
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards being genuinely smart: ‘isothermally-responsive’ polymers as versatile, programmable scaffolds for biologically-adaptable materials
Daniel J.
Phillips
a and
Matthew I.
Gibson
 *ab
*ab
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: m.i.gibson@warwick.ac.uk
bWarwick Medical School, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK
First published on 5th December 2014
Abstract
Responsive polymers have found diverse application across polymer, biomaterials, medical, sensing and engineering fields. Despite many years of study, this has focussed mainly on those polymers which undergo thermally-induced changes – either a lower or upper critical solution temperature. To rival the adaptability of Nature's macromolecules, polymers must respond in a ‘smarter’ way to other triggers such as enzymes, biochemical gradients, ion concentration or metabolites, to name a few. Here we review the concept of ‘isothermal’ responses where core thermoresponsive polymers are chemically engineered such that they undergo their useful response (such as coil-globule transition, cell uptake or cargo release) but at constant temperature. This is achieved by consideration of their phase diagram where solubility can be changed by small structural changes to the end-group, side-chain/substituents or through main chain modification/binding. The current state-of-the-art is summarised here.
 Daniel Phillips | Daniel Phillips graduated from the University of Warwick (MChem) in July 2011, which also included a 12 month period of Industrial Training in the Analytical Science department of MSD looking at the development of novel biorelevant dissolution technologies. He is currently pursuing Doctoral studies under the supervision of Dr Matthew I. Gibson with research largely focussed on the design, synthesis and characterisation of responsive polymer-based systems for therapeutic and diagnostic applications. |
 Matthew I. Gibson | Dr Matthew I. Gibson graduated from the University of Durham (MChem) in 2003, followed by a PhD at the same institute under the supervision of Prof. Neil Cameron (2007). Following a postdoctoral period with Prof. Harm-Anton Klok, Matthew was appointed as an Independent Research Fellow in the Department of Chemistry at the University of Warwick and then as an Assistant Professor in 2012. Matthew leads a diverse research team, addressing challenges in healthcare using polymer science tools. In particular, the design of biologically adaptable polymers, antifreeze glycoprotein mimetic polymers and the study of glycopolymers, especially related to infection and diagnostics. In recognition of his work, Matthew was received several prizes including the 2012 MacroGroup UK Young Researchers medal and more recently, a prestigious ERC starting grant. |
1. Introduction: increasing the complexity of thermally-responsive systems
Research into the fundamental understanding and applied nature of stimuli-responsive or “smart” materials has exploded in recent years. The ability to programme and manipulate small structural changes at the molecular level such that a dramatically enhanced macroscopic response can be achieved has placed the exciting field of responsive materials at the forefront of many areas of modern research. Of the variety of stimuli which can be applied, including pH, redox, light, magnetic fields, electric fields, mechanical stress, enzymes and metal ions, temperature remains a particularly popular option.1–5 So-called thermo-responsive polymers are typically characterised by a change in aqueous solubility; either the precipitation of a polymer solution at a lower critical solution temperature (LCST) or the solubilisation of a polymer precipitate at an upper critical solution temperature (UCST).Arguably the true value of a responsive system, particularly as our understanding of polymer synthesis develops, lies in the art of combining multiple stimuli to prepare increasingly complex materials. This is particularly relevant for biological applications given the host of microenvironments such as pH, redox potentials, metal ions and salts that exist within living systems. It thus follows that a macroscopic response may be achieved in parallel with, or sequentially following a second stimulus.6 To exemplify a thermo-responsive system, the solubility switch associated with the LCST may be controlled with an additional stimulus without the need for a temperature gradient, i.e. isothermally. Employing this, a thermally-responsive system can be used as a generic “scaffold” upon which the action of other stimuli can be targeted whilst maintaining the macroscopic thermo-responsive effect. This approach offers the chance for small environmental changes to have a profound effect on a polymer by only affecting small parts of its structure.
Considering the above, this review seeks to firstly provide a brief overview of key concepts and applications related to thermo-responsive systems. Secondly, a discussion into methods in which thermo-responsive “scaffolds” can be functionalised such that a host of stimuli can be combined to achieve more than simple temperature-driven precipitation is provided. Strategies including changes in end-group, side chain/substituents and main-chain functional units will be discussed.
(i) Thermally-responsive polymers: key concepts
The LCST phenomenon is best understood by considering how the “favourability” of mixing changes with variations in temperature, as derived from the Helmholtz equation of free energy (eqn (1)).7 Binary mixing of two species A and B (ΔFmix) is dependent on both entropic and enthalpic phenomena, where φ is the volume fraction of A, Nx is the number of lattice sites occupied by species A or B and χ is the Flory interaction parameter.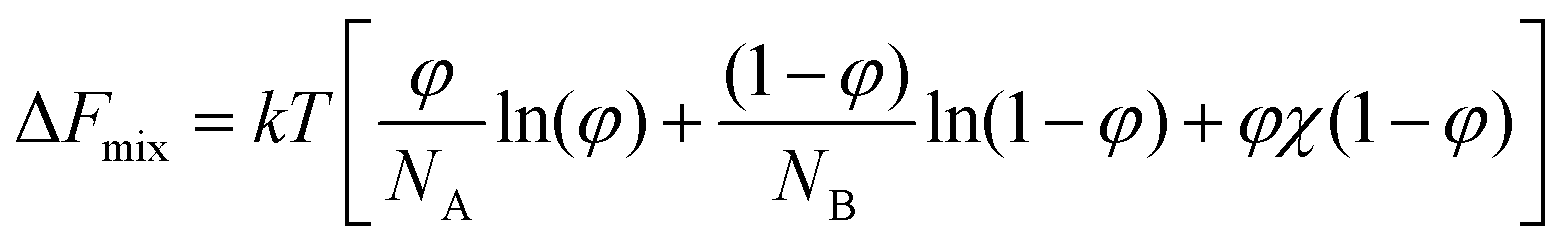 | (1) |
Whilst entropy always acts in favour of mixing, the enthalpics of mixing depend heavily on the Flory–Huggins interaction parameter, itself comprised of an entropic term (A) and a temperature-dependent enthalpic term (B) (eqn (2)). Generally, when B is negative χ < 0, mixing is favoured; when B is positive, χ > 0, and therefore mixing is disfavoured.
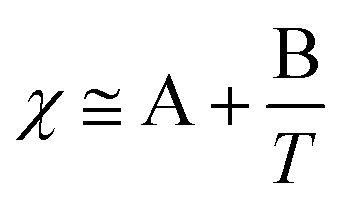 | (2) |
When B has a negative value χ can be rendered more positive by increasing the temperature and hence negating the ‘B’ term, giving rise to the LCST behaviour. In practice, polymers exhibiting LCST-type behaviour exist in flexible, extended coils when dissolved in aqueous solution due to extensive hydrogen bonding interactions with the surrounding water molecules playing a dominating role. As the temperature is then increased, this bonding is disrupted allowing intra- and inter-molecular hydrogen bonding between polymer molecules, together with hydrophobic interactions to become significant. Consequently, the polymer chains hydrophobically collapse and aggregate in a globule conformation (Scheme 1).8
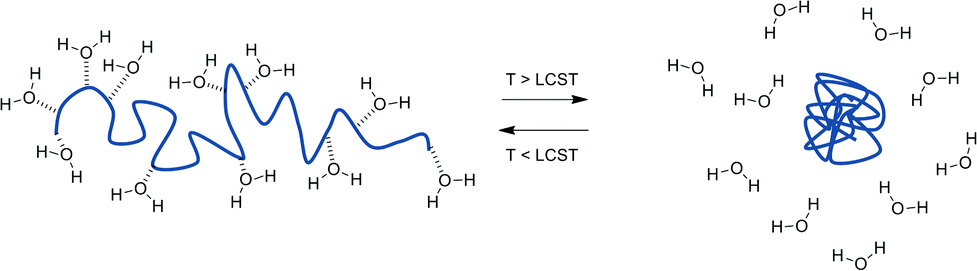 | ||
| Scheme 1 Schematic demonstrating the change in polymer conformation (coil – globule) observed when a polymer solution is heated through its LCST. | ||
It should also be highlighted that transition temperatures quoted in the literature are not always the absolute LCST. This single temperature is represented as the lowest point on a temperature vs. composition phase diagram (Fig. 1). In the absence of such a diagram, the term “cloud point” is a more suitable term, describing the temperature at which a solution transitions from transparent to opaque at a given solution composition (or concentration).9 In the interests of consistency with the original published works discussed herein, the two terms will (incorrectly) be used interchangeably throughout this review as it is not possible to distinguish between the two based within all primary literature.
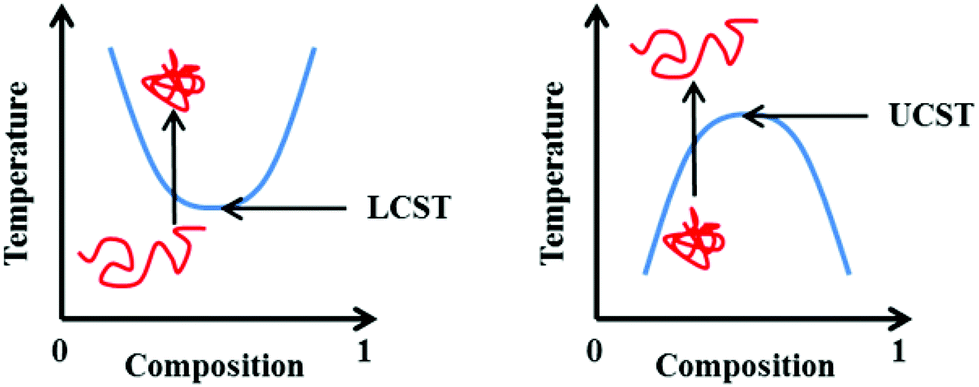 | ||
| Fig. 1 Representation of the phase transition associated with the LCST (left) and UCST (right). Blue line represents the phase separation boundary at which a cloud point is observed. | ||
(ii) Thermally-responsive polymers: applications
One of the earliest studies reporting a thermal transition was discussed by Klotz and co-workers on a poly(N-isopropylacrylamide), pNIPAM, system.10 They measured the kinetics of hydrogen-deuterium exchange in D2O solutions and observed precipitation when the temperature of a 2% aqueous polymer solution (molecular weight ≈ 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) reached 31 °C. Although the most commonly used thermally responsive polymer is still pNIPAM,11 a wide range of other polymers have been introduced including poly(N-substituted (meth)acrylamide)s, poly(N-vinylalkylamide)s, poly(lactam)s, poly(pyrrolidone)s, poly(alkoxide)s, and poly(2-alkyl-2-oxazoline)s and poly(oligo ethylene glycol) methyl ether (meth)acrylates, pOEG(M)A.12 The LCST response can be translated onto alternative structures such as to install polymer and inorganic nanoparticles with a responsive corona.13–15 Flat substrates have also been functionalised such that the resulting polymer brushes can exist in extended or collapsed states depending on the temperature of the system16,17 and desired properties have been convened on polymer-protein conjugates.18,19
000 g mol−1) reached 31 °C. Although the most commonly used thermally responsive polymer is still pNIPAM,11 a wide range of other polymers have been introduced including poly(N-substituted (meth)acrylamide)s, poly(N-vinylalkylamide)s, poly(lactam)s, poly(pyrrolidone)s, poly(alkoxide)s, and poly(2-alkyl-2-oxazoline)s and poly(oligo ethylene glycol) methyl ether (meth)acrylates, pOEG(M)A.12 The LCST response can be translated onto alternative structures such as to install polymer and inorganic nanoparticles with a responsive corona.13–15 Flat substrates have also been functionalised such that the resulting polymer brushes can exist in extended or collapsed states depending on the temperature of the system16,17 and desired properties have been convened on polymer-protein conjugates.18,19
The concept of an LCST transition holds a myriad of attractive applications such as to control cell culture and adhesion,20,21 to influence catalytic activity,22 and as a purification tool.23,24 In a biological context, the hydrophilic–hydrophobic switch can be used to enhance a polymer's interaction with biological membranes. In this manner, the cell uptake of thermo-responsive architectures can be enhanced by employing polymers in their hydrophobic, collapsed state.25 For instance, Saaka et al. have demonstrated that when held above their LCST, pOEGMAs are sufficiently lipophilic to insert into, or adhere to, lipid bilayers.26 Edwards et al. have demonstrated that gold nanoparticles capped with thermo responsive pOEGMAs can cross reversibly between a water–oil interface which was used as a basic mimic of a biological surface.27 Gold nanoparticles functionalised with a pNIPAM-co-acrylamide sample possessing an LCST at 37 °C have also been used by Alexander and co-workers to drive uptake unto human breast adenocarcinoma MCF7 cells. When heated at 40 °C (above LCST), an 80-fold greater uptake was observed compared to when the same cells were heated at 34 °C (below LCST).28 This group has also prepared polymer particles comprising a paclitaxel-containing, biodegradable poly(lactide-co-glycolide) core and a thermo-responsive, PEG-based shell. A significantly enhanced uptake into MCF7 cells and paclitaxel-based cytotoxicity was observed when incubated above the particle thermal transition temperature (Fig. 2).29
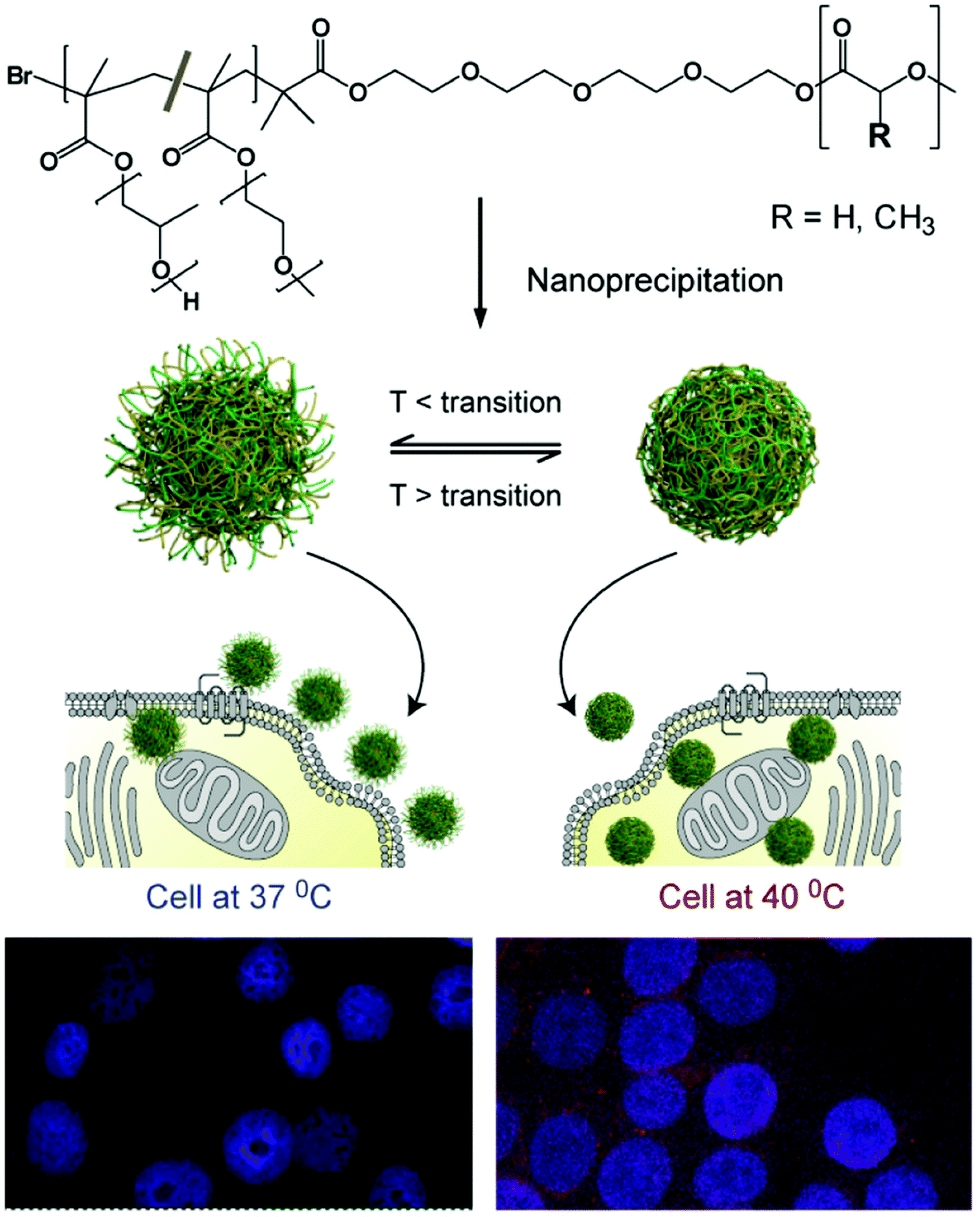 | ||
| Fig. 2 Formulation of thermoresponsive nanoparticles and the proposed enhancement in cellular uptake due to change in surface corona with thermal response: an increased signal from nanoparticle-encapsulated fluorescent dye is indicative of increased particle uptake above their thermal transition temperature in cells at 40 °C (bottom right) compared to 37 °C (bottom left). Reproduced from ref. 29. | ||
Okano and co-workers have prepared andriamycin-loaded polymeric micelles comprising pNIPAM and the hydrophobic poly(butylmethacrylate). Higher cytotoxicity towards bovine aortic endothelial cells was observed above the micelle LCST with the temperature-induced hydrophobicity triggering drug release and/or enhanced adsorption to cells mediated by hydrophobic interactions.25 The same group has further explored the potential for thermo-responsive micelles prepared from fluorescently tagged p(NIPAM-co-dimethylacrylamide)-b-p(D,L-lactide) as drug delivery vehicles. The polymers were shown to form large aggregates when held above the LCST which enhanced intracellular uptake into bovine carotid endothelial cells. This was proposed to be either the result of unique hydrophobic interactions between the cell membranes and hydrophobic micelle cores promoted by a collapse of the thermo-responsive corona, or, due to the pNIPAM corona which can regulate micelle adhesion to cell surfaces and sustain intracellular uptake. Interestingly, the linear polymers devoid of a p(D,L-lactide) block (and hence unable to form micelles) exhibited no additional uptake when held above the LCST.30,31 Chilkoti and co-workers have however demonstrated a greater accumulation of pNIPAM-co-acrylamide inside solid tumours when heated at above its cloud point at 42 °C, albeit not to the extent of a thermally responsive elastin-like polypeptide where a two-fold increase in accumulation was observed.32
2. Modulating thermal transitions through structure manipulation: the “isothermal response”
The application of a thermal-stimulus has found multiple uses, especially for triggered cell uptake/release of cargo. For instance, many cancers are characterised by mild hyperthermia (1–2 °C above healthy tissues) meaning materials can be tuned to enhance selective delivery to tumours.32–34 Induced hyperthermia or thermotherapy using an external heat generator has therefore been applied previously as a cancer treatment.35 However, a temperature response is not always relevant, nor practically useful. Indeed, the ability to respond to biochemical gradients or triggers without the need for external intervention is arguably both more desirable and also ‘smarter’. As our material understanding improves, an obvious extension is to therefore prepare substances capable of responding to multiple stimuli, either in a parallel nature, or via serial interplay whereby the impact of one response directly affects another.6 It follows that for thermally-responsive systems the hydrophilic–hydrophobic balance could be manipulated isothermally, using changes in the local environment as the primary trigger. For example, where in vivo applications are to be considered, gradients in ion concentration, pH, redox strength or the presence of enzymes are all indicative of different disease states.36–42 These may therefore be targeted to increase compound specificity, minimise side-effects and control therapeutic release.43–45A schematic representation of this hypothesis is shown in Fig. 3. Rather than changing the temperature (i.e. by holding the system at a fixed T1), an additional stimulus can be applied to shift the polymers’ phase separation boundary. This can reduce/increase the apparent LCST such that the polymer now sits in a different region and hence undergoes its coil-globule (Fig. 3A) or globule-coil (Fig. 3B) transition.
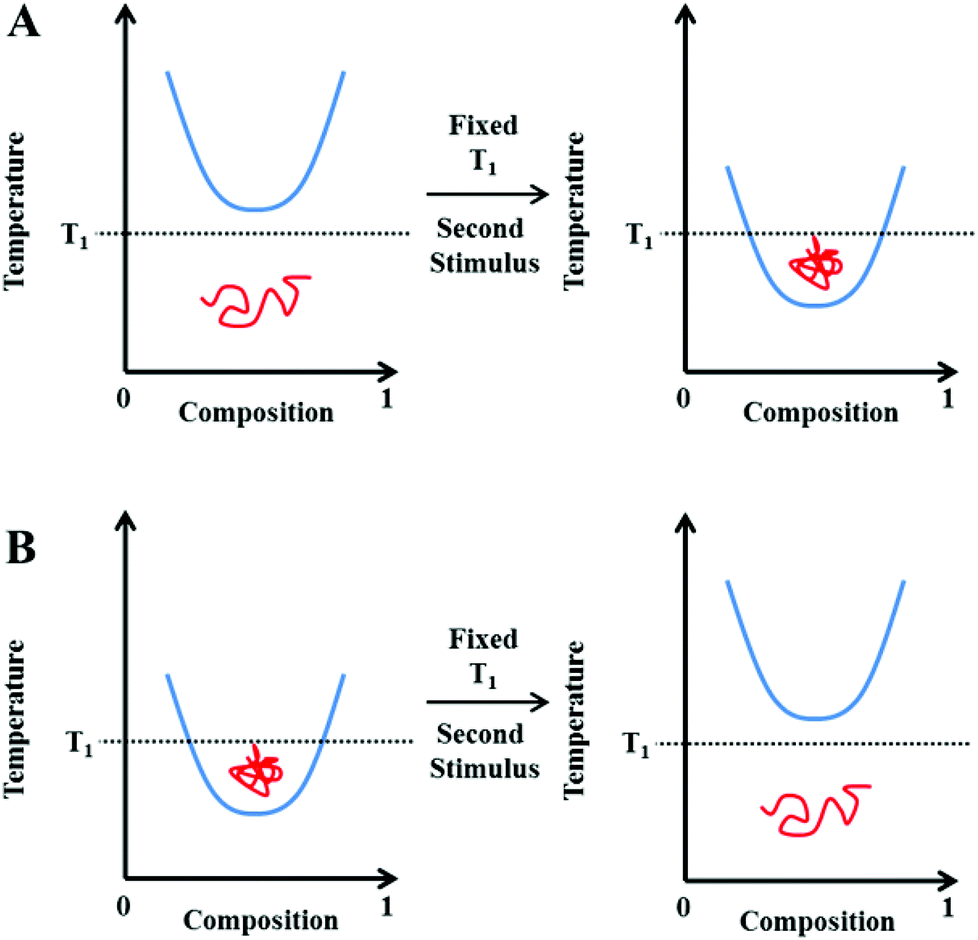 | ||
| Fig. 3 Representation of the phase diagrams associated for a polymer exhibiting an isothermal LCST transition at a fixed temperature, T1. Blue line represents the phase separation boundary at which a cloud point is observed. Red line represents a polymer coil (open chain) or globule (compact chain). | ||
To respond to such environments, a carefully designed synthetic structure is required. Analysis of a typical polymer suggests four main areas which could be manipulated to alter the overall hydrophilic–hydrophobic balance of the compound and hence the transition temperature of an aqueous polymer solution. These are the (i) backbone; (ii) end-group(s); (iii) side-chain/substituents or (iv) local aqueous environment (i.e. presence of additives) (Fig. 4).
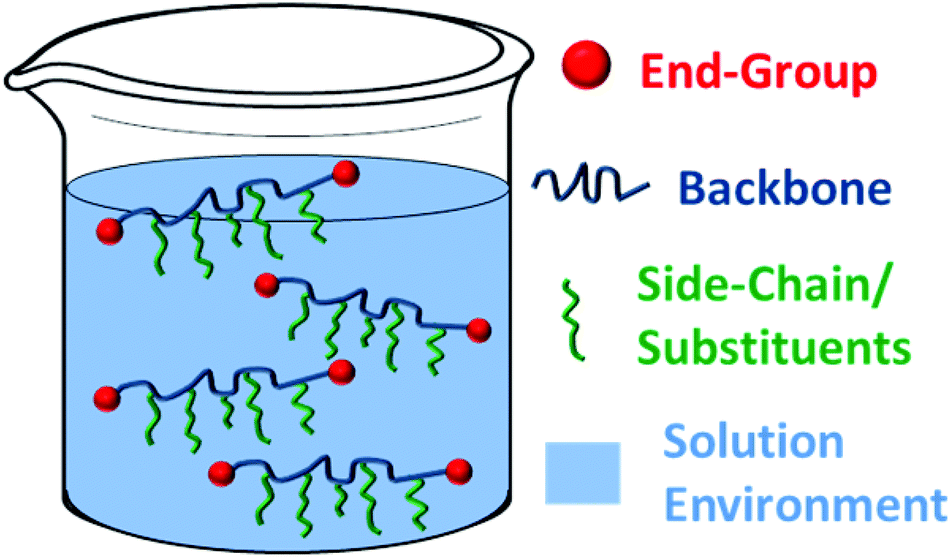 | ||
| Fig. 4 Sections of a polymer structure and its solution which may be modified to tune the macroscopic behaviour. | ||
The remainder of this review therefore seeks to highlight some of the published reports describing the use of multiple stimuli to achieve shifts in polymer LCST by influencing these areas of a polymer chain. It should be noted that some cases merely demonstrate the ability to change the system LCST; not all utilise it to explicitly trigger an isothermal property change.
(i) Triggering an “isothermal” response via backbone modification
To introduce a responsive element to polymer backbones, functionality beyond the carbon–carbon backbones intrinsic to many polymerisation processes is required. Poly(esters) or poly(amides), prepared by ring-opening polymerisation techniques, offer a route towards this where degradation by hydrolytic and/or enzymatic means is possible. Moreover, the development of further functionalised backbones upon which further chemistry can be performed remains an important area of study.46–51 Poly(sulfides) provide an option for situations in which an oxidative response, such as those found in wound sites, is required52,53 and poly(disulfides) have potential application given the highly reducing environment found within cells compared to the systemic circulation.54–56 Considering the latter, Gibson and coworkers have developed a bio-reducible system based on homopolymers of pNIPAM containing a pyridyl disulfide moiety at the α-terminus. Subsequent aminolysis of the ω-terminal dithioester/trithiocarbonate resulted in an in situ polycondensation-type polymerisation, driven by the release of pyridine thione, to produce a poly(disulfide).57 These disulfide linkages were selectively degradable in the presence of cellular levels (mM) of glutathione (the main in vivo anti-oxidant) whilst remaining stable in the presence of extracellular (μM) levels. Moreover, given the inversely proportional relationship that exists between pNIPAM molecular weight and LCST, degradation of the large disulfide-linked chains, to smaller chains was accompanied by an increase in cloud point providing a route towards isothermal LCST behaviour (Fig. 5).58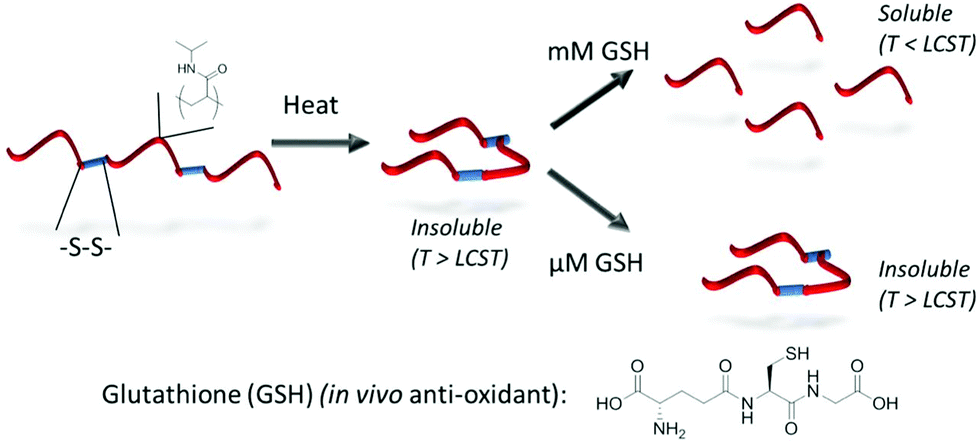 | ||
| Fig. 5 Isothermal transitions triggered by polymer backbone reduction: in vivo glutathione concentration gradient can be used to trigger selective degradation of disulfide-linked pNIPAM species. Reproduced from ref. 57. | ||
There are few other reports describing backbone modulation to modulate the LCST. One report which has utilised a functional polymer chain, namely poly(ethylene glycol), has been described by Choi et al. who showed the polymer LCST to be sensitive to the gases dissolved in the solution. A reversible LCST was observed between the temperatures of 24.5 °C to 26.0 °C when dissolved carbon dioxide was replaced by oxygen. This was accounted for by considering differing degrees of PEG dehydration and differences in the intermolecular interactions in the presence of the two gases.59
(ii) Triggering an “isothermal” response via end-group modification(s)
The development of controlled radical polymerisation (CRP) processes has increasingly afforded polymers with high levels of structural control. Importantly, the functionality of both chain-ends is now routinely accessible following careful selection of the chain transfer agents/initiators employed, and/or the use of post-polymerisation modification methodologies.60–62 The functionality of end-groups is known to have a pronounced effect on the thermal transition temperature with hydrophilic/hydrophobic moieties generally seen to increase/decrease the overall LCST respectively.63–65 Moreover, the effect of the end-group is more pronounced with decreasing molecular weight on account of it contributing to a higher percentage of the total structure.66,67Theato and co-workers prepared thermally responsive systems based on pOEGMA in which the hydrophilic–hydrophobic balance could be modulated by irradiation with light. Samples were prepared by the RAFT methodology and azobenzene units installed at α- and ω-termini using a combination of functional CTAs and aminolysis-based post polymerisation processing. The LCST of the polymers decreased with increasing azobenzene incorporation and with decreasing polymer chain length. Moreover, azobenzene units undergo trans–cis conformational change upon application of UV light which was shown to increase the LCST due to a change in dipole moment. This transition could also be reversed by irradiating with visible light (Fig. 6).68
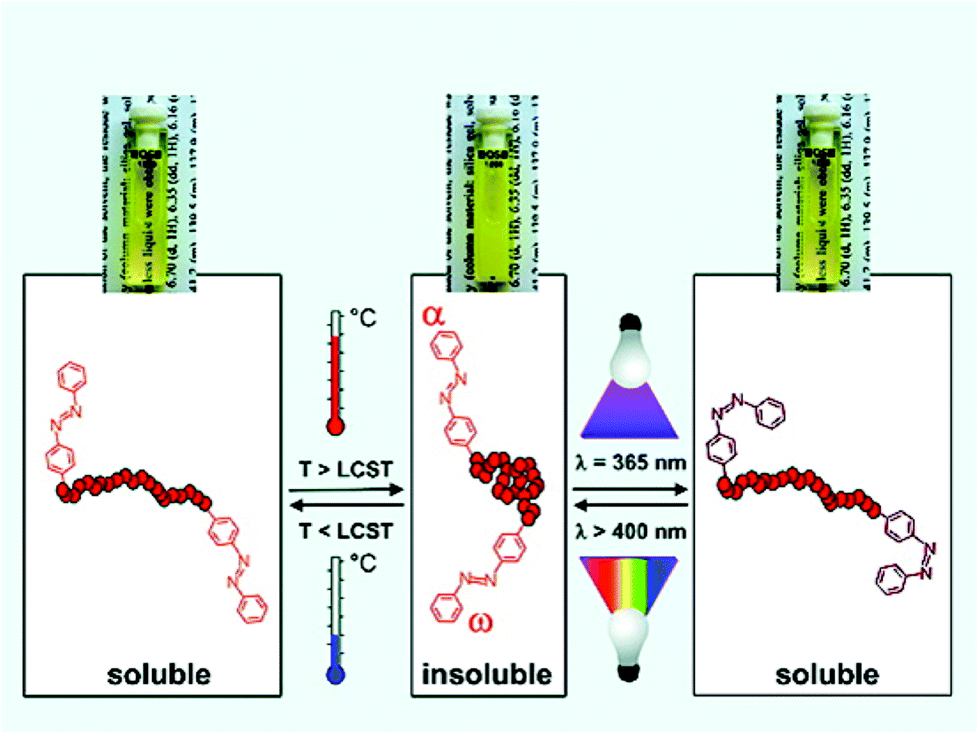 | ||
| Fig. 6 The application of different wavelengths of light to alter the conformation of α- and ω-terminal azobenzene units and hence the LCST, as described by Theato and co-workers. Reprinted with permission from Macromolecules, 2009, 42, 7854–7862. Copyright (2014) American Chemical Society. | ||
Hyperbranched polymers provide an attractive option for end-group modulations given the inherently large number associated with their structure. Rimmer and co-workers have exploited this to prepare imidazole-terminated hyperbranched pNIPAM which exhibit higher LCSTs in the presence of Cu(II) due to an increased hydrophilicity of the polymer chains.69 Similarly, when functionalised with vancomycin or polymyxin end-groups, a selective ligand for Gram-positive/negative bacteria respectively, incubation with Staphylococcus aureus or Pseudomonas aeruginosa induced a coil-to-globule phase transition. Subsequent cooling of the aggregated bacterium–polymer mixture below its LCST released the bacterium.70,71
pH is an attractive stimulus given the range found throughout the body and in diseased tissues. For instance, in vivo pH ranges from 1–8.2, whilst chronic wounds and cancer tumours have a different pH to healthy tissues.9 Typical monomers that are pH responsive are those containing carboxylic acid or tertiary amine functional groups which can become polyelectrolytes when deprotonated or protonated respectively.72 Work by Stayton and co-workers using carboxylic acid-terminated pNIPAM-co-propylacrylic exemplifies this, where alkaline conditions prompted an increase in LCST due to ionisation of the chain-end.73
Gibson et al. have taken a rational reductionist approach to show that the inclusion of a single chain-end disulfide is sufficient to switch polymer solubility. The substitution of a hydrophobic pyridyl disulfide terminus with more hydrophilic compounds such as thioglycerol was shown to trigger polymer re-solubilisation which could be reversed by re-employing a more hydrophobic chain-end.74 This concept has been extended to prepare polymer nanoparticles, using the nanoprecipitation approach, containing a model hydrophobic guest molecule. Glutathione-triggered disulfide reduction was used to prompt particle disassembly and cargo release, whilst also increasing the cloud point of the constituent polymer chains such that complete solubilisation of the nanoparticle structure was obtained (Fig. 7).75
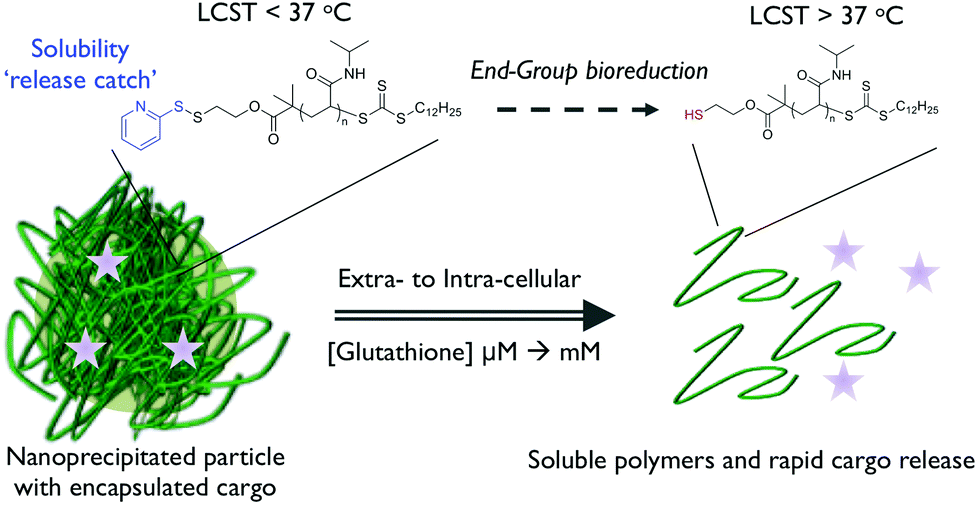 | ||
| Fig. 7 Isothermally-responsive polymer nanoparticle concept: glutathione reduction of the end-group shifts the LCST to induce a solubility switch and cargo release. Reproduced from ref. 77. | ||
The same group has employed a metal ion binding approach to manipulate the thermal response. This work was inspired by the action of siderophores, compounds secreted by bacteria to abstract iron from its mammalian host. Hence, pNIPAM samples containing a 3,4-dihydroxybenzene (catechol) end-group were prepared and the LCST shown to decrease selectively upon incubation of ferric iron (Fe3+).76 Such an approach is attractive given a range of neurodegenerative disorders such as Alzheimer's and Parkinson's disease are characterised by atypical concentrations of this metal.77
(iii) Triggering an “isothermal” response via side-chain/substituent modification
The side-chain/substituents remain the most commonly accessed way of influencing the macromolecular properties of a polymer given the wide-range of monomers accessed by C(L)RP and easy modification of the resulting structure. For example, the ability to tailor the polymer response by co-polymerising with hydrophilic or hydrophobic monomers is well known.78–80 Jochum and Theato have prepared thermo-responsive acrylamides containing salicylideneaniline groups via postpolymerisation modification of a poly(pentafluorophenyl acrylate) precursor. Salicylideneaniline is known to isomerise upon UV irradiation from the enol form to the keto form, with the accompanying difference in dipole moment capable of influencing the polymer LCST.81 Light has also been used by Shimoboji et al. to regulate substrate access and enzyme activity of endoglucanase 12A. This was achieved by varying the wavelength of light irradiated on copolymers of N,N-dimethylacrylamide and azophenyl-containing monomers held at a fixed temperature. Changes in size and hydration of the polymer chain were photo-induced to regulate enzyme activity.82 Light-triggered solution self-assembly of amphiphilic co-polymers comprising NIPAM, ethylene oxide and azobenzene-functional acrylamide blocks has been described by Liu et al. The co-polymer LCST decreased with increasing azobenzene inclusion up to 11 mol%, before unexpectedly increasing above this. Moreover, a surprising decrease in LCST upon irradiation with UV light was observed, implying the azobenzene units in their more polar (cis) form exhibit poorer aqueous solubility, in contrast to that typically expected. This was shown to correlate with the formation of inter-chain assemblies in solution, highlighting the potential of hydrophobic clustering as a tool for tailoring the thermal properties of these polymer systems.83 In addition to azobenzene and salicylideneaniline, studies utilising fulgimides84 and spiropyran85 as light-responsive units have also been reported.Another popular motif used to influence a systems’ solubility is based on compound binding/sequestration. Yin and co-workers have developed an isothermal, thermochromic sensor based on an ABC triblock copolymer consisting of pNIPAM, poly(methacrylic acid) and poly(2-hydroxyethyl methacrylate) which was modified to contain tetra(4-carboxylatophenyl)porphyrin. In water at 32 °C, the polymer has a transparent, red-brown colour which becomes turbid and orange when heated above its LCST due to porphyrin aggregation. When held below its cloud point, a variety of colours are produced depending on the metal cation added to the system. The sensor also displayed thermochromic characteristics in the temperature range of 35–61 °C depending on the metal ion used (Fig. 8).86
 | ||
| Fig. 8 Metal-ion driven thermochromic sensor in the absence of metal ions (left) and upon introduction of various metal ions (right) developed by Yan and co-workers. Reproduced from ref. 86. | ||
Wischerhoff and co-workers have used protein binding to influence the thermoresponsive behaviour of biotin-containing pOEGMA samples. The LCST was observed to increase proportionally with added avidin (up to a maximum of 9 °C at 60 μmol L−1) suggesting the resulting polymer-biotin complex was more hydrophilic than the polymer alone. The specificity of this binding process was demonstrated by a reduction in LCST upon the addition of free biotin due to competition with polymer-bound avidin.87
Redox cycling motifs have been applied by a variety of groups to modulate the LCST of thermo-responsive systems. For example, Fu et al. have synthesised acrylamide co-polymers containing N-isopropyl and redox-sensitive 4-N-amino-2,2,6,6-tetramethylpiperidin-1-oxyl-4-yl (TEMPO) groups. The incorporation of 5–10% TEMPO groups in the copolymer structure was sufficient to afford the material with redox-responsive LCST behaviour: at a polymer concentration of 10 mg mL−1, reduction with 19.2 mM ascorbic acid increased the LCST by 14 °C whilst re-oxidation with 48 mM K3[Fe(CN)6] largely reversed the change.88 Simple redox activity of the ferrocene/ferrocenium ion pair has been used by Kuramoto and Shishido to shift the cloud point of a variety of thermo-responsive polymers isothermally.89,90 Likewise, Peng and co-workers has used host–guest complexation between ferrocene and β-cyclodextrin as a means of modulating the LCST of a thermo-responsive co-polymer comprising N,N-dimethylacrylamide and ferrocene. When in its reduced form, ferrocene could interact with β-cyclodextrin (β-CD), increasing the LCST due to disruptions in hydrophobic associations between ferrocene side groups. However, minimal change in LCST was observed when the ferrocene was oxidised to the ferrocenium ion due to a weaker interaction with β-CD.91 The same group has developed an approach to self-tuneable thermosensitive behaviour using the Belousov–Zhabotinsky reaction. Redox-triggered dynamic complex formation between NIPAM-containing co-polymers and a terpyridine-ruthenium complex was sufficient to cycle the system between soluble and insoluble states.92
The application of pH to trigger hydrophilic–hydrophobic changes in the side-chain has been explored by several groups. Xiao et al. demonstrated a significant effect of pH on the LCST of tertiary amine-functionalised poly(L-glutamates). As the N-substituted groups became more hydrophobic, the LCST was observed to increase in acidic conditions due to the increased hydrophilicity imparted by the protonated amino group.93 Müller and co-workers have employed a similar strategy to change the cloud point of star and linear polymers of poly(N,N-dimethylaminoethyl methacrylate), pDMAEMA.94 Several other examples describing the protonation of tertiary-amine functionalised side-chains as a way of altering LCST have also been reported.95,96
The incorporation of acid-labile groups has proven attractive where a response to low pH is required. For example, Huang et al. have prepared polymers from N-(2-ethoxy-1,3-dioxan-5-yl) methacrylamide where hydrolysis of the cyclic orthoester to hydroxyl groups allowed complete resolubilisation of previously hydrophobic polymers at 37 °C – an acid-catalysed isothermal transition.97 Similarly, Heath et al. have used trimethoxy benzene-linked acetal-functional polymers to influence the solution self-assembly of NIPAM-based materials isothermally. This was achieved due to increased system hydrophilicity upon acid-catalysed cleavage of an acetal to diol.98 Acetals have also been employed by Zhang et al. to raise the cloud point of tri(ethylene glycol) acrylate-based co-polymers upon cleavage. In this case hydrolysis was used to promote micelle disassembly at pH 4, release the encapsulated Nile Red within 200 hours, and re-solubilise the polymer materials.99 Monteiro and co-workers have devised a clever hydrolysis-based degradation strategy to disassemble nanoparticles within a pre-determined timeframe (Fig. 9). Micelles were prepared by heating a solution of diblock copolymers comprising a hydrophilic pDMA block and a random copolymer block of (dimethylamino) ethyl acrylate, DMAEA, butyl acrylate, BA, and NIPAM at 37 °C (above the NIPAM LCST). As DMAEA hydrolysed to acrylic acid, the LCST of the diblock increased above 37 °C resulting in micelle disassembly. The time taken for disassembly to start was controlled by the number of BA units whilst the time taken for complete disassembly was controlled by the number of DMAEA units in the polymer structure.100
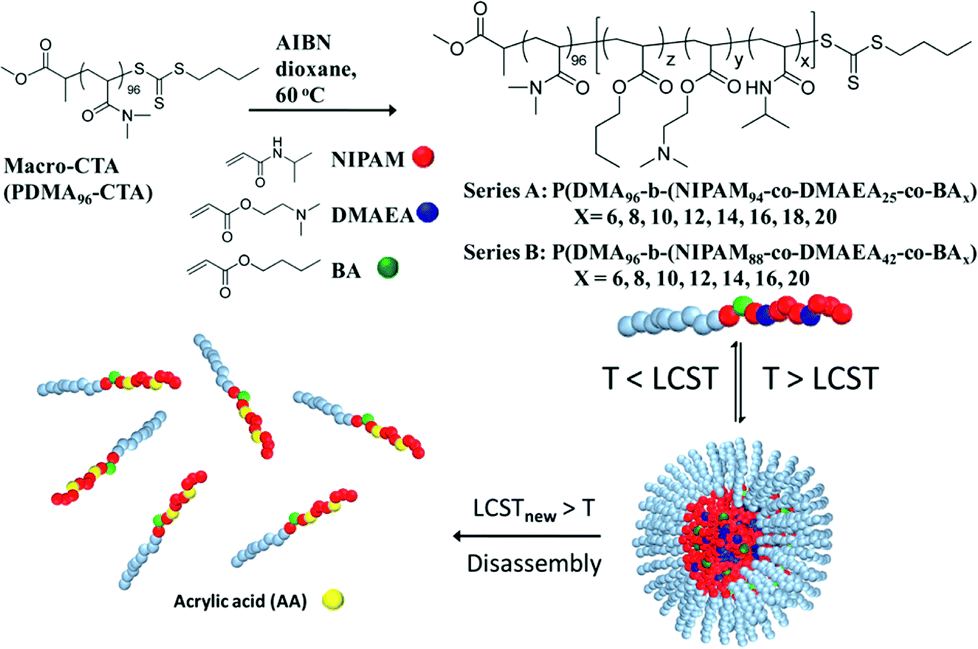 | ||
| Fig. 9 Synthesis and self-assembly of thermoresponsive and self-catalysed degradable polymers described by Monteiro and co-workers. Acid-catalysed degradation of DMAEA to acrylic acid shifts the polymer LCST and triggers micelle disassembly. Reprinted with permission from Biomacromolecules, 2013, 14, 3463–3471. Copyright (2014) American Chemical Society. | ||
In addition to those described above, a variety of other stimuli have been exploited to trigger an “isothermal”-type transition by altering side-chain/substituent integrity including ultrasound, dynamic covalent bond formation, dissolved gases and enzymes (vide infra). Ultrasound, a longitudinal pressure wave with frequency above 20 kHz, has been used for a variety of medical applications including tissue ablation and to release drugs from a polymer matrix.101,102 Using this trigger, Xuan et al. prepared micelles from a diblock copolymer comprising the water soluble poly(ethylene oxide), pEO, and the thermoresponsive poly(2-(2-methoxyethoxy)ethyl methacrylate). The latter block was modified to contain a small amount of high-intensity focused ultrasound (HIFU)-labile 2-tetrahydropyranyl methacrylate. Hence, ultrasound irradiation-triggered hydrolysis of the THPMA groups to methacrylic acid increased the LCST resulting in isothermal micelle disassembly.103
Bon and co-workers have exploited hydrazide-aldehyde chemistry to control the solubility of pNIPAM-based microgels. Here, the addition of aldehydes to a methacryloyl hydrazide containing co-polymer chain furnished dynamic hydrazone bonds. Importantly, the microgel solubility, and hence volume phase transition, could be modulated by careful selection of aldehydes with varying hydrophilicities.104
Guo et al. prepared an amidine-based polymer which underwent a hydrophilic-hydrophilic transition using CO2 as a stimulus. When added to a biphasic system comprising water and chloroform, the polymer initially resided within the organic layer. However, upon bubbling with CO2, the amidine groups converted into amidinium bicarbonates producing a charged, hydrophilic polyelectrolyte which transferred into the aqueous phase. Moreover, the reversible hydrophilic–hydrophobic transition was observed to be reversible at 60 °C upon the bubbling of CO2 or N2 respectively.105
Finally, the ability to alter solubility isothermally through enzymatic activity has obvious application in vivo yet there are few examples in the literature. Thayumanvan and co-workers tethered varying numbers of pOEGMA chains to oligoamine scaffolds also containing a hydrophobic, methyl ester-terminated alkyl chain. Upon increasing the number of pOEGMA chains involved from one (monomer) to six (hexamer), a systematic decrease in the LCST was observed. Incubation with porcine liver esterase converted the methyl esters to a more hydrophilic carboxylic acid, significantly increasing the LCST onset at pH 7.4. To verify the propensity for ester hydrolysis, the authors also measured the cloud point without enzyme at various pH values. Whilst the cloud point gradually increased at pH 10.8 due to hydrolysis and subsequent deprotonation of the carboxylic acid group, no change was observed between pH 5.0 and pH 8.5, confirming an esterase-mediated change in LCST.106
(iv) Triggering an “isothermal” response via the solution environment
The phase separation properties of an aqueous polymer solution are well-known to be influenced by the presence of a variety of additives. For example, salts are able to act as water “structure-makers” (kosmotropes) or “structure-breakers” (chaotropes), thus affecting the polymer hydration shell and the resulting transition temperature.107–109 This is largely dictated by the Hofmeister series which originates from the ability of ions to precipitate egg white proteins.110 Surfactants have been shown to improve the solubility of polymer chains and hence increase the transition temperature as they adsorb onto the polymer by means of their hydrophobic tails, either individually or as micelles.111,112 Ionic liquids have been shown to have an impact depending on their hydrophilicity; the LCST of poly(N-vinylcaprolactam), for example, increases in the presence of hydrophobic ionic liquids, but remain largely unaffected in the presence of hydrophilic alternatives.113 Other additives such as saccharides114 and alcohols115 have also been reported to have an effect. It therefore stands to reason that alterations in the solution in which the polymer is dissolved can be used to isothermally change its solubility. To date, most reports have used salts as additives to achieve this. Alexander and co-workers prepared co-polymers containing OEGMAs with either 3 or 8 PEG units. A linear increase in LCST was observed with increasing proportions of the more hydrophilic, larger PEG-containing OEGMA. Incubation of these polymers with NaSCN (a strong chaotrope), NaCl and Na2SO4 (a strong kosmotrope) greatly affected the cloud point with NaSCN and Na2SO4 prompting an increase and decrease respectively. “Hybrid” co-polymers were then prepared by adding an additional 8-PEG containing OEGMA homoblock to the pre-existing statistical co-polymer. As the two “blocks” had different LCSTs, heating at a temperature above the LCST of the statistical block, but below that of the homoblock generated micelles, in which the dye carboxyfluorescein was encapsulated. When these micelles were held at 37 °C, minimal dye release was observed. The addition of NaCl however lowered the cloud point of the statistical block to trigger micelle disassembly and dye release. Sharper release was observed when Na2SO4 was added as a result of a salting-out effect.116 Wang et al. have used the influence of salts to prepare ionic strength-mediated “schizophrenic” micelles. Double hydrophilic block co-polymers comprising the weak polybase poly(N-(morpholino)ethyl methacrylate), pMEMA and the zwitterionic poly 4-(2-sulfoethyl)-1-(4-vinylbenzyl)pyridinium betaine, pSVBP were shown to exhibit variations in self-assembly behaviour dependent on the concentration and types of salt added.117Bloksma et al. have also demonstrated the Hofmeister effect on poly(2-oxazoline)s observing an ionic response that was strongly dependent on the hydrophilicity of the polymer. Here, the LCST of the most hydrophilic polymer, poly(2-ethyl-2-oxazoline), could be tuned over almost the whole temperature range of water under atmospheric pressure whilst the LCST of the more hydrophobic poly(2-isopropyl-2-oxazoline) and poly(2-n-propyl-2-oxazoline) varied to a lesser extent. Comparisons between linear and comb polymers highlighted the architecture did not significantly influence the effect of the Hofmesiter ions.118 Finally, Sharma and Srivastava have prepared amphiphilic random copolymers based on biodegradable polyaspartamides that respond to temperature, pH and metal-ions. Reversible thermosensitivity was achieved by the attachment of the hydrophobic 1-propylimidazole or hydrophilic, dimethylpropylammonium pendants. The anions of the Hofmeister series were found to affect the thermosensitivity. Modulation of the LCST was also achieved by varying the pH or by including metal ions in the solution (Fig. 10).119
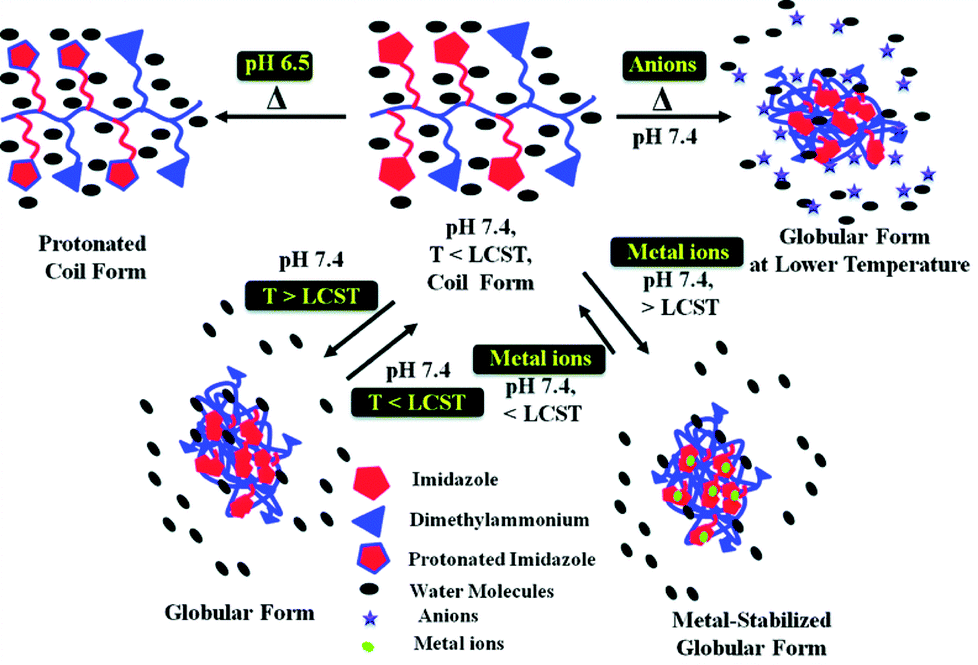 | ||
| Fig. 10 Multi-responsive copolymers prepared by Sharma and Srivastava have demonstrated a response to temperature, pH, anions and transition metals. Reproduced from ref. 119. | ||
3. Conclusion and future perspective
As our ability to precisely control polymer structure continues to improve, the toolbox available for the synthetic chemist to make increasingly complex assemblies which mimic biological systems also expands. The diverse field of stimuli-responsive polymers has been dominated by thermoresponsive polymers which are synthetically accessible and widely employed, but limited in their application scope. In this review we have shown that the core thermoresponsive polymer scaffolds can be transformed into far more versatile polymers capable of generating response to a huge range of (bio)chemical stimuli through subtle structural modifications. Indeed, the ultimate goal is to generate complex polymer assemblies which can rival natural macromolecules for their complexity, and ability to both respond and/or convey information about their local environment. It is thus hoped that this review will spark the development of new chemistries and structural platforms from which next-generation materials can be produced which begin to rival Mother Nature's complexity.Acknowledgements
MIG was a Birmingham Science City Interdisciplinary Research Fellow funded by the Higher Education Funding Council for England (HEFCE). DJP thanks the University of Warwick for a Postgraduate Scholarship and the Institute of Advanced Study (Warwick) for an Early Career Fellowship.Notes and references
- J. F. Mano, Adv. Eng. Mater., 2008, 10, 515–527 CrossRef CAS.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3–23 CrossRef CAS PubMed.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- R. Tong, L. Tang, L. Ma, C. Tu, R. Baumgartner and J. Cheng, Chem. Soc. Rev., 2014, 43, 6982–7012 RSC.
- P. Schattling, F. D. Jochum and P. Theato, Polym. Chem., 2014, 5, 25–36 RSC.
- M. Rubinstein and R. H. Corby, Polymer Physics, Oxford University Press, Oxford, 2003 Search PubMed.
- E. Ruel-Gariépy and J.-C. Leroux, Eur. J. Pharm. Biopharm., 2004, 58, 409–426 CrossRef PubMed.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- J. S. Scarpa, D. D. Mueller and I. M. Klotz, J. Am. Chem. Soc., 1967, 89, 6024–6030 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC.
- M.-C. Jones and J.-C. Leroux, Eur. J. Pharm. Biopharm., 1999, 48, 101–111 CrossRef CAS.
- M. I. Gibson, D. Paripovic and H.-A. Klok, Adv. Mater., 2010, 22, 4721–4725 CrossRef CAS PubMed.
- N. S. Ieong, K. Brebis, L. E. Daniel, R. K. O'Reilly and M. I. Gibson, Chem. Commun., 2011, 47, 11627–11629 RSC.
- J. Wang, M. I. Gibson, R. Barbey, S.-J. Xiao and H.-A. Klok, Macromol. Rapid Commun., 2009, 30, 845–850 CrossRef CAS PubMed.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS PubMed.
- P. S. Stayton, T. Shimoboji, C. Long, A. Chilkoti, G. Ghen, J. M. Harris and A. S. Hoffman, Nature, 1995, 378, 472–474 CrossRef CAS PubMed.
- M. W. Jones, M. I. Gibson, G. Mantovani and D. M. Haddleton, Polym. Chem., 2011, 2, 572–574 RSC.
- S. Dey, B. Kellam, M. R. Alexander, C. Alexander and F. R. A. J. Rose, J. Mater. Chem., 2011, 21, 6883–6890 RSC.
- H.-Y. Tsai, K. Vats, M. Z. Yates and D. S. W. Benoit, Langmuir, 2013, 29, 12183–12193 CrossRef CAS PubMed.
- G. Chen and A. S. Hoffman, Bioconjugate Chem., 1993, 4, 509–514 CrossRef CAS.
- H. Kanazawa, J. Sep. Sci., 2007, 30, 1646–1656 CrossRef CAS PubMed.
- C.-W. Chang, T. H. Nguyen and H. D. Maynard, Macromol. Rapid Commun., 2010, 31, 1691–1695 CrossRef CAS PubMed.
- J. E. Chung, M. Yokoyama, M. Yamato, T. Aoyagi, Y. Sakurai and T. Okano, J. Controlled Release, 1999, 62, 115–127 CrossRef CAS.
- Y. Saaka, R. C. Deller, A. Rodger and M. I. Gibson, Macromol. Rapid Commun., 2012, 33, 779–784 CrossRef CAS PubMed.
- E. W. Edwards, M. Chanana, D. Wang and H. Möhwald, Angew. Chem., Int. Ed., 2008, 47, 320–323 CrossRef CAS PubMed.
- S. Salmaso, P. Caliceti, V. Amendola, M. Meneghetti, J. P. Magnusson, G. Pasparakis and C. Alexander, J. Mater. Chem., 2009, 19, 1608–1615 RSC.
- S. R. Abulateefeh, S. G. Spain, K. J. Thurecht, J. W. Aylott, W. C. Chan, M. C. Garnett and C. Alexander, Biomater. Sci., 2013, 1, 434–442 RSC.
- J. Akimoto, M. Nakayama, K. Sakai and T. Okano, Biomacromolecules, 2009, 10, 1331–1336 CrossRef CAS PubMed.
- J. Akimoto, M. Nakayama, K. Sakai and T. Okano, Mol. Pharm., 2010, 7, 926–935 CrossRef CAS PubMed.
- D. E. Meyer, B. C. Shin, G. A. Kong, M. W. Dewhirst and A. Chilkoti, J. Controlled Release, 2001, 74, 213–224 CrossRef CAS.
- M. R. Dreher, W. Liu, C. R. Michelich, M. W. Dewhirst and A. Chilkoti, Cancer Res., 2007, 67, 4418–4424 CrossRef CAS PubMed.
- S. R. Abulateefeh, S. G. Spain, J. W. Aylott, W. C. Chan, M. C. Garnett and C. Alexander, Macromol. Biosci., 2011, 11, 1722–1734 CrossRef CAS PubMed.
- N. Tirelli, Curr. Opin. Colloid Interface Sci., 2006, 11, 210–216 CrossRef CAS PubMed.
- D. Janigro, Epilepsia, 2012, 53, 26–34 CrossRef CAS PubMed.
- M. M. Chaumeil, J. Valette, C. Baligand, E. Brouillet, P. Hantraye, G. Bloch, V. Gaura, A. Rialland, P. Krystkowiak, C. Verny, P. Damier, P. Remy, A.-C. Bachoud-Levi, P. Carlier and V. Lebon, J. Cereb. Blood Flow Metab., 2012, 32, 771–779 CrossRef CAS PubMed.
- G. Perry, R. J. Castellani, K. Hirai and M. A. Smith, J. Alzheimer's Dis., 1998, 1, 45–55 CAS.
- K. Sugamura and J. J. F. Keaney, Free Radical Biol. Med., 2011, 51, 978–992 CrossRef CAS PubMed.
- S. M. Dhanasekaran, T. R. Barrette, D. Ghosh, R. Shah, S. Varambally, K. Kurachi, K. J. Pienta, M. A. Rubin and A. M. Chinnaiyan, Nature, 2001, 412, 822–826 CrossRef CAS PubMed.
- P. Meers, Adv. Drug Delivery Rev., 2001, 53, 265–272 CrossRef CAS.
- J. D. Raffetto and R. A. Khalil, Biochem. Pharmacol., 2008, 75, 346–359 CrossRef CAS PubMed.
- C. d. l. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- B. P. Timko, T. Dvir and D. S. Kohane, Adv. Mater., 2010, 22, 4925–4943 CrossRef CAS PubMed.
- H. Priya James, R. John, A. Alex and K. R. Anoop, Acta Pharm. Sin. B, 2014, 4, 120–127 CrossRef PubMed.
- R. W. Lenz and R. H. Marchessault, Biomacromolecules, 2004, 6, 1–8 CrossRef PubMed.
- M. I. Gibson and N. R. Cameron, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2882–2891 CrossRef CAS.
- M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 CrossRef CAS PubMed.
- J. Undin, A. Finne-Wistrand and A.-C. Albertsson, Biomacromolecules, 2014, 15, 2800–2807 CAS.
- D. P. Sanders, D. J. Coady, M. Yasumoto, M. Fujiwara, H. Sardon and J. L. Hedrick, Polym. Chem., 2014, 5, 327–329 RSC.
- G. G. Hedir, C. A. Bell, N. S. Ieong, E. Chapman, I. R. Collins, R. K. O'Reilly and A. P. Dove, Macromolecules, 2014, 47, 2847–2852 CrossRef CAS.
- S. T. Reddy, A. Rehor, H. G. Schmoekel, J. A. Hubbell and M. A. Swartz, J. Controlled Release, 2006, 112, 26–34 CrossRef CAS PubMed.
- P. Carampin, E. Lallana, J. Laliturai, S. C. Carroccio, C. Puglisi and N. Tirelli, Macromol. Chem. Phys., 2012, 213, 2052–2061 CrossRef CAS.
- Y. Lee, H. Mo, H. Koo, J.-Y. Park, M. Y. Cho, G.-w. Jin and J.-S. Park, Bioconjugate Chem., 2006, 18, 13–18 CrossRef PubMed.
- C.-C. Chang and T. Emrick, Macromolecules, 2014, 47, 1344–1350 CrossRef CAS.
- D. J. Phillips and M. I. Gibson, Antioxid. Redox Signaling, 2014, 21, 786–803 CrossRef CAS PubMed.
- D. J. Phillips and M. I. Gibson, Chem. Commun., 2012, 48, 1054–1056 RSC.
- D. J. Phillips and M. I. Gibson, Biomacromolecules, 2012, 13, 3200–3208 CrossRef CAS PubMed.
- J. Y. Choi, J. Y. Kim, H. J. Moon, M. H. Park and B. Jeong, Macromol. Rapid Commun., 2014, 35, 66–70 CrossRef CAS PubMed.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS.
- M. A. Tasdelen, M. U. Kahveci and Y. Yagci, Prog. Polym. Sci., 2011, 36, 455–567 CrossRef CAS PubMed.
- C. Boyer, M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 551–595 CrossRef CAS.
- J. E. Chung, M. Yokoyama, T. Aoyagi, Y. Sakurai and T. Okano, J. Controlled Release, 1998, 53, 119–130 CrossRef CAS.
- Y. Xia, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2006, 39, 2275–2283 CrossRef CAS.
- X. Jiang and B. Zhao, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3707–3721 CrossRef CAS.
- Q. Duan, Y. Miura, A. Narumi, X. Shen, S.-I. Sato, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1117–1124 CrossRef CAS.
- W. Steinhauer, R. Hoogenboom, H. Keul and M. Moeller, Macromolecules, 2010, 43, 7041–7047 CrossRef CAS.
- F. D. Jochum, L. zur Borg, P. J. Roth and P. Theato, Macromolecules, 2009, 42, 7854–7862 CrossRef CAS.
- S. Carter, B. Hunt and S. Rimmer, Macromolecules, 2005, 38, 4595–4603 CrossRef CAS.
- J. Shepherd, P. Sarker, K. Swindells, I. Douglas, S. MacNeil, L. Swanson and S. Rimmer, J. Am. Chem. Soc., 2010, 132, 1736–1737 CrossRef CAS PubMed.
- P. Sarker, J. Shepherd, K. Swindells, I. Douglas, S. MacNeil, L. Swanson and S. Rimmer, Biomacromolecules, 2010, 12, 1–5 CrossRef PubMed.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449 RSC.
- X. Yin, A. S. Hoffman and P. S. Stayton, Biomacromolecules, 2006, 7, 1381–1385 CrossRef CAS PubMed.
- M. J. Summers, D. J. Phillips and M. I. Gibson, Chem. Commun., 2013, 49, 4223–4225 RSC.
- D. J. Phillips, J. P. Patterson, R. K. O'Reilly and M. I. Gibson, Polym. Chem., 2014, 5, 126–131 RSC.
- D. J. Phillips, I. Prokes, G.-L. Davies and M. I. Gibson, ACS Macro Lett., 2014, 3, 1225–1229 CrossRef.
- N. C. Andrews, N. Engl. J. Med., 1999, 341, 1986–1995 CrossRef CAS PubMed.
- H. Feil, Y. H. Bae, J. Feijen and S. W. Kim, Macromolecules, 1993, 26, 2496–2500 CrossRef CAS.
- M. M. Ali and H. D. H. Stöver, Macromolecules, 2004, 37, 5219–5227 CrossRef CAS.
- C. Diehl and H. Schlaad, Macromol. Biosci., 2009, 9, 157–161 CrossRef CAS PubMed.
- F. D. Jochum and P. Theato, Macromolecules, 2009, 42, 5941–5945 CrossRef CAS.
- T. Shimoboji, E. Larenas, T. Fowler, S. Kulkarni, A. S. Hoffman and P. S. Stayton, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16592–16596 CrossRef CAS PubMed.
- Y.-J. Liu, A. Pallier, J. Sun, S. Rudiuk, D. Baigl, M. Piel, E. Marie and C. Tribet, Soft Matter, 2012, 8, 8446–8455 RSC.
- F. D. Jochum, F. R. Forst and P. Theato, Macromol. Rapid Commun., 2010, 31, 1456–1461 CrossRef CAS PubMed.
- A. E. Ivanov, N. L. Eremeev, P. O. Wahlund, I. Y. Galaev and B. Mattiasson, Polymer, 2002, 43, 3819–3823 CrossRef CAS.
- Q. Yan, J. Yuan, Y. Kang, Z. Cai, L. Zhou and Y. Yin, Chem. Commun., 2010, 46, 2781–2783 RSC.
- J. Buller, A. Laschewsky, J.-F. Lutz and E. Wischerhoff, Polym. Chem., 2011, 2, 1486–1489 RSC.
- H. Fu, D. M. Policarpio, J. D. Batteas and D. E. Bergbreiter, Polym. Chem., 2010, 1, 631–633 RSC.
- N. Kuramoto, Y. Shishido and K. Nagai, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1967–1972 CrossRef CAS.
- N. Kuramoto and Y. Shishido, Polymer, 1998, 39, 669–673 CrossRef CAS.
- F. Zuo, C. Luo, X. Ding, Z. Zheng, X. Cheng and Y. Peng, Supramol. Chem., 2008, 20, 559–564 CrossRef CAS.
- H. Zhou, E. Liang, Y. Pan, X. Ding, Z. Zheng and Y. Peng, RSC Adv., 2013, 3, 2182–2185 RSC.
- C. Xiao, Y. Cheng, Y. Zhang, J. Ding, C. He, X. Zhuang and X. Chen, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 671–679 CrossRef CAS.
- F. A. Plamper, M. Ruppel, A. Schmalz, O. Borisov, M. Ballauff and A. H. E. Müller, Macromolecules, 2007, 40, 8361–8366 CrossRef CAS.
- M. Mertoglu, S. Garnier, A. Laschewsky, K. Skrabania and J. Storsberg, Polymer, 2005, 46, 7726–7740 CrossRef CAS PubMed.
- D. Fournier, R. Hoogenboom, H. M. L. Thijs, R. M. Paulus and U. S. Schubert, Macromolecules, 2007, 40, 915–920 CrossRef CAS.
- X. Huang, F. Du, D. Liang, S.-S. Lin and Z. Li, Macromolecules, 2008, 41, 5433–5440 CrossRef CAS.
- F. Heath, A. O. Saeed, S. S. Pennadam, K. J. Thurecht and C. Alexander, Polym. Chem., 2010, 1, 1252–1262 RSC.
- Q. Zhang, N. Vanparijs, B. Louage, B. G. De Geest and R. Hoogenboom, Polym. Chem., 2014, 5, 1140–1144 RSC.
- N. T. D. Tran, Z. Jia, N. P. Truong, M. A. Cooper and M. J. Monteiro, Biomacromolecules, 2013, 14, 3463–3471 CrossRef CAS PubMed.
- K. W. Ferrara, Adv. Drug Delivery Rev., 2008, 60, 1097–1102 CrossRef CAS PubMed.
- I. Lentacker, S. C. De Smedt and N. N. Sanders, Soft Matter, 2009, 5, 2161–2170 RSC.
- J. Xuan, O. Boissière, Y. Zhao, B. Yan, L. Tremblay, S. Lacelle, H. Xia and Y. Zhao, Langmuir, 2012, 28, 16463–16468 CrossRef CAS PubMed.
- Y. Chen, N. Ballard, O. D. Coleman, I. J. Hands-Portman and S. A. F. Bon, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1745–1754 CrossRef CAS.
- Z. Guo, Y. Feng, Y. Wang, J. Wang, Y. Wu and Y. Zhang, Chem. Commun., 2011, 47, 9348–9350 RSC.
- F. Wang, A. Klaikherd and S. Thayumanavan, J. Am. Chem. Soc., 2011, 133, 13496–13503 CrossRef CAS PubMed.
- I. Nwankwo, D. W. Xia and J. Smid, J. Polym. Sci., Part B: Polym. Phys., 1988, 26, 581–594 CrossRef CAS.
- T. Baltes, F. Garret-Flaudy and R. Freitag, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2977–2989 CrossRef CAS.
- Y. Yang, F. Zeng, Z. Tong, X. Liu and S. Wu, J. Polym. Sci., B: Polym. Phys., 2001, 39, 901–907 CrossRef CAS.
- F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, 24, 247–260 Search PubMed.
- L.-T. Lee and B. Cabane, Macromolecules, 1997, 30, 6559–6566 CrossRef CAS.
- K. Van Durme, H. Rahier and B. Van Mele, Macromolecules, 2005, 38, 10155–10163 CrossRef CAS.
- W. Li and P. Wu, Polym. Chem., 2014, 5, 761–770 RSC.
- S. Belbekhouche, V. Dulong, L. Picton and D. Le Cerf, Colloids Surf., A, 2013, 428, 25–31 CrossRef CAS PubMed.
- R. Hoogenboom, H. M. L. Thijs, D. Wouters, S. Hoeppener and U. S. Schubert, Soft Matter, 2008, 4, 103–107 RSC.
- J. P. Magnusson, A. Khan, G. Pasparakis, A. O. Saeed, W. Wang and C. Alexander, J. Am. Chem. Soc., 2008, 130, 10852–10853 CrossRef CAS PubMed.
- D. Wang, T. Wu, X. Wan, X. Wang and S. Liu, Langmuir, 2007, 23, 11866–11874 CrossRef CAS PubMed.
- M. M. Bloksma, D. J. Bakker, C. Weber, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2010, 31, 724–728 CrossRef CAS PubMed.
- A. Sharma and A. Srivastava, Polym. Chem., 2013, 4, 5119–5128 RSC.
| This journal is © The Royal Society of Chemistry 2015 |