
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling monomer-sequence using supramolecular templates
Niels
ten Brummelhuis
Department of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany. E-mail: niels.ten.brummelhuis@hu-berlin.de
First published on 10th December 2014
Abstract
The transcription and translation of information contained in nucleic acids that has been perfected by nature serves as inspiration for chemists to devise strategies for the creation of polymers with well-defined monomer sequences. In this review the various approaches in which templates (either biopolymers or synthetic ones) are used to influence the monomer-sequence are discussed.
 Niels ten Brummelhuis | Dr ten Brummelhuis received his PhD-degree in 2011 at the Max Planck Institute of Colloids and Interfaces in Potsdam, Germany, under the supervision of Prof. Dr Markus Antonietti and Prof. Dr Helmut Schlaad. He spent two years as a Postdoc at the Molecular Design Institute, Department of Chemistry of the New York University, where he worked with Prof. Dr Marcus Weck. In 2013 he returned to Germany where he now heads an independent research group at the Department of Chemistry of the Humboldt-Universität zu Berlin, investigating novel methods towards the synthesis of complex, controlled monomer-sequences using chain-growth polymerisation techniques. |
Introduction
The monomer sequences in biopolymers such as nucleic acids and proteins are strongly linked to their function, and even small errors can yield non-functional macromolecules, or worse. Nature therefore expends significant resources on the (nearly) flawless synthesis of these polymers.1 For example, DNA is replicated using DNA polymerases, which achieve error rates as low as 1 in 106 nucleotide additions, a rate which is further reduced by several orders of magnitude by proof-reading and repair mechanisms.1 Presently, DNA is the primary information carrier used in nature.2 RNA, which can be prepared by the transcription of DNA by RNA polymerases, in most cases serves as a middleman, conveying information from DNA to amino acid sequences in proteins (e.g. as messenger or transfer RNA; mRNA and tRNA), though RNA can also perform catalytic functions itself, and it has been suggested that RNA played a much bigger part in emerging life before the advent of proteins.3–5The wide variety of functions that biopolymers perform with a limited number of monomers (e.g. the 21 proteinogenic amino acids in proteins) has led polymer chemists to conclude that an increased control over the monomer sequence could similarly imbue complex properties upon synthetic polymers, allowing them to be used for a plethora of novel applications.6–13
Various methods towards this ultimate goal of sequence control have been suggested, ranging from solid-phase synthesis, the coupling of presynthesised oligomers, ring-opening polymerisation of multifunctional ring systems and kinetic control.7–11 Perhaps the most extensively investigated method, i.e. template polymerisation, is closer to the procedures perfected by nature though. Replication, transcription and translation are all based on templated synthesis of biopolymers; the monomer sequence is strictly predetermined by the template molecule (DNA in replication and transcription, mRNA in translation). In this review the use of various types of template polymerisation for the control over monomer sequence in synthetic polymers are discussed.
Template polymerisation
The term template polymerisation is widely used in literature to describe any polymerisation where a template is used to influence properties of the daughter polymer. Molecular weight, dispersity and tacticity can for example be influenced strongly by their polymerisation in the presence of a template (macro)molecule.14–18Supramolecular interactions (such as Coulombic interactions or hydrogen bonding) between the template and monomers and/or the growing polymer are required for a template to exert control over any property of the polymer. In literature, the term templating is occasionally also used for systems where covalent bonds are used to influence the properties of polymers (in an intramolecular polymerisation), but these will not be considered here.
The nature of the template varies depending on the polymer characteristic that one wishes to influence. For the control of the molecular weight and dispersity of polymers, template polymers with well-defined molecular weight and dispersity are used. Ideally, daughter polymers with identical dispersity and degree of polymerisation are obtained.14,16 Both small molecules and macromolecules can be used as templates to influence the tacticity of polymers. Small molecules (or metal-ion complexes) are used to supramolecularly influence the relative positions of monomers,17 placing them in positions that promote e.g. iso- or syndiotactic incorporation of the monomers. Alternatively, a small molecule can be used to effectively make a monomer bulkier, thereby prohibiting the isotactic incorporation of monomers and yielding preferentially syndiotactic polymers.17
The best example of a macromolecular template for the control of the tacticity of polymers is based on the formation of stereocomplexes,17e.g. between isotactic and syndiotactic poly(methyl methacrylate), where either one induces the formation of the complementary polymer.
This review focusses on the use of templates for the control of monomer sequence in synthetic polymers. This is not the only reason why template polymerisations have been studied though. Template directed control of monomer sequences has also been studied extensively to understand the fundamental requirements for the transfer of information between (bio-)macromolecules, and how this relates to the origin of life.19 Reproduction is arguably the most crucial feature of living systems, and reproduction in nature is purely a templated process. Even before self-replicating nucleic acid systems arose, templating by clay crystals20,21 or lipid membranes22 could very well have catalysed the formation of nucleic acid containing polymers.
The questions chemists have been trying to answer about the origin of self-replicating nucleic acid systems, and about later, more complex, biological systems, are becoming increasingly relevant to material chemists in their quest for routes towards sequence-controlled polymerisations, since a thorough understanding of the replication, transcription and translation processes might allow for the adoption of these principles for the preparation of synthetic polymers with similarly well-defined monomer sequences. Efforts to adopt aspects of template polymerisation to synthetic polymers range from polymerisations that are strongly connected to biological systems to fully synthetic approaches. In the following sections the various approaches have been divided into (1) systems where biopolymers (i.e. nucleic acids) are used as template molecules and (2) fully synthetic systems.
Biopolymer templates
The use of biopolymers (or close analogues thereof) as templates is the most commonly used method in the sequence control of synthetic polymers. A number of very different methods based on biopolymers have been proposed. For clarity, they have been categorised as methods akin to replication/transcription or translation in natural systems, or as approaches fully unrelated to biopolymer synthesis.Template polymerisation akin to replication and transcription
Scientists have extensively used nature's machinery for the creation of fully or partially synthetic polymers. In these approaches enzymes (DNA/RNA polymerases) are used to catalyse the transcription of a template nucleic acid polymer. This strategy is limited in its scope since the natural systems are highly specialised and only effectively catalyse the synthesis of nucleic acids that are similar in structure to DNA/RNA.23,24 Fortunately many sources of polymerases are available and the specificity of the various enzymes varies greatly. Some examples of nucleic acids that can be polymerised using polymerases are shown in Fig. 1. Both the (deoxy)ribose group (Fig. 1a) and the phosphodiester group can be exchanged (Fig. 1b).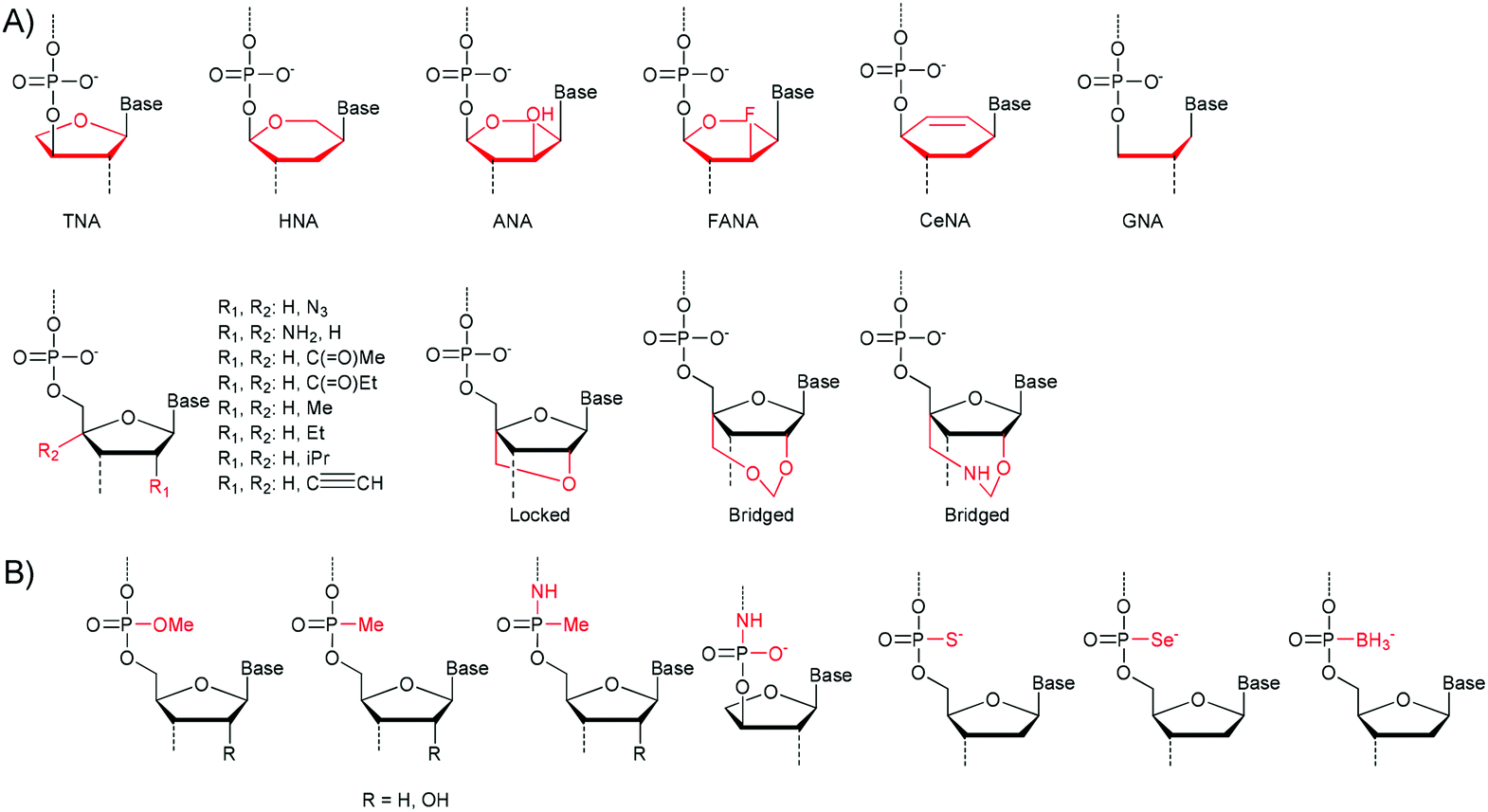 | ||
| Fig. 1 (A) Derivatives of the (deoxy)ribose group which can be incorporated by polymerase enzymes. TNA: threose NA, HNA: 1,5-anhydrohexitol NA, ANA: 1,5-anhydroatritol NA, FANA: 2′-deoxy-2′-fluoroarabino NA, CeNA: cyclohexenyl NA, GNA: glycerol NA. (B) Modification of the phosphodiester backbone. PNA: peptide NA.23,24 | ||
For TNA, HNA, ANA, FANA, CeNA and LNA heredity and evolution could even be shown, supporting the assumption that other nucleic acids besides RNA and DNA could have been involved in the rise of early life-forms.25
Several groups have attempted to extend the scope even further by modifying the polymerases to allow for a broader scope of monomers. In many cases though these efforts also result in less specific incorporation of the appropriate base pair.26,27
New functions can potentially be incorporated into nucleic acids by introducing functional groups. This is most reliably achieved by placing functional groups in the major and/or minor groove of the double-stranded helix. To avoid interference with the recognition between bases, any changes need to be performed on sites on the opposite side from the H-bonding face (Fig. 2a). A notable exception is presented by Szostak and co-workers.28 They showed that the use of 2-thiothymidine (2-TT) instead of thymidine (T) lead to the suppression of mismatched pairs through stronger binding between 2-TT and adenine (A) and weaker unspecific binding to cytosine (C) and guanine (G), and therefore yields lower error rates in the enzyme-free replication of phosphoramidate DNA.
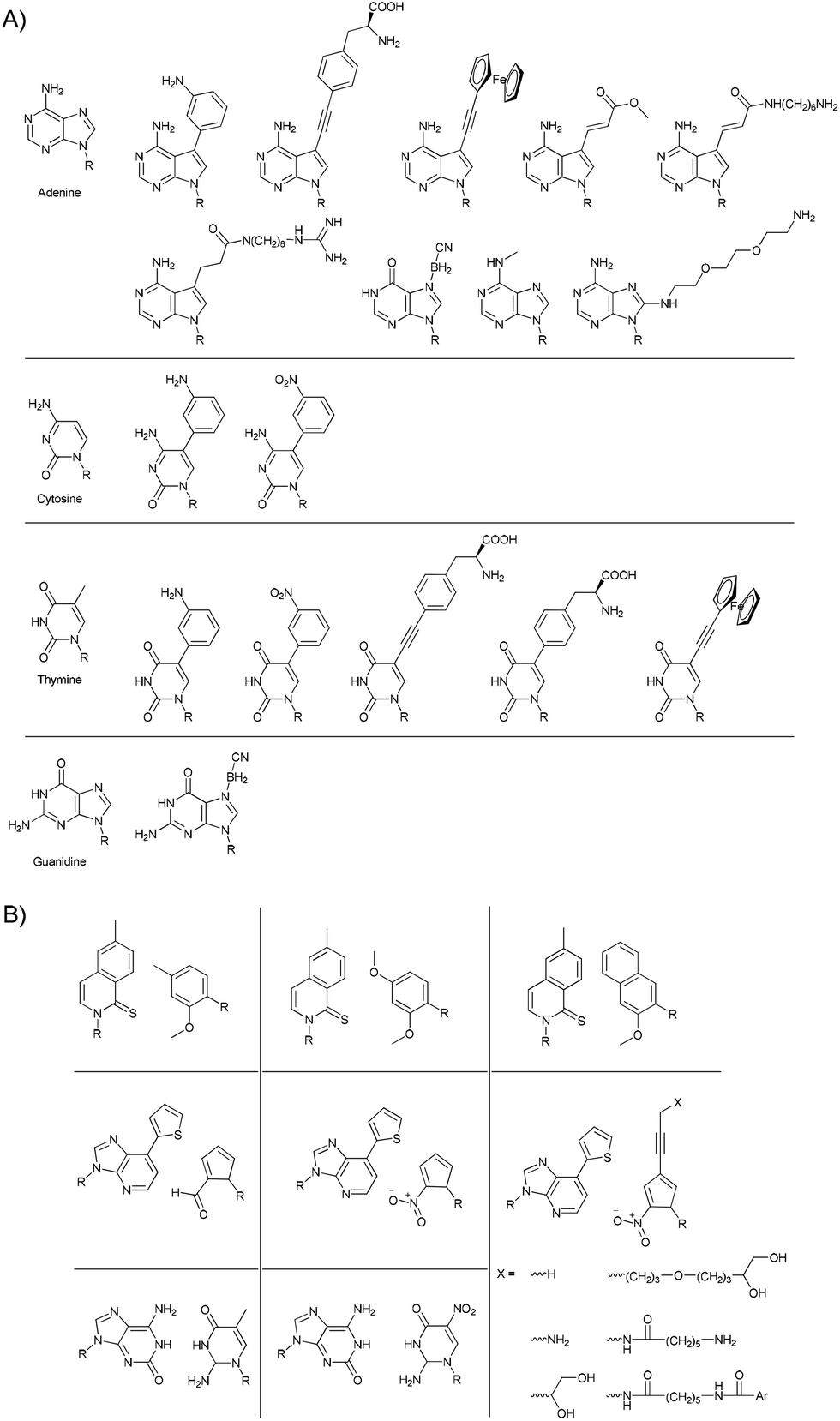 | ||
| Fig. 2 (A) Some examples of modified bases used to introduce functional groups into nucleic acids.23 (B) Examples of artificial base pairs that are capable of being selectively incorporated into nucleic acids by PCR.29 | ||
Besides using modified versions of the four natural bases, in which recognition is limited to two base pairs, efforts have been made to devise completely new artificial recognition pairs which are orthogonal to the A/T and G/C recognition.29 The base pairs shown in Fig. 2b could even be shown to be compatible with the polymerase chain reaction (PCR) procedure, in which they were specifically and efficiently incorporated into the nucleic acid chain.
The incorporation of functional groups and unnatural bases is still limited by the selectivity of the polymerases that are used. Liu and coworkers suggested using DNA ligases rather than polymerases for the transcription of a ssDNA template.30 They were able to show that T4 DNA ligase was able to efficiently couple 5′-phosphorylated trinucleotides in the presence of a suitable single-stranded DNA (ssDNA) template. The presence of functional groups on the 5′-base did not effect the efficiency of the ligase significantly. Eight different functional groups could be incorporated in a sequence specific way using this strategy, and 10 to 50 three-nucleotide codons could be transcribed efficiently (full-length product yields >80%).
Enzyme-free replication and transcription
Replication and transcription have also been extensively studied without the use of the highly optimised enzymes that nature has given rise to,23,24 which has the advantage that also backbones very different from natural nucleic acids can be used, provided the template nucleic acid strand and the daughter polymer efficiently hybridise.Enzyme-free replication and transcription of nucleic acids has been far more difficult than was originally assumed. Pioneering work by Orgel and co-workers showed that the template polymerisation of activated phosphate moiety containing nucleotides could be used to promote the formation of a small number of phosphoesterifications between mono-, di-, tri- or oligonucleotides, though mostly in rather poor yields.31–39 Also, recognition between cytosine and guanine were shown to be far more effective as templates than adenine and thymine (or uracil), presumably because of the stronger interaction between C and G.
Using mononucleotides the incorporation of monomers was very error prone: error rates as high as 30% for G/C and >50% for A/T base pairing were found.40 Enzyme-free template polymerisation with some accuracy and efficiency has become possible only using oligonucleotides. Similar observations were made when using non-natural nucleotides (e.g. PNA, ANA). Though a large body of work relating to the template catalysed ligation of two oligomers can be found in literature, using a wide variety of different chemical reactions and polymer backbones, these works do not provide a real control over monomer sequence and have been reviewed extensively elsewhere41,42 and will therefore not be covered here.
A good example where oligonucleotides were coupled together was provided by Liu and co-workers.43,44 They utilised PNA tetramers42 to recognise four-base codons on a DNA template. Various 5′-amine (which serves as the initiator) DNA hairpin template molecules with predominantly AGTC codons and a single codon with a different sequence (ten codons in total) were used. The position of the differing codon was varied. In the presence of only NH2-GACT-CHO PNA tetramer primarily the truncated product, with a length dependent on the position of the mismatched codon, were obtained. Even the presence of all possible PNA tetramers, except for the one complementary to the second codon, primarily yielded the truncated product. Only in the presence of NH2-GACT-CHO and the PNA tetramer complementary to the second codon were full length products obtained in good yield.
In an extension of this work it was shown that 12 different PNA pentamers could be polymerised in a sequence-specific manner.45 A suitable set of PNA tetramers which polymerize efficiently, selectively (e.g. with minimal out of frame hybridisation) and exhibit sufficient complexity could not be found. The authors used this system to show that functional PNA sequences can be selected from a library. This is done by displacing the PNA strand from the DNA template using a DNA polymerase to create the corresponding double stranded (ds) DNA, thereby allowing the PNA strand to interact with moieties in solution.
The group also investigated the influence of various side-chains on the α- or γ-carbon atoms of PNA pentamers.43 20 different groups could be placed on one or several of the PNA monomers in the NH2-GGATT-CHO PNA pentamers that were used, and in most cases the full-length product was obtained in high yields (70–85%). Only the use of monomers (functionalised on the underlined thymidine residue: NH2-GGA![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) T-CHO) functionalised with D-stereochemistry resulted in very low yields (∼10%). The authors show this as a first proof that functional groups can be incorporated in PNA polymers. The work is summarised graphically in Fig. 3.
T-CHO) functionalised with D-stereochemistry resulted in very low yields (∼10%). The authors show this as a first proof that functional groups can be incorporated in PNA polymers. The work is summarised graphically in Fig. 3.
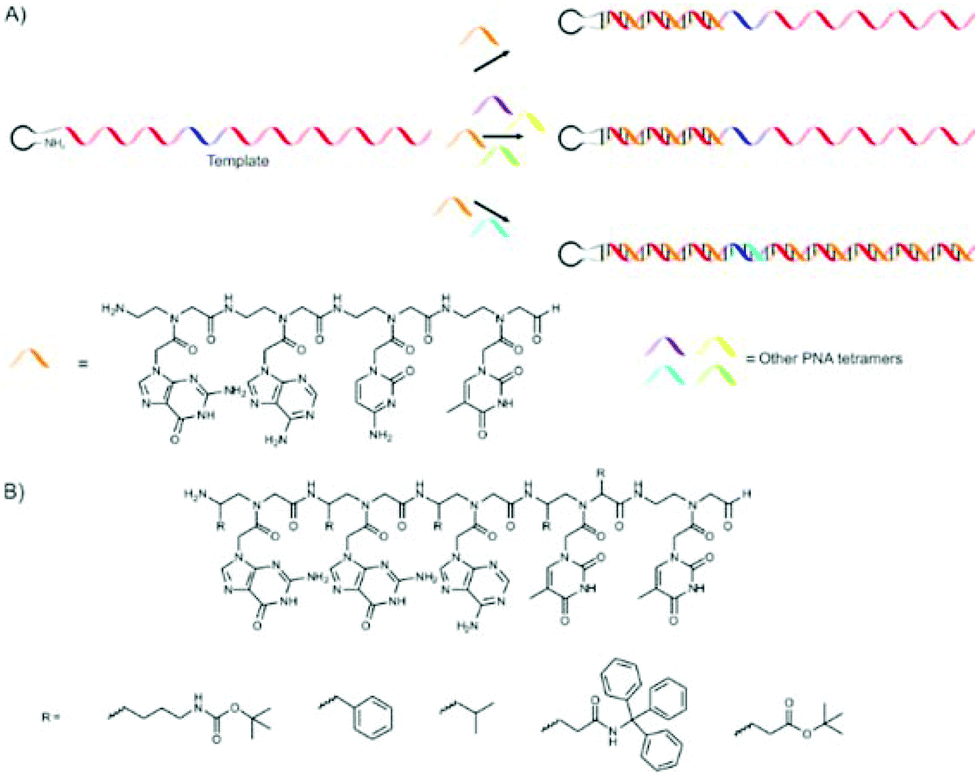 | ||
| Fig. 3 (A) Template polymerisation of PNA tetra- or pentamers using 5′-amine functionalised DNA as a template. Truncated products are mainly formed in the absence of the PNA aptamer complementary to all codons present in the template. (B) PNA aptamers in which functional groups were introduced in the positions marked with ‘R’. In most cases a single functional group was introduced into an aptamer, but multiple functional groups could also be used.43,44 | ||
The overwhelming majority of investigations into enzyme-free template polymerisation using nucleic acids as templates have used the linkage of monomers/oligomers to control the monomer sequence of the daughter polymer. A novel approach was described by Ura et al., who used DNA templates for the selective post-polymerisation functionalisation of peptides containing cysteine at every second position.46 They used bases with a thioester linker that, through a transesterification were linked to the free thiol groups of the peptides. The presence of a DNA template molecule (e.g. C20 or T20) was shown to promote the incorporation of the complementary base, with a selectivity of nearly 90%. Removal of the template and addition of another led to the enrichment of the newly complementary base (Fig. 4). Though no real control over the sequence of bases on the cysteine moieties could be shown yet, the methodology certainly allows for the incorporation of a functional group at every second amino acid, and the nature of the intermediary amino acids (AAs) (between the cysteine residues) in the precursor polymer could be varied.
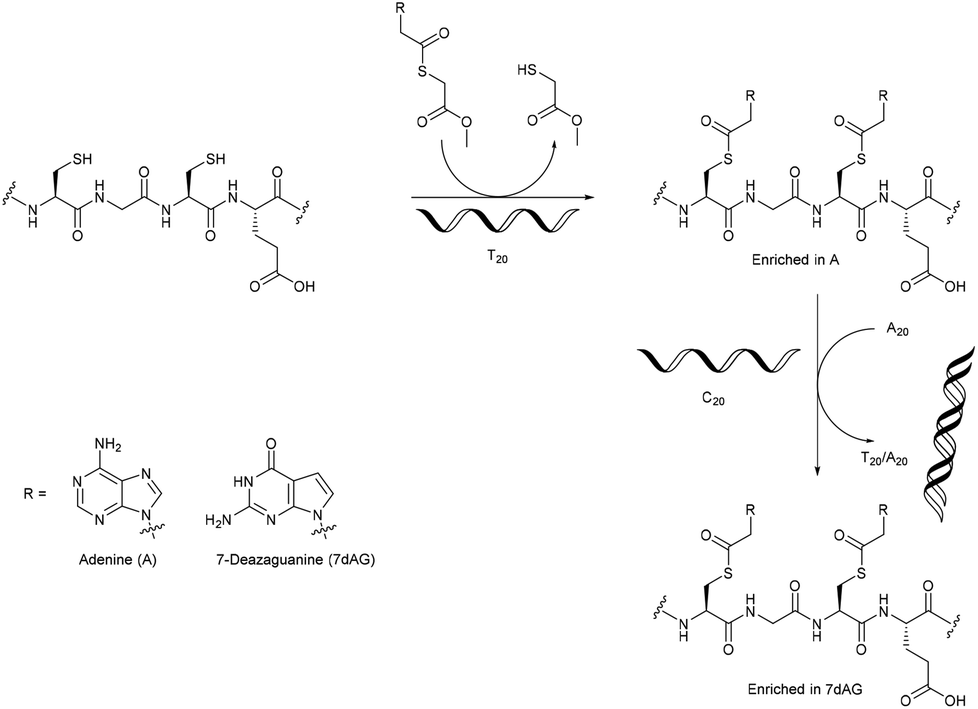 | ||
| Fig. 4 The DNA-template directed post-polymerisation functionalisation of cysteine-rich peptides.46 | ||
Template polymerisation akin to translation
The translation of the genetic information carried by mRNA to amino acid sequences performed in ribosomes is a highly complex procedure, which is nicely illustrated by the large number of components that constitute a functional ribosome (57 proteins and three ribosomal RNA strands (rRNA) for bacterial ribosomes, and even more for eukaryotic ribosomes).2 Nevertheless, ribosomes can also be used to produce synthetic polymers. The most common procedure is the incorporation of non-canonical amino acids into proteins.47–51Early approaches towards the incorporation of non-canonical AAs used non-canonical aminoacyl tRNAs based on the desired AA and a recombinant tRNA to suppress a nonsense stop codon. In this way only a small number of non-canonical AAs could be incorporated in vitro. More advanced approaches, which allow for the incorporation of larger numbers of non-canonical monomers, uses a reconstituted translation system including purified ribosomes and a complex mix of recombinant proteins and cofactors instead of a cell lysate. The fact that various components, e.g. canonical aminoacyl-tRNAs, can be omitted, eliminates the risk of undesirable incorporation of the amino acid for which a given codon naturally codes, thereby allowing an alternative designation of codons to monomers.
Obviously, amino acids with altered side-chains can be incorporated, but in a few cases also the nature of the peptide backbone was changed. N-methylation, which in nature is often performed post-translationally, can also be realised using N-methylated amino acids during ribosomal translation.52–56 Though the incorporation of longer alkyl chains was also attempted, the success was very limited. Besides the L-amino acids that are typically used in nature, also D-amino acids can be incorporated under the right conditions.57,58 Scientist have even gone so far as to replace the amide bond in the peptide backbone with an ester bond.59,60 Ohta et al., following up on observations by Fahnestock and Rich,61 were able to change the meaning of up to eight codons (seven α-hydroxy acids and formylated methionine assigned to the AUG start codon) using a flexizyme62 system, and used these to prepare polyester sequences with lengths of up to 12 monomers (Fig. 5). Additionally, also lysine, aspartic acid and tyrosine could be incorporated in the polymer chain.
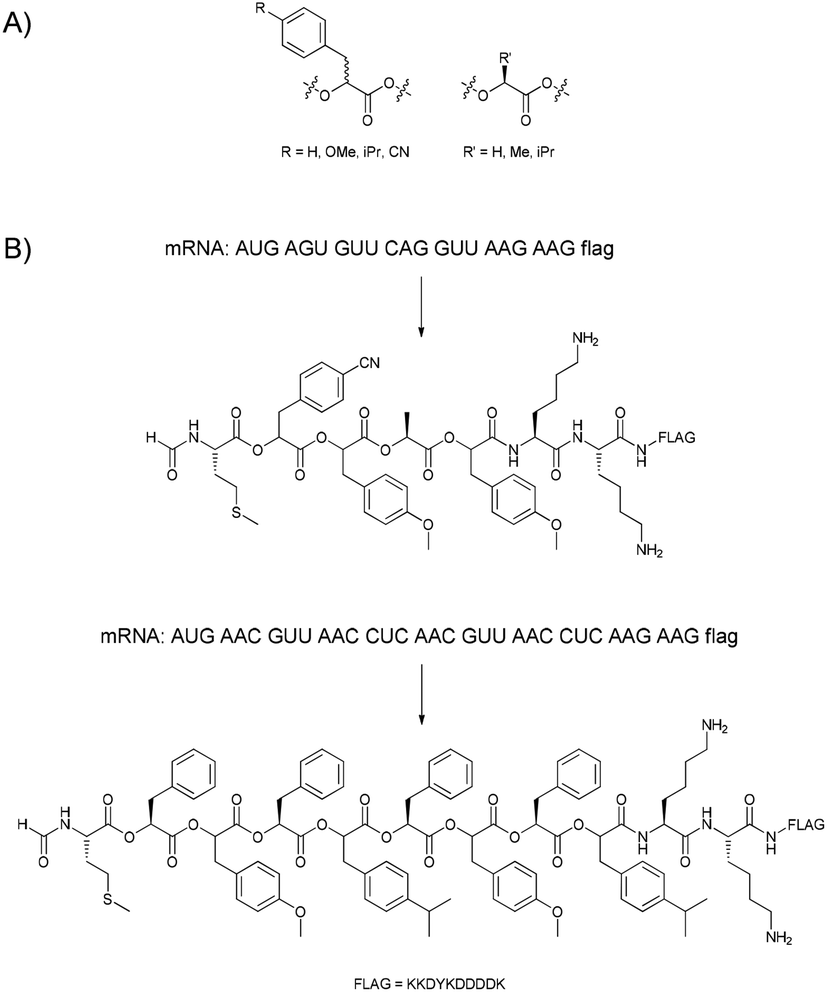 | ||
| Fig. 5 (A) The α-hydroxy acids used by Suga and co-workers. (B) Two examples of the in vitro translation of mRNA sequences into polyesters.59,60 | ||
The incorporation of non-canonical amino acids (or, more generally, monomers) is limited by the specificity that ribosomes exhibit. Some groups have attempted to modify the ribosome in such a way that more monomers can be incorporated in a well-defined manner, but the complexity of the ribosome makes this endeavour challenging.55 Similarly, the incorporation of non-canonical AAs in peptides synthesised by nonribosomal peptide synthases can be facilitated by point mutations in these enzymes.63
Liu and co-workers recently presented a more synthetic procedure to translate nucleotide sequences in DNA to a sequence of polymer building blocks.64 They used a PNA adapter covalently linked with polymer building blocks at the α- and ω-ends to recognise nucleotide codons. The formation of covalent bonds (copper-catalysed azide alkyne cycloaddition proved most efficient) between the polymer building blocks led to the extension of the hairpin DNA with up to 16 polymeric building blocks, though the yield of full-length product was reduced with longer templates. Homotelechelic monomers, i.e. monomers having two alkyne or two azide moieties at both ends, were used since heterotelechelic monomers yielded undesired side products due to cyclisation. The use of homotelechelic building blocks limits the number of possible products from a given number of PNA adapters though. Cleavage of the disulphide linkers between the PNA adapter and the polymer building blocks yielded the final polymer (Fig. 6).
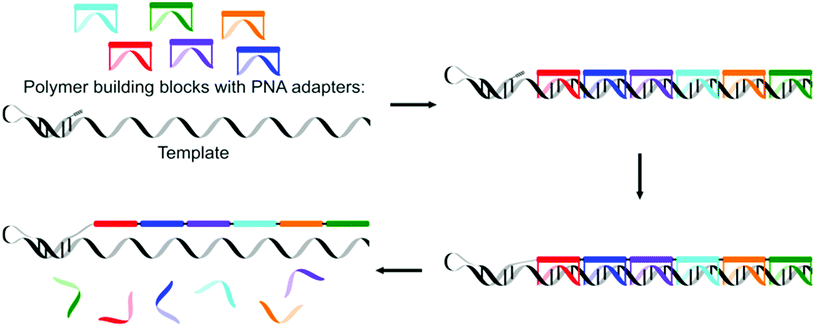 | ||
| Fig. 6 The use of a DNA template for the synthesis of sequence defined polymers by using a PNA adapter as an artificial tRNA.64 | ||
Novel approaches
The hybridisation of two complementary single stranded DNA (ssDNA) strands can be used to place moieties (e.g. monomers) into close proximity to each other. Liu and co-workers, and more recently the combined groups of Turberfield and O'Reilly, used this principle extensively to create sequence-defined oligomers.The first approach by Liu and co-workers used a DNA template with a 5′-amine group which consisted of three coding domains.65 A DNA decamer, complementary to bases 21–30 of the template, bearing a 3′-amino acid on a base labile sulfone linker and a 5′-biotin moiety, was added. A peptide bond was formed, using EDC as the coupling reagent, between the terminal amine group of the template DNA and the carboxylic acid group on the DNA decamer. After breaking of the sulfone linker using basic conditions and removal of the DNA decamer using streptavidin-linked magnetic nanoparticles, another decamer, complementary to bases 11–20, or in the last step to bases 1–10, was added and the cycle was repeated to yield a tripeptide (Fig. 7a). The yield of the additions ranged from 52 to 82%, so that the overall yield of the tripeptide was rather poor.
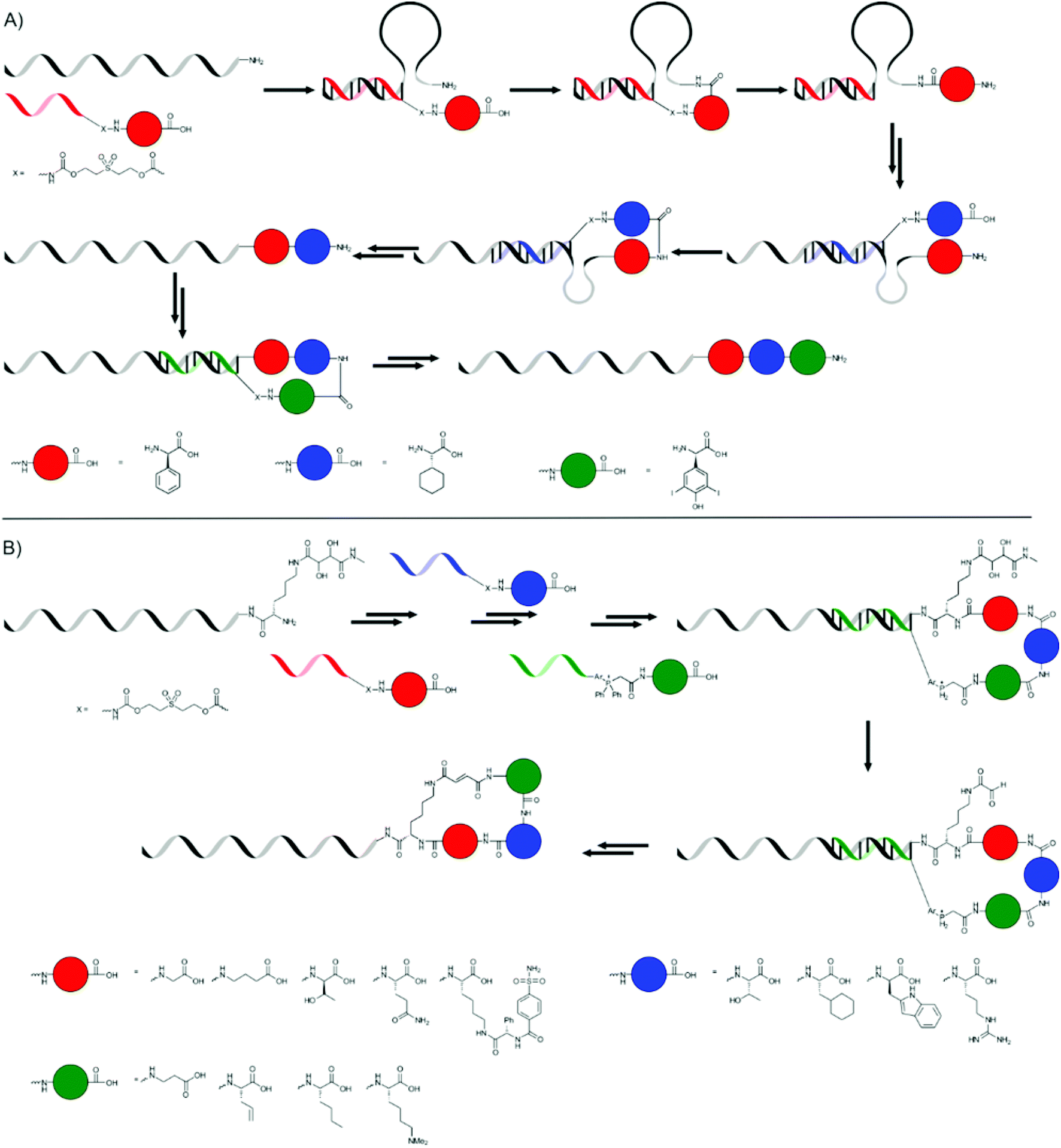 | ||
| Fig. 7 The synthesis of tripeptides (A) and peptide macrocycles (B) presented by Liu and co-workers.65,66 | ||
In a second publication they used this same strategy to produce macrocycles using an extra Wittig olefination reaction for ring closure.66 An amino acid bearing a diol group on the 5′-terminus of the template DNA was used to provide the primary amine instead of a simple 5′-amine group. After three successive couplings, the last of which incorporated a phosphonium group, an aldehyde was generated by the oxidative cleavage of the diol group, making the Wittig olefination possible (Fig. 7b). The authors were able to produce a library of different macrocycles, and it even proved possible to prepare four different macrocycles in one pot using four different templates.
Liu and co-workers also showed that masking of the template using non-functional ssDNA of different lengths could be used to facilitate the one pot synthesis of tripeptides.67 The template DNA bearing a 5′-amine was hybridised with two ssDNA masks of different lengths, thereby masking two of the three coding regions. Subsequently, three ssDNA fragments 3′-functionalised with non-natural amino acids or biotin were added to the mixture at 4 °C. Since only one of the coding regions (the shortest one) is left unmasked, only the first of the ssDNAs can bind and only one amino acid is transferred to the template DNA. The hybridisation of both the spent first ssDNA and the shorter mask with the template is selectively disrupted by increasing the temperature to 37 °C, using the low melting temperatures of this hybridisation. This allows for binding of the second ssDNA to the second coding region and the subsequent addition of the second amino acid. A further increase of the temperature to 62 °C leads to the release of the last mask and the second spent ssDNA fragment, so that hybridisation of the final and longest ssDNA is made possible and addition of the biotin moiety can take place, yielding the desired product in an overall yield of 38% (Fig. 8). The formation of oligomers in the absence of the masks yielded a mixture of all possible products. In addition to the formation of peptide bonds also Wittig olefination could be used as the coupling reaction in this approach.
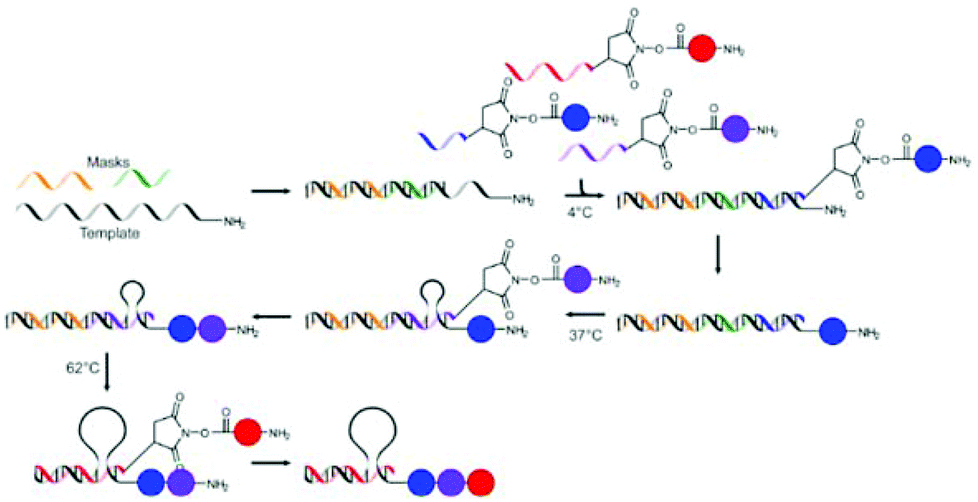 | ||
| Fig. 8 The stepwise addition of monomers is achieved through masking of part of the template ssDNA and stepwise removal of the masks using increasing temperature.67 | ||
Using ssDNAs of increasing lengths, all complementary to the same template molecule, also results in the replacement of the previous ssDNA through stronger binding (Fig. 9a). In this way hexamers could be prepared in an overall yield of 35%.68
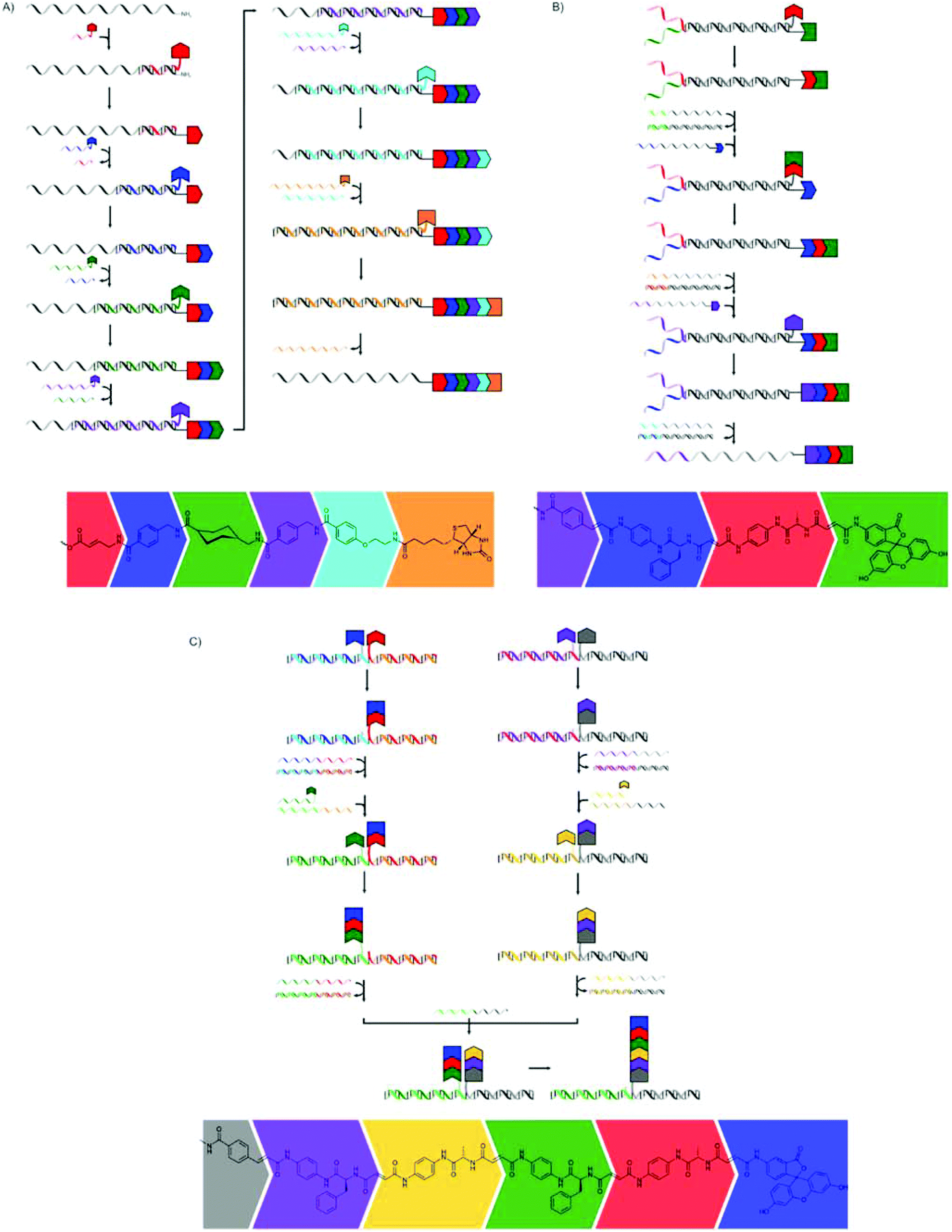 | ||
| Fig. 9 Various stepwise methods using the hybridisation of nucleotides for the synthesis of sequence-defined oligomers. (A) The use of ssDNAs of increasing lengths can be used to prepare hexamers.68 (B) The use of partially complementary ssDNA strands for the subsequent addition of monomers in Wittig reactions and removal of the empty strands using fully complementary ssDNA strands.69,70 (C) Binding of two ssDNA strands with monomer residues to a series of template ssDNA strands.71 | ||
The groups of Turberfield and O'Reilly used a strategy where two end-group functionalised ssDNA fragments are hybridised leaving a non-complementary section.69,70 After reaction of the monomers in a Wittig reaction the spent ssDNA is hybridised with a (non-functionalised) fully complementary ssDNA strand, thereby effectively removing it from the mixture, leaving the remaining ssDNA free to again be hybridised with an end-group functionalised ssDNA strand which is only partially complementary. Through repetition of such cycles a sequence defined polymer (up to decamers were shown) is formed (Fig. 9b).
The same groups also used the hybridisation between template ssDNA with two shorter, complementary end-group functionalised ssDNA strands in a stepwise DNA template coupling.71 One of the shorter strands carried a monomer residue at the 5′-terminus, while the other carried another monomer at the 3′-terminus. The addition of the template ssDNA brings the monomers (or, in the first step, a monomer and a monofunctional fluorescein end-group, FAM) close together, allowing the transfer of one of the monomers to the other chain by the Wittig reaction between an aldehyde and ylide functional group. Removal of the template and empty ssDNA, followed by addition of another template and ssDNA with terminal monomer group, results in the addition of another monomer. Such a stepwise procedure can be sped up by coupling oligomers (Fig. 9c).
The system currently closest to the strategy that nature uses in ribosomal translation was presented by the group of Liu, who used a single DNA strand as a template in a system that mimics the process that takes place in the ribosome.72 As tRNA mimics they used several single stranded RNAs 5′-functionalised with the NHS ester of an amino acid or another small molecules (which will later be the end groups). A section of the tRNA mimic is complementary to a codon section on the template (which contains four codons) while the rest contains a central recognition site for an RNA-cleaving DNAzyme. One of the tRNA mimics (initiator) serves to recognise the start codon, is shorter and does not include a cleavable site or amino acid. The final component is a DNA walker with a 3′-terminal amine group. The 5′-section of the DNA walker is complementary to the RNA of the tRNA mimic closest to the recognition section, followed by a DNAzyme sequence and another section complementary to the final section (closest to the amino acids) of the tRNA mimic. The formation of the oligomers using this procedure is shown in Fig. 10. The template and tRNA mimics self-assemble by hybridisation. The DNA walker and the initiator are added after self-assembly of the other components, ensuring translation starts at the right position. The 3′-section of the walker DNA remains free to hybridise with the 5′-side of the first tRNA mimic. The amine end-group on the walker DNA is at that stage in close proximity to the first activated amino acid group and the first amide bond is formed. The 5′-section binds equally strongly to a section of the first tRNA mimic and moves to that location, bringing the DNAzyme in the DNA walker close to the cleavable site in the tRNA mimic. The cleavage frees up the 3′-section of the DNA walker, which can again bind to the next tRNA mimic. This cycle repeats itself several times.
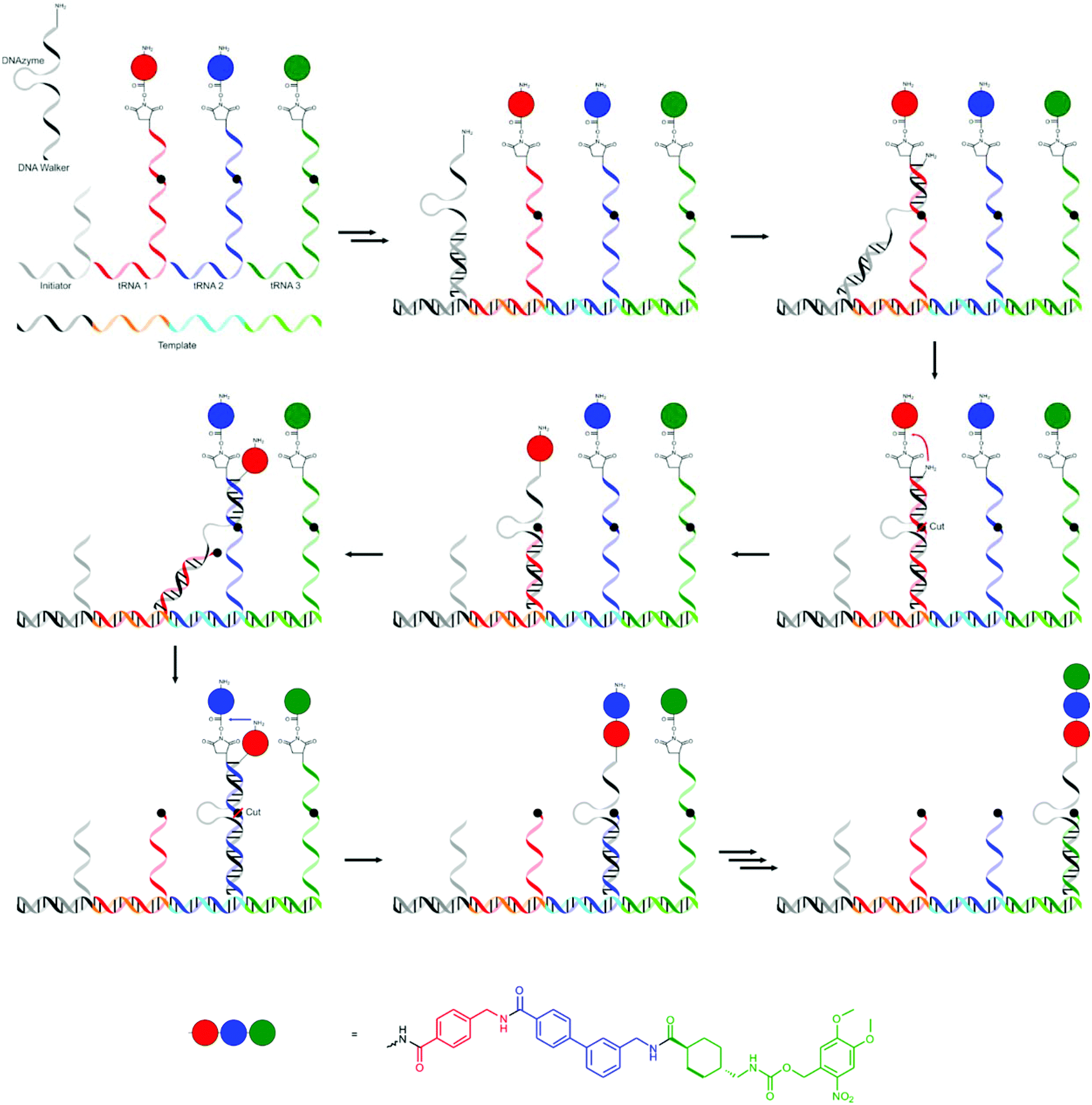 | ||
| Fig. 10 The strategy developed by Liu and co-workers mimicking the process of translation in ribosomes.72 | ||
Though the procedures described above only yield oligomers rather than true, high-molecular weight polymers, and similar oligomers can be prepared in much more straightforward procedures than the ones described above, e.g. through solid-phase synthesis methods, the work by the groups of Liu and the two groups of Turberfield and O'Reilly provide first steps toward novel methods for the synthesis of sequence defined polymers.
Schuster and co-workers used the hybridisation of (base modified) DNA strands for the preparation of sequence-defined linear and cyclic conducting oligomers,73 continuing earlier studies where conducting oligomers were prepared using similar strategies without introducing sequence control.74–76
Two different strategies are presented. The first uses two complementary DNA strands in which some of the cytosine bases are modified with aniline (ANi) or 2,5-bis(2-thienyl)-pyrrole (SNS) moieties. Hybridisation of the strands yields complexes where ANi and SNS moieties are in appropriate distances to each other to allow for the oxidative polymerisation of the moieties using horseradish peroxidase (HRP) and hydrogen peroxide (Fig. 11a).71 The relatively large size of the SNS groups requires their separation by one base,73 while ANi moieties are located on neighbouring bases.72 This strategy could be used to prepare block copolymers (or oligomers) and AABB-periodic copolymers of ANi and SNS residues.
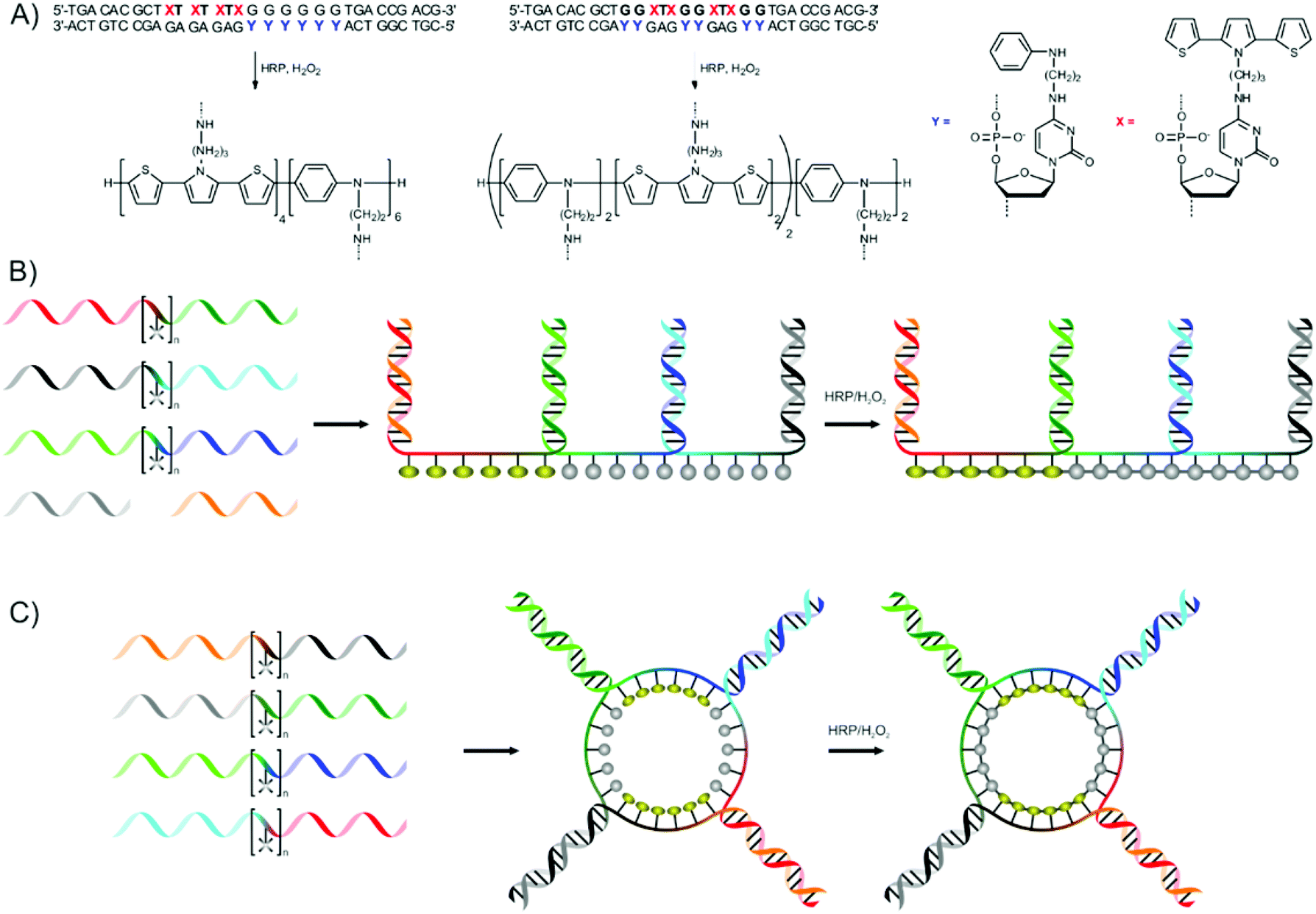 | ||
| Fig. 11 (A) Preparation of sequence-defined conducting polymers using the hybridisation of modified cytosine bases presented by Schuster and co-workers. X and Y represents 2,5-bis(2-thienyl)-pyrrole (SNS) and aniline (ANi) modified cytosine bases respectively. (B) Strategy for the preparation of linear and (C) cyclic sequence-defined conducting copolymers of SNS and ANi.73 | ||
A more modular strategy, based on the orthogonal hybridisation of flanking stretches of ssDNA on both sides of a central segment bearing SNS or ANi moieties, is also presented.71 Linear polymers become accessible by using three ssDNA building blocks together with two ‘caps’ (Fig. 11b). The central segment contains five ANi or six SNS moieties. By using different ssDNA building blocks a range of possible conducting homo- and copolymers could be prepared (ANi15, ANi10-b-SNS6, ANi5-b-SNS6-b-ANi5, SNS6-b-ANi5-b-SNS6, SNS12-b-ANi5 and SNS18) in good yields. Cyclic polymers are prepared using either three or four ssDNA building blocks which form cyclic assemblies (Fig. 11c shows the assemblies formed from four ssDNA building blocks). A range of cyclic polymers could be prepared by the oxidative polymerisation using HRP and H2O2, with different compositions and block sequences. Finally, it was shown that a longer central segment (bearing ten instead of five ANi moieties) could also be used to prepare cyclic copolymers. The spectroscopic properties of both linear and cyclic (co)polymers was shown to be dependent on the monomer sequence, and experiments using longer central segments bearing 10 ANi moieties showed that the spectroscopic properties of the conducting polymers were not influenced by the presence of DNA. A final step in which the DNA template is removed has not yet been reported by the authors, and the absence of hydrolysis sites might make this difficult. Nevertheless, the template-directed synthesis of conducting copolymers presents a novel method of creating well-defined materials for electronic- and optical applications.
Synthetic templates
The number of purely synthetic systems where template polymerisation is used to control monomer sequences is still severely limited. The earliest examples where a certain influence over the monomer sequence distribution can be obtained rely on the presence of homopolymers that interact specifically with one of the monomers.14,16 One example is from the work of Bamford et al., who investigated the N-carboxyanhydride (NCA) copolymerisation of the NCAs of L-glutamic acid ethyl ester or phenylalanine with the NCA of sarcosine.77 If a poly(sarcosine) homopolymer was used as the initiator, hydrogen bond interaction between poly(sarcosine) and the NH-groups in the NCAs of L-glutamic acid ethyl ester or phenylalanine led to the preferred incorporation of these monomers over the monomers of sarcosine, which does not interact with its homopolymer through H-bonding. Similarly, poly(2-vinylpyridine) as a template was shown to yield small blocks of poly(L-alanine) in copolymers of L-alanine and sarcosine (polymerised by NCA polymerisation).78 The same effect was observed during the copolymerisation of styrene and methacrylic acid in the presence of a poly(2-vinylpyridine) template in radical polymerisations.79The group of Sawamoto has more recently started exploring the use of template polymerisation for the selective incorporation of monomers as a route towards sequence-defined polymers in chain-growth polymerisations. Though their efforts have thus far focused on the specific incorporation of only a single monomer80,81 or on templated homopolymerisation,82 a number of different interactions have been shown to successfully yield selective addition of the complementary monomer.
The systems investigated by Sawamoto and co-workers are based on a bifunctional initiator capable of initiating cationic polymerisation and atom-transfer radical polymerisation. The two functional groups are placed on the ortho-positions of a phenyl group, thereby ensuring their proximity. A templating group (monomer or homopolymer) is added to the site designated for cationic polymerisation, whereas metal-catalysed atom-transfer radical addition is used to selectively add a complementary monomer. The authors have thus far shown the incorporation of methacrylic acid monomers in the presence of primary amine groups and the incorporation of sodium methacrylate using a 15-crown-5-ether template. By comparison, the addition of methyl methacrylate, benzyl methacrylate (in case of the amine template) or methacryloyloxyethyltrimethylammonium chloride (when using a crown ether template) did not proceed efficiently because of the absence of (or far weaker) interactions between the monomers and the template (Fig. 12).
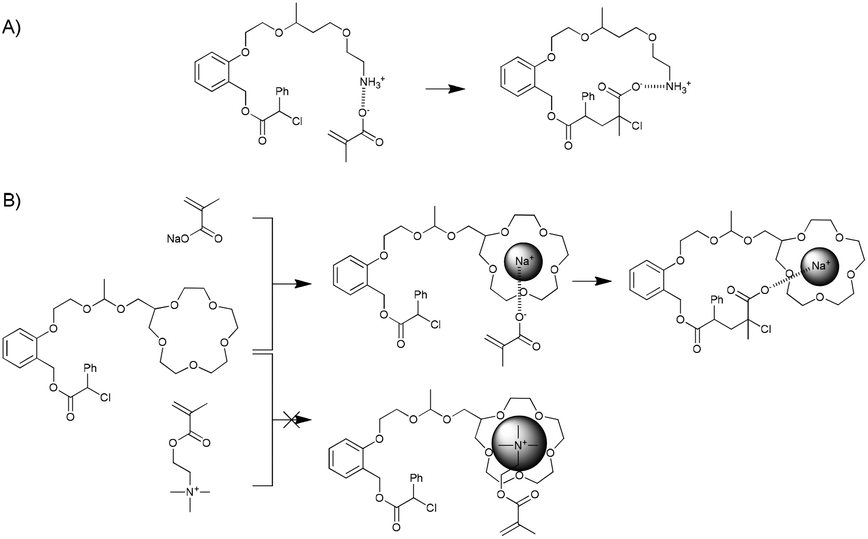 | ||
| Fig. 12 Templated addition of single monomers presented by Sawamoto and co-workers.80,81 | ||
Though it is debatable whether it should be considered a template polymerisation, supramolecular interactions between monomers can be used to influence monomer sequence. Early examples by Salamone et al. e.g. showed that attractive Coulombic forces between monomers can result in an increased degree of alternation in copolymers.83–86 Combinations of protonated 4-vinyl pyridine and p-styrenesulfonate, 2-methacryloyloxyethyltrimethylammonium and 2-methacryloyloxyethanesulfonate or 3-acrylamido-3-methylbutyltrimethylammonium 2-acrylamido-2-methylpropanesulfonate were used (Fig. 13). Similarly, hydrogen bonding between monomers can lead to the formation of highly alternating monomer sequences.87
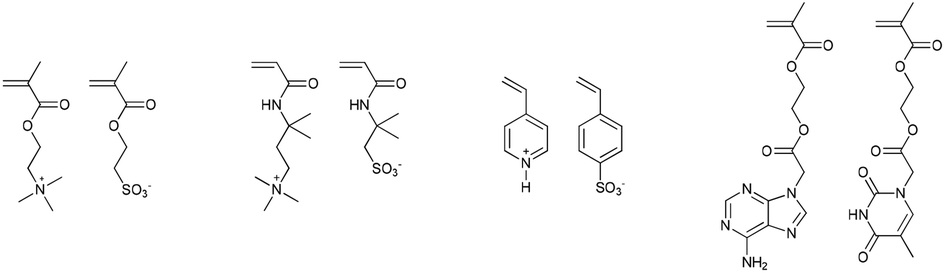 | ||
| Fig. 13 Monomer pairs where supramolecular interactions between the monomers lead to the formation of predominantly alternating monomer sequences.83–87 | ||
Jana et al. used metal–ligand complexation to create chiral metal complexes incorporating two polymerisable moieties ((meth)acrylates), Zn(II) and (−)-sparteine for the preparation of main-chain optically active polymers.88,89 Racemic diads spaced by a single different monomer are needed to obtain optically active polymers.90 The coordination of two (meth)acrylic acid monomers and optically active spartein to Zn(II), which is tetracoordinating, yielded a complex, the copolymerisation of which with aryl containing monomers (e.g. styrene or 2-vinylnaphthalene) led to the preferential formation of such triads and therefore optically active polymers, though the overall composition of the polymers indicated that the acrylic acid or methacrylic acid diads were mostly separated by more than one aryl containing monomer (Fig. 14a).
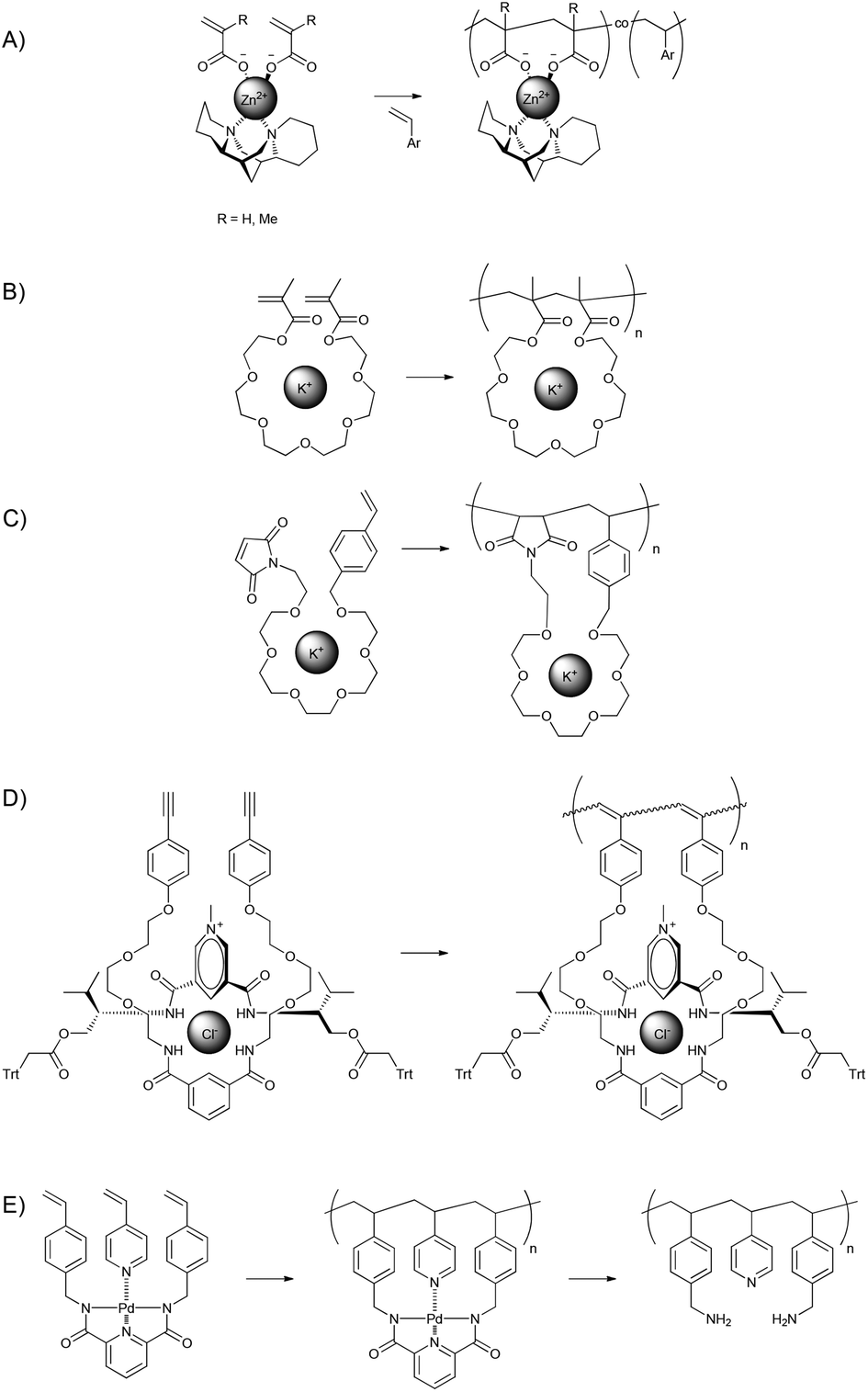 | ||
| Fig. 14 (A) Cyclocopolymerisation of a supramolecular complex of two (meth)acrylic acid monomers with aryl monomers.88,89 (B) Promotion of cyclopolymerisation of ethylene glycol containing bivalent monomers using potassium-cations.92 (C) Promotion of the cyclopolymerisation of a heterobifunctional monomer containing both a styrene and a maleimide moiety using a potassium-cation.93 (D) Promotion of cyclopolymerisation by the binding of chloride in a ternary complex.94 (E) Preparation of periodic copolymers with ABA-monomer sequences using a supramolecular complex of 4-vinylpyridine, palladium and a bivalent pincer containing monomer.95 | ||
The work of Jana et al. employs a synthetic pathway similar to cyclopolymerisation. In cyclopolymerisations bifunctional monomers are polymerised in such a way that the two polymerisable groups polymerise subsequently (intramolecularly), thereby yielding linear polymer chains with cyclic side-chains.91 A requirements for bifunctional monomers to polymerise in a cyclopolymerisation is that the polymerisable groups need to be in close proximity to each other. If this requirement is not fulfilled, branching or cross-linking can result. Therefore the linkers between the polymerisable groups are typically fairly short and promote their colocalisation using bulky groups.
A templating approach can be used to induce cyclopolymerisation for monomers with longer linkers though. If the polymerisable groups, i.e. methacrylate groups in this case, are linked by oligo-ethylene glycol (EG) linkers, ions can be used as templates.92 Crown-ether-like complexes are formed between the linkers and metal ions (Fig. 14b). A potassium cation was used to form complexes with dimethacrylate monomers with EG5, EG6 and EG8 monomers, though the highest degree of cyclisation was found for EG6. By promoting cyclopolymerisation, the intramolecular polymerisation of the two functional groups from one molecule was promoted, and thereby also a control over the monomer sequence is exerted. This same principle was utilised by Zou et al. for heterobifunctional monomers that contains both a styrene moiety and a maleimide moiety separated by a EG6 or EG7 spacer. Normally styrene and maleimide monomers copolymerise in an alternating fashion, but templating using a potassium-cation ensured that cyclopolymerisation took place and cross-linking was prevented.93 Isono et al. used the ternary complexation of a bivalent monomer, a chloride ion and a cationic, anion coordinating axle compound to promote the cyclopolymerisation of a bivalent monomer (Fig. 14d).94
Sawamoto and co-workers used a combination of metal–ligand complexation and cyclopolymerisation to create periodic copolymers (ABA sequences).95 A tridentate bifunctional monomer was used to complex Pd(II) while a 4-vinylpyridine monomer served as a monodentate ligand. The alignment of the polymerisable groups, which was additionally supported by the H-bonding of bulky fluoroalcohols to the carbonyl groups of the bifunctional monomer, yielded the desired ABA-sequences with a high degree of accuracy. Removal of the templating Pd(II) and hydrolysis of the bifunctional monomer yielded primary amine and pyridine side groups (Fig. 14e).
Conclusions and outlook
Template polymerisation as a method towards sequence-controlled synthetic polymers is a very useful pathway, though at present predominantly for the synthesis of structures closely related to biomacromolecules. Accordingly, the biggest successes have thus far been achieved using the machinery that nature has given rise to. The transcription and translation processes that biology uses could be employed for the incorporation of non-natural monomers with high accuracy by utilising polymerases or ribosomes, for example by using non-natural nucleic acids and non-canonical amino acids (or α-hydroxy acids). The accuracy achieved using nature's machinery also limits the scope of this approach, since only a limited number of monomers can be incorporated in this way, and modifications of the polymerisation systems remains highly challenging, while often selectivity is traded against accuracy. A wider variety of monomers can be incorporated using enzyme-free template polymerisations, even though the accuracy is a far cry from that achieved using specialised proteins, even when oligomers (rather than single monomers) are linked together. Nevertheless the formation of stable double stranded helices between the template and the daughter polymer is still required, a limitation that can be circumvented using various stepwise processes based on the hybridisation of nucleic acid strands, using which in theory any type of step-growth polymerisation can be developed into a sequence-defined polymerisation. Realistically, these methods are only suitable for the preparation of sequence-controlled oligomers though, and the synthesis is still very tedious and inefficient.The potential of template polymerisations using templates unrelated to nucleic acids is illustrated by the influence that can already be exerted over other properties of polymers, such as the molecular weight, dispersity and tacticity. Though some simple systems have been described where synthetic templates are used to control monomer sequence, this area is still in its infancy. One of the challenges has been that chain-growth polymerisations are often targeted, which are far harder to control than step-growth polymerisations due to the reactive nature of the ions and radicals that are used. Nevertheless promising results have already been obtained which no doubt will lead the way for novel strategies to also control the monomer sequence in polymers prepared through chain-growth polymerisations.
A second challenge is the availability of suitable templates. Whereas the synthesis of nucleic acids has been developed to a high degree, thanks in no small amount to the relevance of these biomacromolecules for biological and biochemical research, the synthesis of templates for other types of polymers still requires a lot of research since nucleic acids present their binding sites at regular distances not necessarily suitable for other polymerisations. Additionally, also the interactions between the template and monomers or the daughter polymer need to be suitable in order to align monomers and increase their effective concentration.
The effort still required to make control over monomer sequence using template polymerisations feasible, especially for polymers unrelated to biopolymers, is still substantial. In the end the benefits will likely outweigh the costs though, as a plethora of materials with novel properties could come within reach.
Acknowledgements
The author would like to thank the Chemical Industry Fund (FCI Liebig-fellowship) and the German Research Foundation (DFG, project number BR 4363/3-1) for financial support.References
- S. D. McCulloch and T. A. Kunkel, Cell Res., 2008, 18, 148–161 CrossRef CAS PubMed.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, W. H. Freeman and Company, New York, 5th edn, 2002 Search PubMed.
- W. Gilbert, Nature, 1986, 319, 618 CrossRef.
- A. Eschenmoser, Science, 1999, 284, 2118–2124 CrossRef CAS.
- G. F. Joyce, Nature, 2002, 418, 214–221 CrossRef CAS PubMed.
- R. Jones, Nat. Nanotechnol., 2008, 3, 699–700 CrossRef CAS PubMed.
- N. Badi and J.-F. Lutz, Chem. Soc. Rev., 2009, 38, 3383–3390 RSC.
- J.-F. Lutz, Polym. Chem., 2010, 1, 55–62 RSC.
- M. Ouchi, N. Badi, J.-F. Lutz and M. Sawamoto, Nat. Chem., 2011, 3, 917–924 CrossRef CAS PubMed.
- N. Badi, D. Chan-Seng and J.-F. Lutz, Macromol. Chem. Phys., 2013, 214, 135–142 CrossRef CAS.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed.
- J.-F. Lutz, in Sequence-Controlled Polymers: Synthesis, Self-Assembly, and Properties, ed. J.-F. Lutz, T. Y. Meyer, M. Ouchi and M. Sawamoto, ACS Symposium Series 1170, American Chemical Society, Washington, DC, 2014, ch. 1, pp. 1–11 Search PubMed.
- H. Colquhoun and J.-F. Lutz, Nat. Chem., 2014, 6, 455–456 CrossRef CAS PubMed.
- Y. Y. Tan, Prog. Polym. Sci., 1994, 19, 561–588 CrossRef CAS.
- S. Polowiński, Prog. Polym. Sci., 2002, 27, 537–577 CrossRef.
- S. Polowiński, Template Polymerization, ChemTec Publishing, Ontario, 1997 Search PubMed.
- K. Satoh and M. Kamigaito, Chem. Rev., 2009, 109, 5120–5156 CrossRef CAS PubMed.
- P. K. Lo and H. F. Sleiman, J. Am. Chem. Soc., 2009, 131, 4182–4183 CrossRef CAS PubMed.
- L. E. Orgel, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 99–123 CrossRef CAS PubMed.
- J. P. Ferris and G. Ertem, Science, 1992, 257, 1387–1389 CAS.
- S. Miyakawa and J. P. Ferris, J. Am. Chem. Soc., 2003, 125, 8202–8208 CrossRef CAS PubMed.
- F. Olasagasti, H. J. Kim, N. Pourmand and D. W. Deamer, Biochimie, 2011, 93, 556–561 CrossRef CAS PubMed.
- X. Li and D. R. Liu, Angew. Chem., Int. Ed., 2004, 43, 4848–4870 CrossRef CAS PubMed.
- Y. Brudno and D. R. Liu, Chem. Biol., 2009, 16, 265–276 CrossRef CAS PubMed.
- V. B. Pinheiro, A. I. Taylor, C. Cozens, M. Abramov, M. Renders, S. Zhang, J. C. Chaput, J. Wengel, S.-Y. Peak-Chew, S. H. McLaughlin, P. Herdewijn and P. Holliger, Science, 2012, 336, 341–344 CrossRef CAS PubMed.
- A. A. Henry and F. E. Romesberg, Curr. Opin. Biotechnol., 2005, 16, 370–377 CrossRef CAS PubMed.
- R. C. Holmberg, A. A. Henry and F. E. Romesberg, Biomol. Eng., 2005, 22, 39–49 CrossRef CAS PubMed.
- S. Zhang, J. C. Blain, D. Zielinska, S. M. Gryaznov and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17732–17737 CrossRef CAS PubMed.
- I. Hirao, M. Kimoto and R. Yamashige, Acc. Chem. Res., 2012, 45, 2055–2065 CrossRef CAS PubMed.
- R. Hili, J. Niu and D. R. Liu, J. Am. Chem. Soc., 2013, 135, 98–101 CrossRef CAS PubMed.
- O. L. Acevedo and L. E. Orgel, J. Mol. Biol., 1987, 197, 187–193 CrossRef CAS.
- L. E. Orgel, Acc. Chem. Res., 1995, 28, 109–118 CrossRef CAS.
- J. G. Schmidt, L. Christensen, P. E. Nielsen and L. E. Orgel, Nucleic Acids Res., 1997, 25, 4792–4796 CrossRef CAS PubMed.
- J. G. Schmidt, P. E. Nielsen and L. E. Orgel, Nucleic Acids Res., 1997, 25, 4797–4802 CrossRef CAS PubMed.
- I. A. Kozlov, S. Pitsch and L. E. Orgel, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 13448–13452 CrossRef CAS.
- I. A. Kozlov, P. K. Politis, A. van Aerschot, R. Busson, P. Herdewijn and L. E. Orgel, J. Am. Chem. Soc., 1999, 121, 2653–2656 CrossRef CAS.
- I. A. Kozlov, B. de Bouvere, A. van Aerschot, P. Herdewijn and L. E. Orgel, J. Am. Chem. Soc., 1999, 121, 5856–5859 CrossRef CAS.
- I. A. Kozlov, L. E. Orgel and P. E. Nielson, Angew. Chem., Int. Ed., 2000, 39, 4292–4295 CrossRef CAS.
- I. A. Kozlov, M. Zielinski, B. Allart, L. Kerremans, A. van Aerschot, R. Busson, P. Herdewijn and L. E. Orgel, Chem. – Eur. J., 2000, 6, 151–155 CrossRef CAS.
- J. A. R. Stutz and C. Richert, J. Am. Chem. Soc., 2001, 123, 12718–12719 CrossRef.
- J. C. Leitzel and D. G. Lynn, Chem. Rec., 2001, 1, 53–62 CrossRef CAS.
- A. H. El-Sagheer and T. Brown, Acc. Chem. Res., 2012, 45, 1258–1267 CrossRef CAS PubMed.
- D. M. Rosenbaum and D. R. Liu, J. Am. Chem. Soc., 2003, 125, 13924–13925 CrossRef CAS PubMed.
- R. E. Kleiner, Y. Brudno, M. E. Birnbaum and D. R. Liu, J. Am. Chem. Soc., 2008, 130, 4646–4659 CrossRef CAS PubMed.
- Y. Brudno, M. E. Birnbaum, R. E. Kleiner and D. R. Liu, Nat. Chem. Biol., 2010, 6, 148–155 CrossRef CAS PubMed.
- Y. Ura, J. M. Beierle, L. J. Leman, L. E. Orgel and M. R. Ghadiri, Science, 2009, 325, 73–77 CrossRef CAS PubMed.
- L. Wang, J. Xie and P. G. Schultz, Annu. Rev. Biophys. Biomol. Struct., 2006, 35, 225–249 CrossRef CAS PubMed.
- J. Xie and P. G. Schultz, Methods, 2005, 36, 227–238 CrossRef CAS PubMed.
- A. Ohta, Y. Yamagishi and H. Suga, Curr. Opin. Chem. Biol., 2008, 12, 159–167 CrossRef CAS PubMed.
- Z. Tan, A. C. Forster, S. C. Blacklow and V. W. Cornish, J. Am. Chem. Soc., 2004, 126, 12752–12753 CrossRef CAS PubMed.
- T. Passioura and H. Suga, Chem. – Eur. J., 2013, 19, 6530–6536 CrossRef CAS PubMed.
- T. Kawakami, H. Murakami and H. Suga, Nucleic Acids Symp. Ser., 2007, 51, 361–362 CrossRef PubMed.
- T. Kawakami, H. Murakami and H. Suga, Chem. Biol., 2008, 15, 32–42 CrossRef CAS PubMed.
- A. O. Subtelny, M. C. T. Hartman and J. W. Szostak, J. Am. Chem. Soc., 2008, 130, 6131–6136 CrossRef CAS PubMed.
- B. Zhang, Z. Tan, L. Gartenmann Dickson, M. N. L. Nalam, V. W. Cornish and A. C. Forster, J. Am. Chem. Soc., 2007, 129, 11316–11317 CrossRef CAS PubMed.
- A. Frankel, S. W. Millward and R. W. Roberts, Chem. Biol., 2003, 10, 1043–1050 CrossRef CAS PubMed.
- L. M. Dedkova, N. E. Fahmi, S. Y. Golovine and S. M. Hecht, J. Am. Chem. Soc., 2003, 125, 6616–6617 CrossRef CAS PubMed.
- L. M. Dedkova, N. E. Fahmi, S. Y. Golovine and S. M. Hecht, Biochemistry, 2006, 45, 15541–15551 CrossRef CAS PubMed.
- A. Ohta, H. Murakami, E. Higashimura and H. Suga, Chem. Biol., 2007, 14, 1315–1322 CrossRef CAS PubMed.
- A. Ohta, H. Murakami and H. Suga, ChemBioChem, 2008, 9, 2773–2778 CrossRef CAS PubMed.
- S. Fahnestock and A. Rich, Science, 1971, 173, 340–343 CAS.
- Y. Goto, T. Katoh and H. Suga, Nat. Protoc., 2011, 6, 779–790 CrossRef CAS PubMed.
- H. Kries, R. Wachtel, A. Pabst, B. Wanner, D. Niquille and D. Hilvert, Angew. Chem., Int. Ed., 2014, 53, 10105–10108 CrossRef CAS PubMed.
- J. Niu, R. Hili and D. R. Liu, Nat. Chem., 2013, 5, 282–292 CrossRef PubMed.
- Z. J. Gartner, M. W. Kanan and D. R. Liu, J. Am. Chem. Soc., 2002, 124, 10304–10306 CrossRef CAS PubMed.
- Z. J. Gartner, B. N. Tse, R. Grubina, J. B. Doyon, T. M. Snyder and D. R. Liu, Science, 2004, 305, 1601–1605 CrossRef CAS PubMed.
- T. M. Snyder and D. R. Liu, Angew. Chem., Int. Ed., 2005, 44, 7379–7382 CrossRef CAS PubMed.
- Y. He and D. R. Liu, J. Am. Chem. Soc., 2011, 133, 9972–9975 CrossRef CAS PubMed.
- M. L. McKee, P. J. Milnes, J. Bath, E. Stulz, A. J. Turberfield and R. K. O'Reilly, Angew. Chem., Int. Ed., 2010, 49, 7948–7951 CrossRef CAS PubMed.
- P. J. Milnes, M. L. McKee, J. Bath, L. Song, E. Stulz, A. J. Turberfield and R. K. O'Reilly, Chem. Commun., 2012, 48, 5614–5616 RSC.
- M. L. McKee, P. J. Milnes, J. Bath, E. Stulz, R. K. O'Reilly and A. J. Turberfield, J. Am. Chem. Soc., 2012, 134, 1446–1449 CrossRef CAS PubMed.
- Y. He and D. R. Liu, Nat. Nanotechnol., 2010, 5, 778–782 CrossRef CAS PubMed.
- W. Chen and G. B. Schuster, J. Am. Chem. Soc., 2013, 135, 4438–4449 CrossRef CAS PubMed.
- B. Datta, G. B. Schuster, A. McCook, S. C. Harvey and K. Zakrzewska, J. Am. Chem. Soc., 2006, 128, 14428–14429 CrossRef CAS PubMed.
- B. Datta and G. B. Schuster, J. Am. Chem. Soc., 2008, 130, 2965–2973 CrossRef CAS PubMed.
- W. Chen and G. B. Schuster, J. Am. Chem. Soc., 2012, 134, 840–843 CrossRef CAS PubMed.
- C. H. Bamford, H. Block and Y. Imanishi, Biopolymers, 1966, 4, 1067–1072 CrossRef CAS.
- R. A. Volpe and H. L. Frisch, Macromolecules, 1987, 20, 1747–1752 CrossRef CAS.
- H. L. Frisch and Q. Xu, Macromolecules, 1992, 25, 5145–5149 CrossRef CAS.
- S. Ida, T. Terashima, M. Ouchi and M. Sawamoto, J. Am. Chem. Soc., 2009, 131, 10808–10809 CrossRef CAS PubMed.
- S. Ida, M. Ouchi and M. Sawamoto, J. Am. Chem. Soc., 2010, 132, 14748–14750 CrossRef CAS PubMed.
- S. Ida, M. Ouchi and M. Sawamoto, Macromol. Rapid Commun., 2011, 32, 209–214 CrossRef CAS PubMed.
- J. C. Salamone, A. C. Watterson, T. D. Hsu, C. C. Tsai, M. U. Mahmud, A. W. Wisniewski and S. C. Israel, J. Polym. Sci., Polym. Symp., 1978, 64, 229–213 CrossRef CAS.
- J. C. Salamone, C.-C. Tsai and A. C. Watterson, J. Macromol. Sci., Chem., 1979, 13, 665–672 CrossRef.
- J. C. Salamone, L. Quach, A. C. Watterson, S. Krauser and M. U. Mahmud, J. Macromol. Sci., Chem., 1985, 22, 653–664 CrossRef.
- J. C. Salamone, M. K. Raheja, Q. Anwaruddin and A. C. Watterson, J. Polym. Sci., Polym. Lett. Ed., 1985, 23, 655–659 CrossRef CAS.
- Y. Kang, A. Lu, A. Ellington, M. C. Jewett and R. K. O'Reilly, ACS Macro Lett., 2013, 2, 581–586 CrossRef CAS.
- S. Jana and D. C. Sherrington, Angew. Chem., Int. Ed., 2005, 44, 4804–4808 CrossRef CAS PubMed.
- S. Jana, P. A. G. Cormack, A. R. Kennedya and D. C. Sherrington, J. Mater. Chem., 2009, 19, 3427–3442 RSC.
- G. Wulff, Angew. Chem., Int. Ed. Engl., 1989, 28, 21–37 CrossRef.
- G. B. Butler, Cyclopolymerization and cyclocopolymerization, Marcel Dekker, Inc., New York, 1992 Search PubMed.
- T. Terashima, M. Kawabe, Y. Miyabara, H. Yoda and M. Sawamoto, Nat. Commun., 2013, 4, 2321 Search PubMed.
- L. Zou, J. Liu, K. Zhang, Y. Chen and F. Xi, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 330–338 CrossRef CAS.
- T. Isono, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3184–3192 CrossRef CAS.
- Y. Hibi, M. Ouchi and M. Sawamoto, Angew. Chem., Int. Ed., 2011, 50, 7434–7437 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |