistent with the improved Jsc and photovoltaic performance of the BHJ solar cells.26 In particular, the hole mobility increased 10 times as the Mn increased from 9.6 kg mol−1 to 32.0 kg mol−1, while from 32.0 kg mol−1 to 138.9 kg mol−1, the hole mobility only increased by 46%. Furthermore, the P1-72.9k and P1-138.9k polymers displayed very similar hole mobility of 3.7 × 10−5 and 3.8 × 10−5 cm2 V−1 s−1, respectively. These results suggested that the hole mobility was not linearly dependent on the molecular weight, especially in the high molecular weight range. A ‘saturated’ hole mobility can be reached at a certain molecular weight for a specific polymer. Recent studies revealed that low molecular weight conjugated polymers can form solid films with crystalline domains surrounded by a disordered phase, but these domains remain largely disconnected from each other, resulting in charge-transport bottlenecks at the amorphous grain boundaries.46,47 In contrast, in high molecular weight conjugated polymers, even though the long-range order in the crystalline domains is compromised, the interconnectivity is greatly improved as a result of the rigid and sufficiently long chains that link the domains, like bridges that link islands which can improve the mobility.46,47
Morphological studies
To understand the hole mobility change and to study the morphology of the blend films of P1 fractions and PC71BM, tapping mode atomic force microscopy (AFM) studies were conducted (Fig. 4). The P1/PC71BM blend films with polymers of different molecular weight exhibited different topographies, and the root-mean-square roughness (Rrms) increased as the molecular weight increased (Rrms: P1-9.6k, 1.3 nm; P1-17.1k, 2.9 nm; P1-32.0k, 4.1 nm; P1-72.9k, 8.4 nm; P1-138.9k, 9.5 nm). Since the polymer length was in the nanometer scale, random orientation of the polymer backbone could lead to the increase in roughness. The difference in the phase images was even evident. The blend film prepared with the lowest molecular weight polymer, P1-9.6k and PC71BM, showed a clear, phase-separated morphology with finer, fibrous nanoscale domains (Fig. 4f), while this feature was gradually lost as the molecular weight increased (Fig. 4g–j). This could be related to polymer chain conformation and inter-chain interactions of P1. As the polymer molecular weight increased, its solubility decreased and it tended to form aggregates.48 As a result, the domain size became larger. It should be noted that the domain sizes were still comparable to the exciton diffusion length, so the mobility as well as the solar cell performance were not diminished.10,42 | ||
| Fig. 4 AFM images of P1/PC71BM (1:2 weight ratio) films. (a) and (f) P1-9.6 K/PC71BM; (b) and (g) P1-17.1 K/PC71BM; (c) and (h), P1-32.0 K/PC71BM; (d) and (i) P1-72.9 K/PC71BM; (e) and (j) P1-138.9 K/PC71BM. (a)–(e) are height images; (f)–(j) are phase images. All images are 2 × 2 μm. | ||
Meanwhile, less discontinuous phase separation was observed when the Mn increased to 32.0 kg mol−1. The blend films of the P1-72.9k and P1-138.9k showed similar phase images with extended polymer chains promoting interconnectivity between neighbouring fiber domains, reducing grain boundaries and thus facilitating charge carrier transport.49 This result was also consistent with the similar hole mobility of P1-72.9k and P1-138.9k obtained by SCLC measurements.
Conclusions
The influence of the molecular weight on the properties of the donor–acceptor conjugated polymers is a very important consideration when comparing materials. In this work, we optimized the Stille coupling polymerization conditions to synthesize donor–acceptor conjugated polymers of extremely high molecular weight. Our results made possible the comparison of conjugated polymer properties in a large range of molecular weights. In our series of polymers, the absorption maximum red shifted from 651 nm to 658 nm as the Mn increased from 9.6 kg mol−1 to 72.9 kg mol−1 while further increase of the molecular weight did not cause red-shift of the absorption maximum. This phenomenon indicated that a maximum conjugation length was achieved at a certain molecular weight. The BHJ solar cell efficiency increased drastically in the low molecular weight range (from 9.6 kg mol−1 to 32.0 kg mol−1) while in high molecular weight range, the enhancement was less significant. A peak PCE value of 4.25% was achieved at a Mn of 72.9 kg mol−1. The SCLC measurement suggested a ten-fold increase of the hole mobility as the Mn increased from 9.6 kg mol−1 to 32.0 kg mol−1. However, further increase the Mn to 138.9 kg mol−1 only increase the hole mobility by 46%. All in all, the results indicated that there is a finite limit to the benefit of increasing polymer molecular weight on the charge transport properties of the material and BHJ solar cell performance.Experimental
Unless noted, all materials were reagent grade and used as received without further purification. The synthesis of monomers 1 and 2 have been reported in literature.31,50 All the other starting compounds and reagents are commercially available. Experimental methods and instruments and detailed characterization can be found in the ESI.†General procedure for the polymer synthesis
The bis(trimethylstannyl) compound 1 (0.15 mmol), the diiodo compound 2 (0.15 mmol), and the Pd catalyst (the molar ratios of the respective catalysts were listed in Table 1) were added to a 10 mL microwave tube under nitrogen. Dry solvent was then added via a syringe and the reaction mixture was purged with nitrogen for 5 min. The reaction tube was then heated to the reaction temperature (Table 1) for polymerization. After the polymerization was finished, a drop of phenyltrimethyl tin was added as end capping reagent and the end capping process took 2 hours. Bromobenzene was then added and the reaction mixture was stirred for another 2 hours. After cooling to room temperature, the polymer was precipitated by addition of 50 mL methanol, filtered through a Soxhlet thimble. The precipitate was then subjected to Soxhlet extraction with methanol, acetone, hexanes, dichloromethane and chloroform. The polymer was recovered as solid from the chloroform fraction by precipitation from methanol. The solid was dried under vacuum. The molecular weights and yields are listed in Table 1. The IR and 1H NMR spectra can be found in the ESI.†Fabrication of the BHJ polymer solar cells
Polymer solar cells were processed on pre-patterned indium tin oxide (ITO) coated glass substrates with a sheet resistance of 15 Ω per square. First a 30 nm thick layer of ZnO nanopaticles (average size ∼9 nm) was deposited on the ultrasonically cleaned ITO substrates. Active layer was deposited by spin coating an oDCB solution containing 10 mg of polymer and 20 mg of phenyl-C71-butyric acid methyl ester (PC71BM). The films were then transferred to a metal evaporation chamber where MoO3 (10 nm) and Ag (100 nm) were deposited through a shadow mask (active area was 0.1 cm2) at approximately 1 × 10−6 torr. Film thickness was determined by Veeco Dektak 150 Surface Profiler. The current density–voltage measurements of the devices were carried out using a 1 kW Oriel solar simulator with an AM 1.5G filter as the light source in conjunction with a Keithley 2400 source meter. For accurate measurement, the illumination intensity was set to 100 mW cm−2 and was calibrated using a reference silicon solar cell (PV measurements Inc.) certified by the National Renewable Energy Laboratory.Acknowledgements
This work was made possible by support of the Victorian Organic Solar Cell Consortium, with funding provided by the Victorian State Government Department of Primary Industries (Energy Technology Innovation Strategy), Australian Renewable Energy Agency (ARENA Project 2-A018 and Australian Centre for Advanced Photovoltaics) and the Victorian State Government Department of Business Innovation (Victorian Science Agenda). Dr W. W. H. Wong is supported by an Australian Research Council Future Fellowship (FT130100500).Notes and references
- D. Milstein and J. K. Stille, J. Am. Chem. Soc., 1978, 100, 3636–3638 CrossRef CAS.
- Z. Bao, W. Chan and L. Yu, Chem. Mater., 1993, 5, 2–3 CrossRef CAS.
- Z. Bao, W. K. Chan and L. Yu, J. Am. Chem. Soc., 1995, 117, 12426–12435 CrossRef CAS.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed.
- U. Asawapirom, R. Güntner, M. Forster, T. Farrell and U. Scherf, Synthesis, 2002, 1136–1142 CrossRef CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- K. Okamoto and C. K. Luscombe, Polym. Chem., 2011, 2, 2424–2434 RSC.
- Z. B. Henson, K. Mullen and G. C. Bazan, Nat. Chem., 2012, 4, 699–704 CrossRef CAS PubMed.
- R. C. Coffin, J. Peet, J. Rogers and G. C. Bazan, Nat. Chem., 2009, 1, 657–661 CrossRef CAS PubMed.
- T.-Y. Chu, J. Lu, S. Beaupré, Y. Zhang, J.-R. Pouliot, J. Zhou, A. Najari, M. Leclerc and Y. Tao, Adv. Funct. Mater., 2012, 22, 2345–2351 CrossRef CAS.
- I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202–1214 CrossRef CAS PubMed.
- R. J. Kline, M. D. McGehee, E. N. Kadnikova, J. Liu and J. M. J. Fréchet, Adv. Mater., 2003, 15, 1519–1522 CrossRef CAS.
- L. Huo and J. Hou, Polym. Chem., 2011, 2, 2453–2461 RSC.
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed.
- M. Wang, X. Hu, P. Liu, W. Li, X. Gong, F. Huang and Y. Cao, J. Am. Chem. Soc., 2011, 133, 9638–9641 CrossRef CAS PubMed.
- X. Zhan and D. Zhu, Polym. Chem., 2010, 1, 409–419 RSC.
- R. S. Kularatne, P. Sista, H. Q. Nguyen, M. P. Bhatt, M. C. Biewer and M. C. Stefan, Macromolecules, 2012, 45, 7855–7862 CrossRef CAS.
- R. S. Kularatne, F. J. Taenzler, H. D. Magurudeniya, J. Du, J. W. Murphy, E. E. Sheina, B. E. Gnade, M. C. Biewer and M. C. Stefan, J. Mater. Chem. A, 2013, 1, 15535–15543 CAS.
- H. G. Kim, S. B. Jo, C. Shim, J. Lee, J. Shin, E. C. Cho, S.-G. Ihn, Y. S. Choi, Y. Kim and K. Cho, J. Mater. Chem., 2012, 22, 17709–17717 RSC.
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Nat. Photonics, 2012, 6, 591–595 Search PubMed.
- Y. Zhang, S. K. Hau, H.-L. Yip, Y. Sun, O. Acton and A. K. Y. Jen, Chem. Mater., 2010, 22, 2696–2698 CrossRef CAS.
- C. Piliego, T. W. Holcombe, J. D. Douglas, C. H. Woo, P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 7595–7597 CrossRef CAS PubMed.
- J. Hou, M.-H. Park, S. Zhang, Y. Yao, L.-M. Chen, J.-H. Li and Y. Yang, Macromolecules, 2008, 41, 6012–6018 CrossRef CAS.
- W. W. H. Wong, J. Subbiah, S. R. Puniredd, W. Pisula, D. J. Jones and A. B. Holmes, Polym. Chem., 2014, 5, 1258–1263 RSC.
- Z. Xiao, J. Subbiah, K. Sun, S. Ji, D. J. Jones, A. B. Holmes and W. W. H. Wong, J. Mater. Chem. C, 2014, 2, 1306–1313 RSC.
- L. Huo, S. Zhang, X. Guo, F. Xu, Y. Li and J. Hou, Angew. Chem., Int. Ed., 2011, 50, 9697–9702 CrossRef CAS PubMed.
- J. Subbiah, B. Purushothaman, M. Chen, T. Qin, M. Gao, D. Vak, F. H. Scholes, X. Chen, S. E. Watkins, G. J. Wilson, A. B. Holmes, W. W. H. Wong and D. J. Jones, Adv. Mater., 2015, 27, 702–705 CrossRef CAS PubMed.
- Y.-X. Xu, C.-C. Chueh, H.-L. Yip, F.-Z. Ding, Y.-X. Li, C.-Z. Li, X. Li, W.-C. Chen and A. K. Y. Jen, Adv. Mater., 2012, 24, 6356–6361 CrossRef CAS PubMed.
- Y. Zhang, S.-C. Chien, K.-S. Chen, H.-L. Yip, Y. Sun, J. A. Davies, F.-C. Chen and A. K. Y. Jen, Chem. Commun., 2011, 47, 11026–11028 RSC.
- N. Wang, Z. Chen, W. Wei and Z. Jiang, J. Am. Chem. Soc., 2013, 135, 17060–17068 CrossRef CAS PubMed.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., Int. Ed., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- L. Yang, H. Zhou, S. C. Price and W. You, J. Am. Chem. Soc., 2012, 134, 5432–5435 CrossRef CAS PubMed.
- Z. Li, J. Lu, S.-C. Tse, J. Zhou, X. Du, Y. Tao and J. Ding, J. Mater. Chem., 2011, 21, 3226–3233 RSC.
- J. C. Bijleveld, A. P. Zoombelt, S. G. J. Mathijssen, M. M. Wienk, M. Turbiez, D. M. de Leeuw and R. A. J. Janssen, J. Am. Chem. Soc., 2009, 131, 16616–16617 CrossRef CAS PubMed.
- J. Li, Y. Zhao, H. S. Tan, Y. Guo, C.-A. Di, G. Yu, Y. Liu, M. Lin, S. H. Lim, Y. Zhou, H. Su and B. S. Ong, Sci. Rep., 2012, 2, 754 Search PubMed.
- V. Farina and G. P. Roth, Advances in Metal–Organic Chemistry, JAI Press, New York, 1996 Search PubMed.
- M. R. Netherton and G. C. Fu, Org. Lett., 2001, 3, 4295–4298 CrossRef CAS PubMed.
- Z. H. Li, M. S. Wong, Y. Tao and J. Lu, Chem. – Eur. J., 2005, 11, 3285–3293 CrossRef CAS PubMed.
- C. J. Brabec, C. Winder, N. S. Sariciftci, J. C. Hummelen, A. Dhanabalan, P. A. van Hal and R. A. J. Janssen, Adv. Funct. Mater., 2002, 12, 709–712 CrossRef CAS.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- L. Dou, C.-C. Chen, K. Yoshimura, K. Ohya, W.-H. Chang, J. Gao, Y. Liu, E. Richard and Y. Yang, Macromolecules, 2013, 46, 3384–3390 CrossRef CAS.
- I. Osaka, M. Saito, H. Mori, T. Koganezawa and K. Takimiya, Adv. Mater., 2012, 24, 425–430 CrossRef CAS PubMed.
- W. Li, L. Yang, J. R. Tumbleston, L. Yan, H. Ade and W. You, Adv. Mater., 2014, 26, 4456–4462 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- X. Zhang, H. Bronstein, A. J. Kronemeijer, J. Smith, Y. Kim, R. J. Kline, L. J. Richter, T. D. Anthopoulos, H. Sirringhaus, K. Song, M. Heeney, W. Zhang, I. McCulloch and D. M. DeLongchamp, Nat. Commun., 2013, 4, 2238 Search PubMed.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed.
- J. Yuan, Z. Zhai, H. Dong, J. Li, Z. Jiang, Y. Li and W. Ma, Adv. Funct. Mater., 2013, 23, 885–892 CrossRef CAS.
- H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, D. Y. Yoon, R. Graf, W. Pisula, H. W. Spiess and K. Müllen, J. Am. Chem. Soc., 2011, 133, 2605–2612 CrossRef CAS PubMed.
- T. Qin, W. Zajaczkowski, W. Pisula, M. Baumgarten, M. Chen, M. Gao, G. Wilson, C. D. Easton, K. Müllen and S. E. Watkins, J. Am. Chem. Soc., 2014, 136, 6049–6055 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4py01631a |
| This journal is © The Royal Society of Chemistry 2015 |