
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Donor–acceptor block copolymers carrying pendant PC71BM fullerenes with an ordered nanoscale morphology†
Martin
Hufnagel
a,
Matthias
Fischer
b,
Thomas
Thurn-Albrecht
b and
Mukundan
Thelakkat
*a
aApplied Functional Polymers, Department of Macromolecular Chemistry I, University of Bayreuth, Universitaetsstr. 30, 95440 Bayreuth, Germany. E-mail: mukundan.thelakkat@uni-bayreuth.de; Fax: +49921-553109; Tel: +49921-553108
bExperimental Polymer Physics Group, Martin-Luther-Universitaet Halle-Wittenberg, Von-Danckelmann-Platz 3, 06120 Halle, Germany. E-mail: thomas.thurn-albrecht@physik.uni-halle.de; Fax: +49345-5527160; Tel: +49345-5525340
First published on 22nd October 2014
Abstract
We present a straightforward method for the preparation of a novel donor–acceptor block copolymer based on an acceptor block with pendant phenyl-C71-butyric methyl ester (PC71BM) and a regioregular poly(3-hexylthiophene) (P3HT) as a donor. First, a hydroxyl-functionalized polystyrene copolymer with an azide end group was synthesized via nitroxide-mediated radical polymerization (NMRP) and was coupled with alkyne-terminated P3HT using copper(I) catalyzed azide–alkyne cycloaddition (CuAAC). The grafting reaction of phenyl-C71-butyric acid (PC71BA) to the hydroxyl groups of the polystyrene precursor was optimized to yield near-quantitative conversion which is demonstrated for a PC71BM-grafted acceptor copolymer in detail using MALDI-TOF mass spectrometry, thermogravimetric analysis (TGA) and 1H-NMR spectroscopy. Owing to the incorporation of C70, the donor–acceptor block copolymer exhibits enhanced absorption in the entire visible range of 300 to 600 nm. A detailed structural analysis of the block copolymer based on small-angle X-ray scattering in transmission (SAXS) and in grazing incidence geometry (GISAXS) as well as scanning electron microscopy (SEM) gave clear evidence for the formation of a periodic nanostructure of 37 nm in bulk and in thin films.
Introduction
In the last decade polymeric semiconductors have emerged as a leading class of organic materials with great potential in solution processable, flexible and light-weight organic electronic devices, especially organic photovoltaics (OPV).1,2 In the case of OPV the physical mixture of a donor and an acceptor material leading to a bulk-heterojunction (BHJ) delivers the highest performance in polymer devices. The short lifetime of excitons in organic semiconductors typically results in a restricted diffusion length of about 10 nm. Moreover the excitons are strongly bound due to Coulomb forces. Therefore, the morphology of the active layer should provide a large donor–acceptor interface with suitable energy levels and small domain sizes for an efficient charge separation.3 An interpenetrating network of donor and acceptor materials is additionally essential for charge transport to the electrodes. Thus, controlling the nanostructure in BHJ solar cells is the key to achieve high device performance. The blend morphology of a conjugated polymer and a fullerene derivative was extensively optimized using adequate processing conditions and post-preparation thermal treatments.4–6 However, such optimized, kinetically frozen mesostructures are in a non-equilibrium state. It is a big challenge to control the size and long-term stability of such mesostructures, which is essential for long-term operational stability.7,8 These issues become in particular critical for transferring the small area, lab-scale processing of OPV devices to a large scale roll-to-roll fabrication which requires totally different drying or annealing procedures.9An ideal solution to stabilize such a morphology is by the development of nanostructured systems in thermodynamic equilibrium. One of the approaches is by using a single material consisting of both donor and acceptor functions with the capability to self-assemble into the desired nanostructures. Basically, classical coil–coil block copolymers exhibit well-defined equilibrium nanostructures by microphase separation that are tunable in size and shape simply by variation of the degree of polymerization, Flory–Huggins interaction parameter and volume fraction.10 Theoretical studies on block copolymer systems comprising donor and acceptor blocks predict an improved device performance for vertically aligned nanostructures.11 This manuscript deals with the design of fully functionalized donor–acceptor block copolymers with a π-conjugated poly(3-hexylthiophene) (P3HT) and an acceptor block carrying phenyl-C71-butyric methyl ester (PC71BM) fullerenes.
Donor–acceptor block copolymers are attractive candidates for single material solar cells.12,13 Typically, P3HT is employed as the donor block and perylene bisimides (PBI) or fullerenes as acceptor units. We have recently reported well-defined microphase separation in P3HT-b-poly(PBI) active donor–acceptor block copolymers with a lamellar or cylindrical morphology in the range of tens of nanometers.14 Since fullerenes are known to be so far the most efficient acceptor material in OPV, it seems reasonable to integrate fullerenes into donor–acceptor block copolymers, which has been realized by grafting C60 and its derivatives using different synthetic approaches. The very first study on block copolymers carrying a conjugated block and a fullerene pendant block was reported by Hadziioannou et al.15
Perrin et al. showed that the unique electron-acceptor/transporting capability of fullerenes is maintained in C60-grafted polymers.16 Just recently, we could demonstrate a well-defined synthesis method for fullerene-grafted copolymers carrying phenyl-C61-butyric methyl ester (PC61BM) which shows high electron mobilities up to 1 × 10−4 cm V−1 s−1 without the formation of nanocrystals.17
Many of the synthetic strategies which aim at a covalent attachment of fullerenes to the block copolymer rely on reactions with unmodified C60 fullerenes. For example, atom transfer radical addition (ATRA),18 [3+2]-cycloaddition with azides19,20 or routes using tosylhydrazone addition21 to C60 have been reported. Since buckminsterfullerene C60 can act as a multifunctional reactant these methods often lead to polymer crosslinking by multiple additions and result in diminished solubility of the products. More complex monofunctional fullerenes have been attached to block copolymers in an azide–alkyne Huisgen cycloaddition22,23 or Williamson ether synthesis.24 Lee et al. have reported a Steglich esterification procedure using phenyl-C61-butyric acid (PC61BA)25 which is easily accessible from commercially available PC61BM in a straightforward acidic hydrolysis.26 Another concept toward donor–acceptor copolymers with graft-type architecture has been reported by Sivula et al. based on ring opening metathesis polymerization of C60- and P3HT-bearing norbornene monomers.27 Even though different synthetic approaches to realize fullerene containing block copolymers as discussed above have been reported, no long range microphase separation has been observed for C60-grafted donor–acceptor block copolymers so far.28 An additional issue of C60 fullerenes is their low optical density and thus C60 does not contribute substantially to light harvesting. But C70 and its derivatives absorb much more efficiently in the visible range, which considerably improves the photocurrent in solar cells.29 Therefore, PC71BM is the state-of-the-art acceptor material in OPV exhibiting record efficiencies30 and it is desirable to incorporate C70 fullerenes also into donor–acceptor block copolymers. To the best of our knowledge there are no reports regarding the grafting of C70 into fully functionalized donor–acceptor block copolymers.
Taking into account the above facts we address the following questions in the design and structure elucidation of PC71BM-grafted donor–acceptor block copolymers: 1. What are the optimum reactions conditions for an efficient grafting of PC71BM into a polystyrene backbone? 2. Is it feasible to incorporate PC71BM into a donor–acceptor block copolymer maintaining the high solubility and high yield of grafting? 3. Does such a block copolymer self-assemble into a periodic donor–acceptor nanostructure?
In this work we present the high yield grafting of PC71BM to obtain PC71BM-grafted copolymers (PPC71BM) and the controlled synthesis of a P3HT-b-PPC71BM donor–acceptor block copolymer. Furthermore, we study the structure formation of this block copolymer using X-ray scattering methods and scanning electron microscopy.
Results and discussion
Our synthetic strategy for P3HT-b-PPC71BM is based on copper(I) catalyzed azide–alkyne cycloaddition (CuAAC) of two individually designed polymer blocks: a hydroxyl functionalized polystyrene copolymer with an azide end group and a P3HT-alkyne. This was followed by a Steglich esterification procedure using phenyl-C71-butyric acid (PC71BA) to obtain the target donor–acceptor block copolymers (Fig. 1). A major challenge in block copolymer synthesis via CuAAC is the control over the end group functionalization of both polymer blocks. In the case of P3HT-alkyne, high end group fidelity was achieved by termination of the Kumada transfer catalyst polymerization with ethynylmagnesium chloride.31 For the synthesis of the azide-terminated polystyrene precursor a statistical copolymer, poly(4-methoxystyrene-stat-4-tert-butoxystyrene), was used. This was achieved by nitroxide-mediated radical polymerization (NMRP) and a subsequent polymer-analogous introduction of an azide end group. The NMRP copolymers proved to be highly tolerant toward polymer-analogous reactions.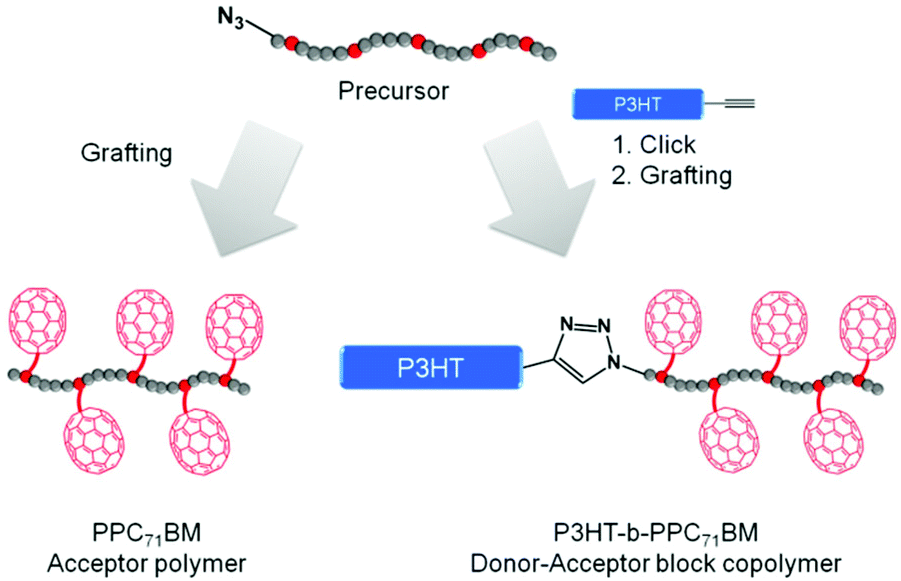 | ||
| Fig. 1 Schematic representation of the synthetic strategies toward PC71BM-grafted acceptor polymers and donor–acceptor block copolymers. | ||
Hydroxyl-functionalized polystyrene copolymers with azide end groups
We examined two synthetic strategies toward azide terminated copolymers using NMRP (Scheme 1): route A is intended to introduce the azide group to the copolymer directly through an azide derivatized alkoxyamine initiator. Route B is based on two steps involving copolymerization with a chloride derivatized initiator and a subsequent polymer-analogous substitution with sodium azide (route B).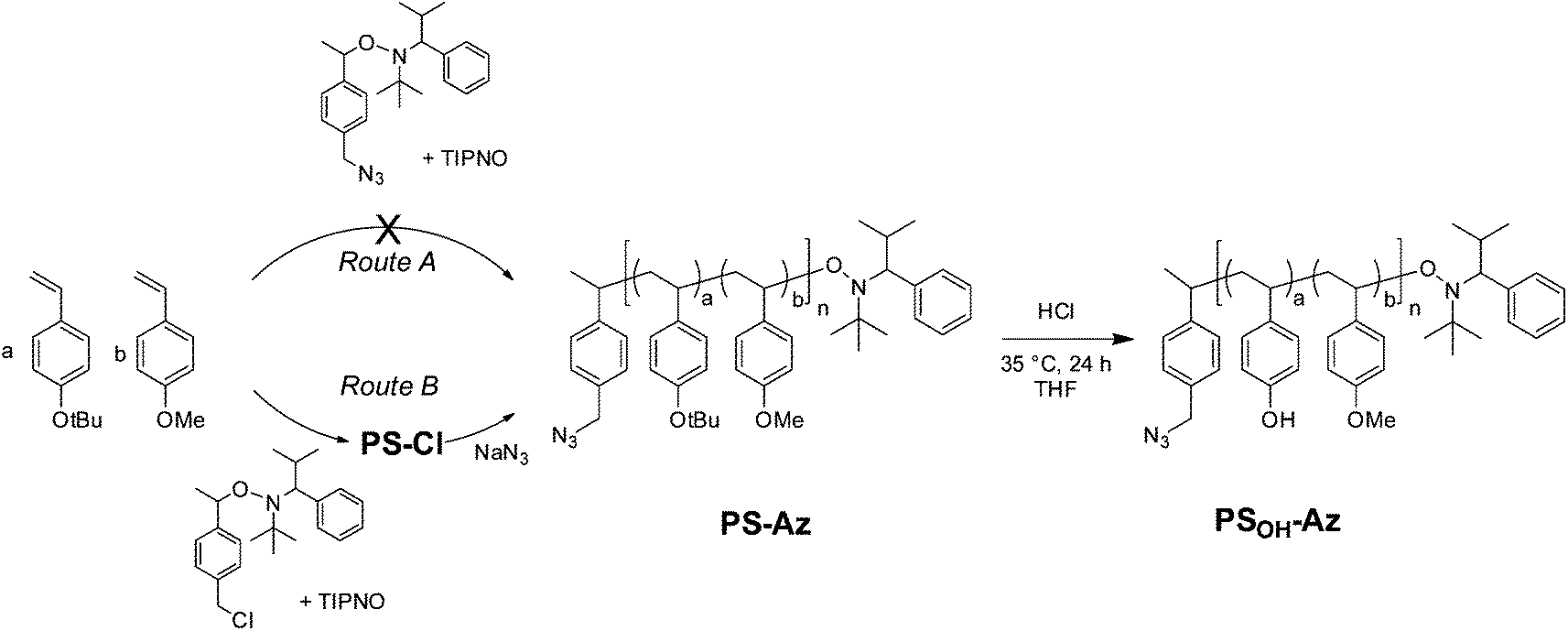 | ||
| Scheme 1 Synthesis strategy toward the azide-terminated precursor copolymer poly(4-methoxystyrene-stat-4-hydroxystyrene). Introduction of the azide end group is attempted by two different methods depicted as routes A and B. The hydroxyl functionality is achieved by hydrolysis of the tert-butyl ether groups in PS-Az maintaining the azide end group to obtain PSOH-Az. | ||
For both routes the feed ratio of 4-tert-butoxystyrene (BS) and 4-methoxystyrene (MS) BS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MS was a:b = 0.08:0.92 and gave the copolymer poly(4-methoxystyrene-stat-4-tert-butoxystyrene) (PS) with a built-in ratio of a:b = 0.10:0.90, which can be extracted from 1H-NMR analysis (Fig. S1†). The particular ratio was selected to realize a ratio of 1:1 w/w of P3HT and PC71BM in the final block copolymer which is reported to be optimum for charge separation and transport.32
:MS was a:b = 0.08:0.92 and gave the copolymer poly(4-methoxystyrene-stat-4-tert-butoxystyrene) (PS) with a built-in ratio of a:b = 0.10:0.90, which can be extracted from 1H-NMR analysis (Fig. S1†). The particular ratio was selected to realize a ratio of 1:1 w/w of P3HT and PC71BM in the final block copolymer which is reported to be optimum for charge separation and transport.32
In route A, the styrene monomers were copolymerized using the azide-functionalized alkoxyamine initiator 2,2,5-trimethyl-3-(1′-p-azidemethylphenylethoxy)-4-phenyl-3-azahexane (Ini-Az) and 2,2,5-trimethyl-4-phenyl-3-azahexane-3-nitroxide (TIPNO) as persistent radical (Scheme 1). The copolymerization was carried out at 125 °C in o-dichlorobenzene (DCB) solution. The copolymerization follows a statistical incorporation of the monomers according to the evaluation of samples which were periodically taken during the copolymerization.
Here, the consumption of both monomer species BS and MS proved to be equally fast. This can be deduced from the 1H-NMR spectra at different polymerization times (i.e. conversions), where the ratio of residual monomers in the reaction mixture remains constant (Fig. S2†). The size-exclusion chromatography (SEC) trace of PS shows a number-averaged molecular weight (Mn) of 19.6 kg mol−1 with a polydispersity (PDI) of 1.19 (Fig. 2a). The slightly broad molecular weight distribution results from a shoulder at roughly double molecular weight and can be assigned to a small amount of radical recombination of polymer chains. To verify the azide end group fidelity, Fourier-transform infrared (FTIR) spectroscopy was performed. This analytical method is highly sensitive for the detection of organic azides since the asymmetric vibration of azides is very strong.33 Unlike the azide initiator Ini-Az, the copolymer from route A does not show a distinct azide vibration at 2095 cm−1 (Fig. 2b). In the literature it has been proposed that the azide end group of the alkoxyamine initiator can react in an azide–alkene cycloaddition during polymerization with the styrene monomers at 125 °C.34 This explains the lack of FTIR azide vibrations in the copolymer from route A. Additionally, this is supported by our observation that the polystyrene copolymers from route A did not form any block copolymer using click chemistry.
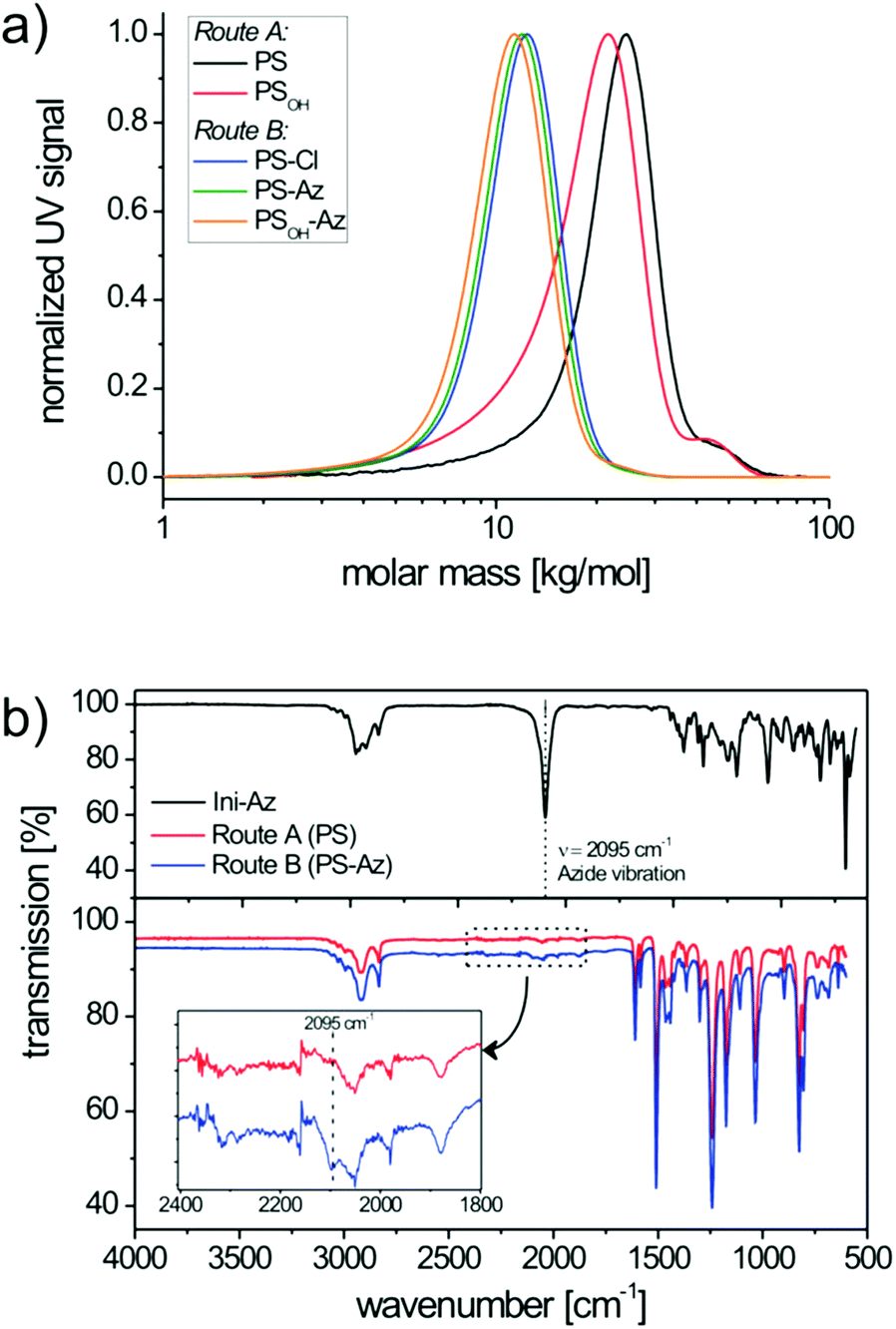 | ||
| Fig. 2 SEC traces (a) of the copolymers from route A (PS and PSOH) and route B (PS-Cl, PS-Az and PSOH-Az) obtained by NMRP. Corresponding FTIR spectra (b) of the copolymers demonstrate the degree of azide end group functionalization. | ||
In route B, the copolymerization was performed in bulk in the presence of the chlorine-functionalized alkoxyamine initiator 2,2,5-trimethyl-3-(1′-p-chloromethylphenyl-ethoxy)-4-phenyl-3-aza-hexane (Ini-Cl) and TIPNO (Scheme 1). Here also the feed ratio of BS:MS was 0.08:0.92 and copolymerization until 40% conversion gave a chlorine-terminated polystyrene copolymer (PS-Cl) with a built-in ratio of BS:MS = 0.10:0.90 (Fig. S3†). Bulk polymerization improved the copolymerization and no high molecular weight shoulder could be observed in SEC analysis (Fig. 2a). This polymerization procedure enables a perfect control of the statistical copolymerization with monomodal molecular weight distribution and a very low polydispersity of 1.10. PS-Cl has a molecular weight of Mn = 9.8 kg mol−1 (Fig. 2a). In a subsequent step, the chloride end group of PS-Cl was replaced in a nucleophilic substitution reaction using an excess of sodium azide. The resulting azide-monoterminated copolymer PS-Az has a molecular weight of Mn = 10.4 kg mol−1 and a narrow polydispersity of 1.10 (Fig. 2a). In contrast to route A, the copolymer PS-Az from route B shows an azide vibration at 2095 cm−1. As expected, the observed signal is weak due to the high dilution of the end group. This indicates a successful end group functionalization with azide via the polymer-analogous substitution in route B. Additionally, the FTIR spectra of PS-Cl and PS-Az are compared in Fig. S4† which also confirm the successful conversion of the halide to azide.
The next step toward the hydroxyl-functionalized poly(4-methoxystyrene-stat-4-hydroxystyrene) precursor copolymer (PSOH-Az) is the deprotection of the tert-butoxystyrene repeating units. Under acidic conditions with hydrochloric acid at a mild temperature of 35 °C, the cleavage of the tert-butylether groups is usually quantitative.17 This was evidenced by 1H-NMR, where the hydroxyl signal at δ = 9.24–8.84 ppm indicates the successful hydrolysis of the tert-butoxystyrene units (Fig. S5†). The obtained copolymer PSOH-Az exhibits a molecular weight of Mn = 9.8 kg mol−1 and a narrow monomodal distribution with a PDI of 1.11 (Fig. 2a). The precursor copolymer carries 10 mol% of 4-hydroxystyrene units which can be used for the esterification with PC71BA.
Optimized fullerene grafting with PC71BA
To find the optimum conditions for the covalent PC71BM fullerene attachment via Steglich esterification, we tested the grafting reaction using a hydroxyl-functionalized polystyrene copolymer PSOH obtained via NMRP having a molecular weight of Mn = 16.7 kg mol−1, PDI = 1.19 and containing 10 mol% hydroxystyrene. As a fullerene derivative we synthesized the monofunctional reactant phenyl-C71-butyric acid (PC71BA) by hydrolysis of the commercially available phenyl-C71-butyric acid methyl ester (PC71BM) in analogy to a protocol from Hummelen et al.26 Generally, toxic solvents such as carbon disulfide (CS2) are used for reactions involving PC61BA. Owing to the increased solubility of PC71BA (1.48 mg mL−1) compared to PC61BA (0.38 mg mL−1) in DCB, we were able to avoid CS2. Even then the grafting yield was very high (Scheme 2).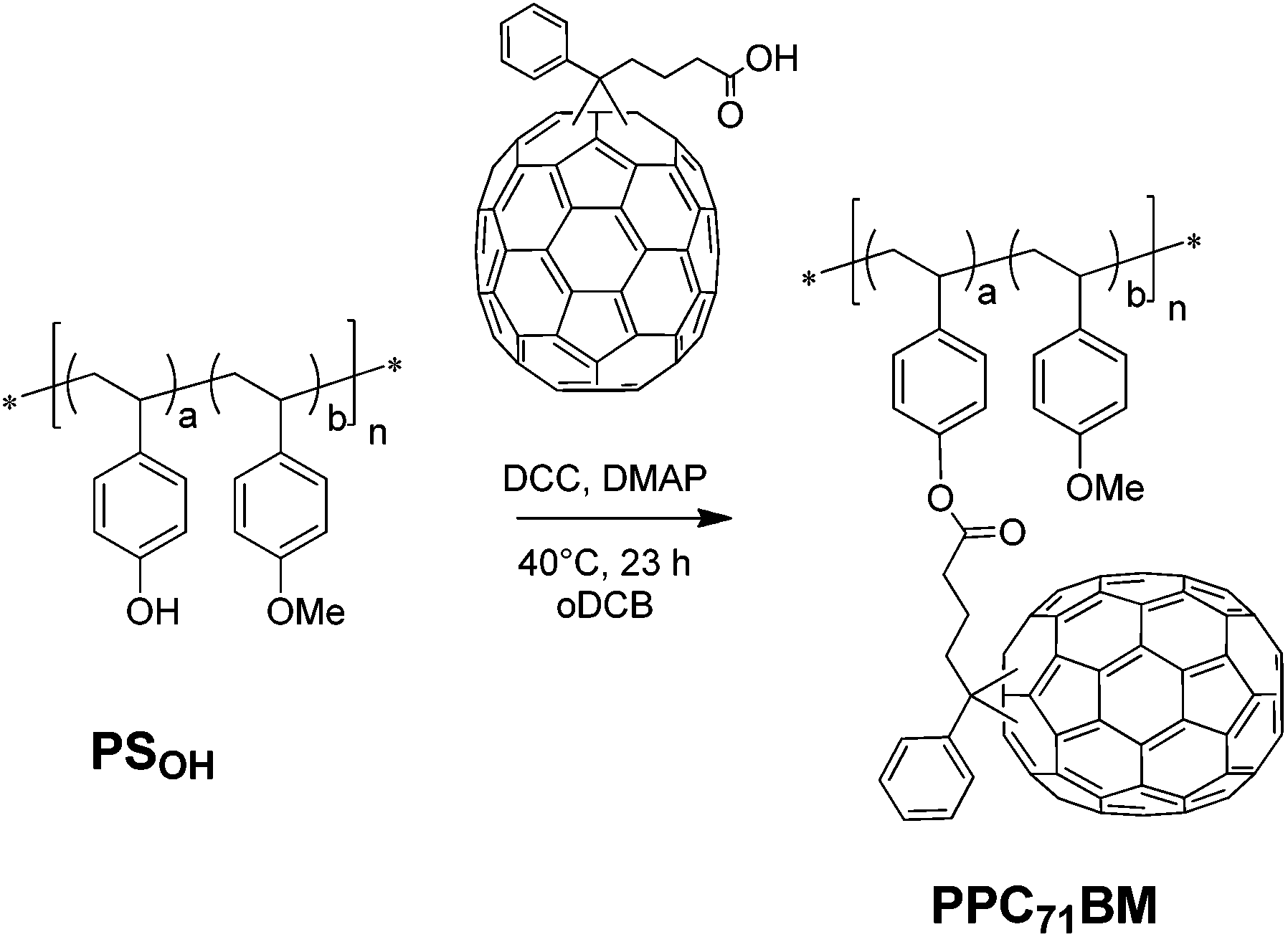 | ||
| Scheme 2 Monofunctional grafting of the precursor copolymer PSOHvia Steglich esterification with PC71BA. | ||
The esterification of PSOH was carried out in DCB at 40 °C for 23 h in the presence of an excess of PC71BA (2 equivalents with respect to the hydroxyl groups) and N,N′-dicyclohexyl carbodiimide (DCC) and 4-dimethylaminopyridine (DMAP). The crude product was purified by an extensive procedure to guarantee that only covalently bound fullerenes remain in the product (see the Experimental part). The low molecular weight fullerene side products (e.g. N-acyl urea derivatives) were removed by several precipitation cycles into mixtures of methanol:toluene (1:2, v:v). The purity of the PC71BM-grafted copolymer PPC71BM was monitored by thin layer chromatography (TLC), where the small molecule impurities appear as distinct spots. Further, the 1H-NMR spectrum (Fig. S6†) verifies the purity of PPC71BM, since any low-molecular weight fullerene impurity would appear as sharp multiplets in the spectrum. The obtained polymer PPC71BM is highly soluble in chlorinated solvents such as chloroform, chlorobenzene and DCB. The grafted product was further analyzed using 1H-NMR to verify the grafting reaction.
The 1H-NMR spectrum of PPC71BM clearly supports a successful grafting reaction since all relevant resonances belonging to the polymer and the pendant fullerenes are present in the product (Fig. S6†). The multiplets of methylene protons of the pendant PC71BM appear as strongly broadened signals indicating a successful covalent linkage to the polymer backbone. SEC analysis in chloroform as an eluent shows a molecular weight of Mn = 20.8 kg mol−1 and a relatively narrow distribution with PDI = 1.27 (Fig. S7†). Note that there is no polymer crosslinking observed in the SEC trace; there is only the shoulder at higher molecular weight which was already present in the precursor PSOH. The peak maximum in SEC increased from 21.7 kg mol−1 for PSOH to 26.3 kg mol−1 for PPC71BM. It is typical of fullerene polymers that the effective molecular weight is underestimated in SEC, most probably due to strong intrachain interactions between the pendant fullerene moieties.15
To quantify the extent of fullerene grafting, we favor matrix assisted laser desorption ionization mass spectrometry with time of flight detection (MALDI-TOF MS) as a very reliable method.17 The mass spectra of the precursor copolymer PSOH and the grafted copolymer PPC71BM are depicted in Fig. 3a. Here, the esterification leads to a significant mass increase of the copolymer due to the pendant fullerenes from Mp = 24.4 kg mol−1 for PSOH to Mp = 42.4 kg mol−1 for PPC71BM. Mp denotes the peak maximum of the molar mass distribution curve. The resulting mass increase, ΔM, is 18.0 kg mol−1 and this corresponds to the amount of attached PC71BA per polymer chain. From Mp of PSOH we can further estimate the average degree of polymerization, which is 181 repeating units per chain. Since the copolymer PSOH contains 10 mol% hydroxystyrene, the number of hydroxyl groups is 18 units on an average. Therefore, a maximum of 18 PC71BA moieties (M = 1016.99 g mol−1) can be grafted per chain which corresponds to a theoretical mass increase of 18.3 kg mol−1. From this, we deduce that the efficiency of grafting is almost quantitative (98%) and yields a copolymer with 42 wt% of pendant PC71BM. Thermogravimetric analysis (TGA) of the polymers PSOH and PPC71BM fully supports these findings (Fig. 3b). Whereas the precursor copolymer PSOH is decomposed almost completely in nitrogen, the fullerene-grafted PPC71BM shows a significant amount of residual char. This residue corresponds to the C70 fullerene core which is thermally very stable and does not decompose until 800 °C. Thus, we can roughly estimate the fullerene weight content of PPC71BM as 49 wt%. This is in very good agreement with the MALDI-TOF MS result since the content of PCBM in TGA is usually overestimated, likely due to incomplete decomposition.17
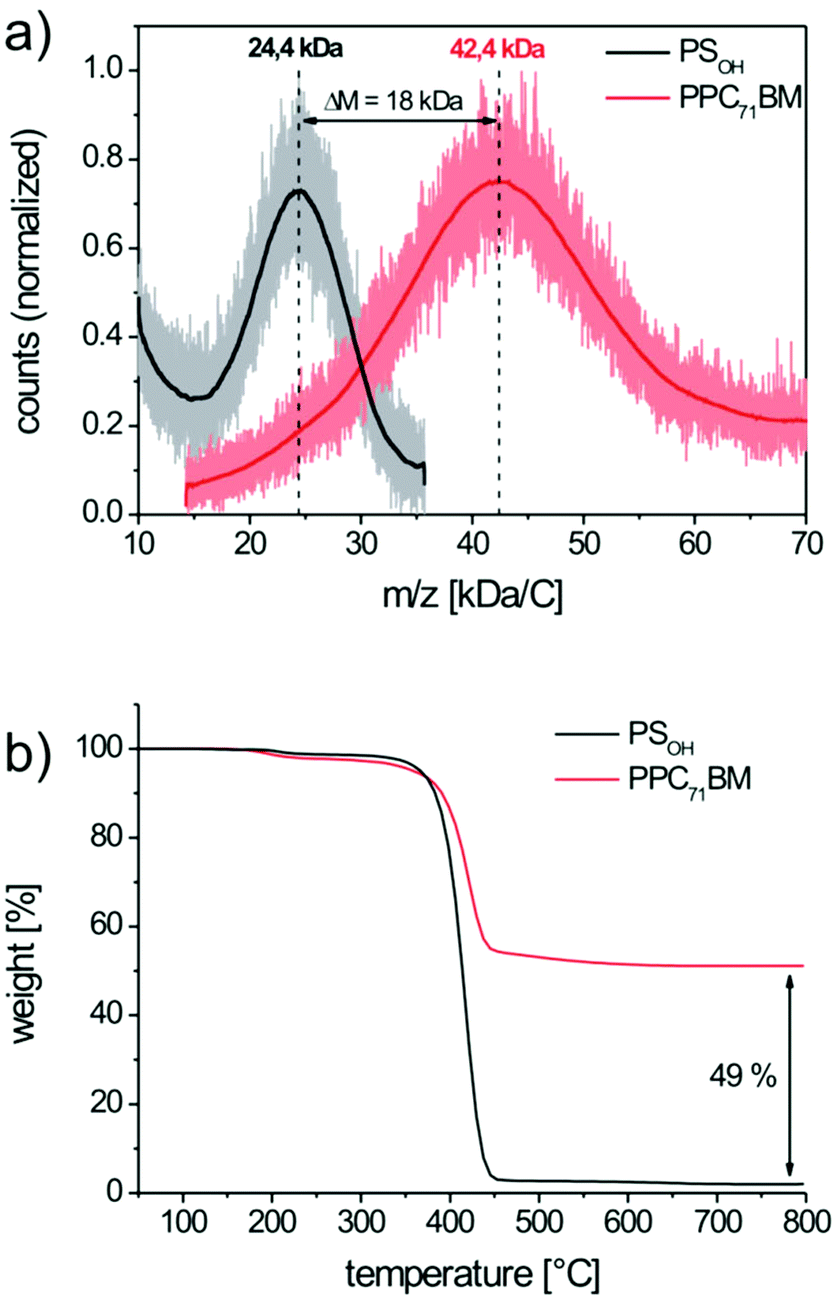 | ||
| Fig. 3 Determination of the PC71BM content in PPC71BM by (a) MALDI-TOF mass spectrometry and (b) by thermogravimetric analysis in nitrogen atmosphere. | ||
The optical absorption of PPC71BM ranges from the UV region to 650 nm and, hence, has an improved light harvesting capability with respect to optical density and wavelength range compared to the known C60 fullerene polymers. This was confirmed by UV-vis spectroscopy in solution (Fig. S8†).
Since PC71BM is a crystalline material, we were also interested in knowing whether the covalent attachment of these fullerene moieties to the copolymer backbone influences the crystallization or not. X-ray diffraction of a powder sample prepared by slow evaporation of a solution of PPC71BM in DCB confirmed the absence of any fullerene crystallites showing only an amorphous scattering signal (Fig. S9†). This is very similar to the observation of amorphous states in PPC61BM polymers.17 This is also an important aspect regarding structure formation in block copolymers comprising a PPC71BM block, because a too strong fullerene interaction may disturb the self-assembly of the block copolymer, which is not the case here.
Synthesis and characterization of the donor–acceptor block copolymer with pendant PC71BM
The target block copolymer was obtained by a polymer–polymer click reaction followed by the grafting of PC71BM onto the PS backbone (Scheme 3). For the click reaction, ethynyl end-capped P3HT having an average molecular weight of Mn = 19.2 kg mol−1 (SEC) and a narrow polydispersity of 1.11 (Fig. 4a) and the PSOH-Az with 9.8 kg mol−1 and polydispersity of 1.11 were used. P3HT with a molecular weight Mn around 20 kg mol−1 (SEC) exhibits optimum charge transport35 and can promote phase separation in block copolymers due to its high degree of polymerization.14 The copolymer PSOH-Az contains 10 repeating units of 4-hydroxystyrene and therefore can be grafted with up to 10 kg mol−1 PC71BM. This yields roughly equal amounts of donor and acceptor compounds in the final block copolymer, which is in analogy to the ratio typically used in blend solar cell devices.32 | ||
| Scheme 3 Synthesis route of the donor–acceptor block copolymer P3HT-b-PPC71BM via polymer–polymer click chemistry and Steglich esterification with PC71BA. | ||
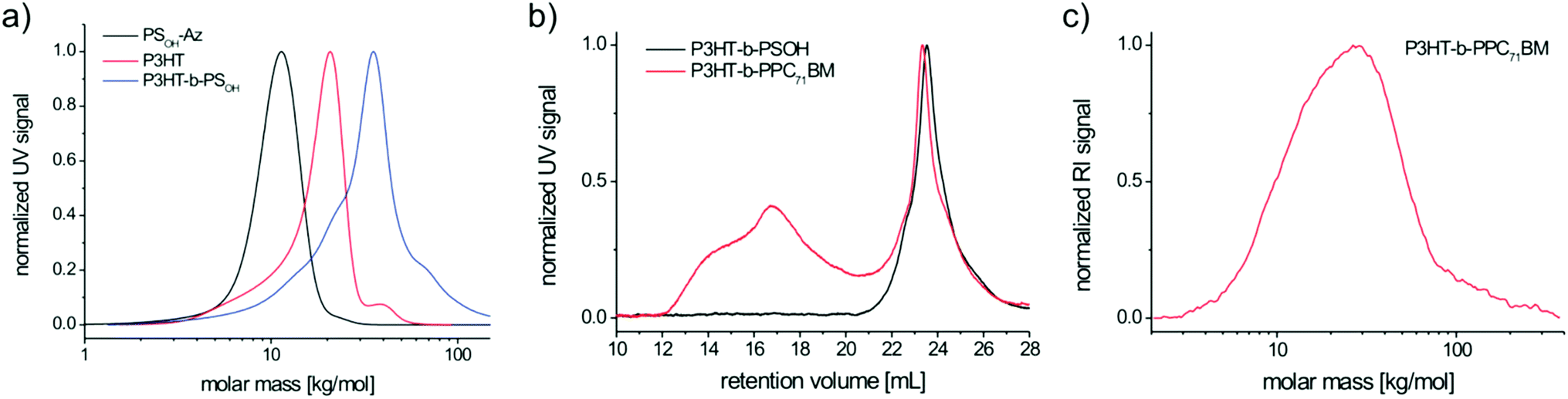 | ||
| Fig. 4 SEC traces of (a) the polymer–polymer coupling by click chemistry (THF, UV detector). Analysis of the block copolymer P3HT-b-PSOH and P3HT-b-PPC71BM via SEC in chloroform (b) and SEC in 1,2,4-trichlorobenzene at 150 °C for P3HT-b-PPC71BM (c). | ||
The CuAAC reaction was carried out with ethynyl-end-capped P3HT and three equivalents of azide-terminated PSOH-Az in the presence of copper(I) iodide/N,N,N′,N′,N′′-pentamethyl-diethylenetriamine (PMDETA) as a catalyst (Scheme 3). The precursor PSOH-Az was used in excess, since it can be separated easily from the block copolymer afterwards by precipitation in a mixture of methanol:acetone (2:1/v:v). We monitored the progress of block copolymer formation by SEC and stopped the reaction after 43 hours, when no further consumption of P3HT could be observed.
From the SEC traces shown in Fig. 4 it is obvious that the polymer–polymer click reaction was successful resulting in the diblock copolymer P3HT-b-PSOH with an average molecular weight Mn of 26.3 kg mol−1 and a polydispersity of 1.35. The broad polydispersity can be accounted for by taking into consideration the residual P3HT homopolymer observed as a shoulder in the SEC trace (Fig. 4a). This can be attributed to either an incomplete click reaction and/or an insufficient ethynyl functionalization of P3HT. The small shoulder at higher molecular weight of the block copolymer SEC trace might be due to aggregation effects in SEC.
The composition of the block copolymer P3HT-b-PSOH can be deduced from the individual blocks that were analyzed by MALDI-TOF MS (Fig. S10†). Here, the mass spectrum of P3HT shows a major peak series corresponding to the ethynyl-P3HT (Fig. S10a†) with an Mp of 12.4 kg mol−1 which is equal to 74 hexylthiophene repeating units (RU). The mass spectrum of PSOH-Az (Fig. S10b†) shows an Mp of 13.4 kg mol−1 which corresponds to 98 repeating units in the copolymer. Thus, for P3HT-b-PSOH we can expect a peak molecular weight of 25.8 kg mol−1. Exactly the same molecular weight is found for the P3HT-b-PSOH block copolymer (25.9 kg mol−1). Therefore, the MALDI-TOF mass spectrum analysis unambiguously proves the success of the polymer–polymer click reaction (Fig. S10c†). To summarize, P3HT-b-PSOH on average contains 74 RU of hexylthiophene and 98 RU of styrene moieties (Table 1). Taking into consideration the composition of the PSOH block (a:b = 0.10:0.90), a total number of ten 4-hydroxystyrene (HS) units per block copolymer chain is present. The composition of the block copolymer P3HT-b-PSOH was also verified using 1H-NMR analysis. We can find all relevant proton resonances in the 1H-NMR spectrum of P3HT-b-PSOH (Fig. S11†). From the integral ratio of the thiophene proton singlet at δ = 7.37 ppm and the broad phenyl proton multiplet at δ = 7.11–6.52 ppm we calculated the composition of P3HT-b-PSOH of m:n = 74:103 repeating units. Thus, the composition of the block copolymers deduced from NMR is in very good agreement with the MALDI-TOF MS data for the individual polymer blocks.
| Polymer | SEC | MALDIc | DP | ||
|---|---|---|---|---|---|
| M n [kg mol−1] | PDI | M p [kg mol−1] | m | n | |
| a SEC in tetrahydrofuran as an eluent, a UV (254 nm) detector, polystyrene calibration. b SEC in chloroform as an eluent, a UV (254 nm) detector, polystyrene calibration excluding the aggregation region. c Matrix-assisted laser desorption/ionization mass spectrometry with time of flight detection. d M p is the peak maximum of the molecular weight distribution curve. e M p determined from the very broad mass distribution. f Degree of polymerization (DP) determined from Mp of the MALDI-TOF mass spectrum. g DP determined from Mn = 12.3 kg mol−1 of MALDI-TOF MS. h DP determined from the 1H-NMR spectrum. | |||||
| PSOH-Aza | 9.8 | 1.11 | 13.4 | — | 98f |
| P3HT-alkynea | 19.2 | 1.11 | 12.4 | 74g | — |
| P3HT-b-PSOHa | 26.3 | 1.35 | 25.9 | 74h | 103h |
| P3HT-b-PPC71BMb | 27.5 | 1.31 | 31.5e | 74h | 102h |
In the final step of polymer-analogous grafting we attached PC71BA to P3HT-b-PSOH by Steglich esterification. Similar reaction conditions using DCC and DMAP in dichlorobenzene as those for PPC71BM were used here. The applied amount of PC71BA with respect to hydroxyl groups in P3HT-b-PSOH was adjusted to approximately 2.5 equivalents to obtain a high degree of grafting. The product was extensively purified by several precipitations into methanol:toluene (1:1, v:v) and pure methanol. The removal of small molecule fullerene side products was thoroughly monitored via TLC. Furthermore, the absence of multiplet resonances for fullerene side products is supported by 1H-NMR in deuterated DCB (Fig. S12†). This NMR solvent turned out to give the most reasonable spectra because aggregation of the P3HT-b-PPC71BM in chloroform is also a challenging problem for NMR analysis. The spectrum exhibits all the expected signals corresponding to both P3HT and PPC71BM blocks. Comparing the ratio of the thiophene protons at δ = 7.36 ppm (1H) with the phenyl protons of the polystyrene block at δ = 7.09–6.51 ppm (4H) we obtain a ratio of repeating units of P3HT and polystyrene of m:n = 74:102, which is very close to the expected ratio of 74:98 from MALDITOF MS analysis. The pendant fullerene proton resonances are rather weak in the aromatic region at δ = 7.94–7.86 ppm and δ = 7.70–7.46 ppm and, additionally, strongly overlap in the case of the methylene resonances.
A significant tendency of P3HT-b-PPC71BM toward aggregation in chloroform is observed in the SEC eluogram of Fig. 4b. While P3HT-b-PSOH exhibits a rather narrow and monomodal eluogram, P3HT-b-PPC71BM is characterized by a sharp peak at 23.34 mL corresponding to the fully dissolved block copolymer and a huge amount of aggregates at very low retention volumes. The aggregation can be suppressed at elevated temperature and by changing the eluent, which is demonstrated by high-temperature SEC in 1,2,4-trichlorobenzene at 150 °C (Fig. 4c). Here, the SEC trace shows a monomodal mass distribution in the expected mass range. Using a polystyrene calibration, the molecular weights of P3HT-b-PSOH and P3HT-b-PPC71BM correspond to Mn = 26.3 kg mol−1 (Mp = 34.3 kg mol−1) and Mn = 27.5 kg mol−1 (Mp = 36.6 kg mol−1) respectively (Table 1). This expected marginal molecular weight shift in SEC is in full agreement with the contractile effects of fullerene polymers (see also PPC71BM).
To quantify the exact composition of the donor–acceptor block copolymer P3HT-b-PPC71BM we applied UV-vis spectroscopy in solution and MALDI-TOF MS. The mass spectrum of P3HT-b-PPC71BM in Fig. S10c,† however, is not suited to derive a clear composition of donor and acceptor blocks. Here, not only a shift toward higher molecular weight indicating a covalent grafting with PC71BA, but also a broadening of the mass distribution is observed.
A very useful method for the estimation of the PC71BM content is UV-vis spectroscopy. It is possible to calculate the content of a chromophore in polymers by comparing the extinction coefficients of polymers and the pure chromophore as a reference in dilute solutions.17,36
To determine the PC71BM content, we used the extinction coefficients of PC71BM and P3HT-b-PPC71BM at 274 nm (Fig. 5a), at which PC71BM shows a shoulder with high optical density. For the calculation, the residual absorption of polystyrene and P3HT at this wavelength was subtracted. Details of the exact calculation of the PC71BM content in the block copolymer are given in the ESI.† According to our calculations, the block copolymer P3HT-b-PPC71BM contains 24 wt% of PC71BM. Taking into consideration the MALDI TOF MS value of P3HT, the composition of P3HT to PC71BM was obtained as 1:0.86. The maximum possible theoretical content of PC71BM can be estimated based on the MALDI molecular weights of the individual polymer blocks (12.4 kg mol−1 for P3HT, 13.4 kg mol−1 for PSOH) and the maximum possible grafting which is supposed to be ten PC71BM units per polymer chain with a total weight of 10.17 kg mol−1. This results in a theoretical PC71BM content of 28 wt%. Comparing the theoretical PC71BM content of 28 wt% in P3HT-b-PPC71BM with the experimental value of 24 wt% from UV-vis, we can conclude a very high grafting efficiency of 86%.
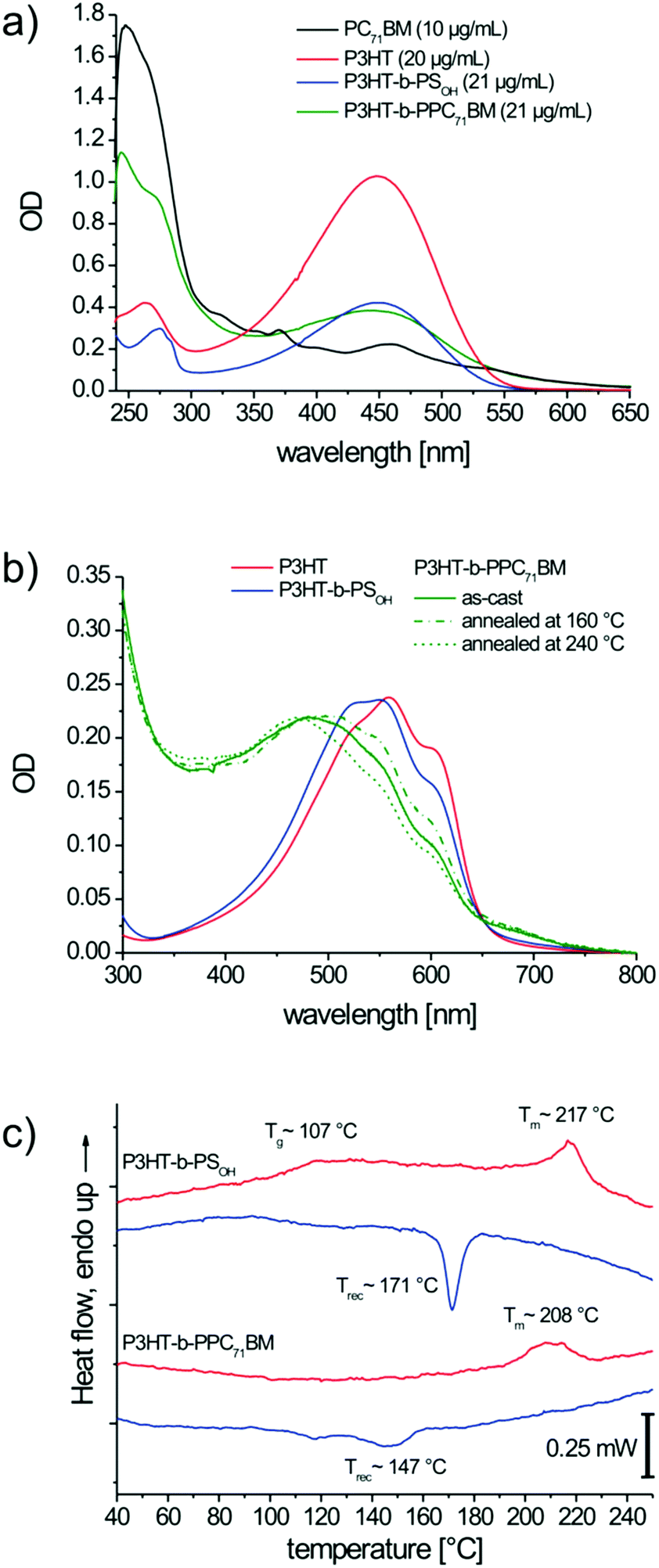 | ||
| Fig. 5 UV-vis absorption spectra (a) taken from solutions in chloroform. The spectra of PC71BM and P3HT are given as references and for the determination of the mass fractions of P3HT and PC71BM incorporated into the block copolymers. UV-vis spectra of thin films (b) after spin-coating (solid lines) and for P3HT-b-PPC71BM additionally after thermal annealing for 2 hours at 160 °C (dash-dot) and after annealing at 240 °C with a subsequent cooling rate of approximately 10 K min−1 (short dash). Differential scanning calorimetry traces (c) of the block copolymers P3HT-b-PSOH and P3HT-b-PPC71BM. The DSC traces for the heating cycles are red and blue for the cooling cycles. | ||
UV-Vis absorption studies of thin films allow for a qualitative estimation of the crystallization of P3HT (Fig. 5b). The neat P3HT shows distinct absorption bands at about 520, 560 and 605 nm which are ascribed to the formation of weak H-aggregates in crystals,37 whereas the amorphous P3HT phase exhibits a non-structured absorption band with a peak maximum at 450 nm. Both the block copolymers P3HT-b-PSOH and P3HT-b-PPC71BM exhibit the typical absorption bands relevant for aggregates or crystallites, but the relative intensities of the aggregate bands are lower compared to that of neat P3HT. On annealing P3HT-b-PPC71BM at 160 °C for 2 hours the aggregate bands appear more pronounced. On the other hand, heating the sample just above the melting point of P3HT (240 °C) and cooling at 10 K min−1 (similar to DSC cooling scan) there is a reduction of intensity of the aggregate bands. This clearly indicates that appropriate annealing procedures need to be developed for the crystallization of P3HT in the block copolymers.
Differential scanning calorimetry (DSC) confirms that P3HT-b-PSOH contains a crystalline P3HT block with a peak melting temperature of 217 °C and a melting enthalpy of 4.6 J g−1 (Fig. 5c). Further, the amorphous polystyrene block exhibits a glass transition Tg of around 107 °C. For P3HT-b-PPC71BM we observe notably broadened peaks showing a melting of P3HT around 211 °C with an enthalpy of 2.3 J g−1. No Tg for the PC71BM-grafted polystyrene was observed. The glass transition in such fullerene-grafted polystyrenes is typically shifted toward higher temperature and often shows a strong broadening.17,38
To obtain an estimation on the crystallinity of the P3HT segment in the block copolymers, we compared the melting enthalpies with that of neat P3HT, which melts at 230 °C with an enthalpy of 15.1 J g−1. Considering the weight fractions of P3HT in P3HT-b-PSOH and P3HT-b-PPC71BM which are 39 wt% and 28 wt% according to UV-vis in solution, we expect a melting enthalpy of 5.9 and 4.2 J g−1 if the same degree of crystallinity as that in the P3HT homopolymer is maintained. The experimentally determined melting enthalpies of 4.6 and 2.3 J g−1 are lower and thus indicate a decreased crystallinity of P3HT in both block copolymers as determined in DSC measurements. Since DSC scans are carried out too fast and therefore do not allow for the full crystallization in confined geometries, it is quite natural that the measured ΔHm values are lower than those expected for the same degree of crystallinity as a homopolymer. This is also in agreement with the varying degree of aggregation depending on annealing conditions observed in UV-vis absorption studies in thin films.
Structural analysis by X-ray scattering and scanning electron microscopy
Structure formation of the donor–acceptor block copolymer P3HT-b-PPC71BM was investigated using temperature-dependent small-angle X-ray scattering (SAXS) in bulk samples. The morphology of thin films was characterized by scanning electron microscopy (SEM) and grazing-incidence small angle X-ray scattering (GISAXS).Fig. 6 shows small angle X-ray scattering data obtained from bulk samples of P3HT-b-PPC71BM as measured at 240 °C in the molten state and at room temperature at which P3HT is in a crystalline state. Both scattering curves show a peak in the SAXS range indicating a periodic nanostructure with a periodicity d of about 37 nm at room temperature (d = 2π/q0, q = 0.168 nm−1 at room temperature, q = 0.16 nm−1 at 240 °C). The fact that the peak is almost unchanged after cooling to room temperature, i.e. after crystallization of the P3HT component, suggests that the nanostructure is already caused by a liquid–liquid phase separation in the melt.14 The weak signal and missing higher order peaks prevent a more detailed analysis of the structure based on X-ray scattering alone. The SEM image shown in Fig. 7 suggests indeed an ordered nanostructure at room temperature of probably cylindrical symmetry. Additionally, there is no indication of the presence of big fullerene aggregates or crystallites. The ordered nanostructure observed in SEM is also consistent with results obtained by GISAXS experiments of thin films as shown in Fig. 8 which show a clear peak indicating a periodic lateral structure within the films. Fig. 8a and b show the corresponding scattering pattern measured in grazing incidence geometry at room temperature for films prepared by drop-casting (Fig. 8a) and by spin-coating with subsequent annealing in the melt (Fig. 8b) respectively. The peak positions in the corresponding horizontal profiles (Fig. 8c) were determined to be q = 0.146 nm−1 (d ≈ 43 nm) for the melt crystallized sample and q = 0.156 nm−1 (d ≈ 40 nm) for the drop casted film. Again the peak positions were determined using an empirical model function consisting of a power law background and a Gaussian to describe the peak (see ESI†). Variation of the fitting parameters revealed an uncertainty of about 0.005 nm−1 for the peak position. We attribute the small difference in the peak position between the melt crystallized sample and the drop casted film to a solvent remaining in the drop casted film which mediates the incompatibility between both blocks and leads to a smaller structure size.39 Both samples have the peak at a similar position as the bulk samples indicating that the bulk morphology is largely preserved in thin films. In detail the peak positions in SAXS and GISAXS measurements show a deviation of about 10% probably due to either calibration errors or confinement effects.
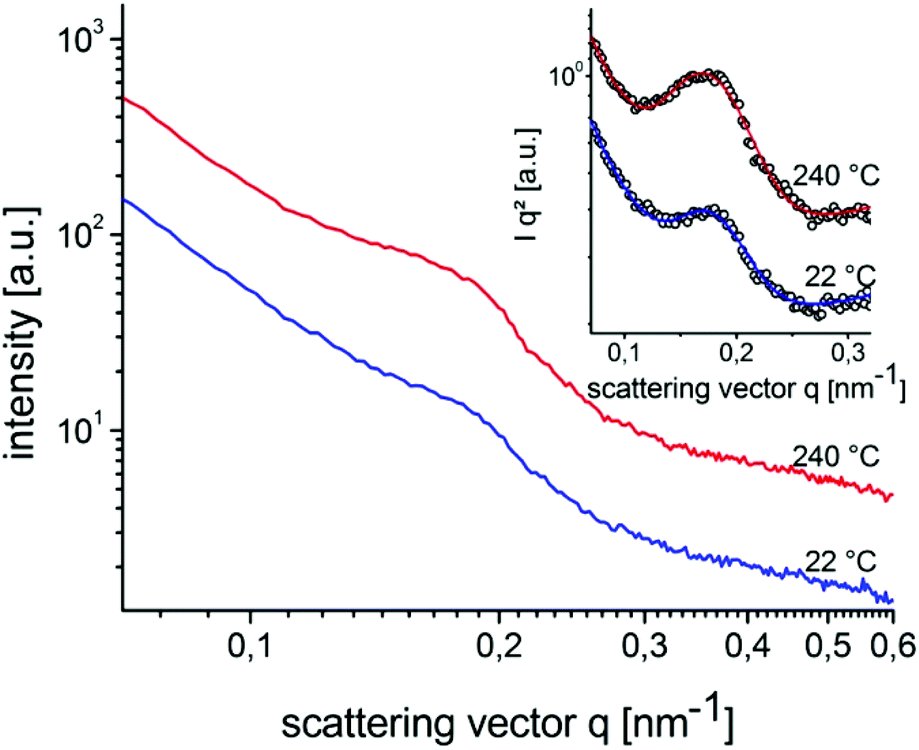 | ||
| Fig. 6 SAXS intensity of P3HT-b-PPC71BM at room temperature (blue) and at 240 °C in the melt (red, data set vertically shifted). The inset shows the same data after Lorentz correction with a fit to a model function consisting of a power law background and a Gaussian used to describe the peak. | ||
 | ||
| Fig. 7 Scanning electron microscopy image of P3HT-b-PPC71BM prepared by drop-casting of a 2 wt% dichlorobenzene solution. The inset shows a magnified section of the film. | ||
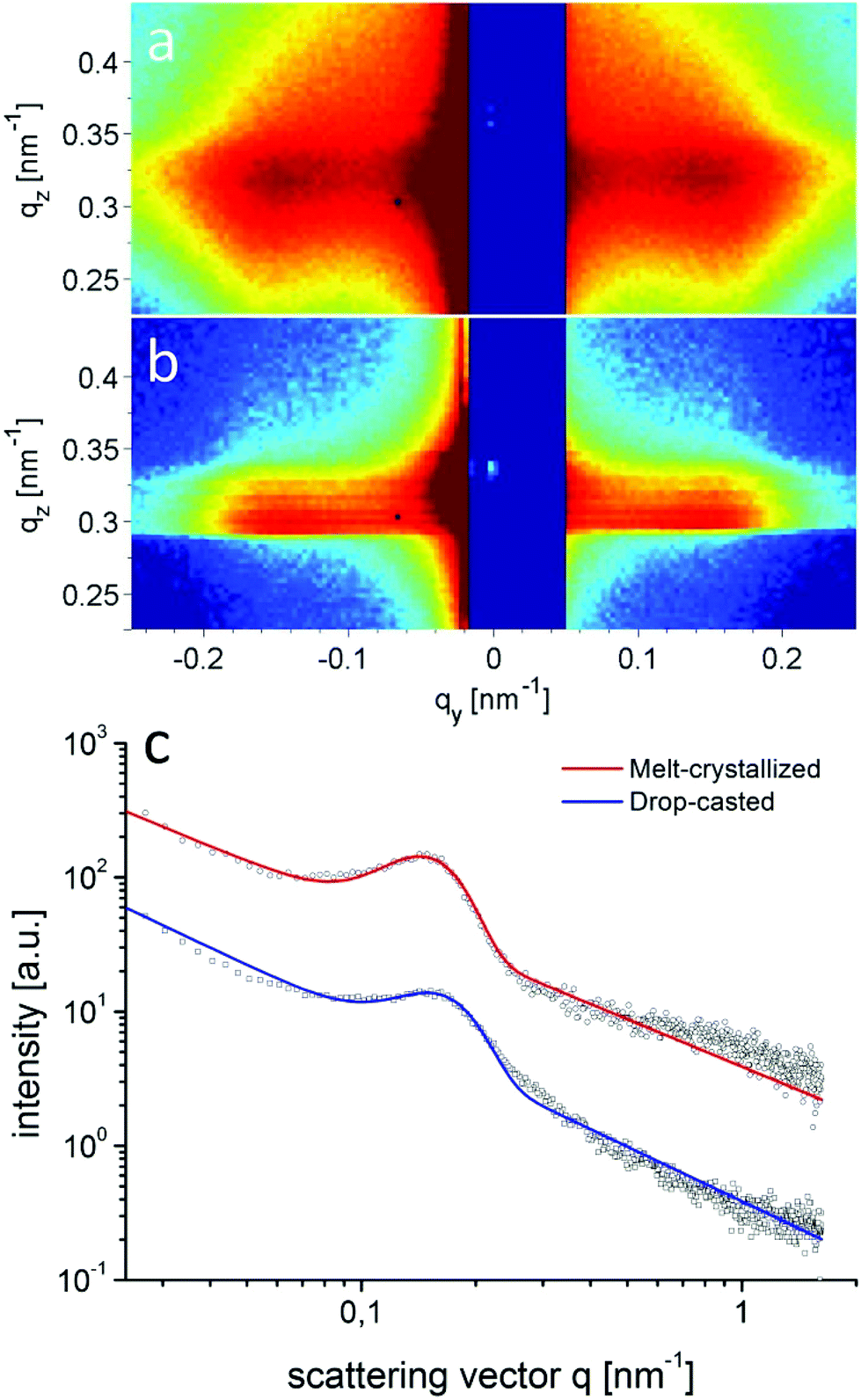 | ||
| Fig. 8 2D-GISAXS data of P3HT-b-PPC71BM around the Yoneda position at room temperature of films prepared by (a) drop-casting and (b) spin-coating with subsequent annealing in the melt. Data in (a) and (b) are displayed on a logarithmic intensity scale. The Yoneda region appears at slightly different nominal qz values due to a somewhat different angle of incidence. The horizontal intensity profiles of these samples, taken along the Yoneda position, are shown in (c) together with an empirical fit. The peak indicates a periodic nanostructure which is consistent with bulk data (data set of the melt crystallized sample vertically shifted). | ||
The aggregation of fullerene moieties was earlier reported to impede the self-assembly of fullerene-grafted block copolymers into a well-defined nanostructured morphology.28 However, the presented block copolymer P3HT-b-PPC71BM does not show aggregation of fullerene moieties. We attribute this favorable situation to a controlled incorporation of monofunctionalized fullerene moieties. Our observation is further supported by reports of Miyanishi et al.22,40 and Lee et al.25 who also utilized a monofunctionalized fullerene for grafting. In the former, a crystallization driven self-assembly of the fullerene-grafted poly(3-alkylthiophene) block copolymers is suggested to result in a fibril-like surface morphology in thin films. In the latter, phase separation of P3HT-b-poly(acrylate) with pendant PC61BM was studied using TEM which indicated phase separation in the range of 10–20 nm. Our findings based on GISAXS and temperature-dependent SAXS clearly show an ordered nanostructure in thin films as well as in the bulk phase of P3HT-b-PPC71BM.
Conclusions
We demonstrated an elegant synthetic route to PC71BM-grafted donor–acceptor block copolymers for the first time using polymer–polymer click chemistry between two tailored polymer blocks followed by a polymer-analogous fullerene grafting. PC71BM grafting was achieved from easily accessible C70 carboxylic acid. The covalent incorporation of PC71BM was achieved in very high yields and improves the optical properties of the block copolymer notably. The donor–acceptor block copolymer shows an ordered morphology on the nanoscale in bulk and in thin films independent of the processing method, i.e. from solution or by melt crystallization. Further, SAXS and GISAXS structural analysis correlates with the domain size observed in SEM. This pioneering work in the field of PC71BM-grafted block copolymers opens new perspectives towards the realization of further fullerene-grafted donor–acceptor block copolymers exploiting the full range of block copolymer morphologies.Experimental
Materials
The reagents for the following synthetic procedures were purchased from Sigma-Aldrich and used as received, unless stated otherwise. Phenyl-C71-butyric acid methyl ester (PC71BM, purity 99%) was purchased from American Dye Source (Canada). Solvents were either distilled at atmospheric pressure utilizing appropriate desiccants or purchased in p.a. (pro analysi) grade. Commercially available anhydrous solvents were purchased from Sigma Aldrich and Acros in sealed bottles with molecular sieves. Prior to polymerization, 4-methoxystyrene (MS) and 4-tert-butoxystyrene (BS) were passed through a column of basic alumina to remove the inhibitor and stored at −18 °C. 2,2,5-Trimethyl-4-phenyl-3-azahexane-3-nitroxide, 2,2,5-trimethyl-3-(1′-p-chloromethylphenylethoxy)-4-phenyl-3-azahexane and 2,2,5-trimethyl-3-(1′-p-azidomethyl-phenylethoxy)-4-phenyl-3-azahexane were synthesized according to the literature.41,42 Ethynyl-terminated poly(3-hexylthiophene) was prepared by Kumada catalyst transfer polymerization followed by end-capping with ethynylmagnesium chloride.31 The stock solution of 0.07 M copper(I) iodide/PMDETA catalyst in tetrahydrofuran was prepared according to a literature procedure.43Instrumentation
1H-NMR spectra were measured with a Bruker Avance AC250 spectrometer at 300 MHz. The obtained spectra were calibrated to the corresponding residual solvent peak (CDCl3δ = 7.26 ppm, DMSO-D6δ = 2.50 ppm). Ultraviolet-visible (UV-Vis) spectra from solutions were recorded on a Hitachi U-3000 spectrophotometer using quartz cuvettes with a path length of 1 cm. UV-vis studies of thin films were carried out on a Hitachi U-3000 spectrophotometer and a JASCO Spectrophotometer V-670. The films were prepared by spin-coating from 2 wt% chlorobenzene solution (P3HT and P3HT-b-PSOH) or 2.5 wt% 1,2-dichlorobenzene solution (P3HT-b-PPC71BM) on glass substrates. Fourier transform infrared (FTIR) spectra were recorded from solids on a Perkin Elmer Spectrum 100 FTIR spectrometer in attenuated total reflection (ATR) mode. Thermal gravimetry analysis (TGA) experiments were performed under a continuous nitrogen stream using a Mettler Toledo TGA/SDTA 851. The measurement range was 30–800 °C with a heating rate of 10 K min−1. The determined decomposition temperatures Tdec are onset temperatures. Size exclusion chromatography (SEC) with stabilized THF as an eluent was performed using a Waters 515-HPLC pump at a flow rate of 0.5 mL min−1. A guard column (Varian, 50 × 0.75 cm, ResiPore, particle size 3 μm) and two separation columns (Varian, 300 × 0.75 cm, ResiPore, particle size 3 μm) are connected in series with a Waters UV detector at 254 nm and 486 nm. 1,2-Dichlorobenzene (DCB) was used as an internal standard. SEC with chloroform as an eluent was carried out at 25 °C with a FLOM Intelligent Pump 301 at a flow rate of 0.5 mL min−1. A guard column and four separation columns of MZ Analysentechnik with a polystyrene solid phase (particle size 5 μm, pore sizes 106, 105, 103 and 102 Å) are connected in series with a Spectra Series UV150 detector at 254 nm and a Shodex RI-71 detector. Toluene was used as an internal standard. High-temperature SEC analysis was carried out on an Agilent (Polymer Laboratories Ltd) PL-GPC 220 high temperature chromatographic unit equipped with DP, RI and LS (15° and 90°) detectors and three linear mixed bed columns of PLgel 13 micrometer (Olexis) with a linear molecular weight operating range: 500–15000000 g mol−1. SEC analysis was performed at 150 °C using 1,2,4-trichlorobenzene as the mobile phase. All SEC data were calibrated in relation to polystyrene standards.
Matrix assisted laser desorption ionization mass spectroscopy with time of flight detection (MALDI-TOF MS) was performed on a Bruker Reflex III using either dithranol or trans-2-(3-(4-tert-butylphenyl)-2-methyl-2-propenylidene)malononitrile (DCTB) as the matrix and, where denoted, silver trifluoroacetate as a cationizing salt. Solutions of the analyte, the matrix and the cationizing salt were prepared, mixed in the given ratio and spotted onto the MALDI target plate. P3HT: chloroform, dithranol (10 mg mL−1), analyte (0.5 mg mL−1), 50:1/v:v. PSOH: THF, DCTB (10 mg mL−1), analyte (10 mg mL−1), AgTFA (10 mg mL−1), 20:5:1/v:v:v. P3HT-b-PSOH: chloroform, dithranol (10 mg mL−1), analyte (5 mg mL−1), 10:2/v:v. P3HT-b-PPC71BM: chloroform, dithranol (10 mg mL−1), analyte (5 mg mL−1), 20:2/v:v.
Small angle X-ray scattering experiments in transmission geometry were performed with a laboratory setup consisting of a Rigaku rotating anode, a focusing X-ray optics device (Osmic confocal max flux), and a Bruker 2D-detector (HI-Star). The optics also served as a monochromator for Cu Kα radiation (λ = 0.154 nm). Aluminum discs with a central hole of 0.8 mm diameter were used as sample holders. The samples were mounted on a Linkam hot stage for temperature control. A heat conducting paste was used to ensure good thermal contact. Grazing incidence small angle X-ray scattering experiments (GISAXS) were performed at the beam line BM 26, DUBBLE, at the ESRF, Grenoble (λ = 0.104 nm). The monochromatic beam was collimated using an assembly of slits and focused on the detector position. An angle of incidence slightly larger than the critical angle of the polymer film and the silicon substrate was used. Rectangular samples had a typical size of 90–140 mm2 and were completely illuminated by the beam. 2D scattering patterns were collected using a PILATUS 1 M detector. The sample to detector distance was set to 3.07 m. Samples for GISAXS measurements were prepared by spin-coating resp. drop-casting. The spin-coated samples were prepared from a 3 wt% solution in 1,2-dichlorobenzene on silicon substrates at 800 rpm, resulting in a thickness of about 125 nm. After spin-coating the samples were annealed under a nitrogen atmosphere for 90 min at 240 °C and slowly cooled down (approximately 5–10 K min−1). The drop-casted films were prepared from a 3 wt% solution in 1,2-dichlorobenzene, covered with a beaker and dried at room temperature. They had a thickness of about 1.4 μm. The calibration of the SAXS and GISAXS data was performed by comparison with the first order ring of silver behenate measured in transmission.
Scanning electron microscopy (SEM) was performed on a Zeiss LEO 1530 (FE-SEM with the Schottky-field-emission cathode; in-lens detector) using an accelerating voltage of 2.0–3.0 kV. The samples were prepared by drop-casting from a 2 wt% solution in 1,2-dichlorobenzene on indium–tin oxide coated glass substrates, covered with a beaker and slowly dried at room temperature. Spin-casting with quick solvent evaporation and a freezing-in of structures yield shorter structural features. For the SEM measurement the samples were mounted on a standard SEM sample holder by a conductive adhesion graphite-pad (Plano) and sputtered with platinum (2 nm using a Cressington HR208 sputter coater and a Cressington mtm20 thickness controller).
Synthesis procedure of PS
To a dry 10 mL Schlenk tube were added 4-methoxystyrene (4.487 g, 33.44 mmol, 410 eq.), 4-tert-butoxystyrene (0.513 g, 2.91 mmol, 36 eq.), 2,2,5-trimethyl-4-phenyl-3-azahexane-3-nitroxide (4 mg, 0.016 mmol, 0.2 eq.) and 2,2,5-trimethyl-3-(1′-p-azidemethylphenylethoxy)-4-phenyl-3-azahexane (31 mg, 0.081 mmol, 1 eq.) and the mixture was dissolved in 4 mL of dry o-dichlorobenzene. The Schlenk tube was sealed with a rubber septum and the reaction mixture was degassed by three freeze, pump and thaw cycles. The polymerization was subsequently started in a preheated oil bath at 125 °C under vigorous stirring. Samples were taken periodically by a syringe to monitor the monomer conversion via1H-NMR spectroscopy. The polymerization was stopped at a conversion of 44% after 550 min by quenching the reaction in liquid nitrogen. The resulting polymer was isolated by precipitation of the reaction mixture three times into 600 mL methanol, filtering the white precipitate and drying under vacuum. Yield: 1.679 g of a white precipitate. 1H-NMR (300 MHz, CDCl3, δ): 7.01–6.09 (br m, 4(a + b)·H, Hphenyl), 3.95–3.49 (br s, 3b·H, HOMe), 2.29–0.97 (br m, 3(a + b)·H, Hbackbone, 9a·H, HOtBu). SEC (THF): Mn = 19.6 kg mol−1, Mp = 24.6 kg mol−1, Mw/Mn = 1.19. MALDI: Mp = 23.5 kg mol−1.Synthesis of PSOH
A 250 mL round bottom flask was charged with PS (1.601 g) and dissolved in 160 mL THF. 20 mL of concentrated hydrochloric acid (37 wt%) were added slowly to the solution. After purging the reaction mixture with argon for 20 min via a glass pipette, it was stirred at 35 °C for 16 h. All volatile components were removed under reduced pressure. The hydrolyzed polymer was isolated by precipitation twice in 500 mL deionized water and freeze-drying from 1,4-dioxane. Yield: 1.29 g of a white precipitate. 1H-NMR (300 MHz, DMSO-d6, δ): 9.25–8.86 (br s, 1a·H, OH), 7.30–5.93 (br m, 4(a + b)·H, Hphenyl), 3.85–3.45 (br s, 3b·H, HOMe), 2.25–0.73 (br m, 3(a + b)·H, Hbackbone). SEC (THF): Mn = 16.7 kg mol−1, Mp = 24.6 kg mol−1, Mw/Mn = 1.19. MALDI: Mp = 24.4 kg mol−1. TGA decomposition Tdec = 389 °C.Synthesis of PPC71BM
A 250 ml Schlenk flask was flame dried at 600 °C under high vacuum. It was charged with PSOH (165 mg, 0.124 mmol hydroxy groups, 1 eq.), phenyl-C71-butyric acid (250 mg, 0.246 mmol, 2 eq.), and 4-dimethylaminopyridine (49 mg, 0.401 mmol, 3.2 eq). The flask was degassed under high vacuum and backfilled with argon twice. 85 mL of dry o-dichlorobenzene was added and the mixture was ultrasonicated for 30 min at room temperature. In a separate 50 mL Schlenk flask under argon, N,N′-dicyclohexylcarbodiimide (178 mg, 0.862 mmol, 7 eq.) was dissolved in 15 mL of dry o-dichlorobenzene. Then the DCC solution was added dropwise to the reaction mixture, which was then stirred at 40 °C for 23 h. After removal of the solvents under reduced pressure at 60 °C, the crude product was redissolved in DCB and filtered. For polymer purification the filtrate was precipitated first in 500 mL methanol, then three times in 500 mL of methanol–toluene (2:1, v:v) and finally in 500 mL methanol. The purity of the obtained polymer was monitored by TLC (SiO2, toluene), where the polymer remains on the start line and the impurities show distinct spots. The precipitate was dried under high vacuum. Yield: 210 mg of a deep brown precipitate. 1H-NMR (300 MHz, CDCl3, δ): 8.11–7.72 (br m, 2a·H, Ph–C–(CH2)3–COOR), 7.65–7.33 (br m, 3·aH, Ph–C–(CH2)3–COOR), 7.08–5.97 (br m, 4(a + b)·H, Hphenyl), 3.99–3.49 (br s, 4b·H, HOMe), 2.79–2.61,2.61–2.44 (br m, 4a·H, Ph–C–CH2–CH2–CH2–COOR), 2.36–0.94 (br m, 2aH, Ph–C–CH2–CH2–CH2–COOR, 3(a + b)H, Hbackbone). SEC (CHCl3): Mn = 20.8 kg mol−1, Mp = 26.3 kg mol−1, Mw/Mn = 1.27. MALDI: Mp = 42.5 kg mol−1. TGA decomposition Tdec = 390 °C.
Synthesis of PS-Cl
To a dry 10 mL Schlenk tube were added 4-methoxystyrene (4.482 g, 33.41 mmol, 260 eq.), 4-tert-butoxystyrene (0.511 g, 2.91 mmol, 23 eq.), 2,2,5-trimethyl-4-phenyl-3-azahexane-3-nitroxide (6 mg, 0.026 mmol, 0.2 eq.) and 2,2,5-trimethyl-3-(1′-p-chloromethylphenylethoxy)-4-phenyl-3-azahexane (48.2 mg, 0.129 mmol, 1 eq.). The Schlenk tube was sealed with a rubber septum and the reaction mixture was degassed by three freeze, pump and thaw cycles. The polymerization was subsequently started in a preheated oil bath at 125 °C under vigorous stirring. Samples were taken periodically by a syringe to monitor the monomer conversion via1H-NMR spectroscopy. The polymerization was stopped at a conversion of 40% after 410 min by quenching the reaction in liquid nitrogen. The resulting polymer was isolated by precipitation of the reaction mixture three times in 600 mL methanol, filtering the white precipitate and drying under vacuum. Yield: 1.593 g of a white precipitate. 1H-NMR (300 MHz, CDCl3, δ): 6.95–6.15 (br m, 4(a + b)·H, Hphenyl), 3.90–3.56 (br s, 3b·H, HOMe), 2.25–0.85 (br m, 3(a + b)·H, Hbackbone, 9a·H, HOtBu). SEC (THF): Mn = 10.7 kg mol−1, Mw/Mn = 1.10.Synthesis of PS-Az
A dry Schlenk flask was charged with PS-Cl (1.30 g, approximately 0.07 mmol, 1 eq.) and sodium azide (0.11 g, 1.69 mmol, 24 eq.) and sealed with a rubber septum. It was degassed under high vacuum and backfilled with argon three times before adding dry N,N-dimethylformamide (20 mL) via a syringe. The suspension was stirred for 70 h at room temperature. The polymer was isolated by diluting the reaction mixture with a small amount of THF and precipitation in 600 mL deionized water twice. After filtering the white precipitate, it was washed with 100 mL methanol and dried in a vacuum at room temperature. Yield: 1.223 g of a white precipitate. 1H-NMR (300 MHz, CDCl3, δ): 6.96–6.14 (br m, 4(a + b)·H, Hphenyl), 3.87–3.59 (br s, 3b·H, HOMe), 2.25–0.83 (br m, 3(a + b)·H, Hbackbone, 9a·H, HOtBu). SEC (THF): Mn = 10.4 kg mol−1, Mw/Mn = 1.10.Synthesis of PSOH-Az
A 250 mL round bottom flask was charged with PS-Az (1.10 g) and dissolved in 110 mL THF. 13.8 mL of concentrated hydrochloric acid (37 wt%) were added slowly to the solution. After purging the reaction mixture with argon for 20 min via a glass pipette, it was stirred at 35 °C for 20 h. For work-up, all volatile components were removed under reduced pressure. The hydrolyzed polymer was isolated by precipitation twice in 600 mL deionized water and freeze-drying from 1,4-dioxane. Yield: 0.92 g of a white precipitate. 1H-NMR (300 MHz, DMSO-d6, δ): 9.28–8.83 (br s, 1a·H, OH), 7.10–5.97 (br m, 4(a + b)·H, Hphenyl), 3.80–3.51 (br s, 3b·H, HOMe), 2.315–0.72 (br m, 3(a + b)·H, Hbackbone). SEC (THF): Mn = 9.8 kg mol−1, Mp = 11.4 kg mol−1, Mw/Mn = 1.11. MALDI: Mp = 13.4 kg mol−1.Synthesis of P3HT-b-PSOH
A dry 20 mL Schlenk tube with a screw cap was charged with P3HT-alkyne (75 mg, approx. 6.1 μmol, 1 eq.) and PSOH-Cl (223 mg, approx. 16.6 μmol, 2.7 eq.). It was degassed under high vacuum and backfilled with argon three times. 20 mL of dry THF were added under argon and the solution was stirred for 20 min at 50 °C until all components were fully dissolved. After purging the mixture with argon via a needle for 20 min, the copper iodide/PMDETA catalyst solution (0.5 mL, 0.07 M, 35 μmol) was added under argon. The Schlenk flask was sealed with the screw cap and cautiously degassed under vacuum and backfilled with argon three times to remove any oxygen traces. The reaction mixture was stirred for 26 h at 50 °C, 0.3 mL of the catalyst solution was added and the solution was stirred for another 17 h. The reaction mixture was passed through a short column of basic aluminium oxide to remove the copper catalyst. The column was washed with 50 mL of THF, the organic fractions were combined and the solvent was removed under reduced pressure. The polymer was isolated by precipitation twice in 300 mL of methanol–acetone (2:1, v:v) from THF and drying the precipitate under vacuum at room temperature. Yield: 163 mg of a black precipitate. 1H-NMR (300 MHz, o-DCB-d4, δ): 7.37 (s, m·1H, Ha), 7.10–6.55 (br m, n·4(a + b)·H, Hphenyl), 4.00–3.66 (br s, n·3b·H, HOMe), 3.20–2.96 (br m, m·2H, Hb), 2.00–1.85 (br m, Hc), 1.68–1.55 (br m, Hd), 1.54–1.38 (br m, He, Hf), 2.38–1.34 (br m, Hbackbone), 1.10 (tr, m·3H, Hg). SEC (CHCl3): Mn = 26.3 kg mol−1, Mp = 35.4 kg mol−1, Mw/Mn = 1.10. MALDI: Mp = 25.9 kg mol−1.
Synthesis of P3HT-b-PPC71BM
A 100 ml Schlenk flask was flame dried at 600 °C under high vacuum. It was charged with P3HT-b-PSOH (50 mg, approx. 1.9 μmol polymer, 19 μmol hydroxy groups, 1 eq.), phenyl-C71-butyric acid (49 mg, 48 μmol, 2.5 eq.), and 4-dimethylaminopyridine (9 mg, 68.6 μmol, 3.6 eq.). The flask was degassed under high vacuum and backfilled with argon twice. 17 mL of dry o-dichlorobenzene were added and the mixture was ultrasonicated for 30 min at room temperature. In a separate 10 mL Schlenk flask under argon, N,N′-dicyclohexylcarbodiimide (33 mg, 160 μmol, 8.4 eq.) was dissolved in 3 mL of dry o-dichlorobenzene. Then the DCC solution was added dropwise to the reaction mixture, which was then stirred at 40 °C for 23 h. After removal of the solvents under reduced pressure at 60 °C, the crude product was redissolved in chloroform and filtered. For polymer purification the filtrate was precipitated first in 300 mL methanol–toluene (1:1, v:v) twice and then in 300 mL methanol. The purity of the obtained polymer was monitored by TLC (SiO2, toluene), where the polymer remains on the start line and the impurities show distinct spots. The precipitate was dried under vacuum at room temperature. Yield: 61 mg of a black precipitate. 1H-NMR (300 MHz, o-DCB-d4, δ): 7.94–7.87, 7.70–7.44 (br m, Hh, Hi), 7.36 (s, m·1H, Ha), 7.09–6.55 (br m, n·4(a + b)·H, Hphenyl), 3.98–3.68 (br s, n·3b·H, HOMe), 3.22–2.95 (br m, m·2H, Hb), 2.01–1.84 (br m, Hc), 1.69–1.55 (br m, Hd), 1.54–1.36 (br m, He, Hf), 2.07–1.24 (br m, Hbackbone), 1.09 (m, m·3H, Hg). SEC (CHCl3): Mn = 27.5 kg mol−1, Mp = 36.6 kg mol−1, Mw/Mn = 1.31. MALDI: Mp = 31.5 kg mol−1.
Acknowledgements
We kindly acknowledge DFG (SPP1355), EU LARGECELLS (Gr. no. 291936) and Bavarian State Ministry of Science, Research, and the Arts for the Collaborative Research Network “Solar Technologies go Hybrid” for financial support of this project. M. Hufnagel also thanks the German National Academic Foundation for a PhD scholarship and the Elite Network Bavaria Macromolecular Science programme at the University of Bayreuth. We also acknowledge Ruth Lohwasser for providing ethynyl-terminated poly(3-hexylthiophene). The GISAXS experiments were performed on the BM26 beam line at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. We are grateful to Guiseppe Portale for providing assistance in using the beam line BM26. Finally, we thank Christian Stelling and Fabian Nutz for their help in polymer synthesis.Notes and references
- K. M. Coakley and M. D. McGehee, Chem. Mater., 2004, 16, 4533–4542 CrossRef CAS.
- J. Peet, M. L. Senatore, A. J. Heeger and G. C. Bazan, Adv. Mater., 2009, 21, 1521–1527 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- J. K. J. van Duren, X. Yang, J. Loos, C. W. T. Bulle-Lieuwma, A. B. Sieval, J. C. Hummelen and R. A. J. Janssen, Adv. Funct. Mater., 2004, 14, 425–434 CrossRef CAS.
- W. Ma, C. Yang, X. Gong, K. Lee and A. J. Heeger, Adv. Funct. Mater., 2005, 15, 1617–1622 CrossRef CAS.
- T. Erb, U. Zhokhavets, G. Gobsch, S. Raleva, B. Stühn, P. Schilinsky, C. Waldauf and C. J. Brabec, Adv. Funct. Mater., 2005, 15, 1193–1196 CrossRef CAS.
- J. U. Lee, J. W. Jung, T. Emrick, T. P. Russell and W. H. Jo, Nanotechnology, 2010, 21, 105201 CrossRef.
- S. Bertho, G. Janssen, T. J. Cleij, B. Conings, W. Moons, A. Gadisa, J. D'Haen, E. Goovaerts, L. Lutsen, J. Manca and D. Vanderzande, Sol. Energy Mater. Sol. Cells, 2008, 92, 753–760 CrossRef CAS.
- F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2009, 93, 394–412 CrossRef CAS.
- M. W. Matsen and F. S. Bates, Macromolecules, 1996, 29, 1091–1098 CrossRef CAS.
- M. Shah and V. Ganesan, Macromolecules, 2010, 43, 543–552 CrossRef CAS.
- P. D. Topham, A. J. Parnell and R. C. Hiorns, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1131–1156 CrossRef CAS.
- M. Sommer, S. Huettner and M. Thelakkat, J. Mater. Chem., 2010, 20, 10788 RSC.
- R. H. Lohwasser, G. Gupta, P. Kohn, M. Sommer, A. S. Lang, T. Thurn-Albrecht and M. Thelakkat, Macromolecules, 2013, 46, 4403–4410 CrossRef CAS.
- U. Stalmach, B. de Boer, C. Videlot, P. F. van Hutten and G. Hadziioannou, J. Am. Chem. Soc., 2000, 122, 5464–5472 CrossRef CAS.
- L. Perrin, A. Nourdine, E. Planes, C. Carrot, N. Alberola and L. Flandin, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 291–302 CrossRef CAS.
- M. Hufnagel, M.-A. Muth, J. C. Brendel and M. Thelakkat, Macromolecules, 2014, 47, 2324–2332 CrossRef CAS.
- B. de Boer, U. Stalmach, P. F. van Hutten, C. Melzer, V. V. Krasnikov, G. Hadziioannou, B. De Boer and P. F. Van Hutten, Polymer, 2001, 42, 9097–9109 CrossRef CAS.
- M. H. van der Veen, B. de Boer, U. Stalmach, K. I. van de Wetering and G. Hadziioannou, Macromolecules, 2004, 37, 3673–3684 CrossRef CAS.
- B. Gholamkhass and S. Holdcroft, Chem. Mater., 2010, 22, 5371–5376 CrossRef CAS.
- C. Yang, J. K. Lee, A. J. Heeger and F. Wudl, J. Mater. Chem., 2009, 19, 5416–5423 RSC.
- S. Miyanishi, Y. Zhang, K. Hashimoto and K. Tajima, Macromolecules, 2012, 45, 6424–6437 CrossRef CAS.
- X.-H. Dong, W.-B. Zhang, Y. Li, M. Huang, S. Zhang, R. P. Quirk and S. Z. D. Cheng, Polym. Chem., 2012, 3, 124–134 RSC.
- M. Heuken, H. Komber, T. Erdmann, V. Senkovskyy, A. Kiriy and B. Voit, Macromolecules, 2012, 45, 4101–4114 CrossRef CAS.
- J. U. Lee, A. Cirpan, T. Emrick, P. Russell, W. Ho, T. P. Russell and W. H. Jo, J. Mater. Chem., 2009, 19, 1483–1489 RSC.
- J. C. Hummelen, B. W. Knight, F. LePeq, F. Wudl, J. Yao and C. L. Wilkins, J. Org. Chem., 1995, 60, 532–538 CrossRef CAS.
- K. Sivula, Z. T. Ball, N. Watanabe and J. M. J. Fréchet, Adv. Mater., 2006, 18, 206–210 CrossRef CAS.
- S. Barrau, T. Heiser, F. Richard, C. Brochon, C. Ngov, K. van de Wetering, G. Hadziioannou, D. V. Anokhin and D. A. Ivanov, Macromolecules, 2008, 41, 2701–2710 CrossRef CAS.
- M. M. Wienk, J. M. Kroon, W. J. H. Verhees, J. Knol, J. C. Hummelen, P. A. van Hal and R. A. J. Janssen, Angew. Chem., Int. Ed., 2003, 42, 3371–3375 CrossRef CAS.
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Nat. Photonics, 2012, 6, 593–597 CrossRef CAS.
- R. H. Lohwasser and M. Thelakkat, Macromolecules, 2012, 45, 3070–3077 CrossRef CAS.
- C. J. Brabec, S. Gowrisanker, J. J. M. Halls, D. Laird, S. Jia and S. P. Williams, Adv. Mater., 2010, 22, 3839–3856 CrossRef CAS.
- E. Lieber, C. N. R. Rao, T. Chao and C. Hoffman, Anal. Chem., 1957, 29, 916–918 CrossRef CAS.
- S. Fleischmann, H. Komber, D. Appelhans and B. I. Voit, Macromol. Chem. Phys., 2007, 208, 1050–1060 CrossRef CAS.
- C. R. Singh, G. Gupta, R. Lohwasser, S. Engmann, J. Balko, M. Thelakkat, T. Thurn-Albrecht and H. Hoppe, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 943–951 CrossRef CAS.
- M. Thelakkat, P. Pösch and H. W. Schmidt, Macromolecules, 2001, 34, 7441–7447 CrossRef CAS.
- F. C. Spano, J. Chem. Phys., 2005, 122, 234701 CrossRef.
- C. J. Hawker, Macromolecules, 1994, 27, 4836–4837 CrossRef CAS.
- A. Knoll, R. Magerle and G. Krausch, J. Chem. Phys., 2004, 120, 1105–1116 CrossRef CAS.
- S. Miyanishi, Y. Zhang, K. Tajima and K. Hashimoto, Chem. Commun., 2010, 46, 6723–6725 RSC.
- A. W. Bosman, R. Vestberg, A. Heumann, J. M. J. Fréchet and C. J. Hawker, J. Am. Chem. Soc., 2003, 125, 715–728 CrossRef CAS.
- W. Binder, D. Gloger, H. Weinstabl, G. Allmaier and E. Pittenauer, Macromolecules, 2007, 40, 3097–3107 CrossRef CAS.
- A. S. Lang and M. Thelakkat, Polym. Chem., 2011, 2, 2213 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR spectra, UV-Vis spectra, SEC characterization, FTIR spectroscopy data and MALDI-TOF mass spectra. See DOI: 10.1039/c4py01357c |
| This journal is © The Royal Society of Chemistry 2015 |