
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, kinetics, and characterization of bio-based thermosets obtained through polymerization of a 2,5-furandicarboxylic acid-based bis(2-oxazoline) with sebacic acid†
Carolus H. R. M.
Wilsens
abc,
Nino J. M.
Wullems
a,
Erik
Gubbels
a,
Yefen
Yao
d,
Sanjay
Rastogi
ce and
Bart A. J.
Noordover
*a
aLaboratory of Polymer Materials, Eindhoven University of Technology, Den Dolech 2, 5600MB, Eindhoven, The Netherlands. E-mail: b.a.j.noordover@tue.nl
bDutch Polymer Institute (DPI), P.O. Box 902, 5600AX Eindhoven, The Netherlands
cDepartment of Biobased Materials, Maastricht University, P.O. Box 616, 6200MD Maastricht, The Netherlands
dShanghai Key Laboratory of Magnetic Resonance, East China Normal University, North Zhongshan Road 3663, 200062 Shanghai, P.R. China
eDepartment of Materials, Loughborough University, England, UK
First published on 13th February 2015
Abstract
The synthesis of renewable 2,5-furandicarboxylic acid-based cross-linked poly(ester amide)s via the polymerization of a 2,5-furandicarboxylic acid based bis(2-oxazoline) monomer (2,5-bis(4,5-dihydrooxazol-2-yl)furan, 2,5-FDCAox) with sebacic acid is reported in this work. It is demonstrated that the amide groups in the 2,5-furandicarboxamide moiety are susceptible to participation in a branching reaction with 2-oxazoline rings. The corresponding enhanced reaction rate decreases the curing times for the preparation of cross-linked polymers compared to systems containing the isophthalic acid based alternative, 1,3-bis(4,5-dihydrooxazol-2-yl)benzene (IAox). The increased tendency to form branches or cross-links in 2,5-FDCAox based systems is attributed to the occurrence of intra-molecular hydrogen bonding of the 2,5-furandicarboxamide moiety. Such an intra-molecular hydrogen bond increases the nucleophilicity of the furanic amide group and makes it more susceptible to participation in an addition reaction with a 2-oxazoline ring. Furthermore, it is demonstrated that the rate of the branching reaction can be enhanced by the addition of triphenyl phosphite as catalyst, resulting in a further decrease of the curing times of the poly(ester amide)s synthesized in this study. Preliminary coating studies indicate that 2,5-furandicarboxylic acid based cross-linked poly(ester amide)s synthesized via the 2-oxazoline ring opening addition reactions with dicarboxylic acids are good candidates for the development of fully renewable cross-linked poly(ester amide)s.
Introduction
The synthesis, ring-opening, and polymerization of oxazoline groups has been a topic of study for many years, as is indicated by the early communications of Wenker,1 Fry,2 and Goldberg3 published more than 60 years ago. The interest in these oxazoline rings originates from their ability to undergo ring-opening addition reactions with species bearing labile protons, such as carboxylic acids, thiols, phenols, amines, and amides.4 For this reason, these versatile compounds are used as monomers in addition or step growth reactions such as cationic polymerizations, enzymatic polymerizations or thermally initiated melt polymerizations.4–8 Over the years, a wide range of applications for poly(2-oxazoline)s have been reported by the groups of Schubert, Nuyken, and Hoogenboom. Examples of these applications include hydrogels,9,10 responsive polymers,11 nanoparticles,12 nanocomposites,13 macroligands,14 or tissue adhesives.15 Besides, bis(2-oxazoline) monomers have been used for chain extension of polymers and end-capping or modification of polymers and for introducing functionalities along the polymer backbone.16–19Especially thermally initiated melt polymerizations or chain extension reactions are of interest for industrial purposes, due to the absence of solvents and solvent recovery systems. Interestingly, besides the synthesis of linear polymer chains, these melt-polymerizations allow for the production of cross-linked polymers in one reaction step. As reported by Sano,20,21 cross-linked poly(ester amide)s can be obtained through a triphenylphosphite (TPP) catalyzed melt-polymerization of bis(2-oxazoline)s with dicarboxylic acids. Néry and coworkers22 reported no detectable branching or cross-linking in systems synthesized in the absence of TPP, indicating that this catalyst is essential for the production of cross-linked polymers via this route. This suggests that the degree of cross-linking, occurring during polymerization of bis(2-oxazoline)s and dicarboxylic acids, can be tailored by using TPP as a catalyst.
Recently, the interest in the development and production of renewable materials such as 2,5-furandicarboxylic acid (2,5-FDCA) has increased significantly. Many examples of the application of 2,5-FDCA as a monomer in polycondensation reactions have been reported over the last years.23 Examples of the synthesis of 2,5-FDCA based polyesters have been reported by Moore and Kelly24–26 and Gandini and coworkers.27,28 These 2,5-FDCA based polyesters generally exhibit different properties compared to their petroleum based analogues containing phenyl based monomers such as terephthalic acid or isophthalic acid. For example, as is reported by Burgess and coworkers, poly(ethylene 2,5-furandicarboxylate) (PEF) has an improved glass transition temperature, improved mechanical properties, reduced oxygen permeability and reduced chain mobility compared to poly(ethylene terephthalate).29 Also 2,5-FDCA based polyamides show properties deviating from the properties of their petroleum based analogues. For example, the low crystallinity observed for 2,5-FDCA based polyamides are in contrast to both the high crystallinities and crystallization rates observed in terephthalic acid based polyamides.30–33 Furthermore, 2,5-FDCA based polyamides exhibit more van der Waals interactions, maintain more rigid structures, and show slower weakening of hydrogen bonds upon heating compared to nylon 4,6, as was recently reported by Yeh and coworkers.34
Although the synthesis of a 2,5-FDCA based bis(2-oxazoline) monomer has been described in literature,35 no reports were found concerning its use as a monomer in melt-polymerization. Therefore, in this study, we describe the synthesis of 2,5-bis(4,5-dihydrooxazol-2-yl)furan (2,5-FDCAox) and its application in polymerizations with sebacic acid (SeA). The behavior of 2,5-FDCAox in this polymerization is compared to that of its isophthalic acid based counterpart, 1,3-bis(4,5-dihydrooxazol-2-yl)benzene (IAox). The branching and cross-linking reactions occurring during the bulk-polymerization of 2,5-FDCAox with SeA are investigated using FTIR spectroscopy and the reaction parameters required for the development of renewable polymer glasses are determined. Lastly, 2,5-FDCA based polymer glasses and coatings are prepared and their thermal properties are evaluated.
Experimental section
Materials
2,5-Furandicarboxylic acid (2,5-FDCA) was obtained from Atomole, China (>99 wt%). Sebacic acid (SeA), 2-chloroethylamine hydrochloride, triphenylphosphite (TPP), thionyl chloride, sodium hydroxide, and potassium hydroxide were purchased from Sigma. Irganox 1330 antioxidant was a kind gift from Ciba Specialty Chemicals. 1,3-bis(4,5-dihydrooxazol-2-yl)benzene (IAox) was purchased from TCI Europe. Deuterated dimethyl sulfoxide (DMSO-d6, 99.9% atom D) was acquired from Cambridge Isotope Laboratories. Methanol, dichloromethane (dried over Al2O3), N,N-dimethylformamide (DMF) and N-methyl-2-pyrrolidone (NMP) were obtained from Biosolve. All chemicals were used as received, unless stated otherwise.Preparation of N,N′-bis(2-chloroethyl)furan-2,5-dicarboxamide
2,5-Furandicarboxylic acid (16.4 g, 0.105 mol) was reacted with an excess of thionyl chloride (27.5 mL, 0.23 mol), in the presence of a catalytic amount of DMF (100 μL) in a 250 mL three-neck flask at 80 °C for 4 hours. A condenser was connected to the flask to allow refluxing of thionyl chloride and the reaction was conducted under an argon rich atmosphere. The released gaseous SO2 and HCl were passed through an aqueous NaOH solution, neutralizing the hydrochloric acid. After 4 hours of reaction time, the mixture was cooled with an ice-bath followed by the application of reduced pressure for 30 minutes to remove any residual thionyl chloride. The obtained 2,5-furandicarboxylic acid chloride was dissolved in anhydrous dichloromethane (150 mL) and dropwise added to an aqueous solution of 2-chloroethylamine hydrochloride (24.4 g, 0.21 mol) and NaOH (16.4 g, 0.41 mol) under vigorous stirring. After 2 hours of reaction time, the organic phase was distilled off using reduced pressure and the water phase was filtered. The product, N,N′-bis(2-chloroethyl)furan-2,5-dicarboxamide, was obtained as white powder and was dried overnight under reduced pressure at 40 °C. Yield = 25.3 g (86.4%). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 7.19 (s, 2H, furan), 3.75 (t, J = 6.0 Hz, 4H, ClCH2), 3.60 (d, J = 6.0 Hz, 4H, NCH2).Preparation of 2,5-bis(4,5-dihydrooxazol-2-yl)furan (2,5-FDCAox)
N,N′-Bis(2-chloroethyl)furan-2,5-dicarboxamide (15.00 g, 0.054 mol) and NaOH (4.8 g, 0.12 mol) were dissolved in 50 mL methanol in a 250 mL round-bottom flask, and were allowed to reflux for 4 hours. During the reaction white crystals precipitated. The formed product was isolated using filtration and washed with water prior to drying in vacuo overnight at 40 °C. Yield = 8.98 g (81.1%). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 7.13 (s, 2H, furan), 4.35 (t, J = 9.6 Hz, 4H, NCH2), 3.92 (t, J = 9.6 Hz, 4H, OCH2). 13C NMR (400 MHz, DMSO-d6, δ, ppm): 155.4 (O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 144.8 (O–CC), 115.9 (C–CC), 68.0 (CH2–O), 55.0 (CH2–N). Elemental analysis for C10H10N2O3, observed (calculated): C = 58.25% (58.19%), H = 4.89% (4.82%), and N = 13.59% (13.58%).
N), 144.8 (O–CC), 115.9 (C–CC), 68.0 (CH2–O), 55.0 (CH2–N). Elemental analysis for C10H10N2O3, observed (calculated): C = 58.25% (58.19%), H = 4.89% (4.82%), and N = 13.59% (13.58%).
General melt polymerization procedure
Reaction mixtures were prepared by weighing and grinding 2,5-FDCAox (1.0 to 2.5 eq.), SeA (1 eq.), Irganox 1330 (1 wt%), and TPP (0 to 7 wt%) using a mortar and pestle. Samples prepared for DMAc-SEC and DSC analysis were loaded into 1 mL glass vials and immersed in an oil-bath at the desired temperature of 160 °C for the desired reaction time (0–60 minutes) prior to analysis. Samples prepared for DMTA analysis were preheated to 160 °C for one minute until a clear casting liquid was obtained. The casting liquid was then poured onto a Teflon film and cured in an oven at 200 °C under a nitrogen rich atmosphere for 10 minutes to 1 hour.Preparation of solvent-borne coatings
2,5-FDCA (2.25 eq.) was cured with SeA (1 eq.) in the presence of 5 wt% TPP using the following procedure. A solution of 2,5-FDCAox (139.3 mg, 0.675 mmol), sebacic acid (60.7 mg, 0.3 mmol) and triphenylphosphite (10 mg, 5 wt%) was prepared in 0.6 mL N-methyl-2-pyrrolidone at 100 °C. This solution was subsequently applied to Q-panels, preheated in an oven at 200 °C, using a coating applicator frame with a spacing of 120 μm. The obtained coatings were cured by heating the coated panels in an oven at 200 °C for 1 hour under a nitrogen rich atmosphere.Characterization methods
1H NMR and 13C NMR spectroscopy was performed on a 400 MHz Brucker AVANCE-III spectrometer. Samples were prepared by dissolving 5–10 mg of monomer or polymer in 0.5–1 mL deuterated dimethyl sulfoxide (DMSO-d6) and were referenced against tetramethylsilane (TMS).N,N-Dimethylacetamide size exclusion chromatography (DMAc-SEC) was performed on a Waters Alliance system equipped with a Water 2413 refractive index detector (40 °C), a Waters 2695 separation module, a Waters 2996 photodiode array detector, a PSS GRAM guard column followed by 2 PSS GRAM columns in series of 100 Å (10 μm particles) and 3000 Å (10 μm particles) at 60 °C. DMAc was used at a flow rate of 1 mL min−1 and molecular weights were calculated against polystyrene standards (Polymer Laboratories, Mp = 580 Da up to Mp = 7.1 × 106 Da). Polymer samples prepared in a concentration of 5 mg mL−1 and were filtered through a 0.2 μm PTFE filter (13 mm, PP housing, Alltech) prior to injection.
Attenuated Total Reflection (ATR) Fourier transform infrared spectroscopy (FTIR) was performed using a Bio-Rad FTS6000 equipped with a Golden Gate, Int. Line WB MCT detector, and KRS-5 polarizer. Polymerization and curing reactions were monitored on-line for one hour at the desired reaction temperature between 150 °C and 230 °C. Spectra were collected every two seconds in the range between 4000 to 650 cm−1 with a spectral resolution of 4 cm−1.
Coating thicknesses of the solvent-borne coatings were measured using a magnetic induction coating thickness gauche (LD0400 by Thermimport Quality Control). Reverse impact tests were performed by dropping a weight of 1 kg from a controlled height of 100 cm on the backside of the coated panels and the pencil hardness test was performed by scratching the obtained coating with pencils of increasing hardness. Solvent resistance was determined by the acetone double rub test in which the material is rubbed back and forth (double rubs) by an acetone drenched cloth 100 times.
The sample preparation procedures and characterization methods of the CP/TOSS 13C NMR spectroscopy, TGA, DSC, and DMTA experiments are supplied in the “Characterization methods” section of the ESI.†
Results and discussion
Polymerization of 2,5-FDCAox with sebacic acid
2,5-Bis(4,5-dihydrooxazol-2-yl)furan (2,5-FDCAox) was successfully obtained after the conversion of 2,5-FDCA to the diacid chloride via its reaction with thionyl chloride, followed by a reaction with chloroethylamine hydrochloride, and consecutive ring closing in methanol (Scheme 1).8 The reaction products obtained after synthesis through this route were easily isolated by filtration and required no additional purification. | ||
| Scheme 1 Synthetic route used in this study to obtain 2,5-FDCAox from 2,5-furandicarboxylic acid. | ||
As is well known from literature, the ring-opening polymerization of a bis(2-oxazoline) with a dicarboxylic acid results in the formation of a linear poly(ester amide) as is shown in Scheme 2a. Furthermore, as is reported by Sano,20,21 the amide group formed in this ring-opening addition reaction can subsequently react with a 2-oxazoline group in the presence of triphenylphosphite (TPP), forming a tertiary amide and a new secondary amide group (branching reaction, Scheme 2b). Generally, the reaction rate of the reaction between a 2-oxazoline ring and carboxylic acid (k1) is significantly higher than the rate of the branching reaction (k2) in the absence of TPP. Nonetheless, if the reaction constant k2 is not zero, tertiary amides are, inevitably, formed during polymerization. These tertiary amides act as branching points in the polymer backbone, and eventually result in the formation of a cross-linked system.
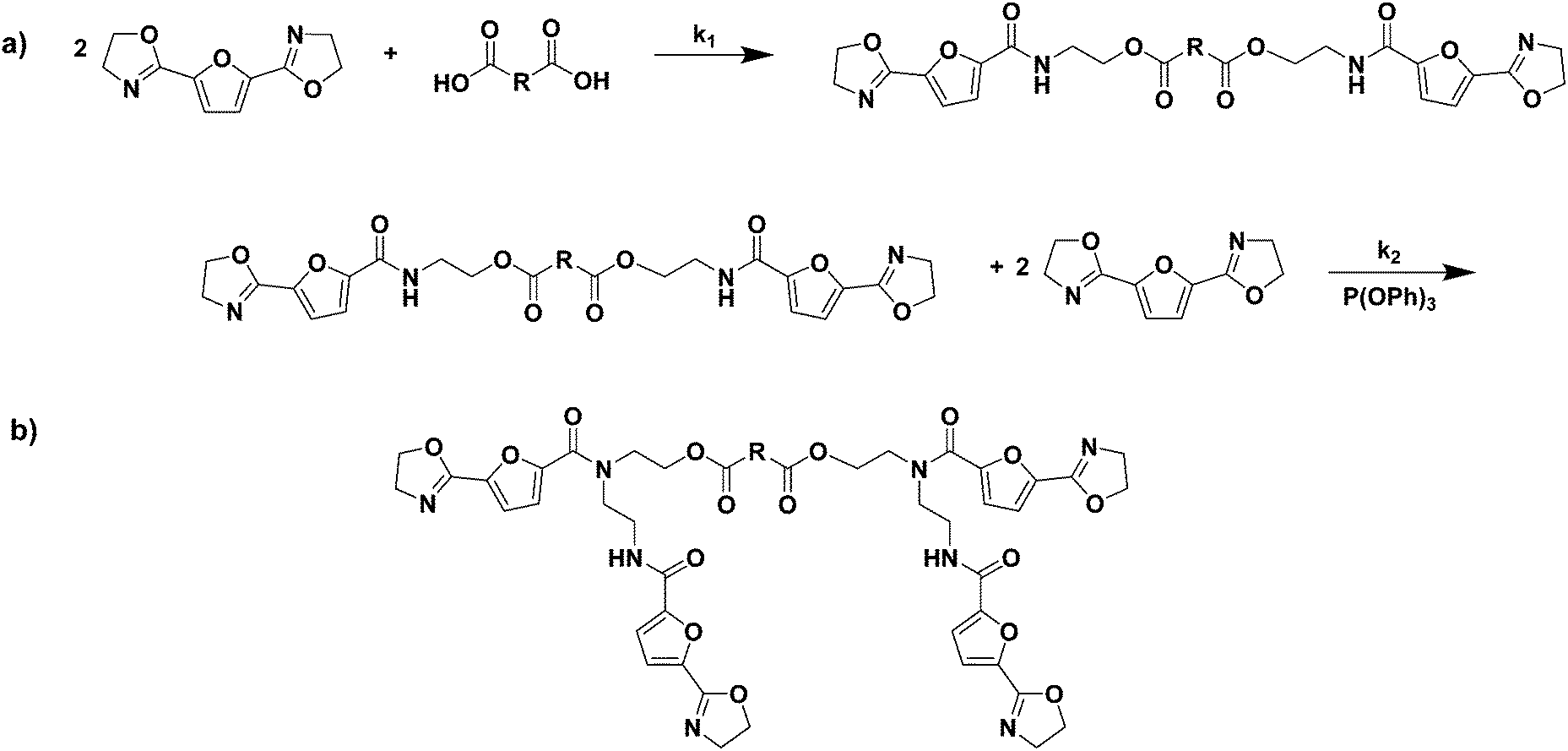 | ||
| Scheme 2 Thermally initiated ring-opening addition reactions occurring during the reaction of 2,5-FDCAox and a dicarboxylic acid. The formation of a linear chain through the reaction of 2,5-FDCAox with a carboxylic acid group is shown in reaction (a), whereas the branching reaction is shown in reaction b. | ||
The reaction kinetics involved during the polymerization of bis(2-oxazoline)s and dicarboxylic acids can be described using differential eqn (1)–(3). To obtain these differential equations, it is assumed that both reactions are irreversible and second order,20 that the 2-oxazoline groups are equireactive,22 and that the secondary amides are equireactive. In differential eqn (1)–(3), the unreacted (2-oxazoline) bulk concentration is denoted as [OX] in mol kg−1. Similarly, the bulk concentrations of the formed ester groups, secondary amide groups, and tertiary amide groups are denoted as [Ester], [Sec. Amide], and [Tert. Amide] respectively in mol kg−1.
 | (1) |
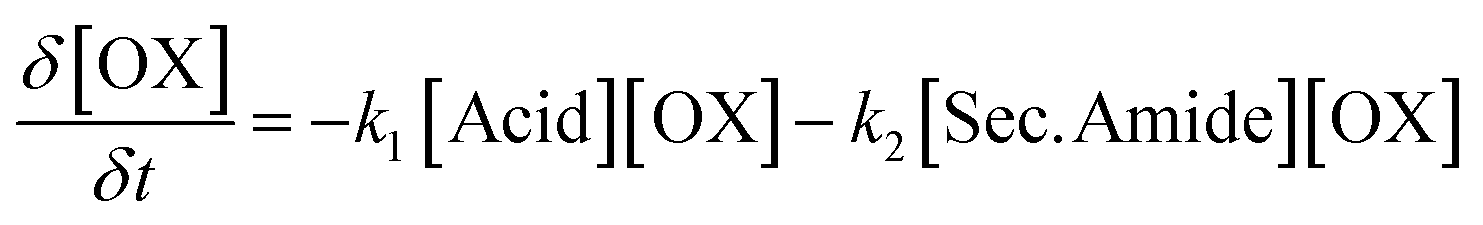 | (2) |
 | (3) |
To study the rates of these reactions occurring during polymerization, the 2-oxazoline conversion was monitored during the polymerization of an equimolar mixture of 2,5-FDCAox and sebacic acid (SeA) at 160 °C. The monomers were mixed in the solid state and heated for a fixed reaction time (tr) using a salt bath. After cooling back to room temperature, the obtained polymers were dissolved in deuterated dimethylsulfoxide (DMSO-d6) and analyzed using 1H NMR analysis. To compare the reaction rate of 2,5-FDCAox with the commercially available 1,3-bis(4,5-dihydrooxazol-2-yl)benzene (IAox), the polymerizations of equimolar mixtures of IAox and SeA were performed and analyzed under the same conditions.
The 2-oxazoline conversion was calculated from the 1H NMR spectra of the products obtained after varying polymerization times (the calculation method is supplied in the ESI†). Next, the 2-oxazoline conversion over time was used for data fitting with differential eqn (1)–(3), using a non-linear regression, to determine reaction constants k1 and k2 (Fig. 1).
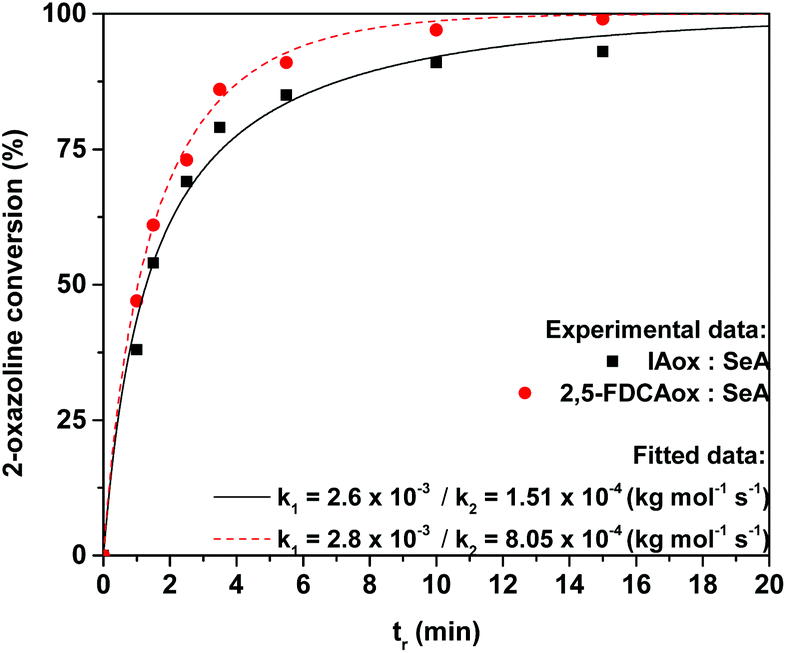 | ||
| Fig. 1 Conversion of 2-oxazoline groups over time during melt-polymerization of equimolar mixtures IAox and SeA (squares) or 2,5-FDCAox and SeA (circles) at 160 °C, as calculated from the 1H NMR spectra. The theoretical curves and reaction constants found after data fitting are shown for the IAox system (continuous line) 2,5-FDCAox system (dotted line). | ||
Interestingly, no accurate fit of the experimental data could be found without taking the k2 reaction constant in account, indicating that the branching reaction also proceeds during polymerization in the absence of TPP and cannot be neglected. This is in contrast to the observations of Néry and coworkers, who detected no branching in the model reaction between IAox and dodecanoic acid.22 It is expected that the absence of branching in the model reaction reported by Néry is a result from the low viscosity of their system. In contrast, the viscosities of the polymerizations reported in this study increase rapidly over time, limiting diffusion and likely promoting the formation of branches.
From the experimental data shown in Fig. 1, it can be clearly seen that the 2,5-FDCAox monomer is consumed faster during polymerization with SeA than the IAox monomer. Interestingly, the observed k1 reaction constant for the polymerization of the system containing IAox (k1 = 2.6 × 10−3 kg mol−1 s−1) and 2,5-FDCAox (k1 = 2.8 × 10−3 kg mol−1 s−1) are comparable, suggesting that both oxazoline-functional monomers have a similar reactivity towards carboxylic acid groups. Furthermore, from Fig. 1 it is observed that the k2 reaction constant found for the polymerization of 2,5-FDCAox with SeA is roughly 5 times larger (8.05 × 10−4 kg mol−1 s−1) than the k2 reaction constant found for the polymerization of IAox with SeA (1.51 × 10−4 kg mol−1 s−1). This indicates that branching is occurring at an enhanced rate in the polymerization of 2,5-FDCAox with SeA.
The results obtained in the 1H NMR analysis suggest that branching occurs at a higher rate in the polymerization of 2,5-FDCAox with SeA than in the polymerization of IAox with SeA. It is anticipated that this increased tendency to form branched polymer chains results in an accelerated increase of the weight-average molecular weight (Mw) and PDI during polymerization, until the polymer starts to cross-link and becomes insoluble. To investigate the effect of this branching reaction on the Mw and PDI, DMAc-SEC was performed on the materials obtained after polymerization of equimolar mixtures of 2,5-FDCAox with SeA and IAox with SeA at 160 °C. Fig. 2a shows an overview of the Mw and PDI obtained for the 2,5-FDCAox and IAox based systems. Fig. 2b and 2c show the SEC chromatograms obtained as a function of reaction time for the 2,5-FDCAox and IAox based systems, respectively.
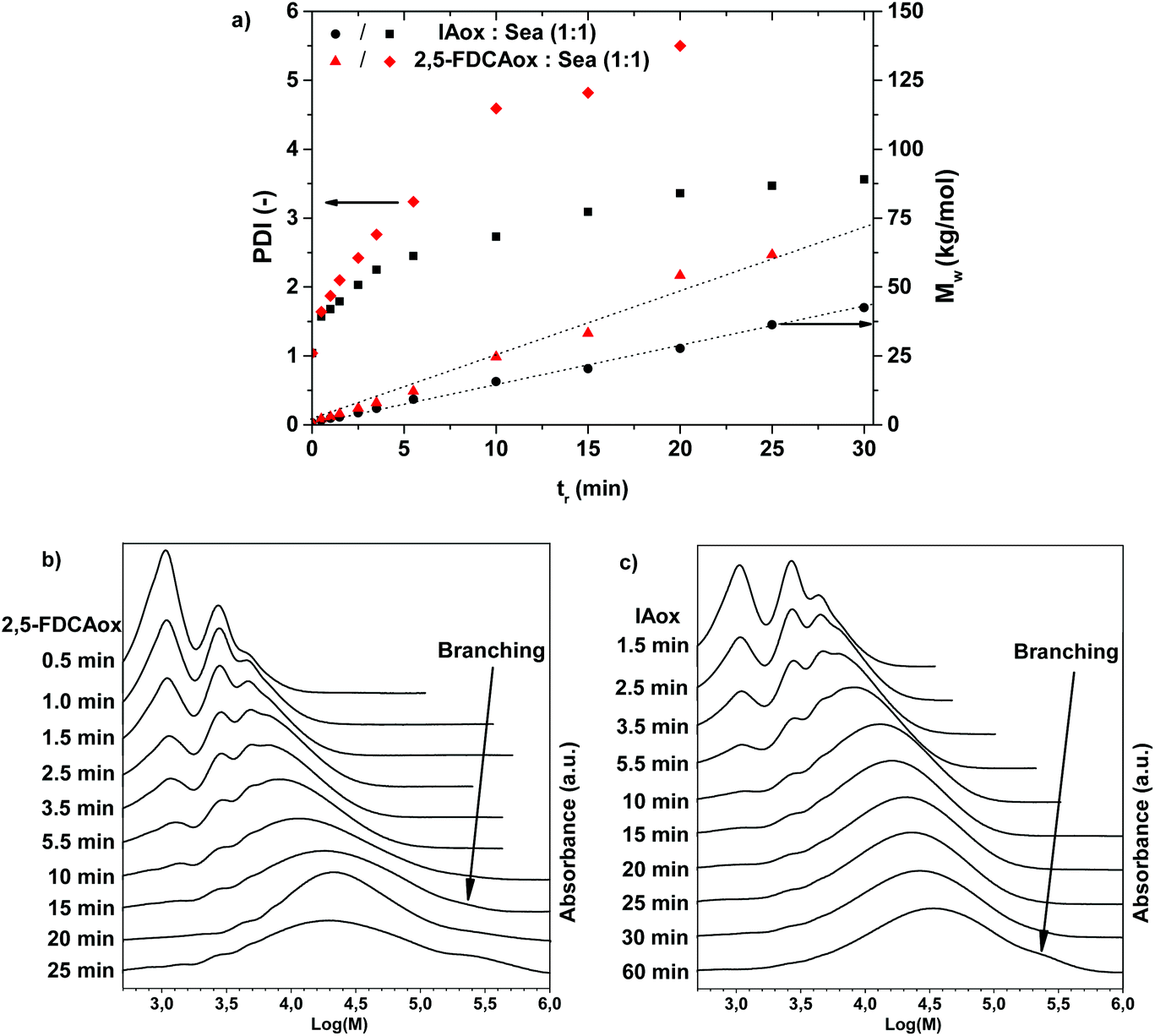 | ||
| Fig. 2 Changes of the weight-average molecular weight (Mw) and PDI over time during the polymerization of FDCAox with SeA and IAox with SeA in an equimolar ratio performed at 160 °C, as observed in DMAc-SEC analysis. (a) an overview of changes in the Mw and PDI of both reactions over time, whereas (b) and (c) show changes in the DMAc-SEC chromatograms observed over time for the systems containing FDCAox and IAox respectively. The dotted lines in (a) are added to guide the eye. | ||
From Fig. 2a, it can be seen that the Mw and PDI increase faster during the polymerization of 2,5-FDCAox with SeA compared to the polymerization of IAox with SeA. For example, a Mw of 33.2 kg mol−1 and a PDI of 4.8 is achieved in the polymerization of 2,5-FDCAox with SeA after 15 minutes of reaction time. In contrast, the isophthalic acid (IA) based polymer has only reached a Mw 20.4 kg mol−1 and a PDI of 3.1 after 15 minutes of polymerization. Such a difference in molecular weight corresponds well to the difference in 2-oxazoline consumption observed in the 1H NMR analysis.
From Fig. 2b, it can be seen that branching of the polymer chains is occurring in the polymerization of 2,5-FDCAox with SeA, as is detected by the elution of a high molecular weight fraction in the DMAc-SEC chromatogram for polymers having a tr of 10 minutes or longer. This high molecular weight fraction, characteristic for branched chains formed through chain coupling, is indicated by the black arrow in Fig. 2b. Interestingly, the continued branching occurring during polymerization results in the formation of a cross-linked and insoluble polymer after 25 minutes of reaction time. In contrast, the polymer obtained after 60 minutes of polymerization of IAox with Sea is fully soluble in DMAc, indicating that the polymer is not cross-linked. However, the detected presence of a high molecular weight fraction after 60 minutes of polymerization does indicate that branching has occurred to some extent (Fig. 2c). The differential scanning calorimetry (DSC) analysis of the products obtained after polymerization for one hour confirm the enhanced branching of the 2,5-FDCAox based system, indicated by a broadened glass transition (ESI†).
Reaction mechanism of the branching reaction
In general, thermally initiated ring-opening addition reactions of a 2-oxazoline groups are reported to proceed through the reaction of a nucleophile bearing a labile proton, such as carboxylic acids, thiols or aromatic phenols.2,3 The presence of this labile proton is considered to be crucial for the ring-opening reaction, since it interacts with the nitrogen atom in the 2-oxazoline ring. The resulting electron deficiency makes the 2-oxazoline ring susceptible for a nucleophilic attack on the carbon next to the oxygen atom. This reaction route explains the high reactivity of the 2-oxazoline ring towards carboxylic acids; the carboxylic acid group donates a proton to the 2-oxazoline ring, followed by a nucleophilic attack of the carboxylate anion on the carbon next to the oxygen atom in the protonated 2-oxazoline ring (Scheme 3a).22 This mechanism also explains the low reactivity of the reaction between amide groups and 2-oxazoline rings occurring in the branching reaction; the amide group is a poor nucleophile due to the resonance stabilization with the carbonyl group.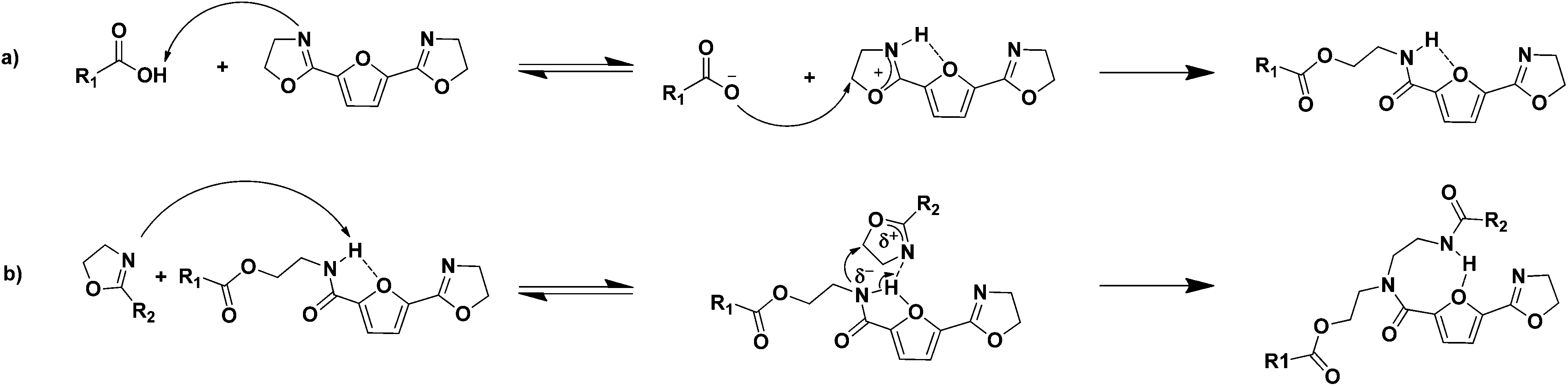 | ||
| Scheme 3 Schematic representation of the weakening of the amide N–H bond via intra-molecular hydrogen bonding occurring during the reaction of a furancarboxamide group with a 2-oxazoline ring. | ||
In the polymerizations studied thus far, we have observed that branching occurs at an enhanced rate in systems containing 2,5-FDCAox compared to systems containing IAox. It is thought that the increased rate of branching observed in the polymerization of 2,5-FDCAox with SeA is a result of the intra-molecular hydrogen bonding occurring in 2,5-FDCA based polyamides (Scheme 3).36 This intra-molecular hydrogen bond is formed through the interaction of the free electron pairs of the oxygen atom of the furan ring with the proton of the amide group. As a result, the N–H bond of the amide group weakens and the nitrogen of the amide group becomes more electron rich and thus becomes a better nucleophile. The increased nucleophilic nature of the amide group enhances its reactivity towards 2-oxazoline rings and explains the increased branching rate occurring in the polymerization of 2,5-FDCAox with SeA. For this reason, we attribute the increased rate of branching observed in the polymerizations involving 2,5-FDCAox to the presence of the furan(di)carboxamide groups rather than to a difference in reactivity of the 2-oxazoline groups in 2,5-FDCAox and IAox.
On-line monitoring of the branching reaction using ATR-FTIR analysis
In the previous section, it was demonstrated that 2-oxazoline rings have an increased reactivity towards furan(di)carboxamide moieties, promoting the rate of formation of branches and cross-links. Although this branching reaction is highly interesting for the development of renewable 2,5-FDCAox based thermosets, chemical analysis of the formed cross-linked products is challenging due to their insoluble nature. For this reason, polymerization reactions of 2,5-FDCAox with SeA were monitored on-line using ATR-FTIR spectroscopy, to obtain more insight into the effect of the reaction temperature and the triphenylphosphite (TPP) concentration on the rate of branching and on the total curing time.The left hand side of Fig. 3 shows the characteristic FTIR spectra between 1800 and 800 cm−1, obtained during a polymerization of 2,5-FDCAox with SeA (ratio = 2.25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 200 °C in the absence of TPP (Fig. 3a) and containing 5 wt% TPP (Fig. 3b). The changes in intensity of different vibrations, normalized on the intensity of the aliphatic CH2 stretch vibration at 2932 cm−1, are shown on the right hand sides of Fig. 3a and 3b. It should be noted that the excess of 2,5-FDCAox used in these polymerization reactions was chosen to promote the cross-linking of the system and to obtain a densely cross-linked material at full conversion.
:1) at 200 °C in the absence of TPP (Fig. 3a) and containing 5 wt% TPP (Fig. 3b). The changes in intensity of different vibrations, normalized on the intensity of the aliphatic CH2 stretch vibration at 2932 cm−1, are shown on the right hand sides of Fig. 3a and 3b. It should be noted that the excess of 2,5-FDCAox used in these polymerization reactions was chosen to promote the cross-linking of the system and to obtain a densely cross-linked material at full conversion.
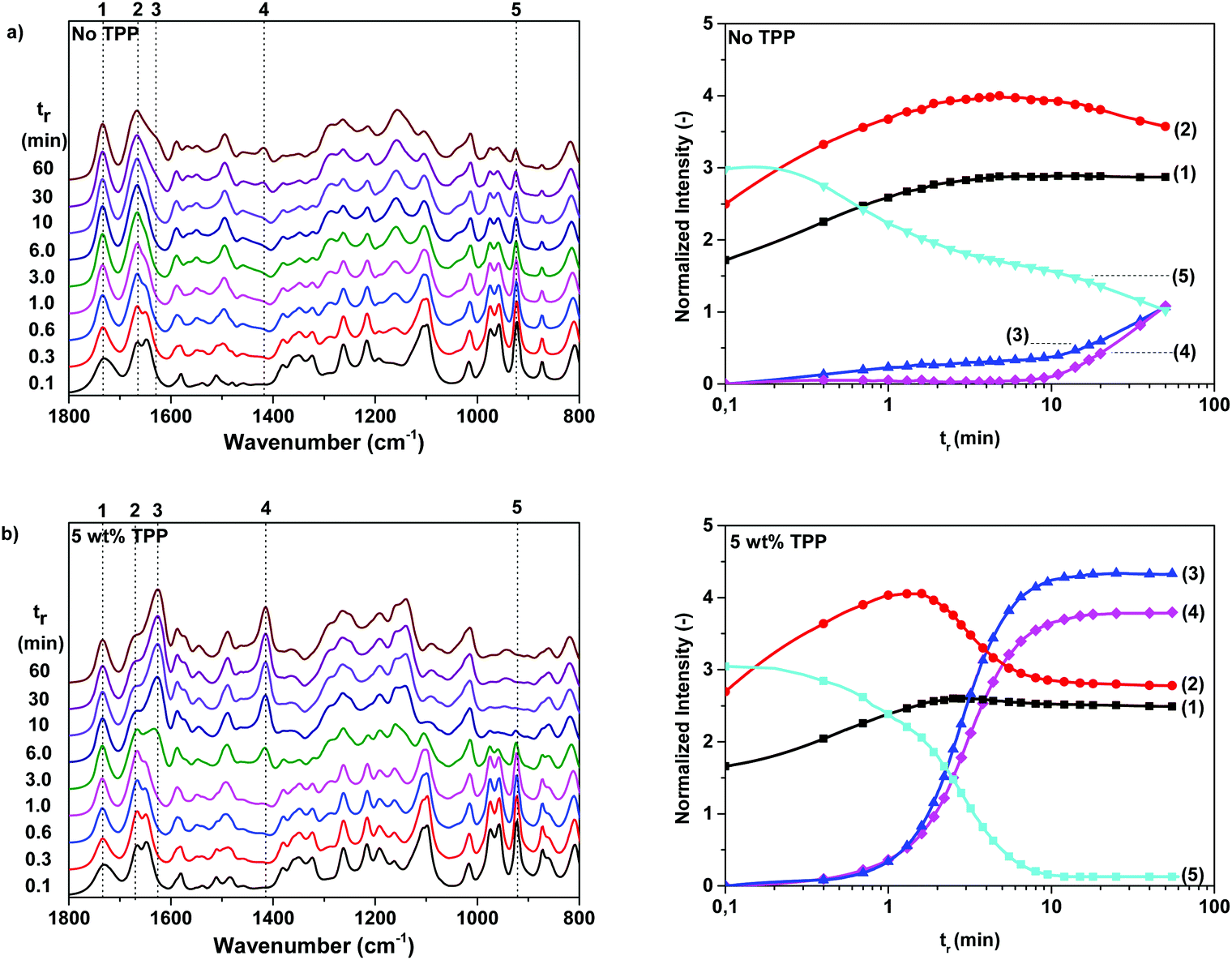 | ||
Fig. 3 Characteristic FTIR spectra between 1800 and 800 cm−1 obtained during the polymerization of 2,5-FDCAox with SeA (ratio = 2.25:1) at 200 °C containing (a) no TPP and (b) 5 wt% TPP. The right two figures show the changes in intensity of the ester carbonyl stretch vibration at 1734 cm−1 (1,  ), the secondary amide carbonyl stretch vibration at 1667 cm−1 (2, ), the secondary amide carbonyl stretch vibration at 1667 cm−1 (2,  ), and the tertiary amide carbonyl stretch vibration at 1625 cm−1 (3, ), and the tertiary amide carbonyl stretch vibration at 1625 cm−1 (3,  ). Furthermore, band 4 at 1417 cm−1 ( ). Furthermore, band 4 at 1417 cm−1 ( ) and band 5 at 922 cm−1 ( ) and band 5 at 922 cm−1 ( ) are attributed to the bend vibrations of the CH2 groups adjacent to the tertiary amide group and the ring vibrations of the 2-oxazoline ring respectively. N.B. the intensity of the vibration bands are normalized against the intensity of the CH2 stretch vibration at 2932 cm−1. ) are attributed to the bend vibrations of the CH2 groups adjacent to the tertiary amide group and the ring vibrations of the 2-oxazoline ring respectively. N.B. the intensity of the vibration bands are normalized against the intensity of the CH2 stretch vibration at 2932 cm−1. | ||
From the FTIR spectra shown in Fig. 3a, it can be seen that the bands corresponding to the carbonyl stretch vibration of the carboxylic acid group at 1734 cm−1 (band 1), the CN stretch vibration of the oxazoline ring at 1637 cm−1, and the vibration of the oxazoline ring at 922 cm−1 (band 5) are present at the start of the reaction. The intensity of the carbonyl stretch vibration increases slightly during the first eight minutes of the reaction, indicating that the groups are converted into ester bonds. Furthermore, a simultaneous decrease of the intensity of the CN stretch vibration band at 1637 cm−1 occurs in the first eight minutes together with an increase of the intensity of the secondary amide carbonyl stretch vibration at 1667 cm−1 (band 2). The rise of band 2 with the simultaneous decrease of the 2-oxazoline CN stretch vibration band confirms that the reaction between the 2-oxazoline groups with carboxylic acid groups results in the formation of secondary amide groups.
After 8 minutes of reaction time, no changes are detected in the intensity of band 1, indicating that all carboxylic acid groups are successfully converted into ester groups. However, band 2 and band 5 decrease slowly over time together with the CN stretch vibration at 1637 cm−1, indicating that the 2-oxazoline concentration is decreasing. As expected, the band of the carbonyl stretch vibration of the tertiary amide at 1625 cm−1 (band 3) and the band of the bend vibrations of the CH2 groups adjacent to the tertiary amide groups at 1417 cm−1 (band 4) rise simultaneously with the decrease of the intensity of vibration bands 2, 5, and the band at 1637 cm−1. The simultaneous rise of bands 3 and 4 and the decrease of band 1 indicate that the concentration of tertiary amides is increasing and thus, branching and cross-linking is taking place in the system. Interestingly, since vibration 5 is still present after 60 minutes of reaction time, it can be concluded that full conversion of the 2-oxazoline moieties was not obtained. For this reason, it is expected that the cross-linking will continue with the extension of the reaction time. Nonetheless, from the FTIR data shown in Fig. 3a, it can be concluded that no catalyst is required to enable the formation of branched and cross-linked structures during polymerization of 2,5-FDCAox and SeA.
As can be seen in differential eqn (1), the concentration of secondary amide groups is only dependent on the reaction of the 2-oxazoline rings with carboxylic acid groups. During the branching reaction, a 2-oxazoline ring reacts with a secondary amide forming a new secondary amide together with a tertiary amide. This implies that the concentration of secondary amides should remain constant after the full conversion of the carboxylic acid groups. However, as can be seen in Fig. 3, the intensity of band 2 slowly decreases over time even after the carboxylic acid groups have reacted into ester groups. It is likely that this change in intensity is a result of the decrease of the CN vibration band at 1637 cm−1 which partially overlaps with the CO stretch vibration of the secondary amide, band 2.
Similar changes in intensity for band 1 to 5 are observed for the TPP catalyzed system shown in Fig. 3b. However, the decrease of the intensity of band 5 and the rise of band 3 and 4 is significantly faster than in the non-catalyzed system. Furthermore, the intensities of bands 3 and 4 are much higher after 60 minutes of reaction in the presence of TPP, indicating that the concentration of tertiary amides is higher in the TPP catalyzed system than in the non-catalyzed system. This clearly indicates that TPP increases the k2 branching reaction constant. No changes in FTIR spectra are observed after 10 minutes of reaction time, indicating that the reaction is close to full conversion. This high conversion of the 2-oxazoline groups is confirmed by the low intensity of band 5 after 60 minutes of reaction time. Overall, from the data shown in Fig. 3, it can be concluded that TPP can be successfully used to enhance the branching and cross-linking reaction of 2,5-FDCA further and to reduce the reaction or curing times.
CP/TOSS 13C NMR spectroscopy was performed on the product obtained after polymerization of 2,5-FDCAox with SeA for 60 minutes at 200 °C in the presence of 5 wt% TPP and confirmed that polymerization and cross-linking had successfully taken place. The obtained CP/TOSS spectra and their interpretation are supplied in the ESI.†
Influence of the reaction temperature and TPP loading on the branching reaction
To obtain more insight in the effect of the reaction temperature and the catalyst loading on the branching reaction, polymerizations of 2,5-FDCAox with Sea (ratio = 2.25:1) were monitored on-line using FTIR spectroscopy. Experiments were performed at different reaction temperatures and at different TPP loadings. The observed change in intensity of the vibration band at 1417 cm−1, indicative of the concentration of branches, during polymerizations at different temperatures is shown in Fig. 4a (no TPP) and Fig. 4b (5 wt% TPP).
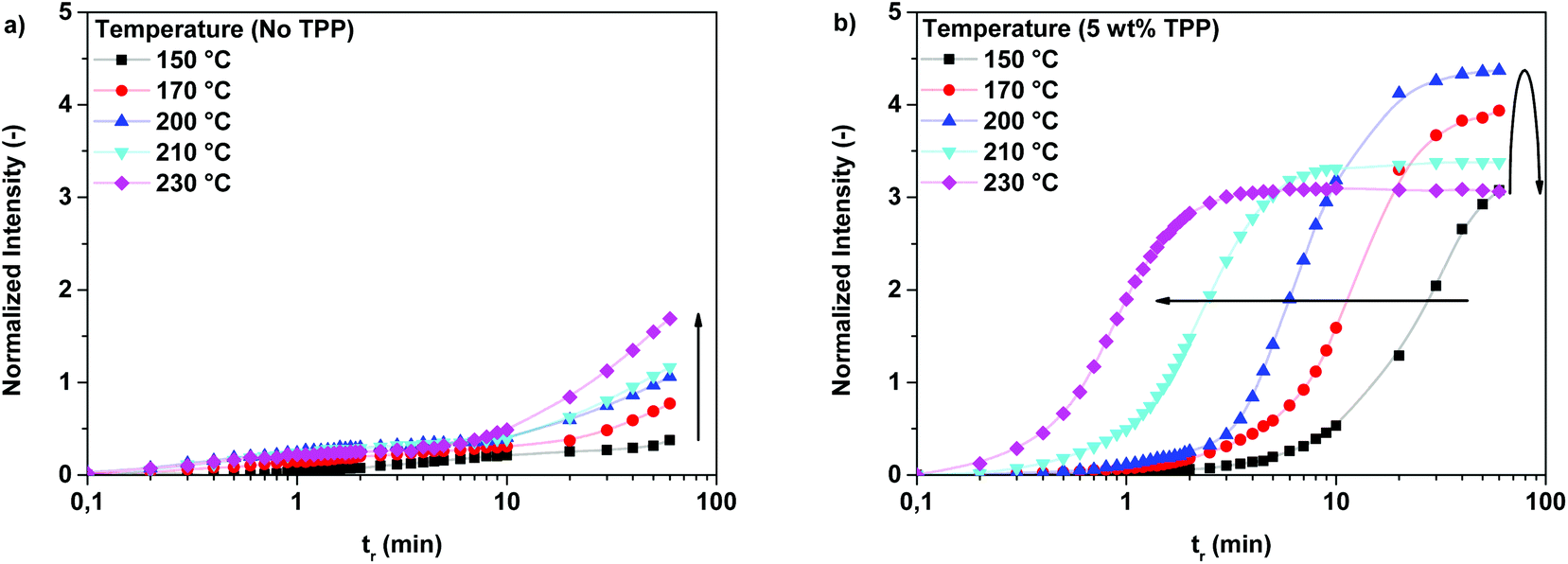 | ||
| Fig. 4 Changes in intensity of the vibration band at 1417 cm−1 during the polymerization of 2,5-FDCAox with SeA (ratio = 2.25:1) at different reaction temperatures containing (a) no TPP and (b) 5 wt% TPP. N.B. The intensity of the vibration band at 1417 cm−1 is normalized against the intensity of the CH2 stretch vibration at 2932 cm−1. | ||
From the data in Fig. 4a, it can be seen that the intensity of the vibration band at 1417 cm−1 increases faster with increasing reaction temperature, indicating that the branching reaction is accelerated by an increase of the reaction temperature. However, it is clear from the low intensity of the vibration band at 1417 cm−1 that the reaction does not reach full conversion within one hour of reaction time. For this reason, it is concluded that, although the increase of the reaction temperature promotes the formation of branches, full conversion is only obtained after polymerization for several hours (over 6 hours at 200 °C). It should be noted that sublimation of 2,5-FDCAox was observed in polymerizations performed above 200 °C, making the performance of experiments at temperatures above 200 °C troublesome, due to the continuous change in stoichiometry during the ongoing reaction.
Similarly, the rate of branch formation increases with increasing reaction temperature in systems containing TPP, as is visible in Fig. 4b. As expected, the rate of branching is significantly higher in systems catalyzed by TPP; full conversion is reached within 5 minutes of reaction time, as is observed for the TPP catalyzed polymerizations performed at 230 °C. Interestingly, the maximum intensity of the normalized absorbance signal of the vibration band at 1417 cm−1 decreases for polymerizations performed above 200 °C. This decrease is attributed to the slow sublimation of 2,5-FDCAox occurring at these temperatures. The resulting ratio of 2,5-FDCAox to SeA after 60 minutes of polymerization is therefore lower than at the start of the reaction, effectively decreasing the maximum number of cross-links at full conversion. For this reason, further polymerizations of 2,5-FDCAox are conducted at 200 °C or lower.
To investigate the effect of the TPP concentration on the branching reaction, experiments were performed at 200 °C (2,5-FDCAox to Sea ratio of 2.25 to 1) with various TPP loadings (Fig. 5a). Similar to the increase of the reaction temperature, the increase of the TPP concentration accelerates the formation of branches. To obtain information regarding the effect of TPP on the reaction constants k1 and k2, differential eqn (1)–(3) were fitted to the data shown in Fig. 5a using a non-linear regression (Fig. 5b). It should be noted that no information was available on the final concentrations of the reactive groups after 60 minutes of polymerization, making it impossible to convert the normalized intensity signal obtained from FTIR to bulk concentration values required for accurate data fitting. For this reason, reaction constants k1 and k2 were calculated using different conversion factors. The conversion factor is defined as the normalized intensity signal divided by the bulk concentration, assuming that correlation between the intensity observed in FTIR spectroscopy and the bulk concentration is linear. In this manuscript, a conversion factor of 0.95 is used to convert the normalized intensity to the bulk concentration. Detailed information on the data fitting process and the selection of the conversion factor is supplied in the ESI.†
 | ||
| Fig. 5 Changes in normalized intensity of the vibration band at 1417 cm−1 during the polymerization of 2,5-FDCAox with SeA (ratio = 2.25:1) at different TPP loadings performed at 200 °C (a). The fit obtained after non-linear regression of the data shown in (a) is shown in (b) (conversion factor = 0.95). A detailed description of the data fitting process is supplied in the ESI.† | ||
Fig. 6 shows the k1 and k2 values obtained after fitting of the data using a conversion factor of 0.95. Interestingly, the effect of the addition of TPP on the k1 and k2 reaction constants was similar, independent of the selected conversion factor; the addition of TPP increases the k2 reaction constant, whereas it does not seem to influence the k1 reaction constant to a great extent. This observation is in good agreement with the statement of Sano, who concluded that TPP catalyzes the branching of IAox, whereas the k1 reaction constant increased only with a factor 1.3 in the presence of 1 wt% TPP.20
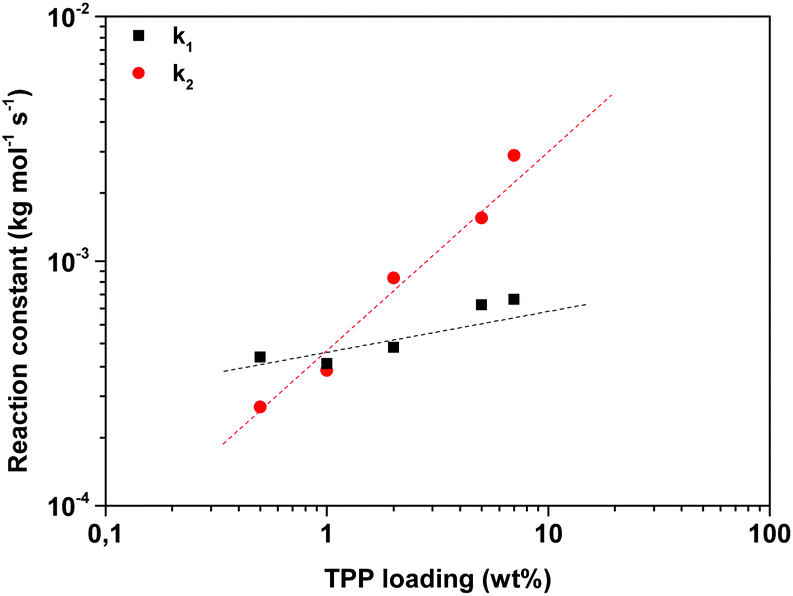 | ||
| Fig. 6 Changes in the observed reaction constants k1 and k2 found after fitting of the FTIR data (conversion factor = 0.95) obtained during polymerizations of 2,5-FDCAox with SeA (ratio = 2.25:1) at 200 °C with varying TPP loadings. N.B. The dotted lines are added to guide the eye. | ||
Dynamic mechanical thermal analysis (DMTA) was employed to determine the glass transition temperature of both the 2,5-FDCA and IA based coatings obtained after one hour of curing. Although the 2,5-FDCA based system could be cured significantly faster, no substantial differences were found in the glass transition temperatures of the two different systems (IAox based: Tg = 155.1 °C; 2,5-FDCAox based: Tg = 156.3 °C). More information regarding the DMTA experiments and correlations between the glass transition of the polymers and the FTIR spectra are supplied in the ESI.†
Preliminary coating performance of solvent-borne coatings
Solvent-borne coatings were prepared using NMP as a solvent and were cured at 200 °C, as is described in the coating procedure in the Experimental section. The obtained coatings had a thickness of approximately 35 μm and were sufficiently cross-linked as was indicated by the solvent resistance test. Furthermore, the coatings were soft, as became evident from the pencil hardness test (2H). This indicates that the materials were not densely cross-linked, which was confirmed through FTIR analysis; the coatings showed an average normalized intensity of 1.6 (the vibration at 1417 cm−1) corresponding to a Tg around 80 °C, as follows from the data in Fig. S7 in the ESI.† This implies that the presence of solvent during the curing process limits the cross-link density, since the bulk cured samples exhibited significantly higher Tg in the range between 140–160 °C. Although the coatings were not densely cross-linked, they showed ductile deformation and showed no crazing or cracking during rapid deformation in reverse impact testing. These preliminary coating results indicate that these 2,5-FDCA based poly(ester amide)s are good candidates for application in both thermally cured and solvent-borne polymer glasses and coatings.Conclusions
2,5-Bis(4,5-dihydrooxazol-2-yl)furan (2,5-FDCAox) was successfully isolated after reaction of 2,5-FDCA with thionyl chloride, followed by reaction with chloroethylamine hydrochloride, and ring closure in methanol. The reaction occurring between a carboxylic acid group and the 2-oxazoline ring and the branching reaction between the formed amide groups with the 2-oxazoline ring were compared. It is demonstrated that the branching reaction occurs more rapidly in the polymerization of equimolar mixtures of 2,5-FDCAox with SeA than in the polymerizations of equimolar mixtures of 1,3-bis(4,5-dihydrooxazol-2-yl)benzene (IAox) with SeA. This enhanced rate of cross-linking in the 2,5-FDCAox based system is attributed to the increased nucleophilicity of the 2,5-furandicarboxamide resulting from intra-molecular hydrogen bonding, causing an enhanced reactivity towards 2-oxazoline rings. FTIR analysis has successfully been employed to monitor this branching reaction on-line and to determine the effect of the reaction temperature and the triphenylphosphite (TPP) concentration on the polymerization. It is shown that TPP can be successfully used to accelerate the branching reaction and to reduce the curing times of the cross-linked poly(ester amide)s synthesized in this study. Furthermore, it is demonstrated that, also in the presence of TPP, the branching reaction proceeds significantly faster in 2,5-FDCAox based systems than in IAox based systems. Although the reaction and curing times vary when using different bis(2-oxazoline) monomers, the glass transition temperatures of the fully cured materials are comparable. Furthermore, preliminary coating studies indicate that these fully renewable 2,5-furandicarboxylic acid based cross-linked poly(ester amide)s are interesting candidates for application in coatings or polymer glasses. Overall, it can be concluded that the decreased curing times and the comparable thermal properties make the 2,5-FDCA based bis(2-oxazoline) monomers interesting candidates for the preparation of fully renewable cross-linked poly(ester amide)s.Acknowledgements
This work is part of the research program of the Dutch Polymer Institute (DPI), #739 BioLCP. The authors thank M.M. de Beer for the support with the fitting of the experimental data.References
- H. Wenker, J. Am. Chem. Soc., 1938, 60, 2152–2153 CrossRef CAS.
- E. M. Fry, J. Org. Chem., 1950, 15, 802–806 CrossRef.
- A. A. Goldberg and W. Kelly, J. Chem. Soc., 1948, 1919–1926 RSC.
- B. M. Culbertson, Prog. Polym. Sci., 2002, 27, 579–626 CrossRef CAS.
- F. E. Du Prez, E. J. Goethals and R. Hoogenboom, Cationic Polymerizations, in Handbook of Polymer Synthesis, Characterization, and Processing, ed. E. Saldívar-Guerra and E. Vivaldo-Lima, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2013, ch. 8, DOI: DOI:10.1002/9781118480793.
- J. Lustoň, J. Kronek and F. Böhme, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 343–355 CrossRef.
- J. Lustoň, J. Kronek, O. Markus, I. Janigová and F. Böhme, Polym. Adv. Technol., 2007, 18, 165–172 CrossRef.
- K. Aoi and M. Okada, Prog. Polym. Sci., 1996, 21, 151–208 CrossRef CAS.
- M. Hartlieb, D. Pretzel, C. Englert, M. Hentschel, K. Kempe, M. Gottschaldt and U. S. Schubert, Biomacromolecules, 2014, 15(6), 1970–1978 CrossRef CAS PubMed.
- M. Hartlieb, D. Pretzel, K. Kempe, C. Fritzsche, R. M. Paulus, M. Gottschaldt and U. S. Schubert, Soft Matter, 2013, 9, 4693–4704 RSC.
- K. Kempe, C. Weber, K. Babiuch, M. Gottschaldt, R. Hoogenboom and U. S. Schubert, Biomacromolecules, 2011, 12, 2591–2600 CrossRef CAS PubMed.
- K. Kempe, A. Vollrath, H. W. Schaefer, T. G. Poehlmann, C. Biskup, R. Hoogenboom, S. Hornig and U. S. Schubert, Macromol. Rapid Commun., 2010, 31, 1869–1873 CrossRef CAS PubMed.
- R. Jordan, N. West, A. Ulman, Y.-M. Chou and O. Nuyken, Macromolecules, 2001, 34, 1606–1611 CrossRef CAS.
- P. Persigehl, R. Jordan and O. Nuyken, Macromolecules, 2000, 22, 6977–6981 CrossRef.
- P. J. M. Bouten, M. Zonjee, J. Bender, S. T. K. Yauw, H. van Goor, J. van Hest and R. Hoogenboom, Prog. Polym. Sci., 2014, 1375–1405 CrossRef CAS PubMed.
- A. Baier, F. Böhme, R. Vogel, H. Martin and D. Leistner, Die Angew. Makromol. Chem., 1995, 228, 117–129 CrossRef CAS.
- E. Taylan and S. H. Küsefoğlu, J. Appl. Polym. Sci., 2012, 124, 3229–3235 CrossRef CAS.
- H. Inata and S. Matsumura, J. Appl. Polym. Sci., 1987, 33, 3069–3079 CrossRef CAS.
- J. Lustoň, F. Böhme, H. Komber and G. Pompe, J. Appl. Polym. Sci., 1999, 72, 1047–1053 CrossRef.
- Y. Sano, K. Arita and I. Masuda, US patent4,474,942, 1984 to Takeda Chemical Industries Ltd Search PubMed.
- Y. Sano, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2749–2760 CrossRef CAS.
- L. Néry, H. Lefebre and A. Fradet, Macromol. Chem. Phys., 2003, 204, 1775–1764 CrossRef.
- A. Gandini and M. N. Belgacem, Prog. Polym. Sci., 1997, 22, 1203–1379 CrossRef CAS.
- J. A. Moore and J. E. Kelly, Polymer, 1979, 20, 627–628 CrossRef CAS.
- J. A. Moore and J. E. Kelly, J. Polym. Sci., Polym. Chem. Ed., 1978, 16, 2407–2409 CrossRef CAS.
- J. A. Moore and J. E. Kelly, Macromolecules, 1978, 11, 568–573 CrossRef CAS.
- A. Gandini, A. J. D. Silvestre, C. P. Neto, A. F. Sousa and M. Gomes, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 295–298 CrossRef CAS.
- M. Gomes, A. Gandini, A. J. D. Silvestre and B. Reis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3759–3768 CrossRef CAS.
- S. K. Burgess, J. K. Leisen, B. E. Kraftschik, C. R. Mubarak, R. M. Kriegel and W. J. Koros, Macromolecules, 2014, 47, 1383–1391 CrossRef CAS.
- H. Hopff and A. Krieger, Makromol. Chem., 1961, 47, 93–113 CrossRef CAS.
- P. M. Heertjes and G. J. Kok, Delft Prog. Rep., Ser. A, 1974, 1, 59–63 CAS.
- O. Grosshardt, U. Fehrenbacher, K. Kowollik, B. Tubke, N. Dingenouts and M. Wilhelm, Chem. Ing. Tech., 2009, 81(11), 1829–1835 CrossRef.
- A. Mitiakoudis and A. Gandini, Macromolecules, 1991, 24(830), 835 Search PubMed.
- I.-C. Yeh, B. C. Rinderspacher, J. W. Andzelm, L. T. Cureton and J. La Scala, Polymer, 2014, 55, 166–174 CrossRef CAS PubMed.
- W. J. Li and S. X. Qiu, J. Heterocycl. Chem., 2010, 47, 1340–1343 CrossRef CAS.
- C. H. R. M. Wilsens, Y. S. Deshmukh, B. A. J. Noordover and S. Rastogi, Macromolecules, 2014, 47, 6196–6206 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4py01609b |
| This journal is © The Royal Society of Chemistry 2015 |