
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochromic and liquid crystalline polycarbonates based on telechelic oligothiophenes†
R.
Stalder
a,
A.
Mavrinskiy
b,
C.
Grand
c,
W.
Imaram
ad,
A.
Angerhofer
a,
W.
Pisula
b,
K.
Müllen
b and
J. R.
Reynolds
*c
aDepartment of Chemistry, Center for Macromolecular Science and Engineering, University of Florida, Gainesville, Florida 32611, USA
bMax Planck Institute for Polymer Research, Ackermannweg 10, D-55128 Mainz, Germany
cSchool of Chemistry and Biochemistry, School of Materials Science and Engineering, Center for Organic Photonics and Electronics, Georgia Institute of Technology, Atlanta, Georgia 30332, USA. E-mail: reynolds@chemistry.gatech.edu
dDepartment of Chemistry, Faculty of Science, Kasetsart University, Bangkok, Thailand
First published on 8th January 2015
Abstract
The triphosgene carbonate synthesis is adapted as a convenient route towards an alternating main-chain rod/coil polycarbonate based on a telechelic sexithiophene oligomer, yielding an electroactive polymer that is a vibrantly electrochromic material despite the conjugation break. This polymer displays morphological behaviour typical of a liquid-crystalline polymer, with stacking distances between chromophores suitable for potential charge transport applications.
The development of pi-conjugated and redox active materials for organic electronic applications relies on chemical structure modifications to tailor each system towards its targeted property, whether it be photovoltaics, field effect transistors (FETs), light emitting diodes or electrochromics. The distinction between discrete small molecules and polymeric compounds as active materials became evident early on: conjugated molecules offer a simple way to study molecular structure/electronic property relationships, and also provide straightforward purification and structural characterisation. They can adopt a wide range of morphologies thanks to their propensity to crystallise,1 but their mechanical integrity on the macroscale does not compete with that of polymeric systems.2 On the other hand, the purification of fully conjugated polymers is more tedious given the ubiquitous transition-metal catalysis that their synthesis requires, and batch-to-batch variations remain a drawback. Since the effective conjugation length of oligo-heterocycles is typically reached within six to ten aromatic rings,3 attempts have been made to link together well-defined, discrete length polyheterocycles of sufficient length for the property at hand via non-conjugated tethers, either with the chromophore pendant off of the polymer chain4 or as part of the main chain.5–12 This approach is attractive because it combines the predictability and purity of conjugated small molecules with the mechanical properties of their polymeric counterparts without significantly altering the electronic properties and perhaps accessing a greater degree of morphological control compared to fully conjugated polymer chains.13,14
Oligothienylene-based molecules have been used to study a wide array of structure–property relationships including the influence of functional groups and side chains on their solid-state morphology.15 Our group has published on a series of thienyl small molecules with various electron-rich and electron-deficient units using the donor–acceptor approach: we showcased systematic tuning of the position of the molecules’ HOMO and LUMO levels to achieve extended absorption across the visible spectrum into the near-IR while displaying vibrant colours.16 These discrete-length small molecules were also end-functionalised with hexanol chains to enable them to further react as diols. In particular, we have previously reported the synthesis of the telechelic sexithiophene T6-diol from its quaterthiophene core and the THP-protected 2-thienylhexanol end-capping unit.17 After removal of the THP group, the telechelic diol was further reacted with acrylate groups to afford crosslinked electroactive polymeric films for electrochromic applications.16,17 To synthesise a soluble and thus processible polymer, this T6-diol could alternatively be reacted with an appropriate linker to afford a main-chain alternating aliphatic-aromatic copolymer. Most main chain polymerisations of telechelic oligothiophenes reported in the literature have afforded polyesters,5–10 with a few other examples still relying on transition metal-catalysed reactions.11,12 While the chemical structure of the linker would determine the nature of the copolymer, it should not impair the electronic properties of the sexithiophene once incorporated in the main chain. It should also be of low molecular weight to avoid “diluting” the electroactive sexithiophene in an electro-optically inactive matrix. To this end, we describe herein the synthesis of the polycarbonate of T6-diol. We investigate the redox and spectroelectrochemical properties of the polymer, analyse its thermal behaviour and employ polarised light microscopy (POM) and two-dimensional wide-angle X-ray scattering (2D-WAXS) to evaluate its ability to yield ordered morphologies potentially suitable for charge transport applications.
We chose triphosgene as a comonomer because of its high reactivity and short length. As a solid, this trimeric form of phosgene can be easily handled for the precise stoichiometric balance required in polymerisation reactions, and it is essentially the shortest linker possible, leading to a polycarbonate (PC) when reacted with diols. Scheme 1 shows the polymerisation of T6-diol using triphosgene, resulting in the polycarbonate T6PC. The polymerisation conditions were a modification of that reported in the literature for the synthesis of polycarbonates from bisphenol A and triphosgene.18 The diol and triphosgene were dissolved in THF at room temperature under inert atmosphere, and stirred until complete dissolution of the reagents. Four equivalents of anhydrous pyridine were subsequently added to the reaction mixture, which started gelling after 1.5 hours of stirring, and was allowed to stir at room temperature for an additional 12 hours.
 | ||
| Scheme 1 Synthesis of the polycarbonate T6PC from the telechelic T6-diol using triphosgene a comonomer. | ||
The extent of polymerisation was easily monitored by 1H-NMR as the triplet at 3.65 ppm corresponding to the methylene next to the hydroxyl group in T6-diol moves downfield to 4.14 ppm after polymerisation, as a result of the withdrawing effect of the newly formed carbonate functionality. Unlike that of T6-diol, the IR spectrum of T6PC shows the carbon–oxygen single and double bond peaks centred at 1260 cm−1 and 1742 cm−1 respectively, corresponding to the carbonate functionality. The reaction mixture was purified by Soxhlet extraction using methanol, hexane and chloroform. The remainder of this study is performed on the polymer sample from the chloroform fraction. The polymer is soluble in THF, toluene and chlorinated solvents. A number average molecular weight (Mn) and polydispersity index (PDI) of 22.7 kDa (PDI = 2.07) was measured for T6PC by size exclusion chromatography in THF against polystyrene standards.
In solution, Fig. 1a displays one symmetrical absorption band centred at 424 nm for T6PC with an absorption onset at 500 nm, which superimposes with that of T6-diol. In thin films sprayed onto ITO-coated glass, the absorption of T6PC is red-shifted with the appearance of local maxima, as shown in Fig. 1b: the peak absorption is shifted to 481 nm, with a higher energy shoulder at 455 nm and a lower energy shoulder at 515 nm. The transition from solution to thin film is thus characterised by a higher vibronic resolution, which could be accounted for by a better ordering of the material in the solid state. With a solid state absorption onset at 556 nm, the optical energy gap of T6PC is calculated to be 2.23 eV, which is about 0.2 eV higher than the bandgap typically reported for the fully conjugated polythiophene P3HT.19
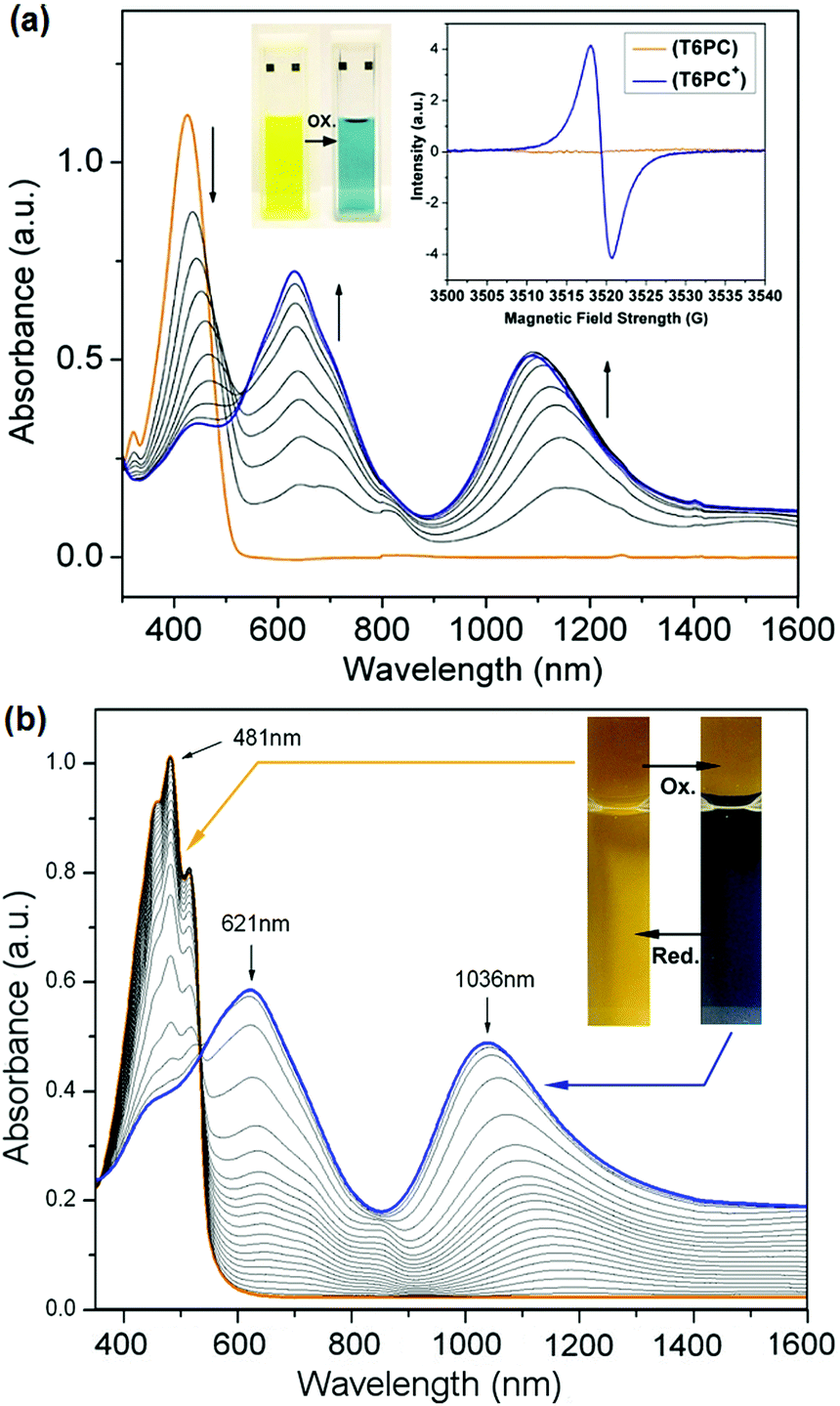 | ||
| Fig. 1 (a) Evolution of the UV-vis spectra of T6PC in DCM solution with increasing amounts of silver triflate, and EPR signals of the neutral (orange) and oxidised (blue) species (inset). Spectroelectrochemistry (b) for a spray-cast film of T6PC on ITO-coated glass, from 0.23 V to 0.54 V versus Fc/Fc+, 10 mV potential increments, recorded in LiBTI/PC solution; the inset shows pictures of the neutral (orange) and oxidised (blue) films. | ||
In order to test the solution redox properties of the polymer, the chemical oxidation of a dichloromethane solution of T6PC performed by the incremental addition of silver triflate as oxidant was monitored in parallel by UV-vis absorption and EPR spectroscopies, as shown in Fig. 1a. As the concentration of oxidant in solution is increased, the neutral state absorption band centred at 424 nm gradually decreases while two new bands centred at 630 and 1090 nm emerge. This translates into the yellow neutral solution switching to a blue colour as the oxidant concentration is increased. While the neutral solution is EPR silent as expected for a diamagnetic sample, the addition of silver triflate oxidant resulted in a broad EPR signal centred at g = 2.005 with a peak to peak width of 2.8 G. The emergence of the two absorption bands upon chemical doping coupled with the appearance of an EPR signal supports the formation of radical cations in solution, and the results are consistent with previous reports of oligothienylene solution doping.17 No additional band was observed when excess oxidant was added in solution.
To investigate the redox properties of T6PC in the solid state, the cyclic voltammograms of T6PC films on Pt button electrodes were recorded in 0.1 M lithium bis(trifluoromethylsulphonyl) imide (LiBTI) in acetonitrile under inert atmosphere. In the 0 V to 0.95 V window, the CV revealed two oxidation processes which were not stable to repeated scans as shown in the ESI.† The first oxidation wave was isolated by confining the CV potential window from 0 V to 0.40 V. With an anodic peak potential at 0.34 V and a cathodic peak potential at 0.21 V, this first oxidation process centred at a half-wave potential of 0.27 V was stable to at least 150 scans from 0 to 0.40 V. From the differential pulse voltammogram (DPV), an onset of oxidation was recorded at 0.20 V, corresponding to a HOMO energy level of 5.30 eV, which is within 0.1 eV of that reported in the literature for P3HT.19 With an optical energy gap of 2.23 eV, the LUMO is calculated to be at 3.07 eV.
Spectroelectrochemical measurements were conducted on thin films of T6PC sprayed onto ITO-coated glass slides in 0.1 M LiBTI in propylene carbonate (PC). A film of T6PC on ITO already subjected to at least 10 CV scans from −0.1 to 0.45 V was oxidised with potential steps from 0.23 V to 0.54 V in 10 mV increments, resulting in the spectra displayed in Fig. 1b. The neutral spectrum (orange line) featuring the peak absorption at 481 nm and some vibronic resolution as described previously gradually decreases as two new bands centred at 621 and 1036 nm appear. This is accompanied by a reversible colour change of the film from orange to blue. This dual-colour electrochromism can be visually appreciated on the oxidised film in the inset or Fig. 1b as both neutral and oxidised areas coexist on either side of the electrolyte solution meniscus. A well-defined isosbestic point can be seen at 533 nm. This is consistent with the polymer repeat unit structure, as the chromophore content in the backbone is monodisperse: spectral change arises from the removal of an electron from π-systems which are all of the same conjugation length. Switching speed experiments displayed in the ESI† show that the contrast (defined as the difference in % transmittance at 454 nm) recorded as a function of the potential step time (ν) decreases from 27% at ν = 10 s to 16% at ν = 0.5 s. The maximum contrast does not compare to that of the best electrochromic polymers because the chromophore design of T6PC does not allow it to become fully transmissive in the first oxidation process.20,21
Upon polymerisation, T6PC acquired film-forming characteristics with sufficient mechanical strength that free standing films were easily obtained by evaporation from a THF solution. A 7.0 cm × 1.5 cm free standing film of T6PC was peeled off of a rectangular Teflon mold, as displayed in Fig. 2a. At room temperature, the film does not stretch, but once heated to 65 °C, the film can be stretched up to approximately three times its initial length prior to mechanical failure. Since thermal treatments of polymeric materials can have a significant impact on the material's morphology, we recorded differential scanning calorimetry (DSC) thermograms of T6PC and compared it to that of T6-diol. Thermogravimetric analysis of the monomer and the polymer shows them to be thermally stable up to 422 °C and 370 °C, respectively (with a 5% weight loss as thermal stability threshold). The DSC thermogram of the T6-diol monomer (Fig. 2b, dashed line) exhibits a sharp melting transition that occurs at 104 °C during the second heating scan. Upon cooling, a crystallisation peak appears at 64 °C. Compared to T6-diol, the DSC thermogram of T6PC shows a broadened melting peak in the heating scan which is shifted to lower temperature at 84 °C, as well as a second peak at 52 °C. Consistent with the macromolecular nature of T6PC, a glass transition temperature (Tg) is observed at 18 °C, but no crystallisation peak was apparent in the cooling scan.
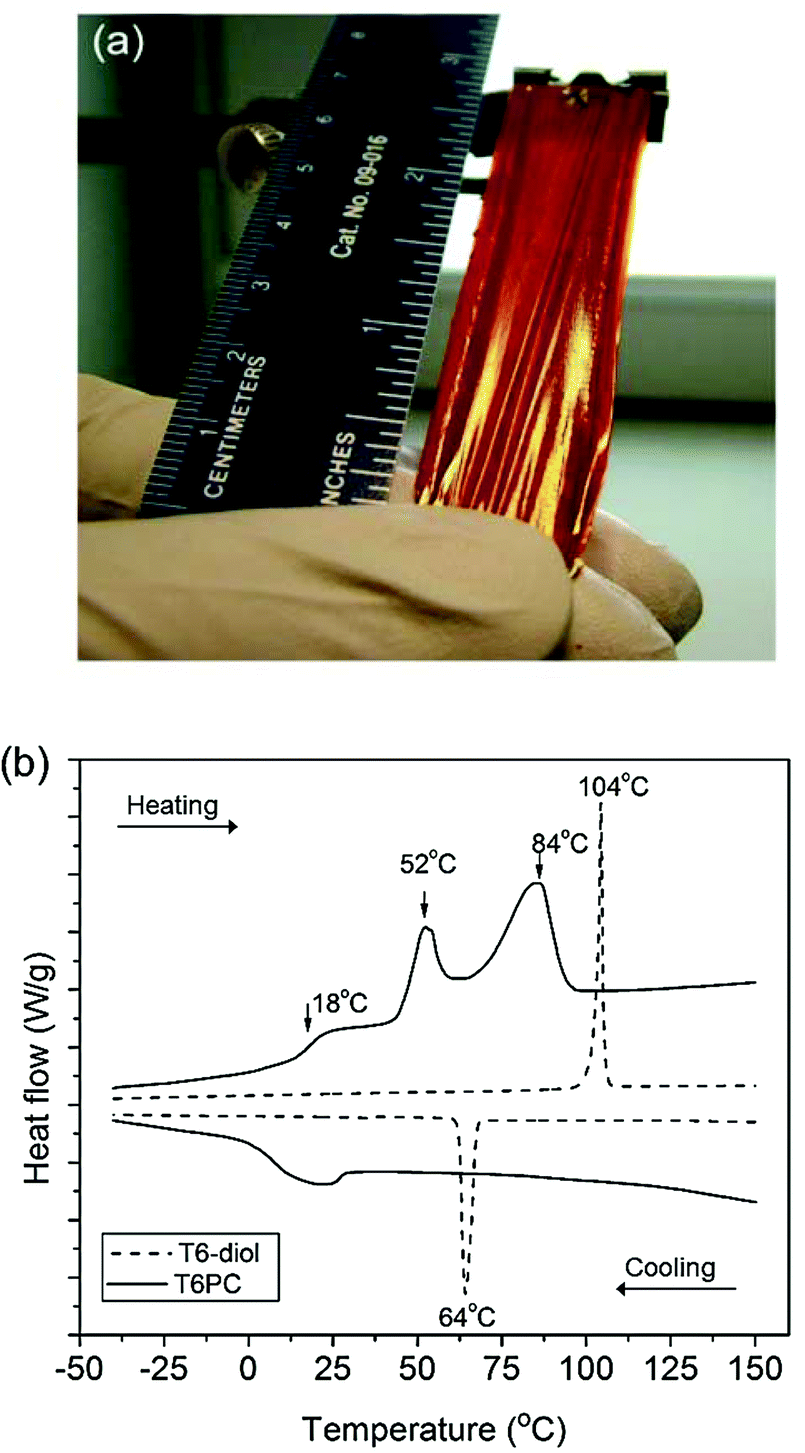 | ||
| Fig. 2 Picture (a) of a 7.0 cm × 1.5 cm free standing film of T6PC and DSC thermograms (b) of T6-diol (dashed line) and T6PC (solid line) recorded at a 10 °C min−1 heating rate. | ||
Annealing experiments were performed at various temperatures between 18 °C and 84 °C to induce phase transformations, as illustrated by Fig. 2b. After erasing the thermal history of T6PC by heating the sample above its melting temperature, the polymer was cooled back to and kept at room temperature (23 °C) for increasingly longer periods of time before recording a second heating scan. We observed that the two endothermic transition peaks intensify the longer the sample is kept at room temperature, as shown in Fig. S8.† These results suggest that annealing T6PC above its Tg offers enough energy for the polymer to undergo reorganisation, which equilibrates at room temperature. This behaviour has been previously reported on samples of poly(3-decylthiophene).22
Since the DSC of T6PC revealed two phase transitions taking place at two different temperatures, we employed polarised optical microscopy coupled to a heating stage to investigate microscopic morphology changes upon thermal treatment and identify phase transitions. Fig. 3 gives the POM images of T6-diol and T6PC. When a sample of T6-diol is heated above its melting point, and then allowed to cool back to its crystallisation temperature, a well-ordered phase with strong birefringence under polarised light emerges (Fig. 3a). The observed Maltese cross patterns are typical of a spherulitic arrangement of the crystallised domains. Specifically, the spherulites could result from the growth of molecular stacks along the π-stacking direction, out of each nucleation site and in an edge-on fashion with respect to the substrate. A radial distribution of the molecular stacks around each nucleation site would explain the observed Maltese cross patterns.23 Spherulites were also found under similar conditions for the self-assembly of phenylene-thienylene oligomers bearing linear alkyl and alcohol terminated chains, as in the structure of T6-diol.24 The molecular arrangement could be explained in terms of amphiphilicity, as a result of mutual exclusion of the alkyl and alcohol groups, along with the sexithiophenes acting as mesogenic cores given their propensity to form π-stacks.
 | ||
| Fig. 3 Polarised light optical microscope images of T6-diol (a) and T6PC (b)/(c), at crossed polariser/analyser. | ||
Upon polymerisation of the diol into T6PC, the oligomers inevitably lose some degree of freedom as a consequence of the covalently formed carbonate functionalities. The polarised-light pattern changes accordingly to a less ordered structure, as shown in Fig. 3b and in higher resolution in Fig. 3c. The birefringence features are on the order of 1 μm.
Optical micrographs showing a micron-scale texture identical to that in Fig. 3c have been described previously in films of random copolyesters of ethylene terephthalate and hydrobenzoic acid.25 Other examples of such texture can be found in reports of liquid crystalline poly(phenylene ethynylene)26 and alternating copolymers containing main-chain quaterphenyl segments.27 These studies describe the microstructures as a Schlieren-type fine nematic structure. For T6PC, the texture slowly appears after resting several hours at room temperature following cooling at a rate of 10 °C min−1 from the isotropic melt above 85 °C, as recorded in the DSC thermogram. No change was observed as the sample is heated through the first transition temperature recorded at 52 °C, until the second transition temperature in the 80 to 85 °C range, after which the sample became optically isotropic.
The slow appearance of the texture at room temperature is consistent with the lack of a clear crystallisation peak in the DSC cooling scan recorded at the same rate. Overall, these observations support the behaviour of T6PC as a thermotropic liquid-crystalline, electroactive polycarbonate.
Quantitative insight into how well T6PC organises when the polymer chains are aligned is provided by 2D-WAXS measurements performed on extruded filaments of the polymer. The sample was prepared as a thin filament of 0.7 mm diameter by heating to 65 °C for extrusion where it becomes plastically deformable. The 2D-WAXS pattern shown in Fig. 4a displays pronounced wide-angle reflections that correspond to a π-stacking distance of 0.37 nm for neighbouring chromophores. Located on the equatorial axis in the wide-angle region, this reflection indicates that the layers of π-stacked chains are aligned along the extrusion direction as depicted in Fig. 4b. Additionally, several pronounced reflections on the equatorial axis appear related to the d-spacings of 1.57 nm, 0.78 nm, 0.52 nm, which can be attributed to the interlayer distance of π-stacked chains. At 75 °C the pattern remains unchanged indicating the same organisation also in the higher temperature phase.
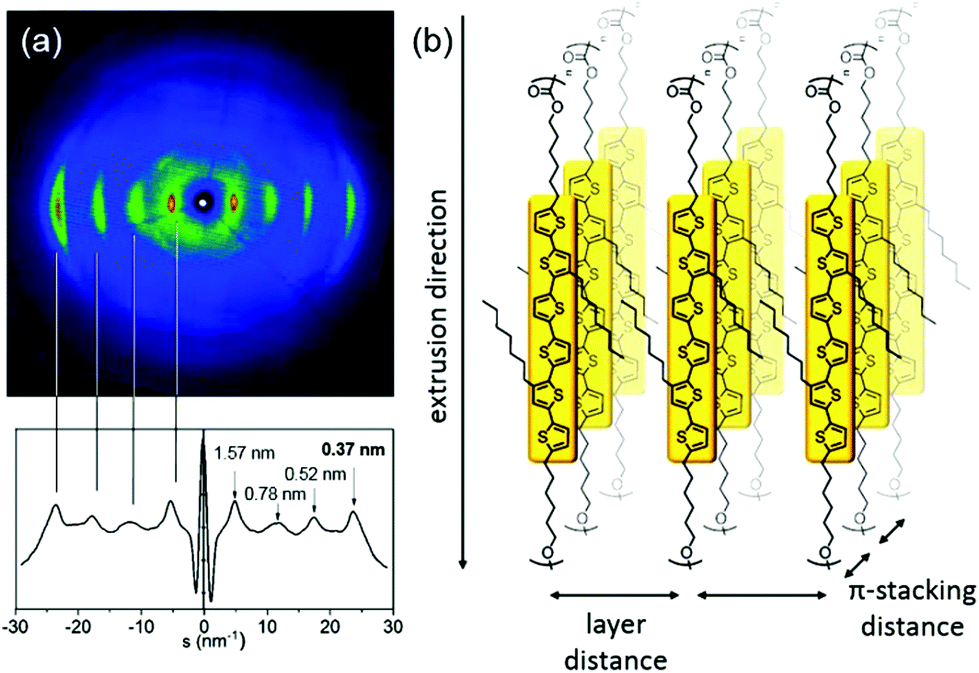 | ||
| Fig. 4 2D-WAXS pattern (a) of T6PC at 30 °C (above) and scattering intensity distribution as a function of the scattering vector (below). (b) Model for the aligned polymer chains in the fibre sample. | ||
Since some of the best polymeric transistors reported in the literature are based on materials for which the π-stacking distances are in the range of 0.36–0.39 nm,28,29 these WAXS results combined with the ability of the polymer to retain locally ordered domains—as evidenced by the POM images—suggest that this main-chain aromatic/aliphatic polycarbonate design could be promising for charge transport applications. To test this hypothesis, FETs using T6PC as active layer were fabricated. Thin films were spin-coated from solution onto a dielectric surface in a bottom contact configuration, and thermally annealed in an attempt to enhance polymer chain ordering within the films. Mechanical ordering of the polymer chains was also investigated by stretching or calendering. Unfortunately, no significant p-type mobility and transistor behaviour were recorded in each case. We investigated the possibility of hole mobility in the direction orthogonal to the casting substrate by fabricating diode devices using spun cast T6PC as the active layer and molybdenum trioxide (MoO3) and gold as hole injection and hole collection electrodes respectively. When sufficient voltage is applied across the active layer, charge carrier transport follows space-charge-limited current (SCLC) model, and the current density-voltage curves (see ESI†) can be fitted in the SCLC region to calculate hole mobility. In thermally annealed active layers, the hole mobility is measured to be 4 × 10−5 cm2 V−1 s−1 across a 80 nm thick T6PC layer, indicating that significant charge carrier transport can occur in the direction perpendicular to the substrate. Previous work by Fréchet and coworkers measured SCLC hole mobility of 3 kDa and 31 kDa regioregular poly(3-hexylthiophene) (P3HT) to be 1 × 10−5 cm2 V−1 s−1 and 3 × 10−4 cm2 V−1 s−1 respectively using a different SCLC device architecture.30 Considering that the contacts for hole injections (MoO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31 in this study and PEDOT:PSS in the study by Goh et al.30) are Ohmic for both studies, the SCLC hole mobility measured for T6PC is comparable to that of low molecular weight P3HT.
31 in this study and PEDOT:PSS in the study by Goh et al.30) are Ohmic for both studies, the SCLC hole mobility measured for T6PC is comparable to that of low molecular weight P3HT.
Conclusions
The polycondensation of T6-diol into its polycarbonate T6PC afforded a polymer which by design contains alternating aliphatic coils and aromatic sexithiophene rods, is fully soluble in a variety of solvents and displays excellent film-forming properties. The results from solution oxidation EPR and UV-vis spectroscopies correlate with thin film spectroelectrochemistry and are consistent with a first stable and reversible oxidation process. The latter corresponds to the formation of energetically discrete radical cations in films of T6PC, characterised by dual-colour electrochromism. Unfortunately, accessing the dicationic species was only possible at higher potentials at which film degradation occurred, but these results suggest that despite the conjugation break, the polymer remains electrochemically active while displaying the mechanical properties typical of polymeric materials. Further chromophore designs could enable access to stable dicationic species and extend the electrochromic performance of such materials. The morphological characteristics induced by the covalent linking of the telechelic oligomers are typical of a nematic structure displaying micron-scale ordering in thin films. Extruded polymer fibres analysed by 2D-WAXS show that chromophores π-stack with a distance of 0.37 nm, which is within the range of high-mobility materials reported in the literature. We thus foresee the chemical strategy reported here to be applicable to a variety of different chromophores, as a new efficient means of combining the desirable characteristics of discrete molecules (vibrant colours, predictable electronic properties, propensity to crystallise, etc…) with the mechanical properties of polymeric materials.Acknowledgements
We acknowledge the Air Force Office of Scientific Research (FA9550-14-1-0271) for funding of this work.Notes and references
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2009, 93, 394–412 CrossRef CAS PubMed.
- H. Meier, U. Stalmach and H. Kolshorn, Acta Polym., 1997, 48, 379–384 CrossRef CAS.
- B. S. Aitken, P. M. Wieruszewski, K. R. Graham, J. R. Reynolds and K. B. Wagener, Macromolecules, 2012, 45, 705–712 CrossRef CAS.
- A. Donat-Bouillud, L. Mazerolle, P. Gagnon, L. Goldenberg, M. C. Petty and M. Leclerc, Chem. Mater., 1997, 4756, 2815–2821 CrossRef.
- N. DiCesare, M. Belletete, A. Donat-Bouillud, M. Leclerc and G. Durocher, Macromolecules, 1998, 9297, 6289–6296 CrossRef.
- N. K. E. Guimard, J. L. Sessler and C. E. Schmidt, Macromolecules, 2009, 42, 502–511 CrossRef CAS PubMed.
- Y. Hong and L. L. Miller, Chem. Mater., 1995, 7, 1999–2000 CrossRef CAS.
- Y. Kunugi, L. L. Miller, T. Maki and A. Canavesi, Chem. Mater., 1999, 9, 1996–1997 Search PubMed.
- S. Wang and F. Brisse, Macromolecules, 1998, 9297, 2265–2277 CrossRef.
- M. T. Hargadon, E. A. Davey, T. B. Mcintyre, D. Gnanamgari, C. M. Wynne, R. C. Swift, J. R. Zimbalist, B. L. Fredericks, A. J. Nicastro and F. E. Goodson, Macromolecules, 2008, 41, 741–750 CrossRef CAS.
- C.-G. Wu, C.-Y. Lai and N.-L. Hsiao, Eur. Polym. J., 2009, 45, 879–887 CrossRef CAS PubMed.
- J. K. Kallitsis and A. K. Andreopoulou, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 2485–2491 CrossRef CAS.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- A. Mishra, C.-Q. Ma and P. Bäuerle, Chem. Rev., 2009, 109, 1141–1276 CrossRef CAS PubMed.
- S. Ellinger, K. R. Graham, P. Shi, R. T. Farley, T. T. Steckler, R. N. Brookins, P. Taranekar, J. Mei, L. A. Padilha, T. R. Ensley, H. Hu, S. Webster, D. J. Hagan, E. W. Van Stryland, K. S. Schanze and J. R. Reynolds, Chem. Mater., 2011, 23, 3805–3817 CrossRef CAS.
- C. B. Nielsen, A. Angerhofer, K. A. Abboud and J. R. Reynolds, J. Am. Chem. Soc., 2008, 130, 9734–9746 CrossRef CAS PubMed.
- D. A. Boyles, T. S. Filipova, J. T. Bendler, G. Longbrake and J. Reams, Macromolecules, 2005, 38, 3622–3629 CrossRef CAS.
- B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS PubMed.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS PubMed.
- C. M. Amb, A. L. Dyer and J. R. Reynolds, Chem. Mater., 2011, 23, 397–415 CrossRef CAS.
- S. V. Meille, V. Romita, T. Caronna, A. J. Lovinger, M. Catellani and L. Belobrzeckaja, Macromolecules, 1997, 9297, 7898–7905 CrossRef.
- M. Kastler, W. Pisula, F. Laquai, A. Kumar, R. J. Davies, S. Baluschev, M.-C. Garcia-Gutiérrez, D. Wasserfallen, H.-J. Butt, C. Riekel, G. Wegner and K. Müllen, Adv. Mater., 2006, 18, 2255–2259 CrossRef CAS.
- A. Mavrinskiy, C. B. Nielsen, J. R. Reynolds, K. Müllen and W. Pisula, Chem. Mater., 2011, 23, 1939–1945 CrossRef CAS.
- A. H. Windle, C. Viney, R. Golombok, A. M. Donald and G. R. Mitchell, Faraday Discuss. Chem. Soc., 1985, 79, 55–72 RSC.
- L. Kloppenburg, D. Jones, J. B. Claridge, H.-C. zur Loye and U. H. F. Bunz, Macromolecules, 1999, 32, 4460–4463 CrossRef CAS.
- M. A. Keegstra, V. Cimrová, D. Neher and U. Scherf, Macromol. Chem. Phys., 1996, 197, 2511–2519 CrossRef CAS.
- P. M. Beaujuge, W. Pisula, H. N. Tsao, S. Ellinger, K. Müllen and J. R. Reynolds, J. Am. Chem. Soc., 2009, 131, 7514–7515 CrossRef CAS PubMed.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- C. Goh, R. J. Kline, M. D. McGehee, E. N. Kadnikova and J. M. J. Fréchet, Appl. Phys. Lett., 2005, 86, 122110 CrossRef PubMed.
- H. T. Nicolai, G. A. H. Wetzelaer, M. Kuik, A. J. Kronemeijer, B. de Boer and P. W. M. Blom, Appl. Phys. Lett., 2010, 96, 172107 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, structural and polymer characterisations, thermal analyses, voltammetry and switching speed results. See DOI: 10.1039/c4py01551g |
| This journal is © The Royal Society of Chemistry 2015 |