
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning thermal properties and microphase separation in aliphatic polyester ABA copolymers†
J. P.
MacDonald
a,
M. P.
Parker
b,
B. W.
Greenland
b,
D.
Hermida-Merino
c,
I. W.
Hamley
b and
M. P.
Shaver
*a
aSchool of Chemistry, University of Edinburgh, Joseph Black Building, West Mains Road, Edinburgh, EH9 3FJ, UK. E-mail: michael.shaver@ed.ac.uk; Tel: +44 (0)131 650 4726
bSchool of Chemistry, Pharmacy and Food Biosciences, University of Reading, Whiteknights, Reading, Berkshire RG6 6AD, UK
cDUBBLE @ ESRF – The European Synchrotron, CS40220, 38043 Grenoble Cedex 9, France
First published on 1st December 2014
Abstract
Four alkyl substituted β-lactones were investigated as monomers in ring opening polymerisation to produce a family of poly(3-hydroxyalkanoate)s. Homopolymers were synthesised using a robust aluminium salen catalyst, resulting in polymers with low dispersity (Đ < 1.1) and predictable molecular weights. ABA triblock copolymers were prepared using poly(L-lactic acid) as the A block and the aforementioned poly(3-hydroxyalkanoate) as the B block via a sequential addition method. Characterisation of these copolymers determined they were well controlled with low dispersities and predictable molecular weight. DSC analysis determined copolymers prepared from β-butyrolactone or β-valerolactone yielded polymers with tunable and predictable thermal properties. Copolymers prepared from β-heptanolactone yielded a microphase separated material as indicated by SAXS, with two distinct Tgs. The polymers could be readily cast into flexible films and their improved tensile properties were explored.
Introduction
Biodegradable polyesters have been of growing interest in recent years with poly(lactic acid) (PLA) and poly(3-hydroxybutyrate) (P(3-HB)) finding use in various specialty and commodity applications.1–4 Ring-opening polymerisation (ROP) of the corresponding cyclic esters, catalysed by either an organocatalyst or a metal complex, has been investigated for improved control over the polymer properties by affording polymers with predictable molecular weight and low dispersity while minimising chain transfer and termination reactions.5–7 These controlled polymerisations also offer the ability to access more complicated macrostructures and facilitates precision polymer synthesis. However, due to the vast majority of reports focusing on a limited number of monomers, namely lactide, glycolide and ε-caprolactone, the range of accessible materials is also quite limited.8–11To grow this range of polymers, aliphatic polyester copolymers have been explored.12,13 One class of particular interest is copolymers of PLA and P(3-HB). Due to the difficulty in polymerising β-butyrolactone (β-BL),4 both the homopolymerisation and copolymerisation of β-BL have seen limited study. The first report of copolymerisations incorporating β-BL was from Hori et al. with a distannoxane complex to make copolymers of L-lactide (L-LA) and (R)-β-BL.14 The product had random monomer incorporation, high molecular weight and a broad dispersity (Đ = 1.7). A subsequent report involved a more detailed investigation of the effects of varying monomer feed ratios.15 The copolymers reported were again high molecular weight with random monomer incorporation and broad dispersities (Đ = 1.8–2.3) and thermal properties that supported a homogeneous polymer sample with no phase separation. Since this report, several groups have developed different systems to synthesise P(LA-co-3-HB).16–20 Our group recently reported the copolymerisation of rac-β-BL and rac-lactide (LA) with aluminium salen (MeAl[salen]BuBu-Pr (1), MeAl[salen]AdMe-Et (2), Fig. 1) and aluminium salan (MeAl[salan]ClClBn-Et (3), Fig. 1) complexes.21
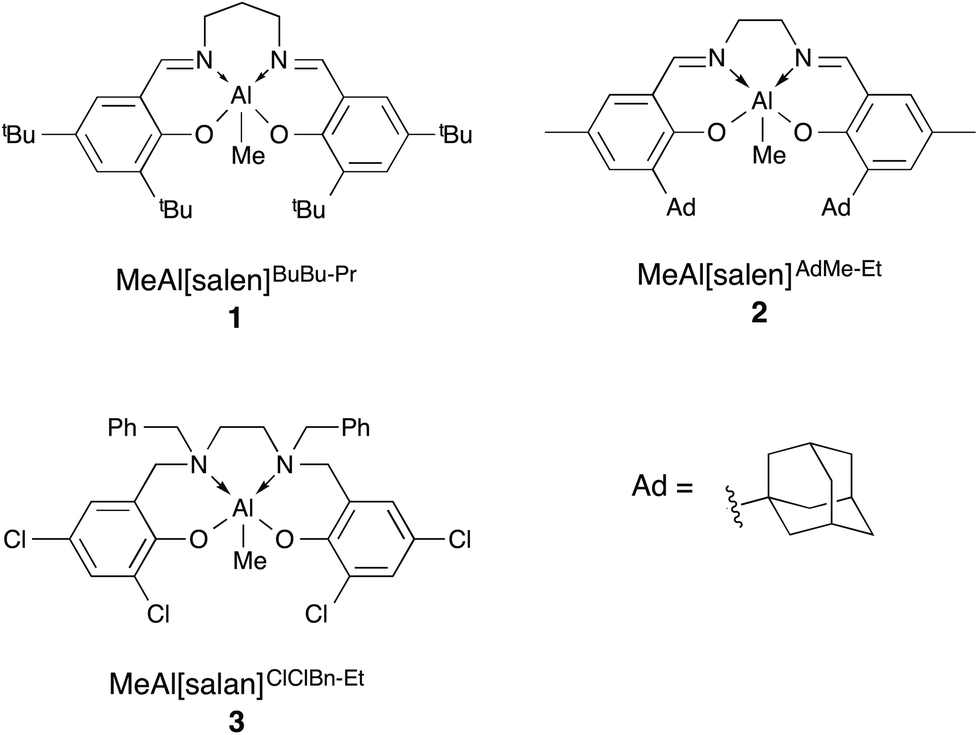 | ||
| Fig. 1 Aluminium salen and salan complexes for copolymerisation of β-BL and lactide. | ||
Interestingly, bulk copolymerisation with 3 at 120 °C, the system favored incorporation of lactide over β-BL despite the rate of β-BL homopolymerisation being significantly higher than for lactide. PLA to P(3-HB) ratios varied from the expected values from 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (initial LA:β-BL = 6:1) to 1:2 (initial LA:β-BL = 1:6). Furthermore, 1H NMR spectra showed strong heterotactic PLA signals, indicative of long uninterrupted PLA segments and a highly gradient copolymer. Although the PLA:P(3-HB) ratios varied from the expected values, dispersities indicated the polymerisation was still very well controlled, with values less than 1.1 in all cases. Copolymerisations carried out at 85 °C in toluene with 1, 2 and 3 were also successful, although 1 gave relatively uncontrolled copolymer (Đ > 1.4).
:1 (initial LA:β-BL = 6:1) to 1:2 (initial LA:β-BL = 1:6). Furthermore, 1H NMR spectra showed strong heterotactic PLA signals, indicative of long uninterrupted PLA segments and a highly gradient copolymer. Although the PLA:P(3-HB) ratios varied from the expected values, dispersities indicated the polymerisation was still very well controlled, with values less than 1.1 in all cases. Copolymerisations carried out at 85 °C in toluene with 1, 2 and 3 were also successful, although 1 gave relatively uncontrolled copolymer (Đ > 1.4).
More recently, advances have been made in the synthesis of block copolymers. Hiki et al. synthesised the first block copolymers of lactide and β-BL based on the method of Kricheldorf.22,23 The ABA block copolymers were synthesised through a multistep synthesis using the bifunctional initiator 1,4-butanediol to first generate the middle P(3-HB) block, followed by growing the end P(L-LA) blocks simultaneously. Interestingly, the thermal properties of the polymers suggested that there was microphase separation between the elastomeric syndiotactic P(3-HB) block and the brittle P(L-LA) blocks. These polymers were the first reported biodegradable thermoplastic elastomers prepared through ROP. Aluthge et al. recently reported the first case of generating ABA block copolymers of PLA and P(3-HB) without the need of a multistep synthesis.24 The dinuclear indium catalyst used generated copolymers with relatively low dispersities (Đ = 1.16–1.32) and were high molecular weight (>100 kDa). The report had focused on using different lactide isomers to control PLA block tacticities.
Expanding the scope of biodegradable ABA copolymer systems outside of PLA and P(3-HB) increases polymer scope and range of accessible properties. The Hillmyer group have reported several systems that are either fully biodegradable or contain biodegradable segments. Their first report of ABA block copolymers containing biodegradable segments was derived from a α,ω-dihydroxyl functionalised polyisoprene as a macroinitiator in the ROP of lactide.25 These copolymers were of low dispersity and exhibited microphase separation.26 More recently, they have reported the synthesis of thermoplastic elastomeric ABA block copolymers derived from renewable plant based monomers, menthide and tulipalin A.27 While the copolymers produced in these reports were of low dispersity with excellent tensile properties, the monomers are relatively expensive and the polymer backbone that was not fully degradable. Bhowmick recently reported the synthesis of an ABA triblock copolymer of from lactide and δ-valerolactone using tin octanoate as a catalyst and 1,6-hexanediol as an initiatior.28 The copolymers synthesised were found to be low molecular weight (>15000 kDa) with have relatively broad dispersities (Đ = 1.2–1.5). Interestingly, the copolymers were found to have two Tgs that were tunable based on the monomer ratios. The system required precipitation and purification of the polymer between homopolymer and ABA triblock copolymer.
While both homopolymerisation and copolymerisation using β-BL are well understood, there has not yet been a study of the effect of changing the nature of the alkyl substituent in β-lactone polymerisation. To address this, we now report our efforts in expanding the synthesis of ABA block copolymers of L-lactide and β-lactones while also using several different alkyl chain lengths (methyl, ethyl and n-butyl) and their relationship to microphase separation. We will also study the effect of molecular weight and monomer feed ratio as well as varying the monomer alkyl group while maintaining full polymer backbone degradability. L-Lactide was chosen to allow formation of more brittle A blocks and a larger difference in Tg of A and B blocks. The catalyst chosen was the aforementioned aluminium salen complex, 1, active in the living and immortal polymerisation of both lactide and β-BL.21 Aluminium salen and salan complexes have shown great promise in controlled ROP, building on the early work from Spassky29–32 and Gibson,33 respectively. We also aimed for a simple synthesis by employing a sequential addition method (Scheme 1).
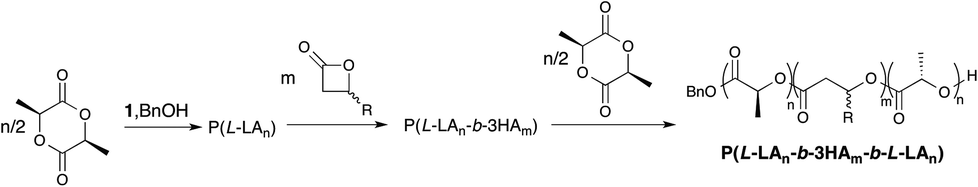 | ||
| Scheme 1 General scheme for aluminium catalysed ABA copolymer synthesis. | ||
Experimental
General methods
All experiments involving moisture- and air-sensitive compounds were performed under a nitrogen atmosphere using an MBraun LABmaster sp glovebox system equipped with a −35 °C freezer and [H2O] and [O2] analysers or using standard Schlenk techniques. Gel permeation chromatography (GPC) was carried out in THF at a flow rate of 1 mL min−1 on a Malvern Instruments Viscotek 270 GPC Max triple detection system with 2 × mixed bed styrene/DVB columns (300 × 7.5 mm). P(L-LA) and P(3-HB) dn/dc values were 0.05034 and 0.065,35 respectively. The dn/dc values for P(3-HP), P(3-HH) and P(3-HTD) were calculated using OmniSEC 4.7 software to be 0.060, 0.059 and 0.059, respectively. 1H NMR spectra were recorded at 298 K with Bruker Avance spectrometers (400 or 500 MHz) in CDCl3 or C6D6. Differential scanning calorimetry (DSC) was carried out using a TA Instruments DSC Q2000 instrument. The samples were heated from −90 °C to 200 °C at a rate of 10 °C min−1 after an initial heating scan to 200 °C (heating rate of 20 °C min−1) to remove any residual solvent. Values of Tg, Tm and Tc were obtained from the 2nd heating scan (−90 °C to 200 °C). Tg values were determined from the midpoint of the transition, while Tm and Tc values were calculated as the peak endotherm or exotherm of the respective transitions. Synchrotron Small-angle X-ray Scattering (SAXS) and Wide-angle X-ray Scattering (WAXS) measurements were performed on BM26B (DUBBLE) at the European Synchrotron Radiation Source, Grenoble, France.36 The sample to SAXS detector distance was 2.095 m using a wavelength λ = 1.033 Å. A Dectris-Pilatus 1 M detector with a resolution of 981 × 1043 pixels and a pixel size of 172 × 172 μm was employed used to record the 2D SAXS scattering patterns. Standard corrections for sample absorption and background subtraction have been performed. The data were normalized to the intensity of the incident beam (in order to correct for primary beam intensity fluctuations) and were corrected for absorption, background scattering. The scattering pattern from rat tail collagen was used for the calibration of the wavenumber (q = 4πsinθ/λ) scale of the scattering curve. The sample was placed in a DSC pan modified with kapton windows and heated using a Linkam hotstage from −50 °C to 150 °C and cooled back to −50 °C and then heated again to 150 °C all at a rate of 5 °C min−1. SAXS data frames were acquired each 30 seconds during this process. Tensile measurements were obtained using a TA XTplus Texture Analyser using Texture Exponent 32 software under ambient conditions. Films were cast by slow evaporation of CHCl3 followed by drying. Samples of 3 mm × 40 mm were cut from the middle of the film to avoid edge defects. To avoid sample slippage or damage from the instrument grips, the top and bottom of the sample were placed between cardboard before loading.
Materials
MeAl[salen]BuBu-Pr,37,38 MeAl[salan]ClClBn-Et33 and ClCr[TPP],39,40 were synthesised via modified literature procedures. Carbon monoxide (99.9%, BOC) was used as received. Benzyl alcohol was dried by refluxing over calcium hydride for 24 h, distilled under inert atmosphere and degassed by three freeze–pump–thaw cycles prior to use. Toluene, hexane, and THF were obtained from an Innovative Technologies solvent purification system, consisting of columns of alumina and copper catalyst. The solvents were degassed by three freeze–pump–thaw cycles prior to use. L-Lactide was purified by three vacuum sublimations and dried under reduced pressure for 18 h prior to use. Epoxides 1,2-epoxybutane, 1,2-epoxyhexane and 1,2-epoxydodecane, were purchased from Sigma-Aldrich and dried by stirring over calcium hydride for 48 h, distilled under inert atmosphere and degassed by three freeze–pump–thaw cycles prior to use. β-Butyrolactone was purchased from Sigma-Aldrich and dried by stirring over CaH2 for 48 h, distilled under inert atmosphere and degassed by three freeze–pump–thaw cycles prior to use.
Representative synthesis of β-lactone
ClCr[TPP] (7 mg, 0.010 mmol), Co2(CO)8 (5 mg, 0.015 mmol) and 1,2-epoxybutane (2.9 g, 40 mmol) in THF (5 mL) was added to an oven dried ampoule. The solution was then transferred to a 40 mL Parr reactor under nitrogen. The reactor was filled with approximately 25 psi CO and allowed to stir for five minutes. The reactor was carefully vented and filled with 500 psi CO and heated to 70 °C. The vessel was heated until CO pressure had stopped decreasing (ca. 3 h). The vessel was carefully vented and the crude mixture was distilled under reduced pressure to afford β-valerolactone (3.9 g, 97%).Representative synthesis of homopolymer
MeAl[salen]BuBu-Pr (23 mg, 0.04 mmol), BnOH (4.4 μL, 0.04 mmol) and β-valerolactone (430 mg, 4.20 mmol) in toluene (4 mL) was added to an oven dried ampoule. The ampoule was then sealed and heated to 85 °C for 6 h. The reaction was then quenched by addition of two drops of MeOH and samples were taken for 1H NMR and GPC analysis. The remainder was added dropwise to cold methanol and upon cooling to −35 °C for two days a colorless solid was separated.Representative synthesis of ABA copolymers
MeAl[salen]BuBu-Pr (38 mg, 0.07 mmol), BnOH (7.2 μL, 0.07 mmol) and L-lactide (100 mg, 0.70 mmol) in toluene (3 mL) was added to an oven dried ampoule. The ampoule was then heated to 85 °C for 3 h. β-Valerolactone (700 mg, 7 mmol) was then added under N2 and the solution was continued to heat for 18 h. L-Lactide (100 mg, 0.7 mmol) in toluene (1 mL) was then added under N2 and stirred for 3 h. The reaction was then quenched by addition of two drops of MeOH and samples were taken for 1H NMR and GPC analysis. The remainder was added dropwise to cold methanol and left in a freezer overnight, filtered and dried under vacuum to constant weight.Results and discussion
Synthesis of β-lactones
While there have been several active metal complexes as catalysts for the synthesis of β-lactones via carbonylation of epoxides,41–45 we chose a chromium porphyrin complex due to its low catalyst loading for terminal alkyl substituted epoxides.46 In a recent report of the in situ formation of the parent complex first reported by Coates et al., β-lactones were synthesised in high yield with short reaction times (Scheme 2).47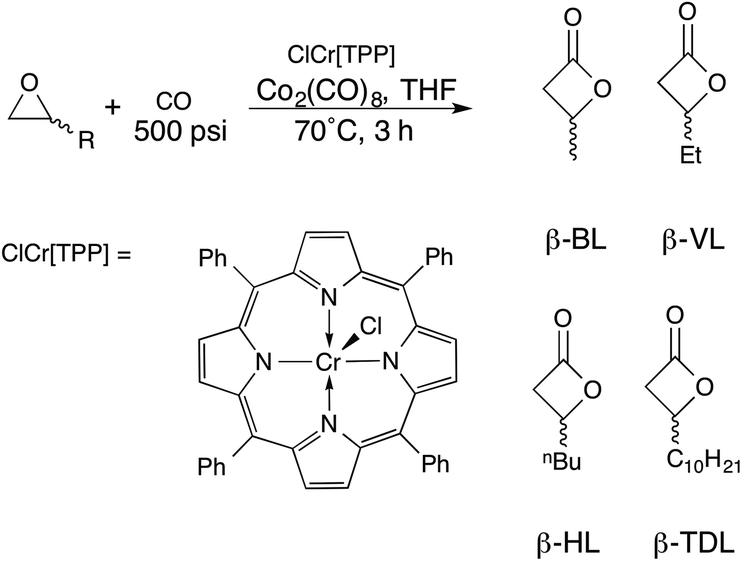 | ||
| Scheme 2 Synthesis of β-lactones by epoxide carbonylation. | ||
Using this strategy, β-valerolactone (R = Et, β-VL), β-heptanolactone (R = nBu, β-HL) and β-tridecalactone (R = C10H21, β-TDL) were prepared. As the alkyl chain increased in length, a higher catalyst loading was necessary to achieve quantitative conversion, maximized at 0.1% catalyst loading for β-TDL synthesis. This is likely due decreasing solubility of the catalyst as the chain length increases. Nevertheless, catalyst loadings were still quite low for each epoxide (Table S1†) and the β-lactones were easily separated from the crude product mixture either by distillation (β-HL, β-VL) or by simple column chromatography (β-TDL).
Homopolymerisation of β-VL, β-HL and β-TDL
Homopolymerisations of β-VL, β-HL and β-TDL were first investigated to gain an understanding of the system. While there have been several reports of homopolymerisation of β-VL48,49 and copolymerisation with β-BL,50–53 no homopolymerisation of β-HL or β-TDL have been reported. It is also worth noting that only a single report of copolymerisation of β-HL and β-BL has been previously reported.49 Homopolymerisations were carried out using 1 and were monitored by 1H NMR spectroscopy (Fig. 2). Complete conversion of β-VL was observed after 16 h at 85 °C. As expected for a bulkier lactone, polymerisation of β-VL showed a significant decrease in rate (kobs = 0.0065 min−1) compared to β-BL. Interestingly, the rate of β-HL polymerisation showed a further decrease in rate with the polymerisation reaching quantitative conversion in just 20 h (kobs = 0.0040 min−1). We decided to investigate whether or not increasing the alkyl chain significantly would still allow polymerisation to occur. Changing the alkyl group to C10H21 in β-TDL still allowed for complete conversion of the monomer, despite requiring 40 h (kobs = 0.0017 min−1). This is the first example of homopolymerisation of β-HL and β-TDL while being one of the first for β-VL.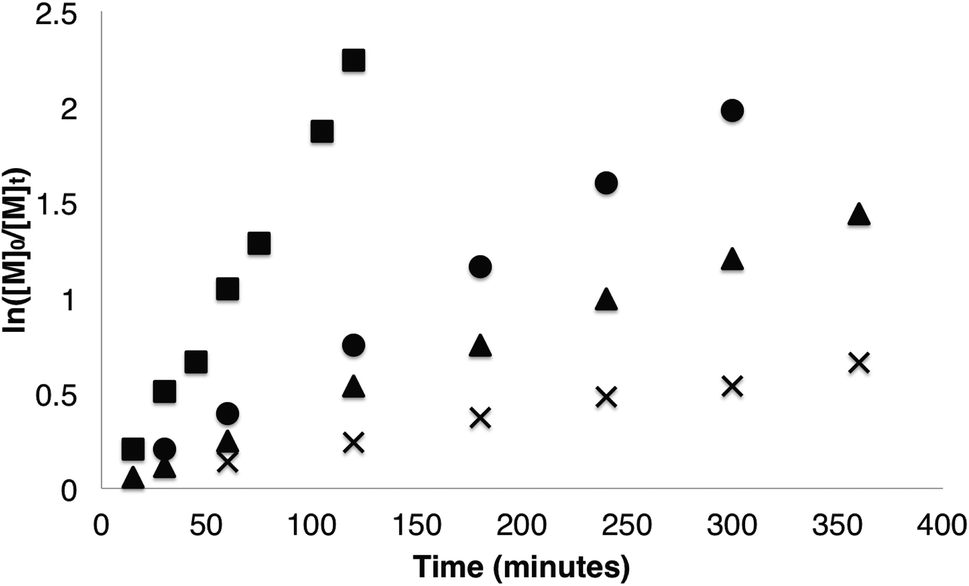 | ||
| Fig. 2 Kinetic plot of β-lactone homopolymerisation (■ = β-BL (0.0194 min−1), ● = β-VL (0.0065 min−1), ▲ = β-HL (0.0040 min−1), × = β-TDL (0.0017 min−1)). | ||
Dispersities were excellent, and ≤1.10 for all cases, while molecular weights were in good agreement with theoretical values. Increasing the temperature to 120 °C gave faster polymerisations yielding polymers with similar molecular weight and dispersity (Table S2†). Tin octanoate (Sn(oct)2) and an aluminium salan catalyst, 3, were also investigated for the homopolymerisation of β-VL and β-HL. Homopolymerisations with Sn(oct)2 were found to be unsuccessful, reaching only low conversion (<20%) after 6 h at 120 °C. Polymerisations at lower temperatures did not yield any polymer. Homopolymerisations using 3 yielded similar rates, molecular weights and dispersities to 1 (Table S3, Fig. S1†). Higher molecular weight P(3-HP) and P(3-HH) can also be synthesised (Table S4†). Dispersities remained low for most samples while reaching high conversion. However, molecular weights were lower than the expected values when [M]0/[Al]0/[BnOH]0 = 500:1.1.
The thermal properties for P(3-HP) and P(3-HH) were investigated by differential scanning calorimetry (DSC). Both samples showed no melt or crystallisation temperatures (Tm, Tc), indicating an amorphous nature, as expected, for atactic P(3-HA)s, especially considering that increasing the substituent length should further disrupt any crystallisation of the polymer. The glass transition temperature (Tg) was found to decrease from that of P(3-HB) to −19.8 °C and −31.8 °C for P(3-HP) and P(3-HH), respectively. This trend is consistent with the expected decrease in likelihood of order in these longer polymer chains.
ABA block copolymer synthesis
Our initial attempt to synthesise ABA block copolymers was by sequential addition at 70 °C. The first L-lactide block reached quantitative conversion after 4 h. β-Lactone was then added and heating was continued for 12 h. The 1H NMR indicated that no β-lactone was converted to polymer. To address this, once β-lactone was added, the reaction temperature was increased to 120 °C for 12 h. While the 1H NMR indicated the presence of poly(3-hydroxyalkanoate), there was also a significant amount of a side product, likely the corresponding carboxylic acid from monomer decomposition. Fortunately, excellent copolymerization could be achieved by conducting the polymerisations at 85 °C. Thus, to simplify the process, all block polymerisations of L-lactide and β-lactone blocks were conducted at 85 °C (Scheme 3). These polymers represent the first examples of block copolymers from β-VL or β-HL. Interestingly, the temperature at which the monomer is added was found to be important to keep dispersities low (Table S5†). Cooling the reaction and adding the monomer at room temperature is ineffective, with the one-pot reaction completed at 85 °C being the preferred synthetic strategy (Table 2). | ||
| Scheme 3 Synthetic route to ABA block copolymers by sequential addition. | ||
1H NMR spectroscopy
After complete conversion of the first P(L-LA) block, confirmed by 1H NMR spectroscopy, β-lactone was added at 85 °C. The solution was then allowed to stir at 85 °C until complete conversion of β-lactone was achieved. The L-lactide for the final block was dissolved in toluene and was again added at 85 °C. The absence of monomer methine protons (∼5.0 ppm for L-lactide, 4.5 for β-lactone) in the 1H NMR spectra of each block indicated >99% conversion for each block (Fig. S2†), suggesting no gradient formation.Overlapping polymer signals make it difficult to directly calculate the ratio of PLA:P(3-HA). In the case of P(3-HB), the methine regions of the two polymers were separated enough to integrate independently. However, for P(3-HP) and P(3-HH), the methine region overlapped with that of PLA. Thus, the total integration for the methine region of PLA and P(3-HA) was compared to the methyl group of the alkyl chain (∼0.85 ppm) to determine the relative ratio of PLA to P(3-HA). In all copolymers, the ratio of PLA to P(3-HA) was within 10% of the theoretical value. Similarly, ABA triblock copolymers were synthesised using L-lactide and β-BL, β-VL or β-HL. Due to the long homopolymerisation time of β-TDL, copolymers were not synthesised.
Gel permeation chromatography
To confirm the polymers were formed as ABA triblock copolymers, GPC analyses were performed after completion of each polymer block (Fig. 3).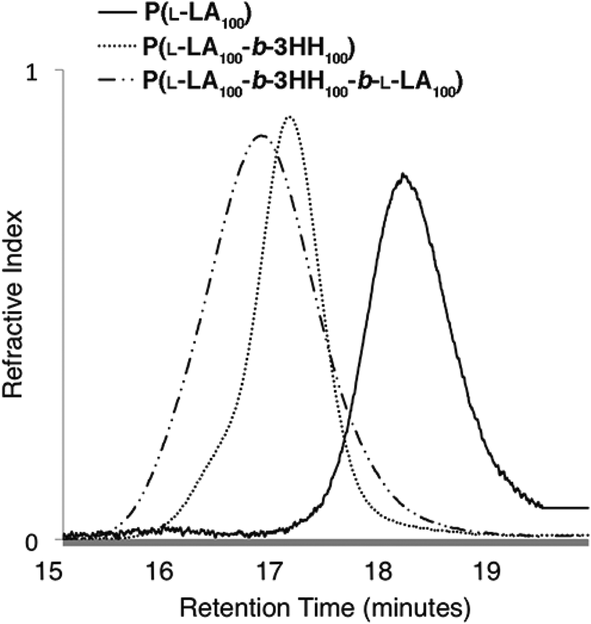 | ||
| Fig. 3 Overlap of GPC traces for P(L-LA100-b-3HH100-b-L-LA100) A block (——, Mn = 6920, Đ = 1.06), AB block (- - -, Mn = 19890, Đ = 1.07) and ABA block (— - -, Mn = 33700 Da, Đ = 1.16) copolymers. | ||
Molecular weights of AB and ABA block copolymers were estimated using averaged dn/dc values based on weight percent of polymers. The dn/dc values of P(L-LA) and P(3-HB) were 0.05034 and 0.065,35 respectively. Values for P(3-HP), P(3-HH) and P(3-HTD) were determined experimentally to be 0.060, 0.059 and 0.059, respectively. The GPC traces showed that there was clear block growth from homopolymer to AB diblock and then again from AB diblock to ABA triblock copolymers. Molecular weight was measured after each block and was again within 10% of the theoretical molecular weight for the homopolymers, AB diblock copolymers and nearly all ABA triblock copolymers. As mentioned, due to the low dn/dc of PLA, low molecular weight PLA A block (n = 10, 20) were not calculated as no light scattering signal could be observed.
Effect of alkyl substituent length
With an effective method to synthesise ABA block copolymers, the effect of alkyl substituent of the β-lactone was investigated. To do this, a series ABA block copolymers were synthesised. While still remarkably well controlled, copolymers synthesised with β-BL demonstrated the highest dispersities. Copolymers with β-VL were better controlled and those with β-HL resulted in the lowest dispersities, correlating with the slower rate of B block formation and allowing a more uniform initiation. As expected, the dispersity increased with the molecular weight of the polymer. This was best demonstrated in the case of β-HL with dispersities increasing from 1.08 (Mn = 15720) to 1.16 (Mn = 33700). GPC traces were found to be monomodal in all cases with only very small shoulders observed after the addition of the B block in samples where n = 10, m = 100.
Thermal analysis of ABA triblock copolymers
To further understand the ABA triblock copolymers, the thermal properties were investigated by DSC analysis. Homopolymers composed exclusively from the studied β-lactone derivatives are amorphous materials and exhibit well defined glass transitions in the range −31 to 5 °C (Table 1). In contrast, the semi-crystalline homopolymer P(L-LA) is known to exhibit a Tg at approximately 60 °C with Tc and Tm at 125 °C and 160 °C (values that can vary depending on Mn and tacticity).54 Within the series of ABA triblock copolymers composed of P(L-LA) and either P(3-HB) or P(3-HP), a single Tg was observed at all compositions. The transition occurred at a temperature that increases linearly with increasing P(L-LA) content between the expected values for each component (Fig. 4).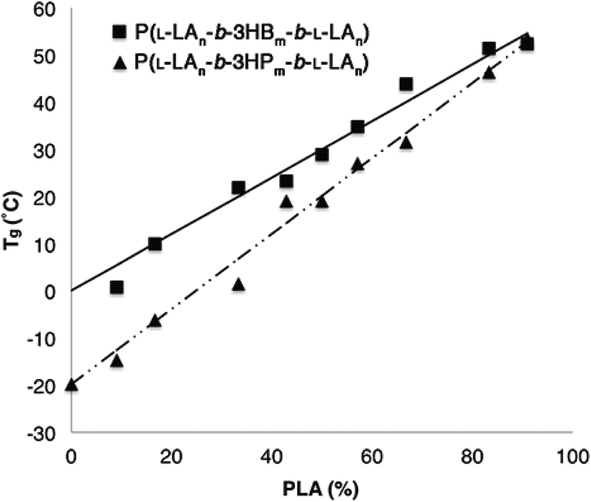 | ||
| Fig. 4 Plot of Tgvs. PLA content for P(L-LAn-b-3HBm-b-L-LAn) (■) and P(L-LAn-b-3HPm-b-L-LAn) (▲). | ||
| Monomer | T (°C) | Time (h) | Conv.b (%) | M n,th | M n | Đ | T g (°C) |
|---|---|---|---|---|---|---|---|
|
a Polymerisations carried out using 1 as a catalyst with benzyl alcohol as an initiator in toluene with monomer/catalyst/initiation ratio of 100:1:1.
b Conversion determined by 1H NMR spectroscopy.
c
M
n,th = (Mw monomer × conversion) + Mw endgroup.
d
M
n and Đ determined by GPC analysis.
e Determined by differential scanning calorimetry.
f n.d. = not determined.
|
|||||||
| β-VL | 50 | 6 | 81 | 8200 | 6570 | 1.10 | −19.8 |
| 85 | 18 | >99 | 10120 |
10080 |
1.09 | ||
| β-HL | 70 | 18 | 91 | 11750 |
11200 |
1.08 | −30.8 |
| 85 | 18 | >99 | 12800 |
12030 |
1.08 | ||
| β-TDL | 85 | 40 | >99 | 21340 |
23700 |
1.05 | n.d.f |
| β-BL | β-VL | β-HL | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | m | M n,th | M n | Đ | M n,th | M n | Đ | M n,th | M n | Đ |
| a Polymerisations carried out in toluene at 85 °C by sequential addition method. | ||||||||||
| 10 | 100 | 10050 |
10560 |
1.16 | 11450 |
11740 |
1.10 | 14260 |
19180 |
1.09 |
| 20 | 100 | 11490 |
12250 |
1.15 | 12890 |
13470 |
1.10 | 15700 |
15720 |
1.08 |
| 50 | 100 | 15820 |
14200 |
1.17 | 17220 |
16100 |
1.10 | 20020 |
22340 |
1.10 |
| 75 | 100 | 19420 |
20300 |
1.18 | 20820 |
19020 |
1.13 | 23630 |
24850 |
1.14 |
| 100 | 100 | 23020 |
22620 |
1.18 | 24420 |
21170 |
1.16 | 27230 |
33700 |
1.16 |
| 100 | 75 | 20870 |
19520 |
1.15 | 21920 |
19720 |
1.17 | 24020 |
25140 |
1.16 |
| 100 | 50 | 18720 |
18200 |
1.16 | 19420 |
18600 |
1.16 | 20820 |
21650 |
1.14 |
| 100 | 20 | 16130 |
16780 |
1.15 | 16420 |
16490 |
1.17 | 16970 |
17850 |
1.14 |
| 100 | 10 | 15270 |
15110 |
1.18 | 15410 |
14760 |
1.15 | 15690 |
15230 |
1.13 |
Samples with the lowest P(L-LA) content (n = 10, 20) did not exhibit either a Tc or Tm, and even materials that contained P(L-LA) blocks where n = 30 showed a significant depression in Tm (approximately 130 °C) compared to that observed for the high PLA content triblock copolymers (n > 80, Tm ≈ 160 °C, Tables S6 and S7†). These data are characteristic of high miscibility between the two blocks of the copolymers as the amorphous nature of the P(3-HA) block disrupts the crystallinity of the P(L-LA) segment. However, the presence of both a Tc and Tm which are close to the values expected for the homopolymer in samples with high P(L-LA) content may indicate a small degree of phase-separation. In samples of P(L-LAn-b-3HHm-b-L-LAn), a different trend is observed (Fig. 5). Samples containing up to 50% P(L-LA) (n < 100, m = 100) content exhibit a single Tg at approximately −25 °C which is essentially invariant of composition and may be compared to the Tg of −31 °C observed for the homopolymer P(3-HH) (Table 1). In contrast, samples with greater than 60% P(L-LA) (n = 100, m < 75) all exhibit a single Tg at approximately 45 °C which is close to that expected for homopolymers of P(L-LA). Melting transitions are not evident in samples below 30% PLA content, but can be observed to increase from 130 °C to plateau at approximately 155 °C at 60% P(L-LA). A similar trend is seen for the Tc measured in this series of materials (Table S8†).
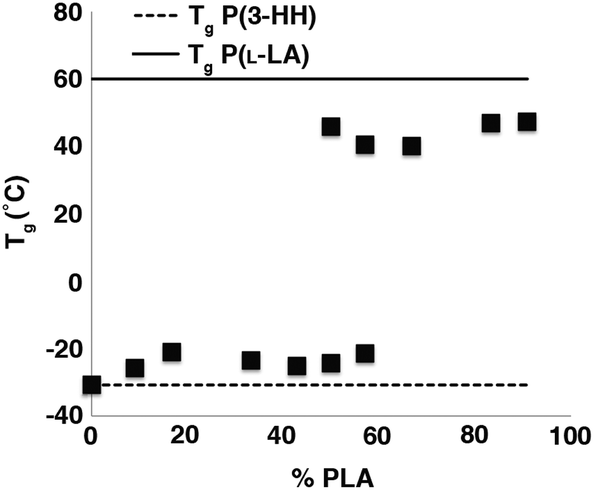 | ||
| Fig. 5 Plot of the most prominent Tg observed vs. PLA content for P(L-LAn-b-3HHm-b-L-LAn). The dashed and solid lines represent the characteristic Tg values for homopolymers of P(3-HH) and P(L-LA) respectively. | ||
For samples containing blocks of similar degrees of polymerisation (n = 100, m = 100 and n = 100, m = 75) two distinct Tgs are observed (Fig. 5 and 6) with the P(L-LA) Tg showing possible evidence of hysteresis.55
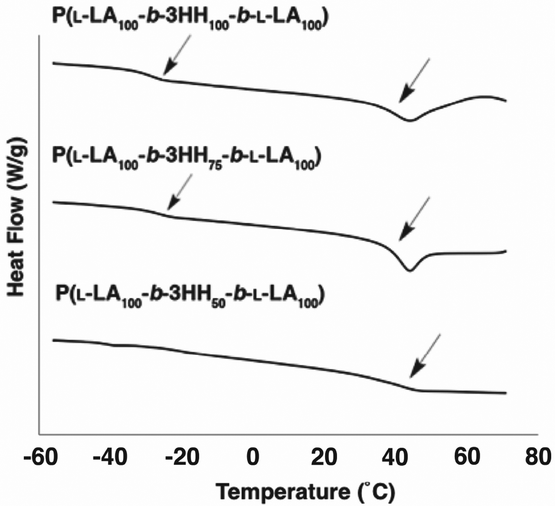 | ||
| Fig. 6 DSC curves of P(L-LA100-b-3HH100-b-L-LA100), P(L-LA100-b-3HH75-b-L-LA100) and P(L-LA100-b-3HH50-b-L-LA100). For full DSC curves see ESI.† | ||
This is characteristic of a phase separated material.56 Thus, predictable control of phase separation can be achieved through varying the length of the aliphatic side group on the polyester component of these triblock copolymers and by control of the relative degree of polymerisation of each block within the copolymer.
Small angle X-ray scattering (SAXS) and wide angle X-ray scattering (WAXS)
SAXS and WAXS measurements may be used to confirm microphase separation in polymeric structures instead of standard TEM/AFM images,57–59 especially if temperature variations are a concern. Fig. 7 provides evidence that both P(L-LA100-b-3HH75-b-L-LA100) and P(L-LA100-b-3HH100-b-L-LA100) are microphase separated at both −50 °C and 25 °C. For the former sample, a peak at q = 0.3 nm−1 is observed, corresponding to a microphase separated structure (lacking a defined morphology, i.e. irregularly microphase separated) with a domain spacing of 21 nm. For P(L-LA100-b-3HH100-b-L-LA100), peaks are observed at q = 0.28 nm−1, q = 0.54 nm−1 and q = 0.9 nm−1 again consistent with microphase separation. The sequence of reflections suggests crystal lamellar order with a period of 22.4 nm.60–62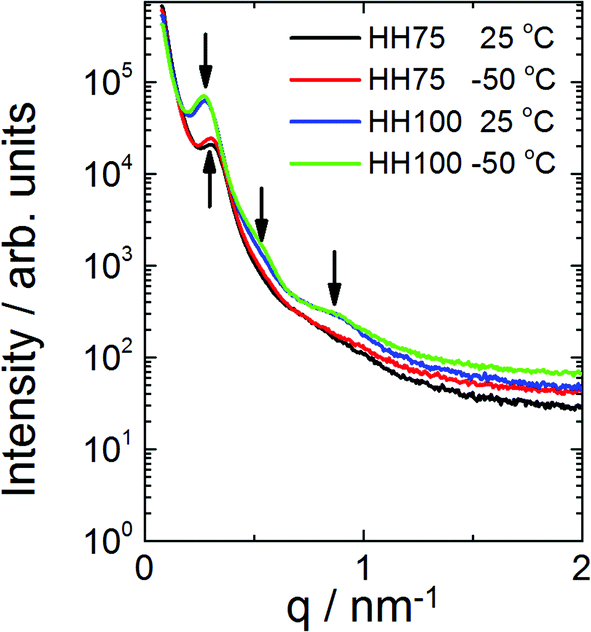 | ||
| Fig. 7 SAXS intensity profiles for P(L-LA100-b-3HH75-b-L-LA100) and P(L-LA100-b-3HH100-b-L-LA100) at the two temperatures indicated (data obtained during second cooling from 150 °C to −50 °C. The arrows point to peaks that arise from microphase separation. | ||
Crystallisation was confirmed by WAXS data obtained concurrently with SAXS (data not shown). For both samples, peaks arising from a microphase separated structure were present across the examined temperature range from −50 °C to 150 °C (with some variation in intensity) indicating the persistence of the microphase separated morphology across the glass transition temperatures.
Tensile properties
To gain a better understanding of the mechanical properties of the ABA triblock copolymers, attempts were made to cast polymer films. Polymer films cast of P(L-LAn-b-3HBm-b-L-LAn) were found to be brittle and opaque with small defects, resulting in films that were not suitable for tensile measurements. Polymers of P(L-LAn-b-3HHm-b-L-LAn) were able to be cast into transparent, peelable films without defects. Samples where m > n were too soft to take tensile measurements. However, P(L-LA100-b-3HH100-b-L-LA100) was cast into flexible films suitable for tensile property measurements (Fig. 8). The tensile strength of the film was found to be 5.5 MPa which is slightly lower than similar films that have been successfully cast of P(L-LAn-b-3HBm-b-L-LAn).22 However, in this report, the middle P(3-HB) block was found to be syndiotactic whereas the middle P(3-HA) block in our sample is atactic. The Young's modulus was calculated to be 80.2 MPa and the elongation at break to be 31.4%. The Young's modulus is significantly lower than that of P(L-LA) as expected for a polymer that is more elastomeric.63 This is shown further by the elongation at break (Table 3). This elongation is higher than reported for high molecular weight P(L-LAn-b-3HBm-b-L-LAn) copolymers reported by Mehrkhodavandiandi24 and lower than the very low molecular weight P(L-LAn-b-3HBm-b-L-LAn) copolymers reported by Kimura.22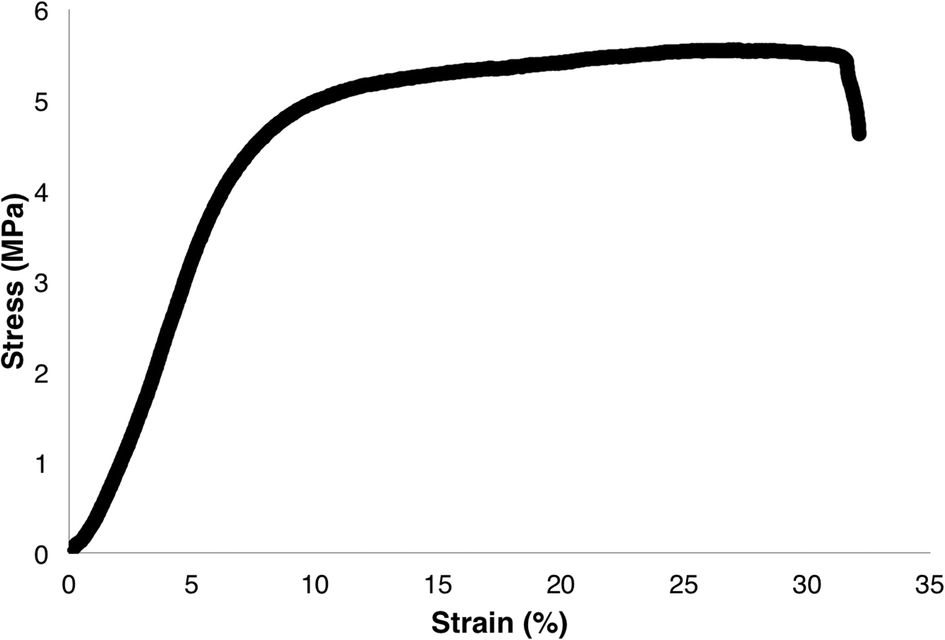 | ||
| Fig. 8 Typical stress-strain curve for P(L-LA100-b-3HH100-b-L-LA100). | ||
| Tensile strength (MPa) | Young's modulus (MPa) | Elongation at break (%) |
|---|---|---|
| 5.5 | 80.2 | 31.4 |
For comparison, efforts were made to cast a film of P(L-LA) (Mn = 22000) without success. This was not unexpected as low molecular weight P(L-LA) is known to be brittle and therefore difficult to form coherent films. However, the tensile properties of higher molecular weight P(L-LA) have been characterised, showing very little elastomeric behaviour (elongation at break ≤3.9%).63
Conclusions
The homopolymerisation of four alkyl substituted β-lactones to produce poly(3-hydroxyalkanoate)s achieved using an aluminium salen catalyst, 1. The polymerisations showed excellent control with predictable molecular weights and low dispersities (Đ ≤ 1.1). The use of 1 as a catalyst allows for the formation of well-defined ABA triblock copolymers with L-lactide through sequential monomer addition. ABA triblock copolymers of L-lactide and either β-butyrolactone or β-valerolactone yielded polymers with tunable thermal properties and a single averaged glass transition temperature. However, when β-heptanolactone is used, microphase separation is observed between the A and B blocks. This was supported by observation of two glass transition temperatures in P(L-LA100-b-3HH100-β-L-LA100) and P(L-LA100-b-3HH75-b-L-LA100) while other samples of P(L-LAn-b-3HHm-b-L-LAn) show a single Tg that remained relatively unchanged by a change in P(L-LA):P(3-HH) ratio. SAXS analysis of P(L-LA100-b-3HH100-b-L-LA100) and P(L-LA100-b-3HH75-b-L-LA100) confirmed that there was in fact microphase separation with the P(L-LA) to P(3-HH) ratio playing a role in the degree of order, the former sample exhibiting well-defined lamellar order. Polymer elastomeric properties are improved relative to PLA. To our knowledge, this is the first report of an ABA triblock copolymer composed of PLA and a PHA with a clear correlation between the nature of the functional group and microphase separation.
Acknowledgements
The authors thank Mr Edward Cross for his initial studies on lactone polymerisations and Dr Laura Allan for helpful discussions and gel-permeation chromatography maintenance. We thank Samuel C. Bizley and Vitaliy V. Khutoranskiy for technical support with tensile testing experiments. This work was supported by the University of Edinburgh and the Marie-Curie Actions Programme (FP7-PEOPLE-2013-CIG-618372).Notes and references
- S. Mecking, Angew. Chem., Int. Ed., 2004, 43, 1078–1085 CrossRef CAS PubMed.
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- H.-M. Müller and D. Seebach, Angew. Chem., Int. Ed. Engl., 1993, 32, 477–502 CrossRef.
- J.-F. Carpentier, Macromol. Rapid Commun., 2010, 31, 1696–1705 CrossRef CAS PubMed.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- C. Thomas and B. Bibal, Green Chem., 2014, 16, 1687–1699 RSC.
- N. Ajellal, J.-F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363–8376 RSC.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- S. Dutta, W.-C. Hung, B.-H. Huang and C.-C. Lin, in Synthetic Biodegradable Polymers, ed. B. Rieger, A. Künkel, G. W. Coates, R. Reichardt, E. Dinjus and T. A. Zevaco, Springer, Berlin Heidelberg, 2012, vol. 245, ch. 156, pp. 219–283 Search PubMed.
- R. Jianming, X. Anguo, W. Hongwei and Y. Hailin, Des. Monomers Polym., 2013, 17, 345–355 CrossRef.
- C.-M. Dong and G. Liu, Polym. Chem., 2013, 4, 46–52 RSC.
- S. Doppalapudi, A. Jain, W. Khan and A. J. Domb, Polym. Adv. Technol., 2014, 25, 427–435 CrossRef CAS.
- Y. Hori, Y. Takahashi, A. Yamaguchi and T. Nishishita, Macromolecules, 1993, 26, 4388–4390 CrossRef CAS.
- H. Abe, Y. Doi, Y. Hori and T. Hagiwara, Polymer, 1998, 39, 59–67 CrossRef CAS.
- L. Liu, Z. Y. Wei and M. Qi, Chin. Chem. Lett., 2007, 18, 744–746 CrossRef CAS PubMed.
- B. J. Jeffery, E. L. Whitelaw, D. Garcia-Vivo, J. A. Stewart, M. F. Mahon, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 12328–12330 RSC.
- E. Brule, V. Guerineau, P. Vermaut, F. Prima, J. Balogh, L. Maron, A. M. Z. Slawin, S. P. Nolan and C. M. Thomas, Polym. Chem., 2013, 4, 2414–2423 RSC.
- M. Basko, A. Duda, S. Kazmierski and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4873–4884 CrossRef CAS.
- Y. Hori, Y. Takahashi, A. Yamaguchi and T. Nishishita, in Studies in Polymer Science, ed. D. Yoshiharu and F. Kazuhiko, Elsevier, 1994, vol. 12, pp. 549–556 Search PubMed.
- E. D. Cross, L. E. N. Allan, A. Decken and M. P. Shaver, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1137–1146 CrossRef CAS.
- S. Hiki, M. Miyamoto and Y. Kimura, Polymer, 2000, 41, 7369–7379 CrossRef CAS.
- H. R. Kricheldorf and S.-R. Lee, Macromolecules, 1995, 28, 6718–6725 CrossRef CAS.
- D. C. Aluthge, C. Xu, N. Othman, N. Noroozi, S. G. Hatzikiriakos and P. Mehrkhodavandi, Macromolecules, 2013, 46, 3965–3974 CrossRef CAS.
- E. M. Frick and M. A. Hillmyer, Macromol. Rapid Commun., 2000, 21, 1317–1322 CrossRef CAS.
- E. M. Frick, A. S. Zalusky and M. A. Hillmyer, Biomacromolecules, 2003, 4, 216–223 CrossRef CAS PubMed.
- J. Shin, Y. Lee, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2012, 13, 3833–3840 CrossRef CAS PubMed.
- N. Kasyapi and A. K. Bhowmick, RSC Adv., 2014, 4, 27439–27451 RSC.
- N. Spassky, M. Wisniewski, C. Pluta and A. Le Borgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 CrossRef CAS.
- M. Wisniewski, A. L. Borgne and N. Spassky, Macromol. Chem. Phys., 1997, 198, 1227–1238 CrossRef CAS.
- A. Le Borgne, V. Vincens, M. Jouglard and N. Spassky, Makromol. Chem., Macromol. Symp., 1993, 73, 37–46 CrossRef CAS.
- V. Vincens, A. Le Borgne and N. Spassky, Makromol. Chem., Macromol. Symp., 1991, 47, 285–291 CrossRef CAS.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688–2689 CrossRef CAS PubMed.
- G. L. Fiore, J. L. Klinkenberg and C. L. Fraser, Macromolecules, 2008, 41, 9397–9405 CrossRef CAS.
- Y. Hori and T. Hagiwara, Int. J. Biol. Macromol., 1999, 25, 237–245 CrossRef CAS.
- G. Portale, D. Cavallo, G. C. Alfonso, D. Hermida-Merino, M. van Drongelen, L. Balzano, G. W. M. Peters, J. G. P. Goossens and W. Bras, J. Appl. Crystallogr., 2013, 46, 1681–1689 CrossRef CAS.
- D. A. Atwood, M. S. Hill, J. A. Jegier and D. Rutherford, Organometallics, 1997, 16, 2659–2664 CrossRef CAS.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, R. I. Pugh and A. J. P. White, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15343–15348 CrossRef CAS PubMed.
- A. D. Adler, F. R. Longo, F. Kampas and J. Kim, J. Inorg. Nucl. Chem., 1970, 32, 2443–2445 CrossRef CAS.
- C. Chatterjee and M. H. Chisholm, Inorg. Chem., 2012, 51, 12041–12052 CrossRef CAS PubMed.
- Y. D. Y. L. Getzler, V. Mahadevan, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1174–1175 CrossRef CAS PubMed.
- Y. D. Y. L. Getzler, V. Mahadevan, E. B. Lobkovsky and G. W. Coates, Pure Appl. Chem., 2004, 76, 557–564 CrossRef CAS.
- J. W. Kramer, E. B. Lobkovsky and G. W. Coates, Org. Lett., 2006, 8, 3709–3712 CrossRef CAS PubMed.
- J. A. R. Schmidt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 11426–11435 CrossRef CAS PubMed.
- H. Wölfle, H. Kopacka, K. Wurst, P. Preishuber-Pflügl and B. Bildstein, J. Organomet. Chem., 2009, 694, 2493–2512 CrossRef PubMed.
- J. A. R. Schmidt, V. Mahadevan, Y. D. Y. L. Getzler and G. W. Coates, Org. Lett., 2004, 6, 373–376 CrossRef CAS PubMed.
- P. Ganji, D. J. Doyle and H. Ibrahim, Org. Lett., 2011, 13, 3142–3145 CrossRef CAS PubMed.
- L. R. Rieth, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 15239–15248 CrossRef CAS PubMed.
- J. W. Kramer, D. S. Treitler, E. W. Dunn, P. M. Castro, T. Roisnel, C. M. Thomas and G. W. Coates, J. Am. Chem. Soc., 2009, 131, 16042–16044 CrossRef CAS PubMed.
- S. Bloembergen, D. A. Holden, T. L. Bluhm, G. K. Hamer and R. H. Marchessault, Macromolecules, 1987, 20, 3086–3089 CrossRef CAS.
- S. Bloembergen, D. A. Holden, T. L. Bluhm, G. K. Hamer and R. H. Marchessault, Macromolecules, 1989, 22, 1656–1663 CrossRef CAS.
- S. Bloembergen, D. A. Holden, T. L. Bluhm, G. K. Hamer and R. H. Marchessault, Macromolecules, 1989, 22, 1663–1669 CrossRef CAS.
- T. Kobayashi, A. Yamaguchi, T. Hagiwara and Y. Hori, Polymer, 1995, 36, 4707–4710 CrossRef CAS.
- J. C. Middleton and A. J. Tipton, Biomaterials, 2000, 21, 2335–2346 CrossRef CAS.
- J. D. Menczel, L. Judovits, R. B. Prime, H. E. Bair, M. Reading and S. Swier, in Thermal Analysis of Polymers, John Wiley & Sons, Inc., 2009, pp. 7–239 Search PubMed.
- D. R. Paul, Polymer blends, Academic Press, New York, 1978 Search PubMed.
- Á. Szabó, G. Szarka and B. Iván, Macromol. Rapid Commun., 2014 DOI:10.1002/marc.201400469.
- R. P. Brannigan, A. Walder and A. P. Dove, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2279–2286 CrossRef CAS.
- S. R. Rathi, E. B. Coughlin, S. L. Hsu, C. S. Golub, G. H. Ling and M. J. Tzivanis, Polymer, 2012, 53, 3008–3016 CrossRef CAS PubMed.
- I. W. Hamley, V. Castelletto, R. V. Castillo, A. J. Müller, C. M. Martin, E. Pollet and P. Dubois, Macromolecules, 2004, 38, 463–472 CrossRef.
- I. W. Hamley, P. Parras, V. Castelletto, R. V. Castillo, A. J. Müller, E. Pollet, P. Dubois and C. M. Martin, Macromol. Chem. Phys., 2006, 207, 941–953 CrossRef CAS.
- R. M. Michell, A. J. Müller, M. Spasova, P. Dubois, S. Burattini, B. W. Greenland, I. W. Hamley, D. Hermida-Merino, N. Cheval and A. Fahmi, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1397–1409 CrossRef CAS.
- J. R. Sarasua, A. L. Arraiza, P. Balerdi and I. Maiza, Polym. Eng. Sci., 2005, 45, 745–753 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data, polymerisation data and thermal data and kinetic plots. See DOI: 10.1039/c4py01459f |
| This journal is © The Royal Society of Chemistry 2015 |