
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Titin-mimicking polycyclic polymers with shape regeneration and healing properties†
Jan-Hendrik
Schuetz
,
Peng
Wentao
and
Philipp
Vana
*
Institut für Physikalische Chemie, Georg-August-Universität Göttingen, Tammannstr. 6, D-37077 Göttingen, Germany. E-mail: pvana@uni-goettingen.de
First published on 25th November 2014
Abstract
Polycyclic polymers based on cyclic (ABC)n-multiblock-copolymers are formed via stepwise polymerization of three individual blocks and exploiting the ring merging reaction of these ring polymers. The so-obtained precursor ring-polymers were interconnected via click reaction. Small blocks within the rings with the ability to form self-complementary hydrogen bonds lead to intra- and intermolecular links between polycyclic polymers. The obtained materials, which mimic nature's paragon Titin, have some extraordinary material properties concerning elasticity and energy dissipation.
Introduction
Tailoring polymeric materials with a variety of desired properties is still a challenging task. However, in order to gain high performance materials it is a crucial point to combine several attributes such as high temperature tolerance with consistent material properties. Concerning mechanical properties it is especially hard to achieve complementary characteristics such as stiffness and tensile strength or elasticity and toughness in one material, due to excluding mechanisms on the molecular level.1Nature has a variety of materials that show enhanced mechanical properties often far better than these of any synthetic material.2 Especially biological fibres often show a good balance between a high tensile stiffness at high tensile length and elasticity and thus the combination of the above mentioned complementary material properties.3,4 Two examples for such biological fibres are spider silk5,6 and muscle fibres.7 Besides the hierarchical structure, which derives mainly from the ability to form supramolecular structures leading to a maximum in mechanical strength at minimum weight,2,3 the mechanical properties also have their origin in their nanoscopic structure in form of the molecular multi-domain architecture of its components.6,8–12
In the case of spider silk, hard semicrystalline regions consisting of antiparallel polyalanin- and poly(alanin-glycine) repeating units are interconnected by soft glycine-rich flexible domains.13 In muscular tissue of mammals, the filament protein titin contained in the sarcomere forming the myofibrils is mainly responsible for the elasticity, stability and the recovery of the resting length of the muscle fibre.14 Titin contains over 300 repeating units composed of hard immunoglobulin (Ig) units mainly consisting of a β-sheet secondary structure showing a compact folding in the tertiary structure interconnected with soft amorphous fibronectin-type-III domains, making it the largest known human protein.15,16 Under mechanical stress, the Ig-domains unfold by breakage of the semicrystalline structure, and refold at stress release.3 Due to the temporary disorder of the tertiary structure, mainly by cleaving H-bonds, the fibre can absorb a high amount of energy at a high tensile length, thus giving the fibre an enormous toughness combined with high elasticity.8,17,18
It is tempting to design synthetic materials with equally outstanding mechanical properties by mimicking such a multi-domain structure using polymer chemistry. Sogah et al. developed several approaches for mimicking the crystalline and amorphous regions of spider silk by means of linking β-sheet-carrying polypeptides made from alanine or alternating glycine and alanine units with phenoxathiin derivatives19 or polyethyleneoxide,20,21 showing microphase separation of the different blocks. Nagapudi et al. used an ABA-type triblock copolymer consisting entirely of biosynthesized polypeptide blocks to mimic the hard and soft segments,22 while Guzmán et al. used connected alternating α-helix and β-sheet blocks to mimic titin's multidomain structure.23 In addition to these biochemical approaches, there were some full-synthetic methods described by Guan and coworkers, who used repeating units based on an ureidopyrimidone motif bearing self-complementary H-bonds24,25 along a polymer chain7 or along connected loop structures26 to mimic the hard blocks in titin. These polymers indeed showed a characteristic stress-strain-behavior with high tensile strength and toughness as well as elastic properties and thus provide a good model system for titin.
In contrast to the functional polymer of the Guan group, which was mainly formed in a multistep organic synthesis approach, we here propose a more modular polymer chemistry approach towards the mimicking of titin's structure, which is easier to implement and provides higher flexibility in terms of polymer properties. Based on the living-like ring-expansion polymerization of thiiranes developed by Nishikubo et al.,27,28 which was investigated intensively by our group,29,30 we synthesized a cyclic triblock-copolymer as precursor, which contains self-complementary hydrogen-bonding moieties that may reversibly unfold (see Scheme 1). Dialing the exact composition of this ring polymer containing three different monomers provided the possibility of tuning the stiffness of the targeted material. The overall ring size of the precursor was, on the one hand, adjusted via the block lengths of the individual blocks; on the other hand, it was taken advantage of the ring-merging mechanism (cross-over ring fusion) occurring in these ring-expansion polymerizations,29,30 by which the amount of doubled macrorings could be controlled. We could thus easily incorporate at least two hydrogen-bonding blocks per ring polymer, which is necessary for the intramolecular interaction within the ring (see Scheme 1). Anchor points (red handles in Scheme 1) located at the original initiator molecule thus increased in number per ring, as well. The precursor polymer rings were subsequently interconnected via azide–alkine click reaction with a bifunctional linker yielding the modular poly(ring-polymer) of high molar mass (see Scheme 1), since the approach of linking numerous cyclic polymers via Click Chemistry was already reported to be successful in the formation of ring-based gels31 and tetracyclic structures.32 The resulting polymer was then tested via tensile testing experiments with respect to its high performance mechanical properties.
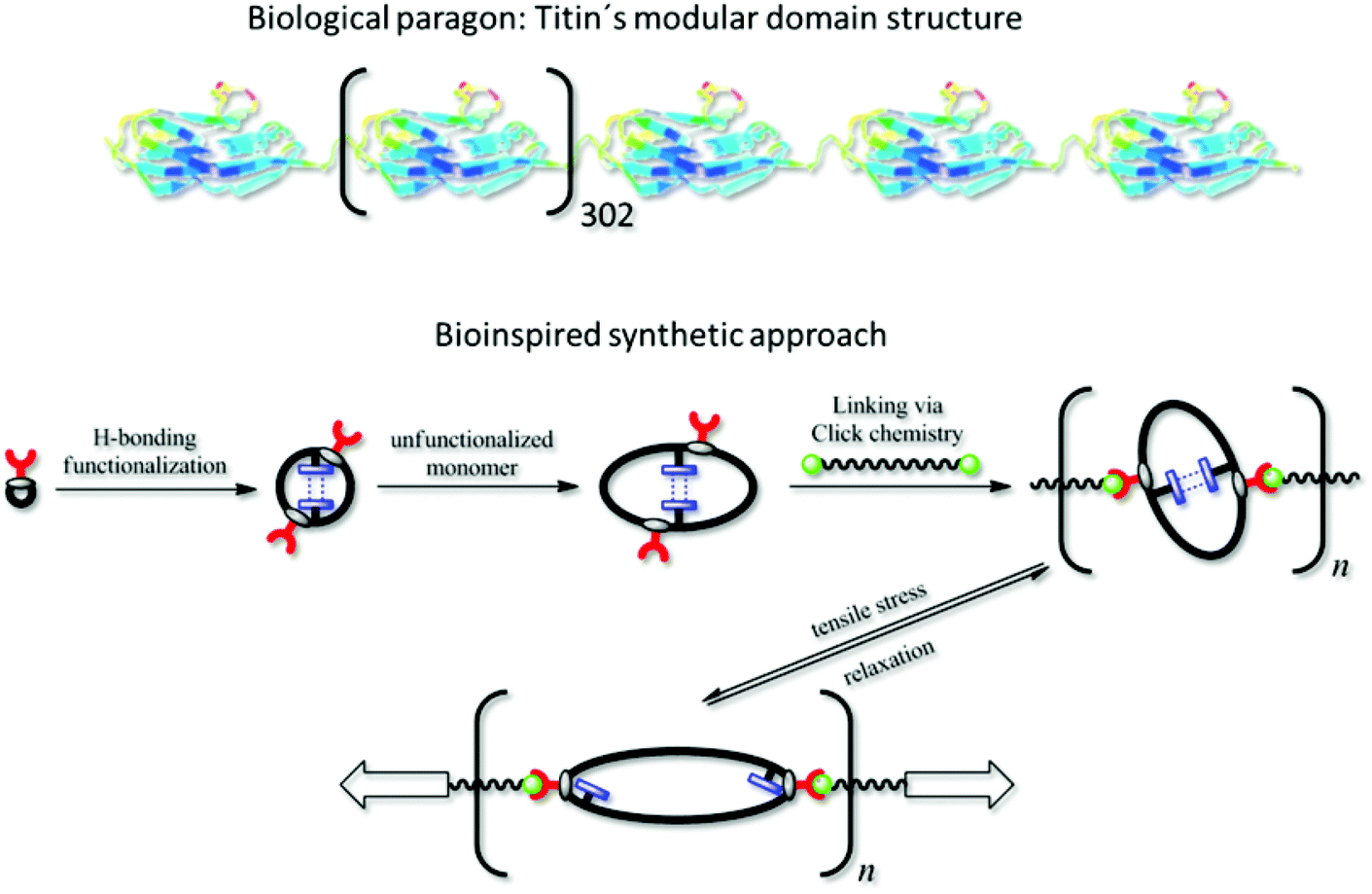 | ||
| Scheme 1 Biological protein Titin7 and applied synthetic approach. | ||
Results and discussion
Explorative experiments
In order to obtain the desired material and structural properties, three different thiirane monomers have been utilized for the formation of the precursor ring polymer. The general ring expansion process used for this synthesis is described in our former work in detail.29,30 To incorporate the necessary H-bonding functionality, a novel monomer 3 (see Fig. 1), which is structurally similar to 2-(phenoxymethyl)thiirane (PMT, 2), which we studied extensively in earlier work, was synthesized in a convenient five-step process (see ESI†). The resulting monomer ethyl-2-(4-thiiran-2-ylmethoxy)benzamido acetate (ETBAA, 3) carries an additional glycine ethyl ester moiety, in which the amide and ester group act as H-bonding donor and acceptor, respectively, in order to allow for self-complementary interaction. PMT 2 and methylthiirane (MT, 1) were used as non-functionalized monomers in order to expand the ring size and to adjust the material stiffness. While MT is commercially available, PMT was synthesized in a one-step process from the corresponding oxirane as previously reported.29 | ||
| Fig. 1 Monomers for the synthesis of the cyclic precursor polymers via ring expansion polymerization: propylene sulfide 1 (PS), 2-(phenoxymethyl)-thiirane 2 (PMT) and ethyl-2-(4-thiiran-2-ylmethoxy)benzamido acetate 3 (ETBAA). | ||
As initiator for the ring expansion polymerization, a slightly modified structure of the previously investigated 2,4-thiazolidinedione30 (TZD) was utilized that carries an alkyne group attached in 5-position to the cyclic TZD scaffold. The resulting compound prop-2-yn-1-yl-2-(2,4-dioxothiazolidin-5-yl) acetate 4 (PTZDA, see Scheme 2) was synthesized in a facile two-step process with good yields of the product (see ESI†).
 | ||
| Scheme 2 Synthesis of the cyclic (ABC)n-multiblock-copolymer precursor polymers CP-01 to CP-03 via ring expansion polymerization of the 2,4-thiazolidinedione-type initiator prop-2-yn-1-yl-2-(2,4-dioxothiazolidin-5-yl) acetate 4 (PTZDA). The different monomer blocks are coloured for better distinction and labelled in their order of polymerization. Block A: ETBAA, block B: MT, block C: PMT. The alkyne-group used for later linkage is indicated in orange. Please note that the ring-merging reaction during the polymerization process leads to n-fold multiples of these ring polymer structures. | ||
In order to form the cyclic precursor block-copolymers, some tests had to be carried out beforehand to clarify three crucial issues: (i) The first one was to find the ideal polymerization order of the three monomers, (ii) the second one was to determine the block-lengths to achieve appropriate material properties for the later produced tensile test specimens, (iii) and the third one was to investigate the degree of ring merging of these cyclic triblock-copolymers, since at least two rings need to merge for having two hydrogen bond forming blocks opposite to each other and for having two coupling moieties per ring for the later applied step-growth reaction leading to ring-(ABC)n-multiblock-copolymers.
Regarding the first issue, the functionalized monomer ETBAA was homopolymerized with either TZD or PTZDA to examine its reactivity and polymerization behaviour. In a first experiment, ETBAA was polymerized with PTZDA in a monomer/initiator ratio of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with 10 mol% tetrabutylammonium chloride as catalyst and a monomer concentration of 0.5 mol L−1. The polymerization yielded 84% conversion of the monomer after 24 h at 60 °C. Size-exclusion chromatography (SEC) measurements exhibited a
:1 with 10 mol% tetrabutylammonium chloride as catalyst and a monomer concentration of 0.5 mol L−1. The polymerization yielded 84% conversion of the monomer after 24 h at 60 °C. Size-exclusion chromatography (SEC) measurements exhibited a ![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n of about 12000 g mol−1 using styrene calibration and thus not showing the true molar mass. The 1H-NMR spectrum showed a monomer/initiator ratio of 35:1, thus indicating that the initiator was not completely consumed during the polymerization. Changing the monomer/initiator ratio to a value of 5:1 at the same monomer concentration resulted in a smaller reaction rate leading to a total monomer conversion of 87% after 96 h. The control over the polymerization, however, was much better as indicated by a smaller dispersity of 1.3 (see Fig. 2).
n of about 12000 g mol−1 using styrene calibration and thus not showing the true molar mass. The 1H-NMR spectrum showed a monomer/initiator ratio of 35:1, thus indicating that the initiator was not completely consumed during the polymerization. Changing the monomer/initiator ratio to a value of 5:1 at the same monomer concentration resulted in a smaller reaction rate leading to a total monomer conversion of 87% after 96 h. The control over the polymerization, however, was much better as indicated by a smaller dispersity of 1.3 (see Fig. 2).
 | ||
| Fig. 2 (a) Molar mass distribution (SEC-distribution) after various times and (b) monomer conversion vs. time in the ring expansion polymerization of ETBAA with PTZDA as the initiator in solution of NMP at 70 °C. Conditions: ETBAA–PTZDA = 5:1, cETBAA = 0.5 mol L−1, cTBAC = 10 mol%. | ||
Compared to the polymerization behaviour of PMT and MT, ETBAA shows a much slower polymerization rate. Consequently, the monomer order for the block copolymerization was chosen to start with the slowest monomer ETBAA, followed by MT, and finished with the most reactive monomer PMT, which counterbalanced the decreasing accessibility of the initiator sites with increasing chain lengths.
Regarding the second issue, it was necessary to estimate the appropriate monomer block-lengths, i.e., the overall copolymer composition, in order to obtain proper materials for mechanical tensile testing, which have a certain dimensional stability – i.e., they keep their shape during production and handling of the tensile test specimens – and, at the same time, are not too rigid so that they would break immediately. For this, some tests were performed: Table 1 shows the composition of various homo- and block-copolymers consisting of PMT, MT and ETBAA with their corresponding glass transition temperature Tg – as determined by Differential Scanning Calorimetry (DSC) – the maximum number of merged rings (see third issue mentioned below), and qualitative mechanical appearance. Samples α, β and γ refer to the homopolymers of MT, PMT and ETBAA, respectively, while samples δ to η refer to (AB)n-multiblock-copolymers with various block lengths and compositions. Since ETBAA is meant to form the smallest block in the resulting polymers, it was omitted in the first three probing copolymerizations. Instead, the first block-polymerization was performed with MT, which forms a very soft polymer with a glass transition temperature at about −36 °C. By adding PMT afterwards forming the second block, with its homopolymer having a Tg of around 18 °C, it was possible to increase the stiffness gradually at wish by adding further portions of monomer (for details see ESI†).
| Sample | Composition | T g/°C | Maximum number of merged rings | Mechanical appearance at room temperature |
|---|---|---|---|---|
| a Depends strongly on actual room temperature. b Could not clearly be evaluated by SEC due to formation of a small shoulder (see Fig. S1 in ESI). c High molar masses observed in SEC which could not be assigned to a distinct polymer species (see text and Fig. S1). | ||||
| α | MT homopolymer | −36 | 2 | Viscous and sticky |
| β | PMT homopolymer | 18 | >4 | Dimensionally stable and deformable/rigida |
| γ | ETBAA homopolymer | 59 | n.d. | Dimensionally stable and rigid |
| δ |
Ring-poly(MT-b-PMT)n![[P with combining macron]](https://www.rsc.org/images/entities/i_char_0050_0304.gif) n,MT/n,PMT = 22:50 n,MT/n,PMT = 22:50 |
0 | 3 | Dimensionally unstable |
| ε |
Ring-poly(MT-b-PMT)nn,MT/n,PMT = 22:97 |
14 | 2–3b | Dimensionally stable and deformable |
| ζ |
Ring-poly(MT-b-PMT)nn,MT/n,PMT = 22:165 |
16 | Multiplec | Dimensionally stable and deformable |
| η |
Ring-poly(ETBAA-b-PMT)nn,ETBAA/n,PMT = 4:31 |
20 | 2 | Dimensionally stable and rigid |
In Fig. 3, the Tg values of several ring-poly(MT-block-PMT) (AB)n-multiblock-copolymers (the multiblock topology stems from ring merging as described below) are plotted against the amount of incorporated PMT (for further information on these block-copolymers, see ESI, Fig. S3 and Table S1†). In all copolymers, only one glass transition – which follows almost a linear increase with increasing relative amount of PMT – occurs in the observed temperature range between −40 and +150 °C, which indicates that this block copolymer is in a disordered state without showing microphase separation. Only two (AB)n-multiblock-copolymers (samples δ and ε in Table 1) exhibit an appropriate dimensional stability with their Tg located between +10 and +16 °C, which is roughly 9 to 15 K below room temperature, where thus a rubber-elastic tensile behaviour (rubber plateau) occurs. In order to examine the impact of ETBAA on the glass transitions as well, an (AB)n-multiblock-copolymer consisting of PMT blocks and short ETBAA blocks having only four ETBAA units (sample η) was produced. Interestingly, the glass transition increased only by 2 K compared to pure ring-poly(PMT) indicating that ETBAA has only a small impact on the Tg of PMT-homopolymer.
 | ||
| Fig. 3 Glass transition temperature Tgversus the molar amount xPMT of PMT in ring-poly(PMT-block-MT) polymer. The regime of sufficient mechanical stability of the (AB)n-multiblock-copolymer at room temperature showing absence of rigid fracture is marked by a red circle. | ||
Regarding the third issue, the observed maximum degree of ring merging for each of the polymers was determined via SEC (see Table 1 for results, see Fig. S2 in the ESI† for SE-chromatograms and method description). As already reported earlier by us, MT-homopolymers lead to doubled rings while PMT-homopolymers show formation of fourfold merged and greater rings.30 Block-copolymerizations exhibited the same behaviour for each of these two monomers and MT-blocks led to doubled rings and the addition of PMT in the second step resulted in formation of up to threefold rings in samples δ and ε corresponding to cyclic (AB)n-multiblock copolymers with up to six blocks. Sample ζ even showed a broad molar mass distribution with much higher degrees of merging which could not be assigned to exactly defined polymer species (see Fig. S2†). These results clearly show the ability of the cyclic block-copolymers to form doubly or multiply merged rings, which is mandatory and highly desired for our purpose.
Synthesis of precursor ring polymers
For the synthesis of the precursor ring polymer samples CP-01, CP-02 and CP-03 (see Table 2), ETBAA was chosen as the first monomer, for reasons mentioned above, with a molar ETBAA-to-initiator ratio of 5:1, which was found to polymerize in a well controlled fashion. Afterwards, MT and PMT were introduced block-wise into the polymer and their block-lengths were varied in order to obtain small (CP-01 and CP-03) and large (CP-02) polymer rings. The complete three-step polymerization approach is depicted in Scheme 2. The reaction conditions, the block lengths, and properties of the resulting polymers are collated in Table 2.
| CP-01 | CP-02 | CP-03 | |
|---|---|---|---|
| a Determined by 1H-NMR spectroscopy. b Here the values apply for unmerged rings only. c The values indicate the molar fractions of ring species (ABC)1 (i.e. unmerged) to (ABC)4 (tetracycles). d Determined by SEC via universal calibration using Mark–Houwink coefficients of ring-polyPMT. e Determined by 1H-NMR spectroscopy using eqn (2). | |||
| Reaction conditions | |||
| Initial molar amounts PTZDA/ETBAA/MT/PMT | 1:5:25:5 |
1:5:50:50 |
1:5:25:10 |
| ETBAA-block (cETBAA = 0.5 mol L−1) | 70 °C for 24 h | 80 °C for 21 h | 60 °C for 24 h |
| MT-block (cMT = 1.0 mol L−1) | 70 °C for 24 h | 80 °C for 51 h | 70 °C for 48 h |
| PMT-block (cPMT = 1.0 mol L−1) | 60 °C for 24 h | 80 °C for 24 h | 70 °C for 24 h |
| Properties | |||
| Found molar amounts PTZDA/ETBAA/MT/PMTa | 1:5:16:5 |
1:6:35:38 |
1:5:20:10 |
| nb |
26 | 79 | 35 |
| (ABC)1/(ABC)2/(ABC)3/(ABC)4c |
77:23:0:0 |
51:28:16:5 |
63:22:15:0 |
| n/g mol−1d |
6 000 | 17200 |
7 300 |
| RP1/g mol−1e |
3 700 | 11700 |
4 800 |
| Đ | 1.5 | 1.7 | 1.4 |
It turned out that ETBAA – in contrast to the small influence of the ETBAA-block in ring-poly(ETBAA-block-PMT)n found in the preliminary experiments (sample η in Table 1) – had a great impact on the occurring interim ring-poly(ETBAA-block-MT)n. ETBAA increased the Tg of ring-polyMT significantly leading to almost dimensional stable polymers. The last block added, consisting of PMT, was thus chosen to be only five units long in order to fine tune the mechanical properties. The final polymeric material was dimensionally stable yet deformable as desired, which shows the great influence even of a small ETBAA-block on the material properties. The final compositions of the resulting polymers slightly differ from the initial monomer composition, possibly due to incomplete conversion of the individual monomers.
Comparing the calculated number average molar masses of a single polymer ring RP1 to the experimentally measured n-values (see Table 2), it is obvious that ring merging took place. This is confirmed by inspection of the molar mass distributions that show a multimodal pattern in each case (see Fig. 4). By application of a multi-Gaussian fit, the molar amounts nRPi of all the ring multiples could roughly be estimated using eqn (1):
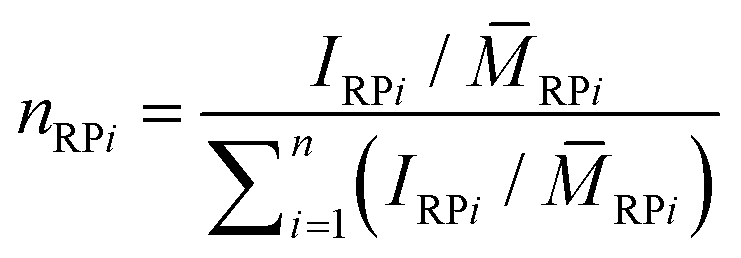 | (1) |
RPi describes the number average molar mass of the ring species RPi, which was determined by the composition of the polymer obtained from 1H-NMR data following eqn (2): | (2) |
 | ||
| Fig. 4 Molar mass distributions (SEC distributions) of samples (a) CP-01, (b) CP-02, and (c) CP-03, which were fitted by several Gaussian functions (orange filled curves) that estimated the individual multiples (ABC)n of the ring merging reaction and residual polymer from the previous polymerization step. Original data: blue curves, sum of Gaussian fits: orange dashed curves. | ||
Synthesis of the polycyclic modular polymers
In order to synthesize the desired polycyclic polymers from the designed precursor polymers, a polyaddition of the precursor ring-block-copolymers with a bifunctional linker was performed. This approach was realized utilizing click-chemistry via a copper-catalyzed Huisgen 1,3-dipolar cycloaddition, which is an easy and versatile method33–35 yielding high conversions quickly at mild reaction conditions. This is especially necessary for reactions of polymers, which exhibit reaction sites with steric hindrance.36,37In order to explore this polyaddition reaction, small scale experiments were performed from CP-01 and the bifunctional linker bis(3-azidopropyl) decandioate 5 (see Scheme 3), of which a system with 0.5 eq. CuI/PMDETA in anhydrous THF proved to be the most appropriate one. The reaction proceeded slowly at room temperature, but could be accelerated significantly by heating to 35 °C, yielding a conversion of more than 95% after 24 h (see Table 3). The reaction progress could easily be monitored by 1H-NMR spectroscopy (see Fig. 5).
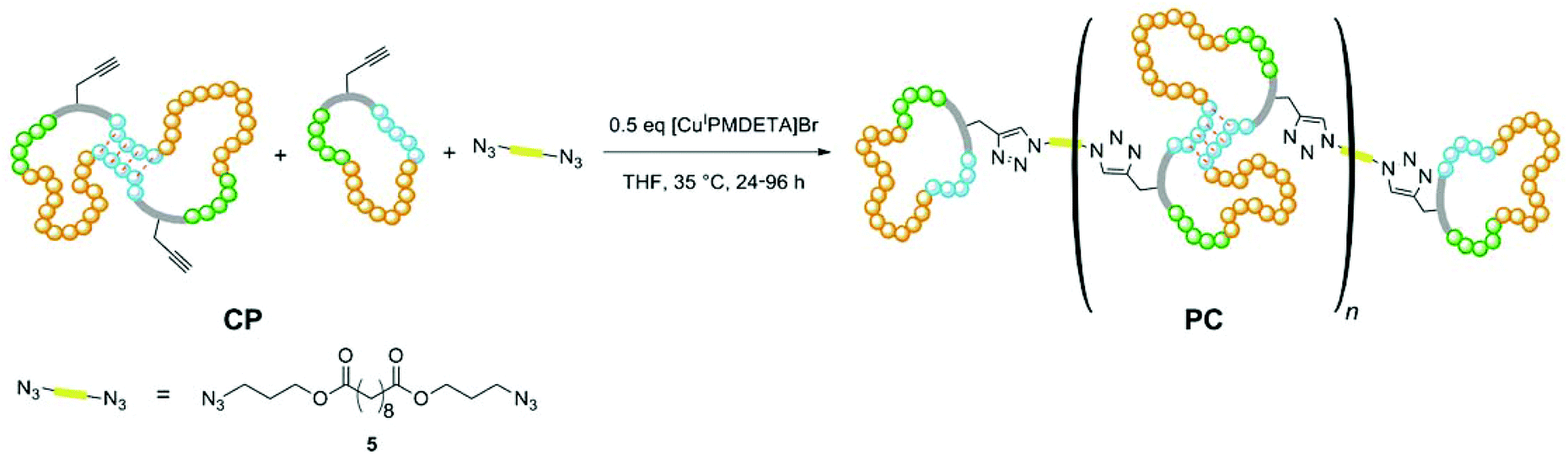 | ||
| Scheme 3 Idealized polyaddition reaction of unmerged and doubled (ABC)n-ring-block-copolymers (CP) with the bifunctional linker bis(3-azidopropyl) decandioate 5via copper-catalyzed 1,3-dipolar cycloaddition yielding polycyclic modular polymer (PC). The colors correspond to the Monomer units of PMT (green), MT (orange), and ETBAA (blue). The formation of intramolecular H-bonds is indicated by red dashed lines. | ||
 | ||
| Fig. 5 1H-NMR-spectra of the bifunctional linker (dark blue), precursor polymer CP-01 (green) and the polyaddition product of both, PC-01 (maroon). The prominent protons indicating the presence of alkyne- (4.68 and 2.53 ppm) and azide groups (3.37 and 1.89 ppm) marked in the reactants disappear or shift to 5.18 and 4.45 ppm, respectively, in the resulting polyadduct PC-01, clearly indicating the formation of a triazole ring. | ||
| PC-01 | PC-02 | PC-03 | |
|---|---|---|---|
| a The conversion could not be determined by means of gravimetry due to insolubility of the dried sample (see text). b Determined by SEC by means of universal calibration with Mark–Houwink coefficients for ring-polyPMT. c Determined before drying and occurrence of physical crosslinking. | |||
| Utilized precursor polymer | CP-01 | CP-02 | CP-03 |
| Click conditions | 92 h, 25 °C +24 h, 35 °C | 65 h, 35 °C | 20 h, 35 °C |
| Conversion/% | 95.5 | 81.0 | n.d.a |
| A maxn | 2.59 | 3.92 | 2.50 |
| Ā n | 4.40 | 1.75 | 2.78 |
| Ā w | 18.5 | 8.83 | 8.87 |
|
n/g mol−1 |
25300b |
17600b |
17200b,c |
| Đ | 5.1 | 10.4 | 4.4 |
| T g/°C | n.d. | 10 | 10 |
| Physical crosslinked | No | No, possibly branched | Yes |
| Young's modulus/MPa | n.d | 5.7 ± 1.0 | 13.3 ± 0.9 |
| Elongation at fracture/% | n.d | (4.6 ± 2.1) × 102 | (3.2 ± 0.6) × 102 |
| Toughness/kJ dm−3 | n.d | 1.2 ± 0.7 | 2.3 ± 0.6 |
| Shape regeneration ability | n.d | Partially | Yes |
| Healing ability | n.d | n.d. | Yes |
After successful performance of the probing reaction yielding PC-01, the other precursor polymers CP-02 and CP-03 were converted on larger scales to polycyclic polymers in order to obtain enough material for the preparation of dog bone samples for tensile testing. The conversion of CP-03 could not be determined by means of 1H-NMR, due to the formation of insoluble material during work-up which will be discussed later. However, by regarding the evolution of molar masses in the SEC before work-up (see Fig. 6c, black and red curves) it could be assumed that monomer conversion was basically complete.
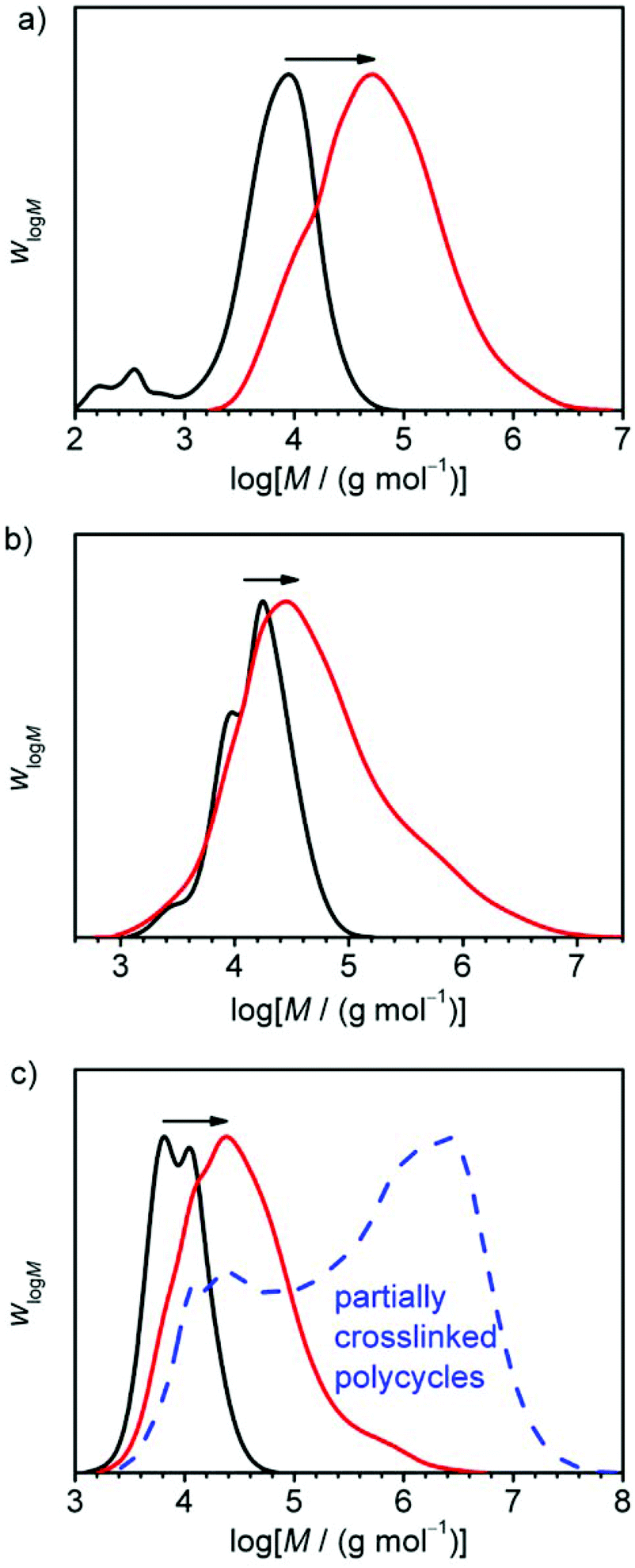 | ||
| Fig. 6 Evolution of molar mass distributions (SEC traces) during the polyaddition reactions of (a) CP-01, (b) CP-02, and (c) CP-03 (black curves) with the bifunctional linker. The polyaddition products (a) PC-01 and (b) PC-02 are shown after precipitation from diethyl ether and drying (red curves). The molar mass distribution of (c) PC-03 was recorded after dissolution of the precipitated material in THF before the drying process (red curve) and after ultrasonic treatment of the re-dissolved material which became insoluble after drying (blue dashed curve), see text for further information. | ||
Due to the different ring species being present in the precursor material, a variety of possible products may emerge from the linking process. As the single rings provide only one linking site, they form 8-shaped structures consisting of two interconnected rings in the case they only react with each other. Alternatively, they form terminal groups of more elongated polycyclic chains. The doubled rings with two reaction sites lead to linear polycyclic chains, whereas triple or quadruple rings may lead to branched structures or even networks at high conversions. Regarding the sample PC-01 containing single and doubled rings only, as seen in the molar mass distribution of the precursor polymer CP-01 (see Fig. 4a), only linear structures may form which are terminated with unmerged rings (see Scheme 3). The other samples having tripled and quadrupled rings, may show branching or even network formation. Under assumption of complete conversion and ideal stoichiometry, a maximum degree of polyaddition Amaxn depending on the molar fraction of unmerged rings xRP1 can easily be calculated by eqn (3):
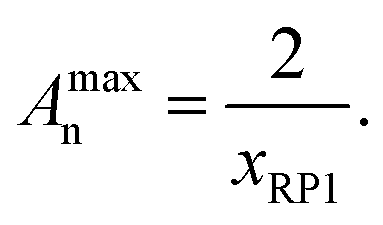 | (3) |
The real number average degree of polyaddition Ān was calculated using eqn (4)
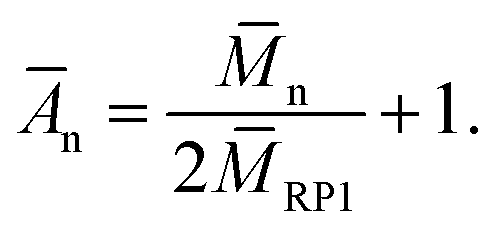 | (4) |
The weight average degree of polyaddition Āw could be calculated similarly viaeqn (5)
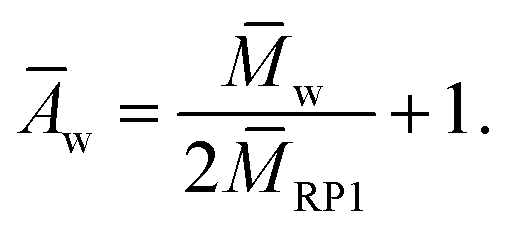 | (5) |
As shown in Table 3, all samples have values of Amaxn that are rather low with values ranging from 2.5 to 4.4. An inherent network formation during the polyaddition, originating from the multiply merged rings, can thus be excluded. PC-03 has an Ān which is very similar to its Amaxn. while PC-02 has an Amaxn of 3.92 which is more than the doubled value of its Ān. This is not surprising regarding its incomplete conversion, and a high conversion is crucial for polyaddition reactions. Opposite to this, PC-01 shows a slightly higher value of Ān than of Amaxn. This most likely occurs from the work-up method by precipitation of the polymer from diethyl ether, which shows a selective precipitation for higher molar masses. This effect is most pronounced with this sample, which contains many of the two interconnected unmerged rings, as mentioned before, which at the same time are relatively small (7 400 g mol−1). These smallest species are larger in PC-02 (23400 g mol−1) and PC-03 (9 600 g mol−1) and are thus naturally less affected by this selective precipitation effect.
The generally small values of Ān differ strongly from the corresponding Āw-values, leading to large dispersities between 4.4 and 10.4. Although the number average values indicate small overall chain-lengths in all polycycles, the mass average value shows that more than half of the material mass and under the assumption of uniform density the same amount of the sample volume consists of polycycles with a length of eight rings or above. The rest, mainly consisting of connected unmerged ring polymer, can essentially be considered as not contributing to the desired material properties.
Although network formation during the linking reaction is not occurring to a considerable extent, polymer PC-03 became insoluble after complete drying of the sample. This finding was attributed to the formation of a physical network built up by hydrogen bonds between the ETBAA-blocks. To prove this, the insoluble sample was ultrasonicated in THF in the presence of a small amount of water in order to break up the hydrogen bonds and substitute them with polymer-water conjugates. After two hours of sonication, the sample could indeed be dissolved again (see Scheme 4) which lead to the conclusion that a physical network was present here. A comparison of the molar mass distribution in the non-crosslinked state before drying and after sonication (see Fig. 6c) shows the appearance of very high molar weight components up to 3 × 107 g mol−1 in the latter case, indicating that the disruption of the physical network was not complete and aggregates held together by hydrogen bonds still are present.
 | ||
| Scheme 4 Photographs and corresponding molecular topology of physically crosslinked sample PC-03 before and after ultrasonic treatment in THF–water. The blue dashed lines display inter- and intramolecular H-bonds. | ||
Mechanical properties of the polycyclic polymers
In order to characterize the synthesized polycyclic polymers with respect to their mechanical properties, uniaxial tensile testing was carried out on dog bone test specimen of PC-02 and PC-03. PC-01 was not tested as it was produced on milligram scale only and was used up for the optimization of the click-reaction conditions, as mentioned before. Due to limited availability of the other sample material, only three specimen per polymeric sample were produced via drop casting (for details see ESI, Fig. S2†). First, a simple tensile testing with elongation until break was conducted with two of the samples. The recorded stress-strain curves with the technical stress σtech (independent of cross section of specimen) plotted versus sample elongation ε gave information about the Young's modulus, the elongation and stress at break, as well as the toughness from the area below the curve (see Fig. 7 and Table 4 for data from tensile-testing). Secondly, cyclic stress-strain curves were recorded with the third specimen giving further information about the hysteresis behaviour between loading and unloading. These curves give information about the elasticity and the ability to dissipate energy. Multiple repetition of the cyclic process additionally provided information about the sample's ability to regenerate its shape in time (see Fig. 8 and 9 and Table 4 for data of the cyclic measurements).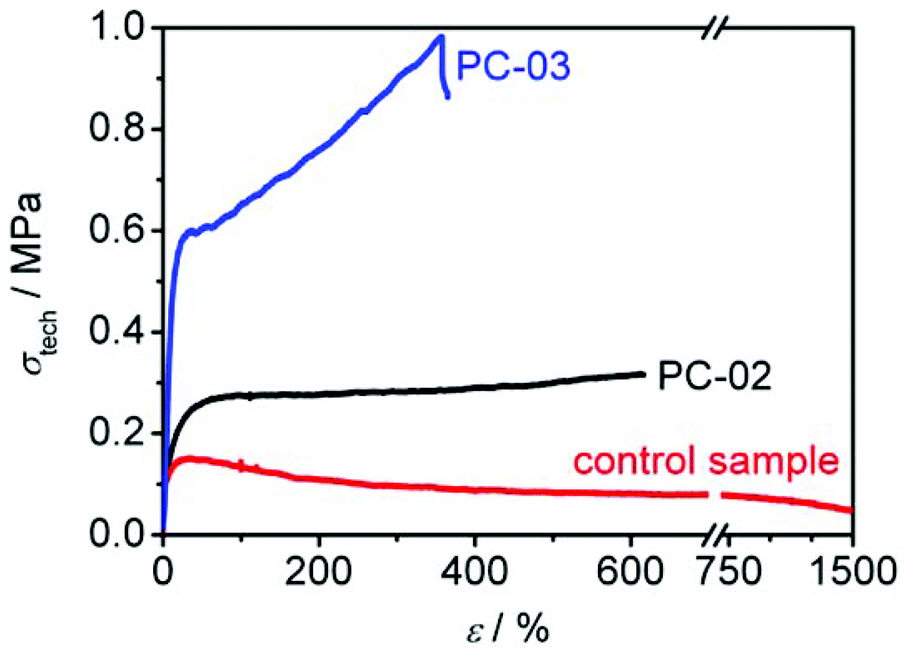 | ||
| Fig. 7 Stress-strain curves of polycyclic samples PC-02 and PC-03 and non-functionalized ring polymer ζ. For reasons of clarity only one curve per material is plotted. | ||
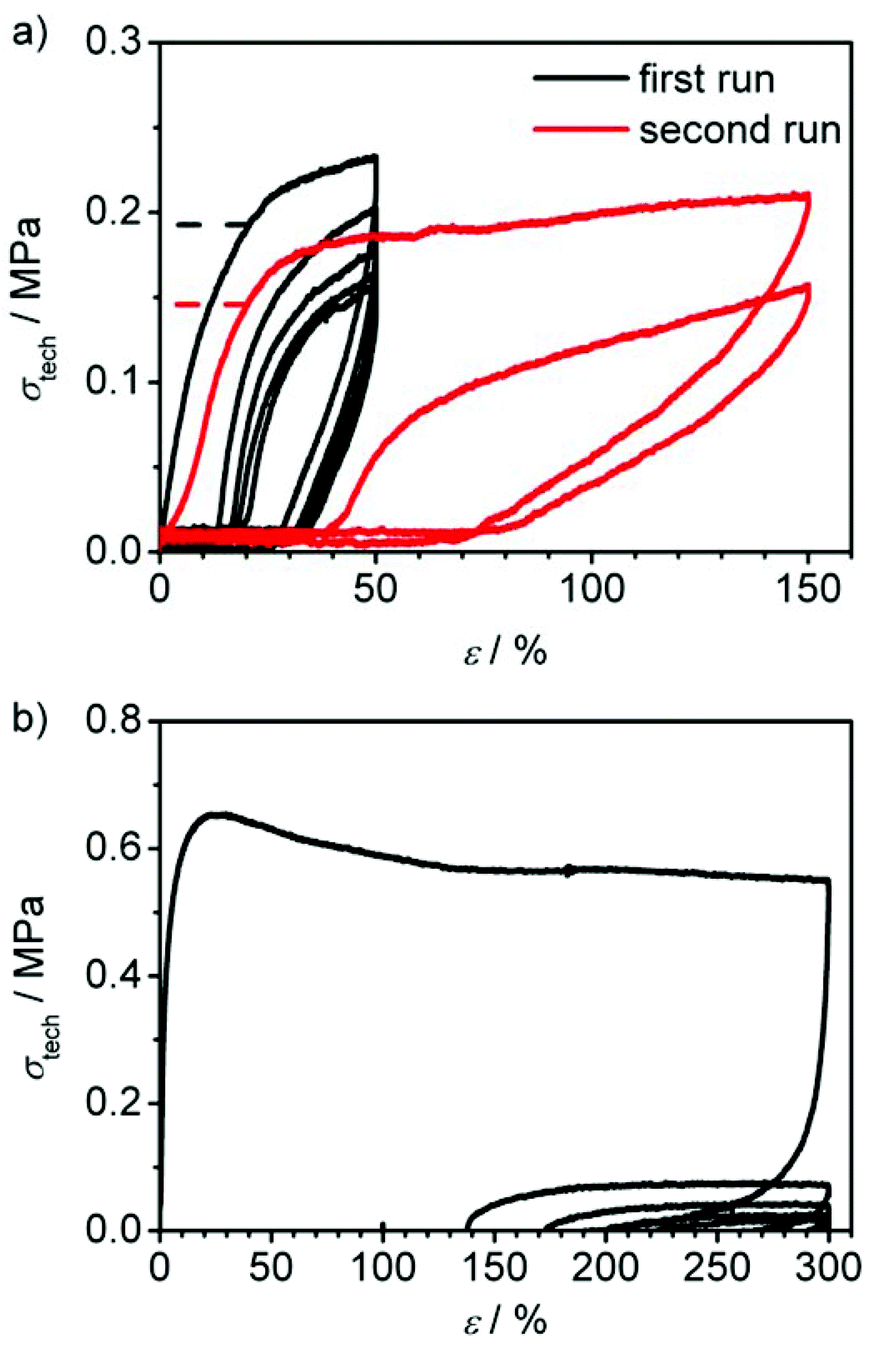 | ||
| Fig. 8 Cyclic stress-strain curves of (a) polycyclic sample PC-02 before (black curve) and after (red curve) thermal regeneration and (b) precursor polymer CP-02. 1 min pause between runs. The dashed lines in a) display the amount of stress applied at 20% strain in the first run (σ020%) and in the second run (σ20%). | ||
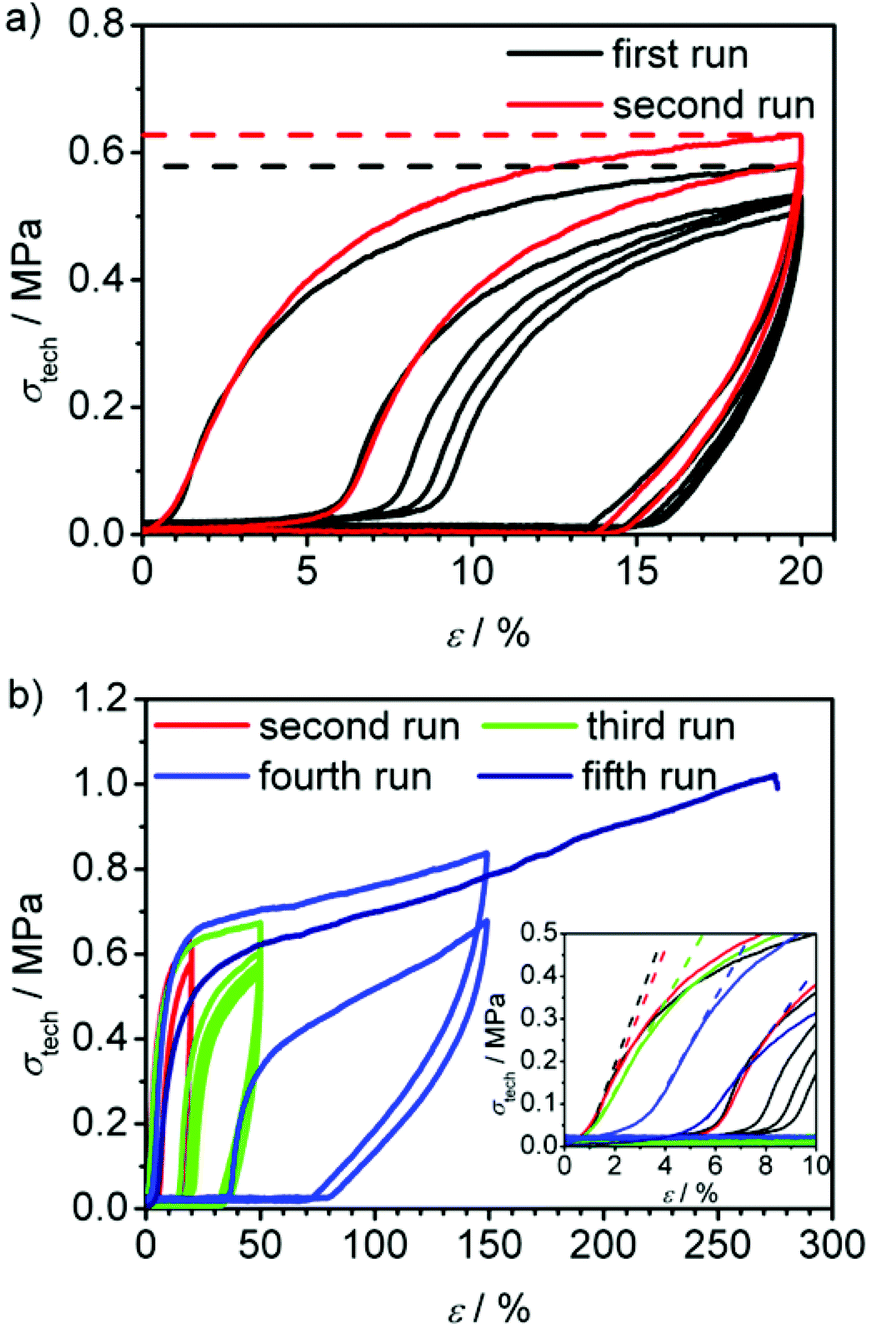 | ||
| Fig. 9 Cyclic stress-strain curves of (a) polycyclic sample PC-03 before (black curve) and after (red curve) first thermal regeneration (first and second run) and (b) second to fifth run. Note that between each run the specimen was thermally regenerated. The dashed lines in a) indicate the stress applied at 20% elongation in the first run (σ020%) and in the second run (σ20%). The small inset diagram in (b) is a zoomed view of the initial stress-strain-region and shows the shift of the curves after each regeneration. | ||
| Sample | Tensile testing conditions | Elongation/% | Fracture stress/MPa | Relative length after regen./% | Young's modulus/MPa | Toughness/dissipation at 1st cycle/kJ dm−3 | σ 20%/σ020%a |
|---|---|---|---|---|---|---|---|
| a σ 020% corresponds to the applied stress at 20% elongation in the first cycle of the first run, whereas σ20% corresponds to the corresponding value in the first cycle of any other run. b Average of two runs. c The specimens were elongated to the instruments maximum deflection without fracture. d Elongation at fracture. e Total energy dissipation until fracture (equals toughness). f Measured values refer to first cycle of each run. | |||||||
| ζ (control) | Until fractureb | 1600c | 0.08 ± 0.06 | (—) | 15.4 ± 0.9 | (—) | (—) |
| PC-02 | Until fractureb | (4.6 ± 2.3) × 102d |
0.28 ± 0.05 | (—) | 5.7 ± 1.0 | 1.2 ± 0.7e | (—) |
| Cyclicf, first run | 50 | (—) | 100.0 | 1.7 | 0.069 | 100.0 | |
| Second run | 150 | (—) | 109.8 | 1.3 | 0.195 | 90.7 | |
| PC-03 | Until fractureb | (3.2 ± 0.6) × 102d |
0.92 ± 0.09 | (—) | 13.3 ± 0.9 | 2.3 ± 0.6e | (—) |
| Cyclic, first run | 20 | (—) | 100.0 | 13.9 | 0.073 | 100.0 | |
| Second run | 20 | (—) | 100.0 | 11.6 | 0.082 | 108.6 | |
| Third run | 50 | (—) | 100.3 | 10.7 | 0.244 | 107.1 | |
| Fourth run | 150 | (—) | 102.4 | 10.7 | 0.805 | 111.0 | |
| Fifth run (fracture) | 276d | 1.02 | 104.6 | 7.8 | 2.07e | 86.2 | |
In Fig. 7, the tensile measurements of PC-02 and PC-03 are depicted. As reference, a control sample made from non-functionalized ring-polymer consisting of PMT and MT only (sample ζ from the preliminary experiments section, see Table 1) was tested as well. The control specimen show a typical behaviour of thermoplastics including a yield point followed by a subsequent decrease of stress with increasing elongation. It could, however, not be fractured even by elongation up to the maximum deflection of the testing machine (see red curve in Fig. 7 and Table 4). The polycyclic test specimens, on the other hand, clearly exhibit a completely different mechanical response behaviour, lacking a distinct yield point. They show a monotonically increasing curve upon elongation until the fracture length was reached. Polymer PC-02 was tested twice showing a Young's modulus of 5.7 ± 1.0 MPa, i.e., it is a rather soft material. It could be elongated to the 4.6 ± 2.3-fold of its original length at 0.28 ± 0.05 MPa until it ruptured (see black curve in Fig. 7 and Table 4). The physically crosslinked sample PC-03 exhibited a higher Young's modulus of about 13.3 ± 0.9 MPa, but could only be elongated to two-third the elongation of PC-02 until it ruptured. It, however, showed a higher fracture stress of 0.92 ± 0.09 MPa (see blue curve in Fig. 7 and Table 4). The continuously rising stress-strain curves of both polycyclic samples differ strongly from the stress-strain behaviour of the control sample and may induced by the continuously occurring cleavage of hydrogen bonds that have formed in the material, which requires an increased energy uptake in order to elongate.
PC-03 shows a larger toughness with 2.3 ± 0.6 kJ dm−3 than PC-02 with 1.2 ± 0.7 kJ dm−3 (see Table 4). The difference may be ascribed to two reasons: Firstly, the molar concentration of ETBAA-units is considerably higher in PC-03 than in PC-02 due to comparable block lengths but smaller ring sizes in PC-03. This leads to a higher concentration of hydrogen bonds that need to be broken in PC-03. Secondly, the presence of stronger physical crosslinks in PC-03 – as described above – hinder the macromolecules to plastically flow at greater elongations, which leads to a higher stress required for elongate the sample.
In Fig. 8a, the cyclic tensile measurements of PC-02 are shown. In a first set of experiments, the sample was strained to 50% elongation and then unloaded again. This cycle was performed five times within one run (see black curve). The loading curve shows the same features as in the previously described tensile-testing, whereas the unloading curve shows a steep fall-off in the beginning, followed by a zero-stress region. In the second cycle, after one minute of regeneration at zero stress, the loading curve shifted, showing an elongation of about 15% of the unloaded sample and a smoothed stress-strain-curve. The hysteresis behaviour indicates a very slow regeneration of the shape and thus no typical rubber-elastic relaxation. Instead, the difference between loading and unloading curve shows a certain energy dissipation by the sample indicated by the large area between both curves. In comparison, cyclic tensile measurements of the precursor polymer CP-02, which contains unlinked macrocycles only and acts as control here, shows no characteristic hysteresis curve but a permanent plastic deformation of the sample (see Fig. 8b). It can be concluded that the polycyclic architecture is a critical feature of these high performance materials. In order to speed up the regeneration of PC-02, the sample was heated carefully to about 100 °C inside the testing machine for several seconds after the first five cycles which lead to a fast regeneration to about 110% of its original length. This shows that the sample has the ability to recover its shape to a great extent and thus has a certain elasticity. This effect is obviously slowed down at ambient temperature, which is only 12 °C above the Tg, which hinders the quick reordering process of the macromolecules. A new tensile testing run with the almost fully regenerated sample up to 150% elongation shows an increased maximum stress compared to the last three cycles before regeneration, however, with a slightly decreased Young's modulus (see red curve in Fig. 8a and Table 4). This indicates a higher amount of dissipated energy in the hysteresis cycle after the thermal regeneration process and thus suggests the partial regeneration of the sample's hydrogen bonds that broke on the first straining.
PC-03 shows a similar loading and unloading behaviour in the first five cycles (see black curve in Fig. 9a and Table 4) compared to PC-02, except the significant larger Young's modulus of 13.9 MPa, which naturally is similar to the observation from the previously mentioned tensile-testing experiments conducted until break (Fig. 7). As expected, heating of the specimen after five cycles of loading and unloading led again to a regeneration of the material. However, this sample recovered its original length completely after a few seconds. Afterwards, another run at the same elongation (red curve in Fig. 9a) led to a 9% higher amount of applicable stress at 20% elongation (σ20%) compared to the first cycle in the first run before regeneration (σ020%); the material was strengthened. However, the Young's modulus showed a slight decrease to 11.6 MPa. The same principle behaviour was observed in a second cycle. Adding a third and fourth run to higher elongations of 50 and 150% (see Fig. 9b, green and blue curves) showed a full recovery up to the third run while in the fourth run an elongation of 2.4% and a decrease of the Young's modulus to 10.7 MPa was observed (see Table 4), whereas σ20%/σ020% stayed in the range of 109 ± 2%, just like in the previous runs. In a last, fifth run the sample could stand a maximum elongation up to 276% until it ruptured and showed a slightly decreased Young's modulus, but again a constant σ20%/σ020% value compared to all previously conducted runs. In general, these results indicate a strong ability to regenerate the broken hydrogen bonds by heat application as well as a good restoration of shape.
Shape regeneration and healing properties
In order to deeper investigate the shape regeneration property of the material, a further experiment was conducted by twisting a fragment of PC-03 and instantly cooling it to −20 °C. This specimen contained its novel shape for several days at a low temperature and recovered its original shape completely within minutes after exposure to 40 °C, which is evidence for a temperature sensitive shape memory behaviour of these polycyclic materials (see Fig. 10). These special materials typically exhibit two states of shape, the original state and a deformed state which is metastable below the sample's glass transition temperature due to the transitionally hindered polymer chains.38–40 Here the shape memory effect is probably caused by the physical crosslinks in the sample which partially break on mechanical deformation of the sample, although a certain amount remains unaltered and thus gives the network a basic stability and structure memory. Thus it has the ability to regenerate its original shape just like in typical elastomers, with the difference of a much higher glass transition temperature, which on one hand leads to a slower regeneration compared to typical rubbers, but on the other hand stabilizes the deformed state at a considerable high temperature. In addition, these new materials are lacking permanent cross-links, which make them potentially processable like conventional thermoplastics. Along with the effective energy dissipation mechanism, which is due to the incorporated hydrogen bonds, this yields an interesting type of shape memory polymer. | ||
| Fig. 10 Series of pictures showing the shape regeneration of a twisted specimen of sample PC-03. The sample was put in a petri-dish and heated to 40 °C in a water bath. | ||
A thermodynamic transition corresponding to a ‘melting’ of the hydrogen bonds, which would lead to a permanent shape deformation, could not be identified in calorimetric DSC measurements (see Fig. S4 in ESI†). Considering the average amount of five ETBAA-monomers per block and thus an expected amount of five to ten hydrogen bonds between two blocks, the bond-dissociation energy may simply be too high to be overcome in the observed temperature range up to 200 °C. With a reasonably estimated value of approximately 30 kJ mol−1 per ETBAA-dimer41 the total dissociation energy is supposed to be around 75 to 150 kJ mol−1, which appears to be too high to be broken thermally up to 200 °C. The degradation temperature of the material is with 300 °C – due to the sulphur-containing polymer backbone of the macrocycles – relatively low (see Fig. S5 in ESI†), which restricts high temperature measurements, where melting of the H-bonds may be observable.
The ability of the material to regenerate its broken hydrogen bonds was examined further by the attempt to heal a completely fractured test specimen by putting the two parts together and heating the fracture site. This was done in a first attempt by simply heating the combined fragments to 100 °C for 15 seconds and cooling down for ten minutes afterwards. The obtained specimen did indeed undergo a partial healing leaving only a small crack at the former fracture site (see Fig. 11 for detailed procedure). Since the fragments did not undergo any visible melting process and completely kept their shape under heating, it can be concluded that the healing process in fact arises from the recombination of broken intermolecular hydrogen bonds. Tensile testing of the healed sample showed a stiffening compared to the unbroken sample (see Fig. 12). The value for σ20%/σ020% increased by about 5%. Due to the remaining crack, the elongation at break decreased to only 26%. A second healing attempt for 15 minutes at 120 °C also led to a partial healing with the sample breaking after 16% of elongation and a slightly decreased stiffness of the sample compared to the run after the first healing process. In summary this material shows a great potential in its healing ability.
 | ||
| Fig. 11 Healing of specimen made from PC-03 via application of heat: (a) fractured specimen, (b) aligning of the fragments in the mold, (c) heating to about 100 °C for 15 s, (d) removal of the specimen after 10 min of cooling to ambient temperature, (e) partially healed specimen. | ||
 | ||
| Fig. 12 Stress-strain curves of PC-03 before rupture (black curve, fifth run of cyclic tensile testing described above) and after first and second rupture including subsequent healing (blue and red curves). | ||
Conclusions
We successfully developed a novel modular synthesis of polycyclic polymers based on cyclic (ABC)n-multiblock-copolymers, consisting of up to twelve separate blocks, by performing stepwise polymerization of three individual blocks and exploiting the ring merging reaction of these ring polymers in order to multiply the ring size and thus functionality. These precursor polymers were successfully interconnected via a Huisgen click reaction in order to form linear and branched polycyclic polymers. A small ETBAA-block within the rings with the ability to form self-complementary hydrogen bonds successfully lead to intra- and intermolecular physical links. The obtained polycyclic materials, which mimic nature's paragon Titin, have some extraordinary material properties concerning elasticity and energy dissipation. Although both tested materials PC-02 and PC-03 are rather soft compared to natural fibres, they show a distinctive stress-strain behaviour that is similar to biological materials. Additionally, the physically crosslinked material PC-03 shows a shape-memory behaviour with good shape recovery after deformation and has healing abilities due to its inherent ability to form hydrogen bonds.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (DFG) for the project VA226/5-2 is gratefully acknowledged. P.V. acknowledges receipt of a Heisenberg-Professorship (DFG).Notes and references
- F. R. Schwarzl, Polymermechanik, Springer-Verlag, Berlin Heidelberg, 1990, p. 416 Search PubMed.
- M. A. Meyers, P.-Y. Chen, A. Y.-M. Lin and Y. Seki, Prog. Mater. Sci., 2008, 53, 1–206 CrossRef CAS.
- Z. Guan, Polym. Int., 2007, 473, 467–473 CrossRef.
- A. M. Kushner and Z. Guan, Angew. Chem., Int. Ed., 2011, 50, 9026–9057 CrossRef CAS PubMed.
- N. Becker, E. Oroudjev, S. Mutz, J. P. Cleveland, P. K. Hansma, C. Y. Hayashi, D. E. Makarov and H. G. Hansma, Nat. Mater., 2003, 2, 278–283 CrossRef CAS PubMed.
- D. Porter and F. Vollrath, Adv. Mater., 2009, 21, 487–492 CrossRef CAS.
- Z. B. Guan, J. T. Roland, J. Z. Bai, S. X. Ma, T. M. McIntire and M. Nguyen, J. Am. Chem. Soc., 2004, 126, 2058–2065 CrossRef CAS PubMed.
- M. Rief, Science, 1997, 276, 1109–1112 CrossRef CAS PubMed.
- M. S. Kellermayer, Science, 1997, 276, 1112–1116 CrossRef CAS PubMed.
- H. Li, a. F. Oberhauser, S. B. Fowler, J. Clarke and J. M. Fernandez, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6527–6531 CrossRef CAS PubMed.
- E. a. Di Marzio and C. M. Guttman, Macromolecules, 1987, 20, 1403–1407 CrossRef CAS.
- P. E. Marszalek, H. Lu, H. Li, M. Carrion-Vazquez, a. F. Oberhauser, K. Schulten and J. M. Fernandez, Nature, 1999, 402, 100–103 CrossRef CAS PubMed.
- M. Xu and R. V. Lewis, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 7120–7124 CrossRef CAS.
- E. Von Castelmur, M. Marino, D. I. Svergun, L. Kreplak, Z. Ucurum-Fotiadis, P. V. Konarev, A. Urzhumtsev, D. Labeit, S. Labeit and O. Mayans, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1186–1191 CrossRef CAS PubMed.
- K. Wang, Adv. Biophys., 1996, 33, 123–134 CrossRef CAS PubMed.
- K. Maruyama, FASEB J., 1997, 11, 341–345 CAS.
- H. Lu and K. Schulten, Biophys. J., 2000, 79, 51–65 CrossRef CAS PubMed.
- H. Lu, A. Krammer, B. Isralewitz, V. Vogel and K. Schulten, in Elastic Filaments of the Cell, ed. H. L. Granzier and G. H. Pollack, Kluwer Academic/Plenum Publ, New York, 2000, vol. 481, pp. 143–162 Search PubMed.
- M. J. Winningham and D. Y. Sogah, Macromolecules, 1997, 30, 862–876 CrossRef CAS.
- O. Rathore and D. Y. Sogah, Macromolecules, 2001, 34, 1477–1486 CrossRef CAS.
- O. Rathore and D. Y. Sogah, J. Am. Chem. Soc., 2001, 123, 5231–5239 CrossRef CAS PubMed.
- K. Nagapudi, W. T. Brinkman, J. Leisen, B. S. Thomas, E. R. Wright, C. Haller, X. Wu, R. P. Apkarian, V. P. Conticello and E. L. Chaikof, Macromolecules, 2005, 38, 345–354 CrossRef CAS.
- D. L. Guzmán, A. Randall, P. Baldi and Z. Guan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1989–1994 CrossRef PubMed.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- a. T. Ten Cate, H. Kooijman, A. L. Spek, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2004, 126, 3801–3808 CrossRef CAS PubMed.
- A. M. Kushner, J. D. Vossler, G. A. Williams and Z. B. Guan, J. Am. Chem. Soc., 2009, 131, 8766–8768 CrossRef CAS PubMed.
- H. Kudo, S. Makino, A. Kameyama and T. Nishikubo, Macromolecules, 2005, 38, 5964–5969 CrossRef CAS.
- H. Kudo, M. Sato, R. Wakai, T. Iwamoto and T. Nishikubo, Macromolecules, 2008, 41, 521–523 CrossRef CAS.
- J.-H. Schuetz and P. Vana, Macromol. Chem. Phys., 2011, 212, 1263–1275 CrossRef CAS.
- J.-H. Schuetz, L. Sandbrink and P. Vana, Macromol. Chem. Phys., 2013, 214, 1484–1495 CrossRef CAS.
- K. Zhang, M. A. Lackey, J. Cui and G. N. Tew, J. Am. Chem. Soc., 2011, 133, 4140–4148 CrossRef CAS PubMed.
- N. Sugai, H. Heguri, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2011, 133, 19694–19697 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- M. V. Gil, M. J. Arévalo and O. López, Synthesis, 2007, 11, 1589–1620 CrossRef.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1–13 CrossRef CAS.
- A. B. W. Brochu, S. L. Craig and W. M. Reichert, J. Biomed. Mater. Res., Part A, 2011, 96, 492–506 CrossRef PubMed.
- F. Herbst, D. Döhler, P. Michael and W. H. Binder, Macromol. Rapid Commun., 2013, 34, 203–220 CrossRef CAS PubMed.
- B. W. Greenland, G. L. Fiore, S. J. Rowan and C. Weder, in Healable Polymer Systems, The Royal Society of Chemistry, 2013, pp. 92–125 Search PubMed.
- C. L. Lewis, K. Stewart and M. Anthamatten, Macromolecules, 2014, 47, 729–740 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Instrumentation and experimental procedures as well as further characterization data of cyclic and polycyclic polymers. See DOI: 10.1039/c4py01458h |
| This journal is © The Royal Society of Chemistry 2015 |