
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polyglycerol coated polypropylene surfaces for protein and bacteria resistance†
Maike C.
Lukowiak
a,
Sascha
Wettmarshausen
b,
Gundula
Hidde
b,
Petra
Landsberger
b,
Viola
Boenke
b,
Karsten
Rodenacker
c,
Ulrike
Braun
b,
Jörg F.
Friedrich
b,
Anna A.
Gorbushina
b and
Rainer
Haag
*a
aInstitut für Chemie und Biochemie, Freie Universität Berlin, Takustr. 3, 14195 Berlin, Germany. E-mail: haag@chemie.fu-berlin.de
bFederal Institute for Materials Research and Testing (BAM), 12200 Berlin, Germany
cHelmholtz Zentrum München, German Research Center for Environmental Health, Institute of Computational Biology, Ingolstädter Landstr. 1, 85764 Neuherberg, Germany
First published on 27th November 2014
Abstract
Polyglycerol (PG) coated polypropylene (PP) films were synthesized in a two-step approach that involved plasma bromination and subsequently grafting hyperbranched polyglycerols with very few amino functionalities. The influence of different molecular weights and density of reactive linkers were investigated for the grafted PGs. Longer bromination times and higher amounts of linkers on the surface afforded long-term stability. The protein adsorption and bacteria attachment of the PP-PG films were studied. Their extremely low amine content proved to be beneficial for preventing bacteria attachment.
Introduction
In light of the rapid technological progress and demographical changes in the civilized world, biofouling has become a serious problem.1–3 Polyolefins are most commonly used in biomedicine for syringes, surgical sutures, mesh implants, and blood bags, as well as for packaging purposes.4–6 Polypropylene's inertness and high stability are interesting features for biomedicine, but its strong hydrophobicity leads to unspecific protein adsorption, which is believed to be the first conditioning step for biofilm formation, infection, and even foreign body reactions.7 Additionally, the chemical inertness of the polymer surface usually requires harsh activation methods to chemically modify the surface,8 for example, by corona treatment, UV-irradiation, or by introducing the recently reported adsorption of proteins to attach functional groups.9 Plasma treatment is also commonly used for activating surfaces.8,10 However, oxygen and ammonia plasma exposure of polyolefins produce a broad variety of different O or N functional groups inapplicable for specific grafting reactions.11 In contrast to that, plasma bromination introduces high concentration of bromine groups selectively with a minimum of side reactions and can be subsequently substituted, e.g., by short linkers or by low molecular weight poly(ethylene glycol) (PEG) by a wet-chemical way.11–15 Protein adsorption and the following biofilm formation are extremely complex processes that remain to be further clarified in the full scope of their interconnectedness.7 Generally, factors like hydrophobic and electrostatic interactions, water-mediated hydration forces, as well as hydrophilic/hydrophobic surface properties, and other factors may play an important role.7 A wide variety of surface coatings were tested for their protein resistant properties.3 Apart from the most prominent candidates like PEG or zwitterionic surfaces, it has been recognized that polyglycerol could achieve high bioinertness as well.7,16–18 Most of these studies, however, were performed on perfect substrates like gold or glass.19–24 Obviously, these studies were very important to generate fundamental knowledge on this kind of surface coating to investigate the antifouling properties. Recently, polyglycerol coatings were also reported on more applicable materials like, e.g., steel, aluminium, and polymeric membranes.25–36 Nonetheless, many real-life situations require additional bioinert coatings on polyolefin surfaces. In this publication we report a simple way to produce polyglycerol (PG) coated polypropylene (PP) surfaces in only two steps by low-pressure plasma bromination and direct grafting of PG. Therefore, we prepared PGs with different molecular weights and amounts of amino functionality and introduced them to surfaces by a grafting-to technique. The characteristics of the different coated films as well as their potential to resist protein and bacteria attachment were also investigated.Results and discussion
Synthesis
This study presents an easy two-step protocol for the production of PG-lated polyolefin surfaces. In the first step, we used plasma bromination to yield reactive bromine functionalities on the surface. In the second step, hyperbranched PG-amines could be directly grafted to the surfaces (Fig. 1).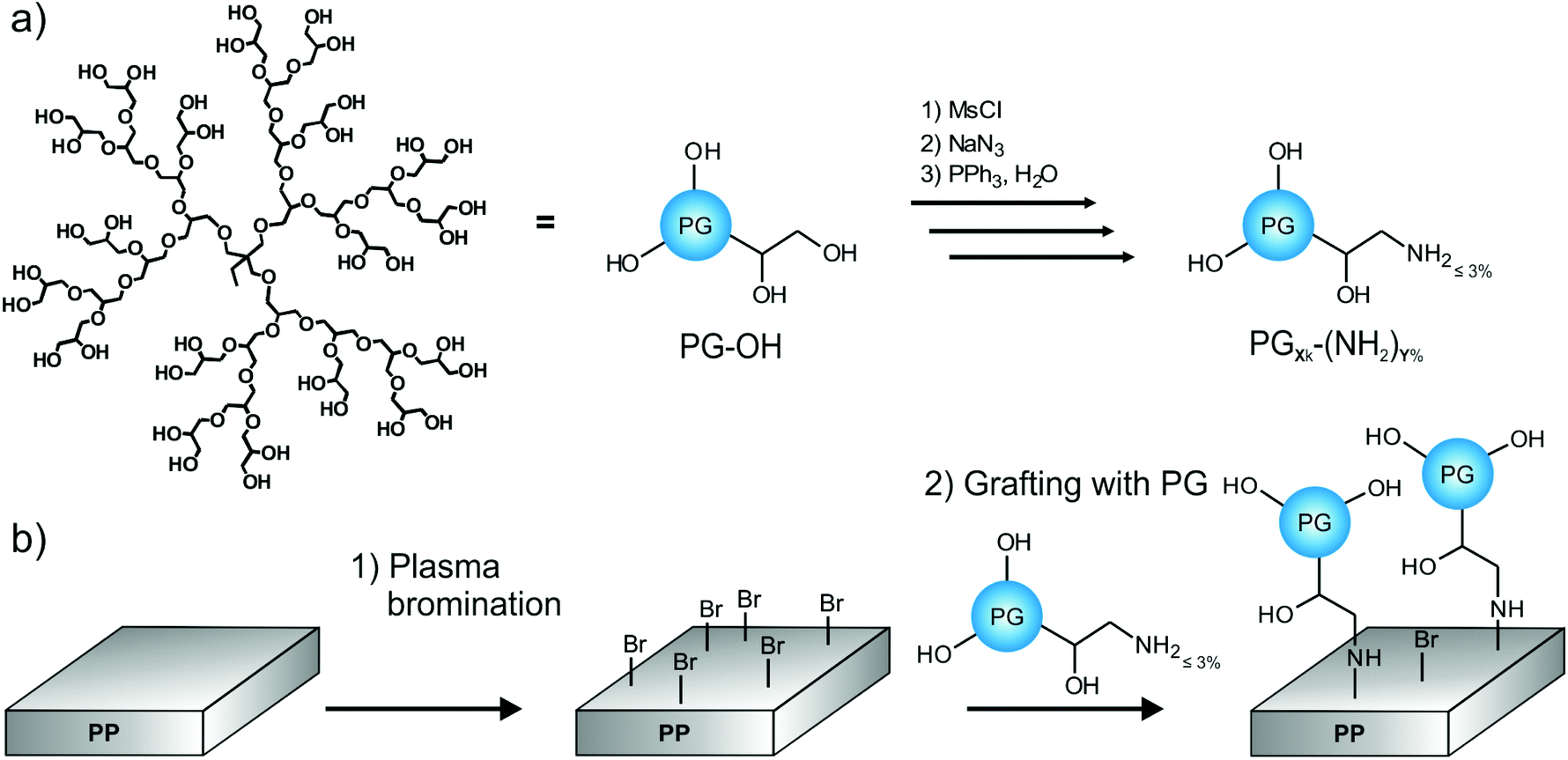 | ||
| Fig. 1 (a) Synthesis of polyglycerols (PG) PGXk-(NH2)Y% with different molecular weights and amounts of amino functionalization. An idealized structure of PG is shown. (b) Schematic representation of the two-step synthesis of PG-lated polypropylene (PP) surfaces by (1) plasma bromination and (2) grafting with PG. | ||
Plasma bromination
Under normal circumstances polyolefin surfaces are chemically inert. The introduction of functional groups on polyolefins requires the substitution of hydrogen and therefore the scission of C–H bonds. Accordingly, harsh activation methods are needed, which unfortunately also lead to the scission of few C–C bonds. Selective grafting is therefore impossible. Plasma treatment is a commonly used technique for polymer surface functionalization.8,37 However, the main challenge remains in the controlled activation, as plasma treatments like oxygen plasmas normally produce a large variety of different functionalities, e.g., hydroxy, carbonyl, ether, ester, etc., that are caused by the high energy level of the plasma process and post-plasma oxidation.11 In contrast, the newly developed plasma bromination introduces exclusively very reactive bromine groups into the topmost surface with high selectivity and yield.12 The high selectivity is caused by the only possible reaction pathway to C–Br. The possible side-reaction to Br− is unimportant because formed HBr is continuously removed by pumping off. As bromine and bromine-containing precursors have a low dissociation energy, the excess of energy and side reactions can be minimized, selectivity improved, undesired radical formation lowered, and therefore the post-plasma oxidation can be nearly eliminated.38 Thus it is possible to generate monotype Br groups suitable for efficient substitution reaction.12,14,39 For these reasons the use of plasma bromination for the production of anchor points was chosen as the first step in this approach. Furthermore, as it has been shown in a previous report, the density of the functional groups can be tuned by varying the plasma time and power.38 To ensure an optimal mode for producing monotype functional group and a minimum of post-plasma oxidation, 3 min plasma treatment time and 100 W power input were chosen to produce PP-Br.39,40 After bromination the PP films were washed intensively with THF to remove unreacted bromine and other low molecular weight material. The success of plasma bromination was checked by X-ray photoelectron spectroscopy (XPS). XPS spectra were normalized per 100 C. Typical elemental compositions of washed PP-Br films ranged around 18 Br/100 C and 5 O/100 C.Grafting-to with PG-amines
It was shown previously that bromine groups at the surface of polypropylene could be easily substituted by amino groups.12 Therefore a simple grafting-to approach was used as second step for the production of PG-lated polyolefin surfaces. For this purpose, a small amount of the hydroxyl functionalities of polyglycerols were converted into amino functionalities by the literature procedure using mesylation, azidation, and Staudinger reduction.41 The reactions were controlled by NMR and IR spectroscopy. It was recently demonstrated in a study on perfect gold as substrate that protein adsorption depends on the amount of amine groups present on PG.22 Polyglycerol still exhibited very good protein resistant behavior with an amount of amino functionalization of up to 9% (of all original OH groups). Moreover, amine groups were accessible for selective ligand and biomolecule attachment.22 In the case of poly(ethylene glycol)s (PEG), higher molecular weight and grafting density resulted in higher protein resistance on smooth surfaces.42,43 However, materials which are more relevant for application, e.g., in the biomedical field are usually rough and the knowledge gained from perfectly smooth substrates might not always be transferable. For instance, rough poly(ether imide) membranes coated with short oligo(ethylene glycol)s performed better than those coated with larger PEGs.44 Therefore more knowledge on potentially applicable surfaces, like polypropylene in this study, is required to design better bioinert surfaces. Grafting of polymers with higher molecular weights is generally more difficult due to steric hindrance. Hence we synthesized hyperbranched PGs with a very low amino functionalization of only up to 3% to obtain the best possible bioinertness for PG. The molecular weights and amino functionalities of the prepared PGs were varied to investigate if adding more binding sites to the surface would overcome the rising steric hindrance upon the increased molecular weight. Three different molecular weights were chosen, namely, 2, 9, and 16 kDa (Mn). For the higher molecular weight PGs, we synthesized two different amounts of amino groups that were comparable to each other. Details of the PG-amines can be found in Table 1.| Name | M n | M w | Degree of NH2-functionalizationa | Average amount of NH2/PGa | Average amount of remaining OH/PGa |
|---|---|---|---|---|---|
| a Calculated from 1H NMR and based on Mw of polyglycerol starting material. | |||||
| PG2k-(NH2)3% | 1.6 kDa | 5.1 kDa | 3% | 2 | 67 |
| PG9k-(NH2)1% | 9.3 kDa | 16.2 kDa | 1% | 3 | 216 |
| PG9k-(NH2)3% | 9.3 kDa | 16.2 kDa | 3% | 7 | 212 |
| PG16k-(NH2)0.4% | 16 kDa | 31 kDa | 0.4% | 2 | 416 |
| PG16k-(NH2)2% | 16 kDa | 31 kDa | 2% | 8 | 410 |
It is important to emphasize that the amount of NH2-groups introduced per PG was very small. For example, this meant that only 3 out of calculated 219 hydroxy groups were converted into amines for the medium molecular weight polyglycerol PG9k-(NH2)1%.
For the grafting-to approach extremely mild reaction conditions were used. PG-amines were reacted in methanol with the brominated polypropylene films PP-Br at room temperature on a shaker. This method allowed larger sized coated films, which is highly important for future application. After reaction overnight, the films were extensively and repeatedly washed to remove any non-covalent bound PG from the surface.
Characterization
Contact angle measurements proved the successful grafting of PGs on the surfaces. While the contact angle of pristine PP was about 100°, and for brominated PP PP-Br still above 90° indicating absence of polar groups, i.e. oxygen-containing groups, the values for PG-lated surfaces could be further reduced to below 20° (Fig. 2). Values in the same range were reported for self-assembled monolayers of PG on gold.19 The resulting water contact angles for the surfaces with different PGs can be also found in Fig. 2.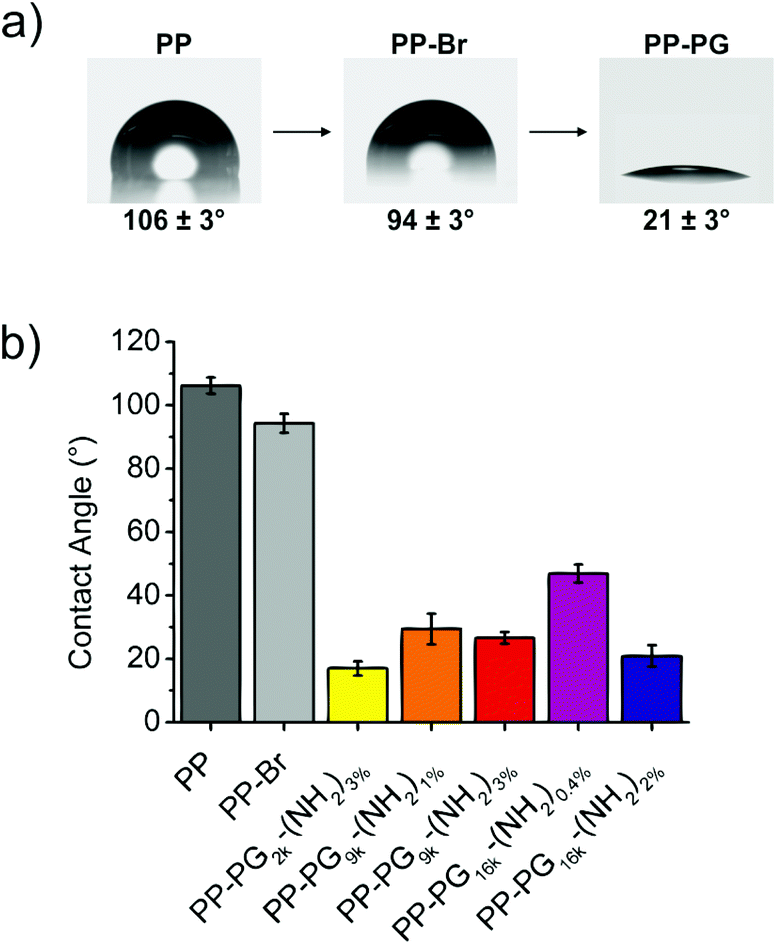 | ||
| Fig. 2 (a) Changes in the contact angle from pristine PP to PP-Br and PP-PG. The picture for PP-PG was chosen as an example for PG16k-(NH2)2%. (b) Different contact angles measured for the different PG-coated PP films. | ||
The lowest contact angle of 17 ± 2° was obtained for the smallest hyperbranched polyglycerol PG2k-(NH2)3%, which is not surprising because lower molecular weight polymers introduce lower steric hindrance in the grafting to surfaces. The next question was if higher molecular weight PGs that have an accordingly higher steric hindrance can still be easily introduced to the PP films by adding more anchoring groups to the surface, which in this case was a higher amount of NH2-groups. For the 9 kDa PG-amine, a contact angle of 29 ± 5° for PG9k-(NH2)1% with an average of 3 NH2/PG and of 27 ± 2° for PG9k-(NH2)3% with an average of 7 NH2/PG was measured. However, the effect became more obvious for bigger PGs. While only contact angles of 47 ± 3° were reached for PG16k-(NH2)0.4% with an average of 2 NH2/PG, the introduction of a higher average amount of linkers of 8 NH2/PG for PG16k-(NH2)2% led to a contact angle (21 ± 3°) which was in the same range as for the small molecular weight PGs. This clearly showed that the steric hindrance resulting from larger polyglycerols could be overcome by introducing a higher number of linkers to the surface. It is assumed that a multivalent surface anchoring also leads to more stable coated polyolefin surfaces.34,45
In the following, the polyglycerol functionalized PP surfaces were analyzed by X-ray photoelectron spectroscopy (XPS), a method that can determine the elemental composition of the surfaces’ top layer. The XPS results are listed in Table 2.
As expected, the amount of oxygen (O/100 C) increased after introducing PG, while the amount of bromine (Br/100 C) decreased. Furthermore, the C1s spectra gave some insight on the binding state of the carbon in the samples. Ether (C–O–C) and alcohol (C–OH) bonds show a peak at a binding energy of approximately 286.5 eV. Due to the polyether structure of hyperbranched polyglycerol such a shoulder in the C1s spectrum of PG-lated PP films was therefore expected at this binding energy. All the different PG functionalized PP surfaces showed the strongly increased O/100 C and reduced Br/100 C amount as well as the expected shoulders in the C1s spectra at 286.5 eV. The percentages for these shoulders in all the C1s spectra are also summarized in Table 2. C1s spectra representing an untreated PP film and a PG grafted PP film are shown in Fig. 3. The oxygen concentration as well as the percentage of the ether/hydroxyl caused shoulder in the C1s peak correlated well with the data from contact angle measurements. The same trend was obtained from the different PG-amines in both cases.
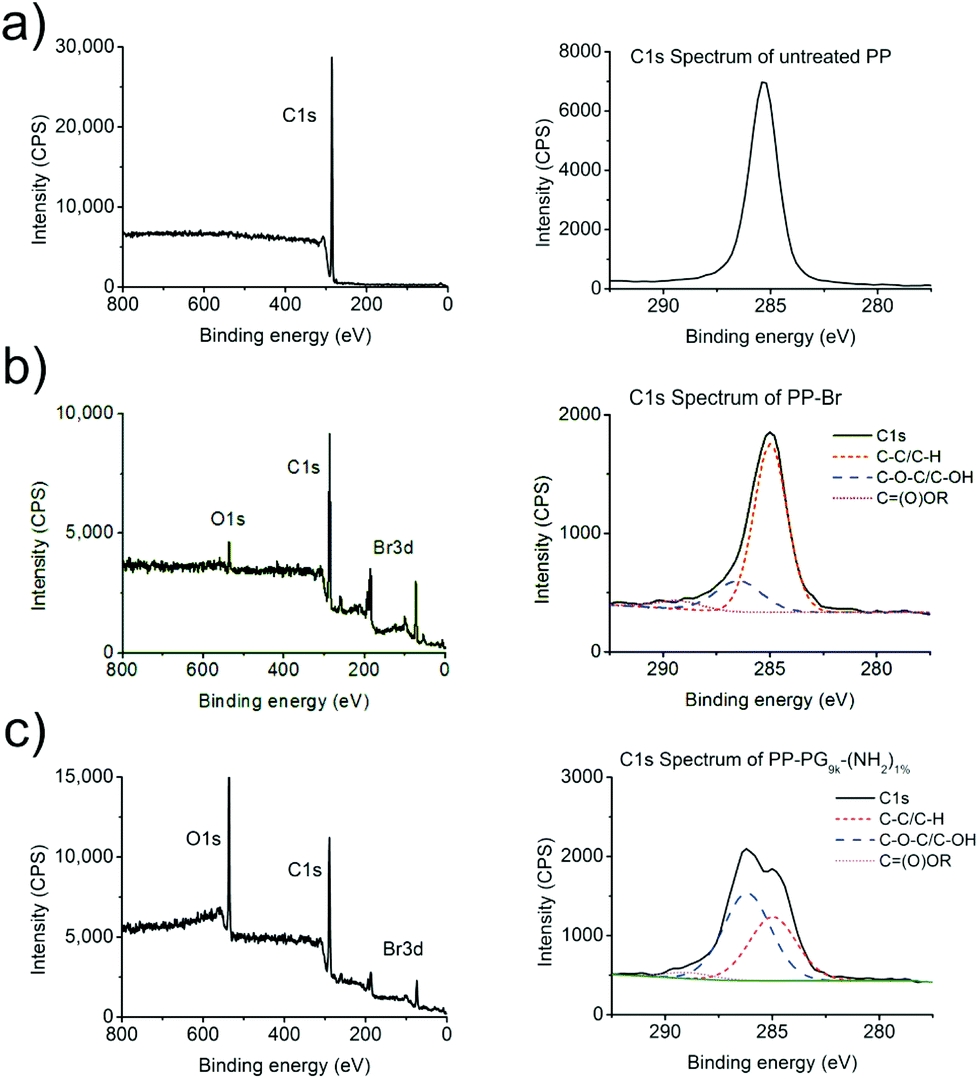 | ||
| Fig. 3 (a) XPS overview (left) and C1s (right) spectra for untreated PP film compared to (b) the XPS overview (left) and C1s (right) spectra of a 3 min plasma brominated PP (PP-Br) film and to (c) the XPS overview (left) and C1s (right) spectra of polyglycerol coated PP (PP-PG9k-(NH2)1%) film which shows the usual shoulder in the peak at 286.5 eV corresponding to ether (C–O–C) and alcohol (C–OH) bonds. | ||
For the medium sized dendritic polyglycerols PG9k-(NH2)1% and PG9k-(NH2)3%, additionally the zeta potential ζ in solution was determined as well as in their functionalized state on the PP surfaces by streaming potential measurements using the SurPASS electrokinetic analyzer (Anton Paar).
For ζ in solution 1.6 ± 0.4 mV for PG9k-(NH2)1% and 5.4 ± 0.3 mV for PG9k-(NH2)3% in phosphate buffered solution at pH 7.4 were measured (Table 3). The higher amount of amino functional groups in PG9k-(NH2)3% led to a higher zeta potential. The values were comparable to previously published data on zeta potential of PG-amines.46
| Sample | Zeta potential ζ (mV) |
|---|---|
| PG9k-(NH2)1% | 1.6 ± 0.4 |
| PG9k-(NH2)3% | 5.4 ± 0.3 |
The zeta potential of the solid samples was analyzed using a SurPASS electrokinetic analyzer. Streaming potential measurements were performed as basic titrations by adding KOH. Even if it might seem surprising, the surface zeta potential of neutral polymer surfaces is usually negative.47,48 The reason for this observation was determined to be the accumulation of hydroxide ions in the immobile Stern layer of the electrical double layer.47,49 Accordingly, the zeta potential of untreated polypropylene surfaces also turned out to be negative over a broad pH range.48,50 The titration curve of pure PP was characterized by a linear decrease with increasing pH value (Fig. 4). This result is in accordance with the model based on a larger double layer of water and hydroxyl ions because of the increased number of ions by titration process.49 The modified films showed a similar decrease but with slightly reduced absolute values between pH 6 to 7. At higher pH values (>pH 8), the zeta potential did not depend on the hydroxyl ions as much, and the zeta potential values for the PG modified films were clearly higher than for PP. The surface of PP3min-PG9k-(NH2)3% apparently adsorbed both the hydroxide ions and presumably water better than PP3min-PG9k-(NH2)1%, whereby PP3min-mPEG absorbed the best (Fig. 4). The zeta potential curves for the 1 min brominated samples were similar and can be found in the ESI (Fig. S1†).
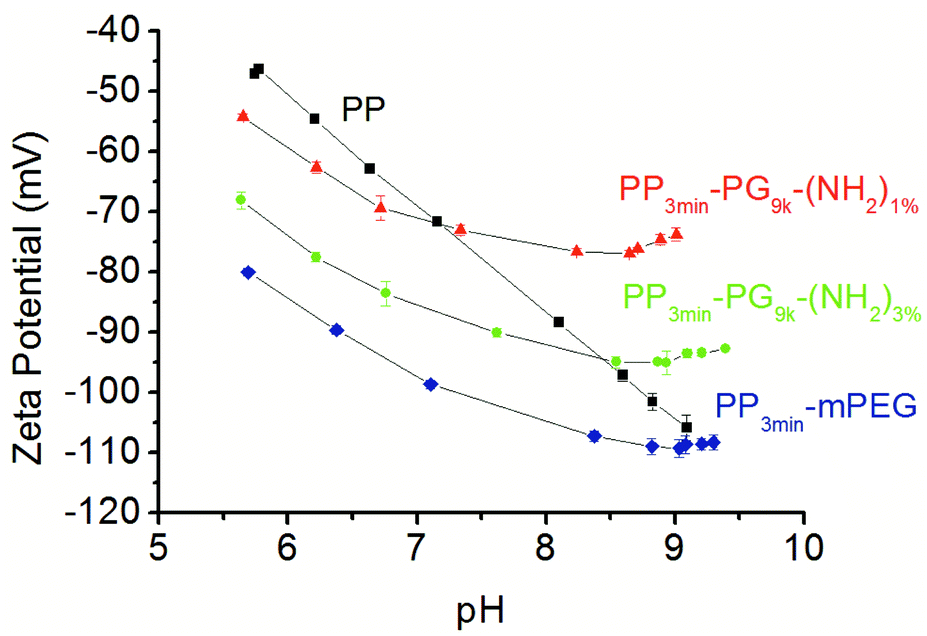 | ||
| Fig. 4 Plot of the surface zeta potential over the pH of bare PP (square, black) and 3 min plasma brominated films PP3min-PG9k-(NH2)1% (triangle, red), PP3min-PG9k-(NH2)3% (circle, green), and PP3min-mPEG control (rhombus, blue) as measured with an electrokinetic analyzer. | ||
The ATR-FTIR spectra of the unmodified and the modified films were dominated by characteristic signals of PP (Fig. 5). Unambiguous signals for the modification, like from ether or hydroxyl functionalities, cannot be detected, but the detailed analysis of the FTIR spectra indicated some modification of the PP films. An increased water adsorption was observed between 1700–1600 cm−1 and 4000–3000 cm−1. The latter signal could also be partly caused by PG or PEG. The O–H stretching band around 3400 cm−1 is dominated by hydrogen bridges between water molecules, whereas H–O–H scissors bending caused the signal at 1635 cm−1. Therefore, the former signal more clearly represented the adsorbed water content.
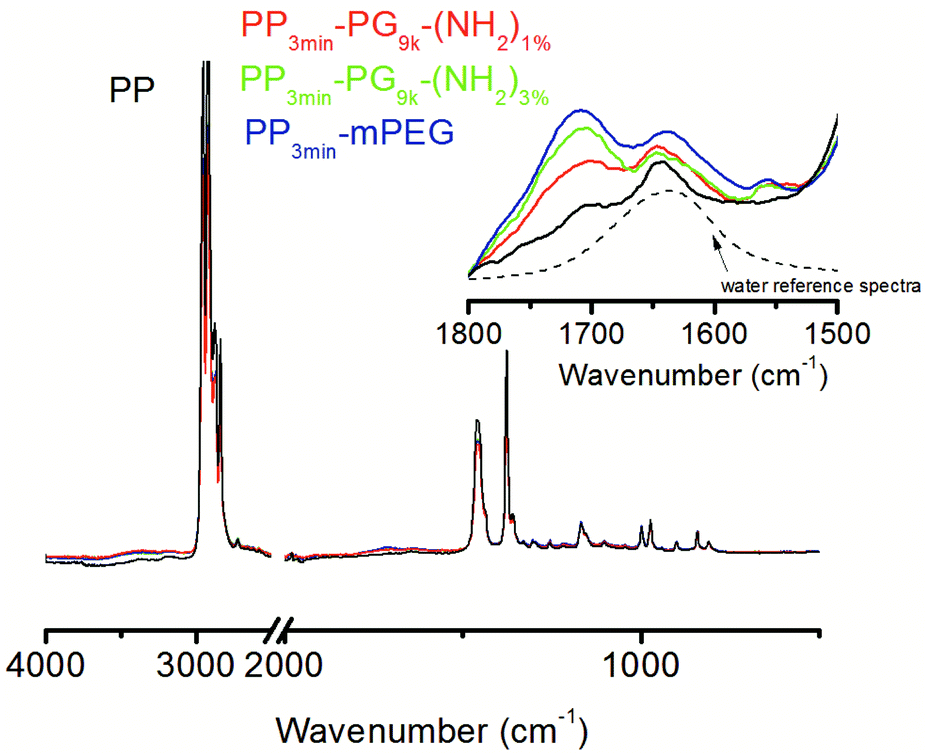 | ||
| Fig. 5 Exemplary ATR-FTIR spectra of PP (black) and modified films PP3min-PG9k-(NH2)1% (red), PP3min-PG9k-(NH2)3% (green), and PP3min-mPEG control (blue) after 24 h of immersion in water with a cut-out of the relevant area. | ||
By evaluating the intensity of the water signal, we intended to find out, whether the modified films were able to absorb more water because of their modification (Table 4). Therefore, the intensity of water signal (1668–1533 cm−1) was evaluated and compared for all materials. In comparison to the unmodified PP, the modified films clearly showed an increase in the water signal. PP3min-PG9k-(NH2)1% and PP3min-PG9k-(NH2)3% showed only slight differences. The water signal for PP3min-mPEG further increased.
Apart from the water adsorption, small signals between 1800–1680 cm−1 were also observed, which were probably caused by carbonyl groups from minor PP damage due to the plasma treatment. One has to keep in mind though that the greater polarizability of the C–O vibration of carbonyls may have overrepresented the signal. Moreover, the FTIR measurement depth is much larger than the XPS one which made the latter method more suitable for our thin films. Nonetheless, we could conclude from the FTIR spectra that the modified films were able to absorb more water.
In addition, we were interested in the stability of the produced films to investigate their applicability. Therefore, we measured the contact angles of PP-PG9k-(NH2)1% and PP-PG9k-(NH2)3% as well as the PP-mPEG control that were either plasma brominated for 1 min or for 3 min after 6 months of storage under air and without any kind of protection. In Fig. 6 the changes in the contact angles after storage were displayed. There was no difference in the 3 min brominated polymer films, but an effect became apparent in the 1 min brominated coated samples, where the polyglycerol functionalized film PP1min-PG9k-(NH2)1% with fewer potential links to the surface showed less stability than the film with more possible bonds to the surface PP1min-PG9k-(NH2)3%. The PP-mPEG film showed the same stability for 1 or 3 minutes of bromination.
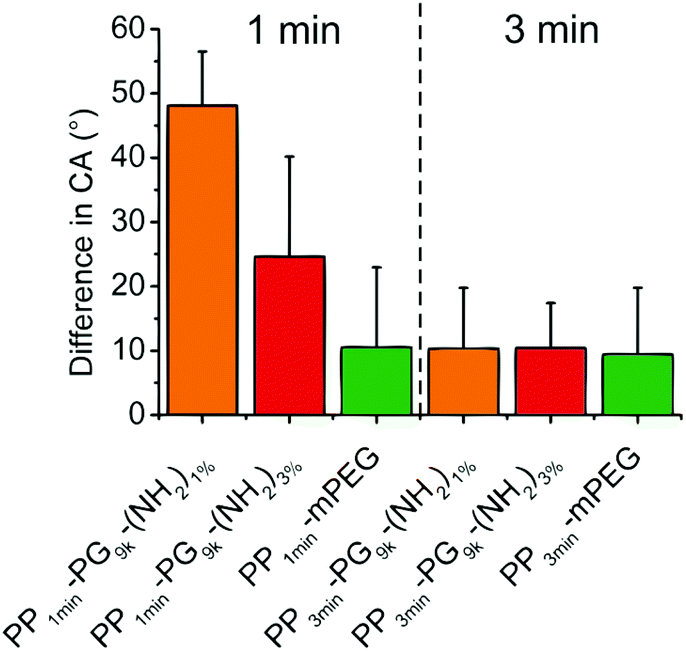 | ||
| Fig. 6 Differences in the contact angles of the demonstrated samples before and after storage without precautions for 6 months are shown. The three bars on the left show the originally 1 min plasma brominated and coated films and the three bars on the right side the 3 min plasma brominated polymer coated PP films. | ||
Protein adsorption study
We investigated the level of bioinertness by measuring the adsorption of two commonly used model proteins, namely, bovine serum albumin (BSA) and fibrinogen (FIB) onto these new PP-PG surfaces. After incubation for 3 h the adsorption of FITC-labeled protein was measured with the help of a fluorescence microscope and was compared to the level of adsorbed protein on untreated polypropylene films. Fig. 7 shows a picture of the bare PP taken with a fluorescence microscope in comparison to a PG-coated PP film. Moreover, the results from the protein adsorption study were displayed in the same figure. The adsorption of BSA-FITC and FIB-FITC could be strongly reduced for all the 3 min plasma brominated and polymer coated samples. Poly(ethylene glycol) coatings still represent one of the gold standards for bioinert surfaces. The performance of the polyglycerol functionalized PP films was as good as or better than the PEGylated control's. The values were all lower than 20% with the lowest at only 2%, but no structure–property relationship could be found for the different coatings. We were also interested to see if the 1 min plasma brominated films reached the same level of bioinertness and found that good protein resistance was detected for the coatings with PG9k. Nonetheless, this was still lower than the 3 min plasma treated films and even more strongly decreased for the mPEG control than for the PG samples. For 1 min brominated films, the sample PP1min-PG9k-(NH2)3% showed a lower average protein adsorption than PP1min-PG9k-(NH2)1%.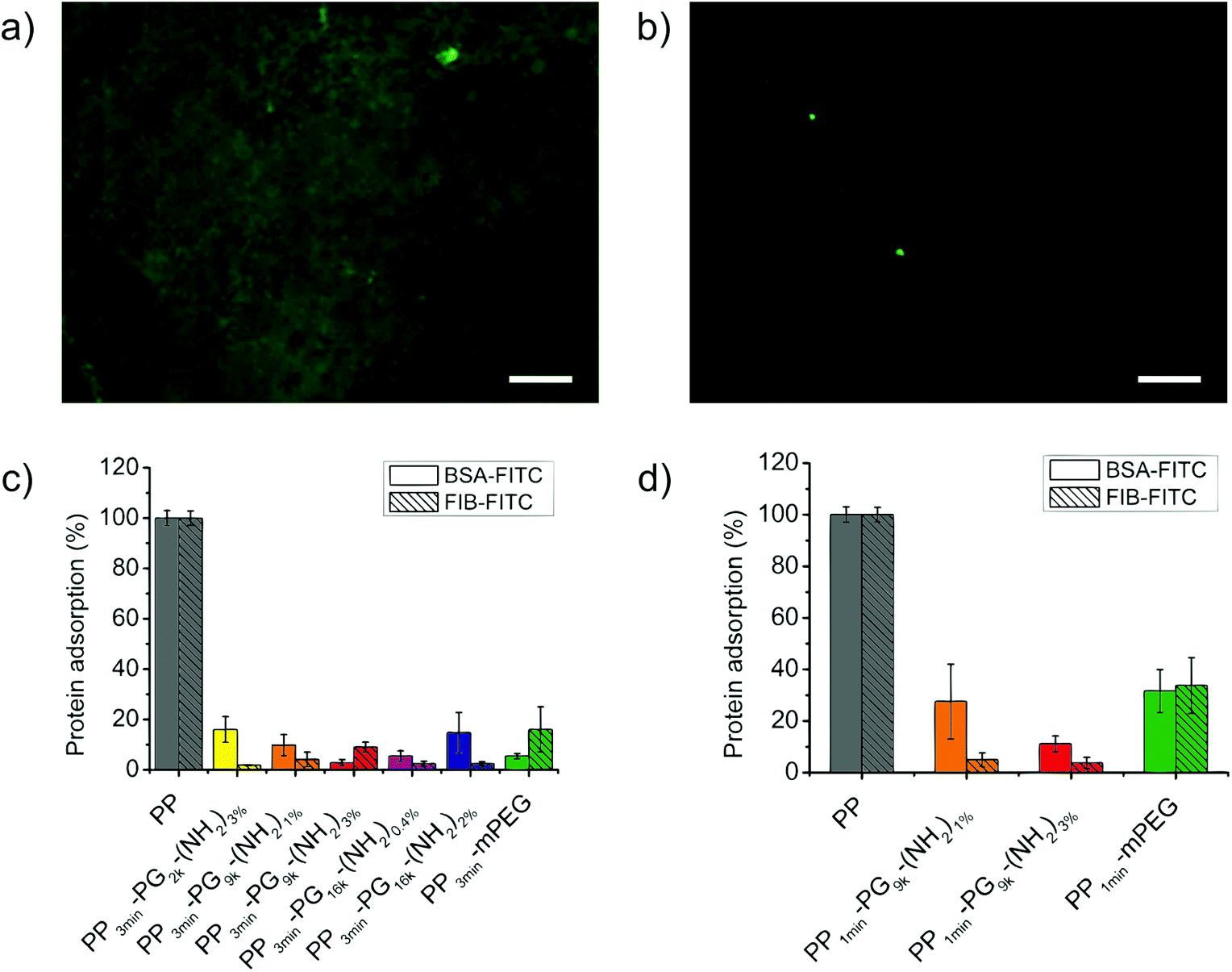 | ||
| Fig. 7 (a) A fluorescence microscopy picture of bare PP film after incubation with FITC-labeled protein, which in this case was FIB-FITC. (b) A fluorescence microscopy picture of PP3min-PG2k-(NH2)3% film chosen as an example after incubation with FIB-FITC. Scale bar: 25 μm. (c) Protein adsorption of 3 min plasma brominated polymer coated PP films in comparison to unmodified PP. (d) Protein adsorption of 1 min plasma brominated polymer coated PP films in comparison to unmodified PP. | ||
Bacteria attachment study
For the medium sized hyperbranched polyglycerol coated PP samples PP3min-PG9k-(NH2)1% and PP3min-PG9k-(NH2)3%, also the material's resistance towards bacterial attachment was tested in comparison to pure PP and the PP3min-mPEG control. In an earlier study on perfect glass coatings, the ability of polyglycerol to prevent bacteria adhesion could be already demonstrated.23 Terfort and co-workers have also recently investigated PG's resistance to bacteria.25,26Previous studies have convincingly shown that tests with only one bacterial strain are not sufficient for a general assessment of material susceptibility to microbial colonization.51 Thus we advocate the use of different reference organisms in bacterial adhesion tests as a mean to reflect and sense different material properties like electrical charge, wettability and surface roughness.51 Varying levels of attachment for different bacterial species are routed in the fact that Gram-positive and Gram-negative bacteria possess characteristic and varying surface properties and macromolecular surfaces and therefore use diverse attachment mechanisms (e.g. surface proteins, flagella and exopolysaccharides). We investigated the bacterial attachment for two different model biofilm-forming bacteria Escherichia coli (E. coli) and Pseudomonas aeruginosa (P. aeruginosa), which are both common pathogens. After an incubation period of 4 h for E. coli or 24 h for P. aeruginosa which corresponded to a single-cell layer biofilm, the samples were repeatedly washed with phosphate buffered saline (PBS) to remove non-adhered bacteria. The firmly attached bacteria were stained with safranine and surface colonization was analyzed by transmission light microscopy and subsequent image analysis. The pictures were sorted into four classes of colonization (low, medium, strong, and very strong colonization) and evaluated statistically. Examples for each of the four classes can be seen in Fig. 8a. The results are illustrated in terms of bacterial attachment in a box plot (Fig. 8b). More detailed plots for all demonstrated samples can be found in the ESI (Fig. S2–S5†). Surprisingly, only PP3min-PG9k-(NH2)1% films showed significant reduction of bacteria attachment. For PP3min-PG9k-(NH2)3% and the PEG control PP3min-mPEG, the bacteria colonization decreased to a certain extent but the samples exhibited a huge variation in values of bacterial attachment. This result clearly indicates that even a very small change in the surface coating can already result in a big impact on the material's performance when in contact with living microorganisms. An additional study was performed on the best coating with only 1 min plasma brominated films PP1min-PG9k-(NH2)1%. Although the material also exhibited reduced attachment of bacteria, it was a little less in comparison to PP3min-PG9k-(NH2)1% (Fig. S6†). Furthermore, no cytotoxic effect was detected for the brominated and polymer coated surfaces (see ESI†).
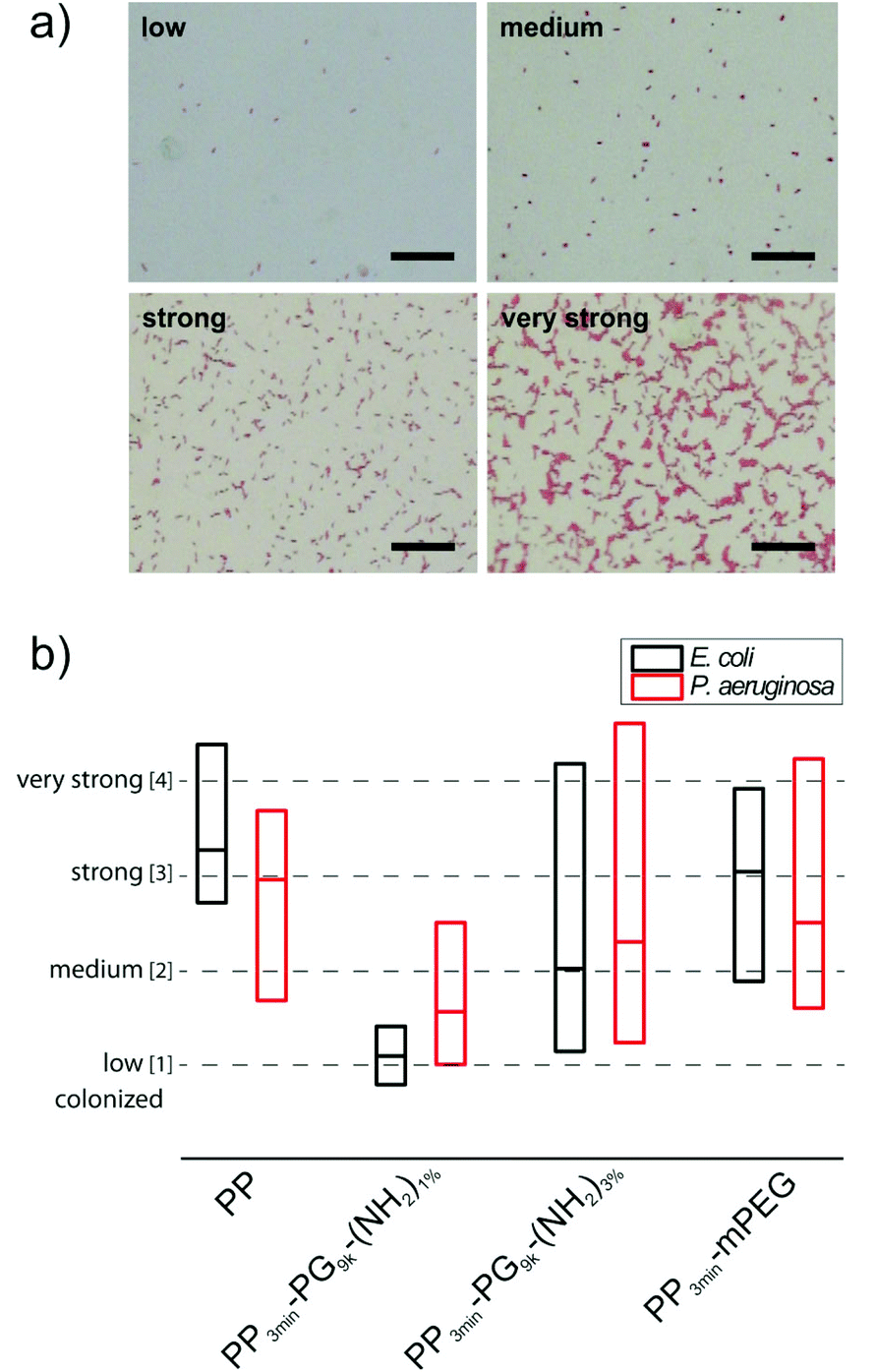 | ||
| Fig. 8 (a) Examples for the four different classes of bacteria colonization of polymer coated PP surfaces (low, medium, strong, and very strong colonization). Scale bar: 25 μm. (b) The box diagram indicates the range of bacteria attachment on the 3 min plasma brominated polymer coated PP surfaces. Top end of boxes: upper quartile (Q3), middle lines: median, lower end of boxes: lower quartile (Q1). Median and quartiles were estimated interpolated by index values 1 to 4. | ||
Experimental section
Materials
All chemicals and solvents were reagent or HPLC grade, used as received, and purchased from Sigma-Aldrich GmbH (Germany) unless stated otherwise. Dialysis was performed in benzoylated cellulose tubes from Sigma-Aldrich (D7884, width: 32 mm, molecular weight cut-off (MWCO) 2000 g mol−1). Polypropylene (PP) films of 100 μm thickness were obtained from Goodfellow GmbH, Germany. Prior plasma treatments the PP films were washed in diethylether for at least 15 minutes to remove residual slip agents, light stabilizers, and antioxidants. Extra pure bromine (99+%) was obtained from Acros Organics (Fisher Scientific GmbH, Germany). Amine functionalized polyglycerols (PG-amine) were prepared according to our previously published procedure41 by mesylation, azidation, and subsequent Staudinger reduction for polyglycerols with different molecular weights and amounts of amino functionalization. Details on the prepared PG-amines can be found in Table 1. Stock solutions of the PG-amines with concentrations of 100 mg mL−1 in methanol were stored in the freezer. α-Methoxy-ω-amino polyethylene glycol (mPEG-NH2, 1210000-2) with a PEG-MW of 10 kDa was obtained from Rapp Polymere GmbH, Germany. Albumin–fluorescein isothiocyanate conjugate (BSA-FITC, A9771) was obtained from Sigma-Aldrich, Germany. Fluorescein labeled bovine fibrinogen (FIB-FITC, 401236) was obtained from LOXO GmbH, Germany. Phosphate buffered saline (PBS; 10× concentrated; 90 g L−1 NaCl, 7.95 g L−1 Na2HPO4, and 1.44 g L−1 KH2PO4, pH 7.4) was purchased from Lonza (Germany) and was diluted to a 1× concentration (10 mM) by Milli-Q water prior to use. For Millipore (Milli-Q) water, the deionized water used was purified using a Millipore water purification system with a minimum resistivity of 18.2 MΩ cm.Plasma bromination of polypropylene PP-Br39,40
All plasma treatments were performed by using a low-pressure capacitively coupled plasma (CCP) in a stainless steel reactor. The working radio frequency (RF) was 13.56 MHz in a continuous-wave mode. Before plasma treatment the chamber was pumped down to a basic pressure of 0.5 Pa. The pressure within the plasma reactor for the bromination treatment was 8 Pa. The power input was fixed at 100 W. Exposure time to the bromine plasma was 1 or 3 min. After plasma treatment and removing of samples from the reactor the PP films were washed intensively with tetrahydrofuran (THF) to remove all low molecular weight material from the plasma treated surface.Grafting of PG-amines to PP-Br surfaces
Brominated polypropylene films (PP-Br) were washed in methanol (MeOH). 1 mL of the corresponding PG-amine stock solution (100 mg mL−1) in MeOH was mixed with 49 mL of MeOH. The mixture was added to a 500 mL Schott wide necked bottle and the PP-Br film was added. Since PP films swim on top in MeOH, the reactive side was turned downwards. Schott bottles were shaken overnight at r.t. with a speed of 100 rpm. Afterwards, the solution was discarded and the films were washed exhaustively with each 50 mL of MeOH (3×), MeOH–Milli-Q H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), Milli-Q H2O, Milli-Q H2O–EtOH (1:1) and EtOH on the shaker for a minimum of 30 min per washing step and then dried in high vacuum in wide necked, round bottomed flasks.
:1), Milli-Q H2O, Milli-Q H2O–EtOH (1:1) and EtOH on the shaker for a minimum of 30 min per washing step and then dried in high vacuum in wide necked, round bottomed flasks.
Instrumentation
1H NMR spectra were recorded on a Jeol ECX-400 spectrometer working at a frequency of 400 MHz at ambient temperature. Solvent calibration was performed according to the literature.52 IR measurements were measured on a Nicolet Avatar 320 FT-IR operating from 4000–400 cm−1. Static contact angle measurements were performed by using a contact angle goniometer (DataPhysics Instruments, Germany) with the sessile drop method. The stated static contact angles are the mean of at least five independent measurements with a drop volume of 2 μL on different locations on the surface applying a Young–Laplace fitting. X-ray photoelectron spectroscopy (XPS) was measured on a SAGE 150 spectrometer (Specs, Berlin, Germany) for C1s, O1s, N1s, Br3d, Br3p, and other specific peaks. The spectrometer was equipped with channeltron and used with non-monochromatic MgKα radiation at 250 W emission power at 11 kV and a pressure of about 1 × 10−7 Pa in the analyzer chamber. XPS spectra were acquired in constant analyzer energy (CAE) mode at a 90° take-off angle. Peak analysis was performed using the peak fit routine from Specs. Results were normalized per 100 carbon atoms (atom/100 C). Zeta potential ζ measurements of PG-amines were conducted using a Zetasizer Nano-ZS (Malvern Instruments, U.K.) with integrated 4 mW He–Ne laser, λ = 633 nm. For zeta potential measurements, the doppler anemometry technique was used, whereby an electric field was applied across the sample solution. All measurements were carried out at 25 °C using folded capillary cells (DTS 1060, Malvern Instruments). The polymer solutions were prepared in 10 mM phosphate buffer (0.795 g L−1 Na2HPO4, and 0.144 g L−1 KH2PO4, pH 7.4) at concentrations of 5 mg mL−1. The stated values are the mean from at least six independent measurements with each automatic number of scans (minimum 12 scans). Data evaluation was performed with Malvern Zetasizer Software 7.01. The surface zeta potential of the films was determined by using a SurPASS electrokinetic analyzer (Anton Paar, Germany). Two films (1 × 2 cm2) were fixed parallel to each other with a gap of 100 μm. Before preparation, the films were stored under water for 24 h. An ionic solution of 1 mmol KBr in Milli-Q water and a maximum pressure of 300 mbar were used. The zeta potential was determined depending on the pH value and obtained by adding a 0.05 molar KOH solution to the start solution until a pH value of ∼9 was reached. The zeta potential was calculated from the mean of four measurements using the Fairbrother-Mastin (FM) equation. All film sample pairs were measured two times. When the derivation of the two curves was more than 5% than the averaged-value curve or when discontinuity was observed, further measurements were performed until the error fell under the critical value. The FTIR spectra were measured using attenuated total reflection (ATR, Smart Orbit Accessory) in a Nicolet 6700 FTIR spectrometer (Thermo Scientific, Germany) with a DTGS KBr detector. To obtain a spectrum, 32 scans were taken at an optical resolution of 4 cm−1. For the ATR-FTIR investigations, the films were pressed on the diamond cell to achieve surface-sensitive test results. For reproducible test results, at least ten measurements were taken for each sample and averaged spectra were determined.Protein adsorption study
Fluorescein labeled protein solutions BSA-FITC and FIB-FITC (1 mg mL−1 in PBS) were freshly prepared and filtered by 0.2 μm cellulose acetate syringe filters (Whatman) prior to use. The polypropylene films were exposed to fluorescent protein solutions for 3 h in a humidity box, and all operations involving the FITC labeled proteins were performed in the dark. Quantification of the amount of adsorbed proteins on the slides was done with epifocal fluorescence microscopy imaging (Zeiss Axio observer Z1 equipped with a Axio Cam MRm Camera, Zeiss, Jena, Germany) applying a 40× objective and a constant excitation time of 3 s throughout all experiments. Fluorescence images were analyzed by the Zeiss software program AutMess, and the gray values obtained from at least five images on randomly chosen areas of a sample were averaged after background subtraction. Non-treated, solvent cleaned bare PP films were used as a reference. Immediately before the protein incubation experiments, the PP films were washed with ethanol and dried in a stream of N2/argon. Afterwards, the slides were subjected to the respective protein solution (20 μL) by using a self-made PDMS (Sylgard 184, Dow Corning, Wiesbaden, Germany) mask isolator (3 wells, Ø 4 mm, height ≥ 3 mm), which was fixed on top of the PP films. After removal of the protein solution PBS (20 μL) was repeatedly added to and removed from the individual wells for a total of three times in order to wash non-adsorbed proteins from the surface. Subsequently, Milli-Q water (20 μL) was added and removed for desalination and the slides were dried in a stream of N2/argon.Bacteria attachment study
Two model biofilm-forming bacterial species E. coli (K-12 W3110) and Pseudomonas aeruginosa (ATCC 13388) were routinely grown at 37 °C either on 3-(N-morpholino)propane sulfonic acid (MOPS) minimal medium (modified)53 or Luria-Bertani (LB) agar plates or for preparatory culture in broth with 130 rpm. A synthetic MOPS minimal medium was used in initial bacterial adhesion experiments for standardizing the cultivation conditions. Bacterial adhesion was tested on polypropylene films submerged in glass flasks with diluted (OD 0.03) bacterial cell suspensions from overnight cultures. After an initial sedimentation/growth phase of 1 h at 37 °C without shaking, the polymer samples were incubated at 37 °C with 60 rpm shaking for 3 h (E. coli) or 23 h (P. aeruginosa). After incubation the test samples were removed and placed into fresh phosphate buffered saline (PBS) for three minutes. Non-adhering bacteria were removed by multiple washing with PBS (room temperature, 8 times). The remaining firmly attached bacteria were fixed with 3.7% formaldehyde (FA) overnight at 4 °C. FA was removed and the material samples were again washed six times with PBS. After drying for 10 minutes at 46 °C, the attached bacteria were stained by safranine and the degree of bacteria attachment of the samples was examined and documented using a Nikon Eclipse Ni-U Microscope with NIS-elements imaging software (Nikon, Japan). Bacterial attachment to all the material surfaces was tested in five or more biologically independent replications for every material type and both reference organisms. Surface colonization by the attached bacteria was documented by transmission light microscopy. A minimum of 30 pictures (size about 0.3 mm2) was taken for every tested material sample (in total ca. 150 pictures per material type). The pictures were examined for colonization and qualitatively divided into four different classes (low, medium, strong, and very strong) of colonization and analyzed statistically according to the distribution of frequencies (Fig. S2–S6†). Median and quartiles were estimated interpolated by index values 1 to 4 (Fig. 8b).Conclusions
A very simple method to produce bioinert PG-lated polypropylene surfaces in only two steps was reported. Initially, plasma bromination was used to introduce quite selectively a high amount of reactive bromine groups for anchoring the inert polyolefin material and with only low general plasma oxidation. The bromine groups were substituted by amine groups from hyperbranched polyglycerol. Steric hindrance of bigger PGs during the very mild grafting-to procedure could be overcome by more initial binding sites on the polymer scaffold. This led to greater stability for the films that were bound with more linkers in a long-term experiment over 6 months. Additionally, films that were plasma brominated for 3 minutes showed more stability than the 1 minute brominated and coated films. The protein adsorption of two commonly used model proteins, BSA and FIB, was reduced for all PG-lated surfaces and was comparable to the mPEG controls. The 3 min brominated PG-coated samples were again superior to the shorter brominated and coated films. Surprisingly, the small difference in amino group concentration of the originally prepared PGs had a major effect on the bacteria attachment. Here, the lower original amino functionalization of PG proved to be beneficial and the results were better than for the mPEG-coated control. For the best PG sample, the reduction in plasma exposure time from 3 min to 1 min still resulted in an increased resistance to bacterial attachment, but with a little less effect.This study presented a simple, easily applicable approach for the coating of polyolefin surfaces with polyglycerols. The new insights that were gained are of great importance for creating more knowledge on real-life bioinert materials that could be practically used – a challenging goal that still needs much further study.
Acknowledgements
This work was supported by the Bundesministerium für Bildung und Forschung (BMBF) through the Poly4Bio grant (1315696B) and the Helmholtz Virtual Institute Multifunctional Biomaterials for Medicine. We thank our colleague Florian Paulus for synthesizing hPG and Cathleen Schlesener for the synthesis of two of the polyglycerol azides. The authors would like to thank Dorothea Thiele and Gisela Kneip for experimental support. We thank Dr. Katharina Achazi and Christina Kühl for the help in performing the toxicity study. Dr. Pamela Winchester is acknowledged for proofreading the manuscript. M. L. thanks Dr. Bala Thota and Dr. Uwe Schedler for helpful discussions.References
- G. D. Bixler and B. Bhushan, Philos. Trans. R. Soc., A, 2012, 370, 2381–2417 CrossRef CAS PubMed.
- J. L. Harding and M. M. Reynolds, Trends Biotechnol., 2014, 32, 140–146 CrossRef CAS PubMed.
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- H. B. Lee, G. Khang and J. H. Lee, in The Biomedical Engineering Handbook, Volume I, ed. J. D. Bronzino, CRC Press LLC and Springer-Verlag GmbH & Co.KG, Heidelberg, Germany, 2000, 2nd edn, pp. 39-8–39-9 Search PubMed.
- A. S. Breitbart and V. J. Ablaza, in Grabb and Smith's plastic surgery, ed. C. H. Thorne, Lippincott Williams & Wilkins, Philadelphia, USA, 2007, 6th edn, pp. 61–62 Search PubMed.
- N. Gomathi and S. Neogi, Appl. Surf. Sci., 2009, 255, 7590–7600 CrossRef CAS PubMed.
- Q. Wei, T. Becherer, S. Angioletti-Uberti, J. Dzubiella, C. Wischke, A. T. Neffe, A. Lendlein, M. Ballauff and R. Haag, Angew. Chem., Int. Ed., 2014, 53, 8004–8031 CrossRef CAS PubMed.
- Y.-F. Yang, L.-S. Wan and Z.-K. Xu, J. Adhes. Sci. Technol., 2011, 25, 245–260 CrossRef CAS PubMed.
- K. K. Goli, O. J. Rojas and J. Genzer, Biomacromolecules, 2012, 13, 3769–3779 CrossRef CAS PubMed.
- P. K. Chu, J. Y. Chen, L. P. Wang and N. Huang, Mater. Sci. Eng., R, 2002, 36, 143–206 CrossRef.
- J. Friedrich, in The Plasma Chemistry of Polymer Surfaces, Advanced Techniques for Surface Design, Wiley-VCH, Weinheim, 2012, pp. 249–297 Search PubMed.
- S. Wettmarshausen, G. Kühn, G. Hidde, H.-U. Mittmann and J. F. Friedrich, Plasma Processes Polym., 2007, 4, 832–839 CrossRef CAS.
- R. T. Chen, B. W. Muir, G. K. Such, A. Postma, R. A. Evans, S. M. Pereira, K. M. McLean and F. Caruso, Langmuir, 2010, 26, 3388–3393 CrossRef CAS PubMed.
- C. Heise, U. Schedler, S. Wettmarshausen and J. F. Friedrich, J. Appl. Polym. Sci., 2014, 131, 40662 CrossRef.
- É. Kiss, J. Samu, A. Tóth and I. Bertóti, Langmuir, 1996, 12, 1651–1657 CrossRef.
- E. Moore, H. Thissen and N. H. Voelcker, Prog. Surf. Sci., 2013, 88, 213–236 CrossRef CAS PubMed.
- M. Calderón, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22, 190–218 CrossRef PubMed.
- C. Schüll and H. Frey, Polymer, 2013, 54, 5443–5455 CrossRef PubMed.
- C. Siegers, M. Biesalski and R. Haag, Chem. – Eur. J., 2004, 10, 2831–2838 CrossRef CAS PubMed.
- M. Wyszogrodzka and R. Haag, Langmuir, 2009, 25, 5703–5712 CrossRef CAS PubMed.
- M. Wyszogrodzka and R. Haag, Biomacromolecules, 2009, 10, 1043–1054 CrossRef CAS PubMed.
- J. I. Paez, V. Brunetti, M. C. Strumia, T. Becherer, T. Solomun, J. Miguel, C. F. Hermanns, M. Calderón and R. Haag, J. Mater. Chem., 2012, 22, 19488–19497 RSC.
- M. Weinhart, T. Becherer, N. Schnurbusch, K. Schwibbert, H.-J. Kunte and R. Haag, Adv. Eng. Mater., 2011, 13, B501–B510 CrossRef.
- P.-Y. J. Yeh, R. K. Kainthan, Y. Zou, M. Chiao and J. N. Kizhakkedathu, Langmuir, 2008, 24, 4907–4916 CrossRef CAS PubMed.
- T. Weber, Y. Gies and A. Terfort, Langmuir, 2012, 28, 15916–15921 CrossRef CAS PubMed.
- T. Weber, M. Bechthold, T. Winkler, J. Dauselt and A. Terfort, Colloids Surf., B, 2013, 111, 360–366 CrossRef CAS PubMed.
- A. Boulares-Pender, A. Prager, S. Reichelt, C. Elsner and M. R. Buchmeiser, J. Appl. Polym. Sci., 2011, 121, 2543–2550 CrossRef CAS.
- X. Li, T. Cai and T.-S. Chung, Environ. Sci. Technol., 2014, 48, 9898–9907 CrossRef CAS PubMed.
- T. Cai, M. Li, K.-G. Neoh and E.-T. Kang, J. Mater. Chem. B, 2013, 1, 1304–1315 RSC.
- E. Moore, B. Delalat, R. Vasani, G. McPhee, H. Thissen and N. H. Voelcker, ACS Appl. Mater. Interfaces, 2014, 6, 15243–15252 CAS.
- Q. Wei, T. Becherer, R.-C. Mutihac, P.-L. M. Noeske, F. Paulus, R. Haag and I. Grunwald, Biomacromolecules, 2014, 15, 3061–3071 CrossRef CAS PubMed.
- O. Z. Higa, H. A. M. Faria and A. A. A. de Queiroz, Radiat. Phys. Chem., 2014, 98, 118–123 CrossRef CAS PubMed.
- M. Khan and W. T. S. Huck, Macromolecules, 2003, 36, 5088–5093 CrossRef CAS.
- A. T. Neffe, M. von Ruesten-Lange, S. Braune, K. Lützow, T. Roch, K. Richau, A. Krüger, T. Becherer, A. F. Thünemann, F. Jung, R. Haag and A. Lendlein, J. Mater. Chem. B, 2014, 2, 3626–3635 RSC.
- E. Moore, B. Delalat, R. Vasani, H. Thissen and N. H. Voelcker, Biomacromolecules, 2014, 15, 2735–2743 CAS.
- Q. Wei, S. Krysiak, K. Achazi, T. Becherer, P.-L. M. Noeske, F. Paulus, H. Liebe, I. Grunwald, J. Dernedde, A. Hartwig, T. Hugel and R. Haag, Colloids Surf., B, 2014, 122, 684–692 CrossRef CAS PubMed.
- S. Holmes-Farley, R. Reamey, R. Nuzzo, T. J. McCarthy and G. M. Whitesides, Langmuir, 1987, 3, 799–815 CrossRef CAS.
- J. F. Friedrich, R. Mix, R.-D. Schulze, A. Meyer-Plath, R. Joshi and S. Wettmarshausen, Plasma Processes Polym., 2008, 5, 407–423 CrossRef CAS.
- J. Friedrich, S. Wettmarshausen and M. Hennecke, Surf. Coat. Technol., 2009, 203, 3647–3655 CrossRef CAS PubMed.
- J. F. Friedrich, S. Wettmarshausen, S. Hanelt, R. Mach, R. Mix, E. B. Zeynalov and A. Meyer-Plath, Carbon, 2010, 48, 3884–3894 CrossRef CAS PubMed.
- S. Roller, H. Zhou and R. Haag, Mol. Diversity, 2005, 9, 305–316 CrossRef CAS PubMed.
- S. I. Jeon, J. H. Lee, J. D. Andrade and P. G. De Gennes, J. Colloid Interface Sci., 1991, 142, 149–158 CrossRef CAS.
- S. I. Jeon and J. D. Andrade, J. Colloid Interface Sci., 1991, 142, 159–166 CrossRef CAS.
- A. T. Neffe, M. von Ruesten-Lange, S. Braune, K. Luetzow, T. Roch, K. Richau, F. Jung and A. Lendlein, Macromol. Biosci., 2013, 13, 1720–1729 CrossRef CAS PubMed.
- Q. Wei, K. Achazi, H. Liebe, A. Schulz, P.-L. M. Noeske, I. Grunwald and R. Haag, Angew. Chem., Int. Ed., 2014, 5, 11650–11655 CrossRef PubMed.
- J. Khandare, A. Mohr, M. Calderón, P. Welker, K. Licha and R. Haag, Biomaterials, 2010, 31, 4268–4277 CrossRef CAS PubMed.
- L. S. McCarty and G. M. Whitesides, Angew. Chem., Int. Ed., 2008, 47, 2188–2207 CrossRef CAS PubMed.
- Z. Kolská, A. Řezníčková and V. Švorčík, e-Polymers, 2012, 12, 960–972 CrossRef.
- R. Zimmermann, S. Dukhin and C. Werner, J. Phys. Chem. B, 2001, 105, 8544–8549 CrossRef CAS.
- P. Slepička, A. Vasina, Z. Kolská, T. Luxbacher, P. Malinský, A. Macková and V. Švorčík, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 2111–2114 CrossRef PubMed.
- P. Landsberger, V. Boenke, A. A. Gorbushina, K. Rodenacker, B. F. Pierce, K. Kratz and A. Lendlein, Mater. Res. Soc. Symp. Proc., 2013, 1569, 85–90 CrossRef.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS.
- F. C. Neidhardt, P. L. Bloch and D. F. Smith, J. Bacteriol., 1974, 119, 736–747 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4py01375a |
| This journal is © The Royal Society of Chemistry 2015 |