
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of branched fullerene-terminated poly(ethylene glycol)s†
Hin Chun
Yau‡
a,
Mustafa K.
Bayazit‡
b,
Piers R. J.
Gaffney
c,
Andrew G.
Livingston
c,
Joachim H. G.
Steinke
a and
Milo S. P.
Shaffer
*a
aDepartment of Chemistry, South Kensington Campus, Imperial College London, London, SW7 2AZ, UK. E-mail: m.shaffer@imperial.ac.uk
bDepartment of Chemical Engineering, University College London, London, WC1E 7JE, UK
cDepartment of Chemical Engineering, South Kensington Campus, Imperial College London, London, SW7 2AZ, UK
First published on 7th November 2014
Abstract
Poly(ethylene glycol) [1, PEG∼4(OH)2, Mn ∼ 200], glycerol ethoxylate [2, PEG∼21(OH)3, Mn ∼ 1000] and pentaerythritol ethoxylate [3, PEG∼15(OH)4, Mn ∼ 797] react directly with phenyl-C61-butyric acid methyl ester (PCBM), in the presence of dibutyltinoxide (DBTO) catalyst at 140 °C, to give a mixture of fullerene [C60] end-capped PEGs via transesterification. Among these PEG linkers, only PEG∼4(OPCB)2 (4a) (OPCB: ester oxygen linked phenyl-C61-butyryl group) was successfully isolated from the crude product mixture in the fully end-capped form. Fully acylated PEG∼21(OPCB)3 (5) and PEG∼15(OPCB)4 (6) could not be separated chromatographically from incompletely reacted species due to the polydispersity in branch lengths. This purification challenge was overcome by using a monodisperse branched core, 1,3,5-tris(octagoloxymethyl)benzene [7, PEG24(OH)3] to give a monodisperse tris-fullerene homostar, PEG24(OPCB)3 (8). The structures of the bis- and tris-fullerene products were confirmed by MALDI-TOF mass spectrometry and 1H NMR spectroscopy with supporting FTIR and UV-vis spectroscopic analysis.
Introduction
Due to its unique optoelectronic properties, and phase behaviour, fullerene[60] is one of the most commonly used materials in the preparation of polymer donor–acceptor bulk heterojunction (BHJ) solar cells.1–4 However, the low solubility and processability of fullerenes in organic solvents has limited their direct use in solution and encouraged the development of a wide range of functionalised derivatives, including methanofullerenes5–9 and fulleropyrrolidines.10,11 These functionalised derivatives offer significant increases in solubility and processability while preserving the important electronic and optical properties of the parent fullerenes. Direct functionalization of fullerenes is a simple option to prepare derivatives; however, multiple addition may occur which can affect optoelectronic properties. Phenyl-C61-butyric-acid-methyl-ester (PCBM) has received considerable attention in solution-processed organic electronic applications, particularly for electron transport in solar cells.12 As a result, this material is readily available commercially, and offers a convenient reagent for the preparation of monofunctionalized fullerene derivatives via transesterification of its methyl ester functional group.8,13,14The performance of fullerene-polymer based bulk heterojunction (BHJ) solar cells depends on the characteristic length-scale and crystallinity of the thin, phase-segregated semiconductor active layer;15–19 this structure is controlled by the dynamics of the segregation process during deposition and annealing,20 and by the intermolecular forces between polymer and fullerene derivatives.16 Recent studies have examined means to modify these interfacial interactions. In particular, the use of poly(ethylene glycol) (PEG),21 or PEG grafted PCBM,22,23 have been explored as additives to control the nano- and microstructure of the active layer, and so to produce highly stable and efficient polymer solar cells. Jeng et al. recently established the role of poly(ethylene glycol) dimethyl ether as an electrode buffer for the fabrication of high performance (poly-3-hexylthiophene, P3HT):PCBM based organic photovoltaics (OPVs).22 PEG-C60 adducts have been variously proposed as nucleants for the fullerene rich electron transporting phase,23 as a means to assemble an active dipole at the electrode interface,21 and as an encapsulant against oxidation.23 However, the uses of short chain, multi-fullerene end-capped linear or branched PEGs, or mixtures thereof, have not yet been explored in this context. Such multiply-bound fullerenes may offer additional opportunities to enhance device performance, potentially acting as more effective nucleants, inherently bringing together groups of fullerenes. At the same time, reducing the proportion of hygroscopic PEG should decrease the sensitivity of the device to moisture.
Synthetically, the preparation of multi-fullerene end-capped PEG dumbbells and stars has already been addressed via azide chemistry.24,25 However, these reactions with fullerenes do not yield pure mono-substituted adducts.26 The possible addition of more than one PEG-azide to a single fullerene creates a mixture of large molecules that is hard to separate. The one-step transesterification of PCBM provides a simple alternative that has not yet been exploited for the preparation of multi-fullerene end-capped PEGs. To date, the mono-fullerene end-capped PEG chains used in BHJ studies have had polymer weights of 2000–4000 Da. The large number of hygroscopic ethylene oxide repeat units per fullerene (ca. one C60 per 45–90 monomers) makes the devices highly moisture sensitive.21–23 Finally, in order to improve control during the BHJ phase segregation, it would be desirable to use pure, monodisperse PEG-PCBM materials as it is anticipated that these will enhance the self-assembly of ordered, well-defined structures.27 The present study develops a synthetic protocol for the preparation of multi-fullerene, end-capped polydisperse linear PEGs, and three- and four-arm PEG stars. Several challenges were encountered during isolation of fully fullerene end-capped PEGs. These difficulties were addressed by the preparation of a monodisperse multi-fullerene end-capped three-arm PEG homostar. Traditionally, such PCBM derivatives have been synthesised by first hydrolysing the methyl ester into carboxylic acid to give [6,6]-phenyl-C61-butyric acid (PCBA), and then converting to the acid chloride for further condensation.28 However, the carboxylic acid derivative (PCBA) is only sparingly soluble in organic solvent (ortho-dichlorobenzene or carbon disulphide) which limits large scale synthesis. Instead, in this paper, the multi-fullerene stars were prepared via a single step catalytic transesterification with dibutyltinoxide (DBTO).29 This approach avoids the multistep synthetic route and more importantly, the low solubility of PCBA during the synthesis.
Experimental
Materials and methods
Poly(ethylene glycol) [1, PEG∼4(OH)2, average molecular weight, Mn ∼ 200], glycerol ethoxylate [2, PEG∼21(OH)3, average Mn ∼ 1000], pentaerythritol ethoxylate [3, PEG∼15(OH)4 average Mn ∼ 797] were bought from Sigma-Aldrich and azeotropically dried by evaporation from anhydrous acetonitrile (99.8%, Sigma-Aldrich) prior to use. 1,3,5-Tris(octagoloxymethyl)benzene [7, PEG24(OH)3] was synthesized according to the procedure recently reported and similarly azeotropically dried.30 Dibutyltinoxide (DBTO, 98%, Sigma-Aldrich) was dried at 200 °C in vacuo for 30 min prior to use. Phenyl-C61-butyric acid methyl ester (PCBM, >99%, Solenne BV) was dried under vacuum at room temperature prior to use. Silica gel for column chromatography [Geduran® Si 60 (40–63 μm), Merck] was neutralised using sodium bicarbonate (VWR). Trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) (>98%, Sigma-Aldrich ) and sodium iodide (99.999%, Sigma-Aldrich) were used as the matrix for MALDI-TOF mass spectrometry. ortho-Dichlorobenzene (o-DCB) (anhydrous, >99.8%, Sigma-Aldrich), trifluoroacetic anhydride (TFAA) (≥99.8%, Sigma-Aldrich), thin layer chromatography plates (TLC Silica gel 60 F254, Merck), toluene, tetrahydrofuran and methanol (HPLC grade, VWR) were purchased and used as received.Characterization
NMR spectroscopy
NMR spectra were acquired using a Bruker AM 400 spectrometer operating at 9.4 T. Samples were dissolved in CDCl3 and all spectra were recorded with 16 scans and D1 = 1 s. All chemical shifts (δ) are given in ppm, where the residual CHCl3 peak was used as an internal reference for 1H NMR (δH = 7.28 ppm), and the CDCl3 peak for 13C NMR (δC = 78.23 ppm). Coupling constants (J) are given in Hz, and chemical splitting patterns are abbreviated as follows: s, singlet; d, doublet; t, triplet; q, quartet; quin, quintet and m, multiplet.UV-vis spectroscopy
UV-visible spectra were collected using a Perkin-Elmer Lambda 950 spectrophotometer. Samples were dissolved in toluene and spectra were recorded from 200 to 800 nm with a resolution of 1 nm, using a 1.0 cm UV quartz cuvette.FTIR spectroscopy
Fourier transform infrared spectra were recorded on powders using a Perkin-Elmer spectrometer 100 equipped with a Pike attenuated total reflectance (ATR) silicon crystal.General procedure for synthesis of fullerene end-capped PEGs
PCBM [1.1 eq. for each PEG(OH)n hydroxyl group] was dissolved in o-DCB (10 mL) under N2 at room temperature, and added to PEG and DBTO [0.11 eq. for each PEG(OH)n hydroxyl group] in a two-necked round-bottomed flask. The solution was then heated at 140 °C for 5 days under N2, after which the solvent was evaporated under a stream of N2 at room temperature, and the resulting dark brown residue fractionated by silica gel column chromatography. Unreacted PCBM eluted with toluene, after which fractions were collected by eluting with toluene–methanol (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v). Depending on their purity, as determined by 1H NMR, the fractions were further purified, either by additional column chromatography and/or preparative TLC until the 1H NMR integral of the peaks remained constant upon further purification.
:1 v/v). Depending on their purity, as determined by 1H NMR, the fractions were further purified, either by additional column chromatography and/or preparative TLC until the 1H NMR integral of the peaks remained constant upon further purification.
Synthesis of PEG∼4(OPCB)2, 4
Polydisperse bis-C60 linear PEG 4 was synthesized from PEG∼4(OH)2 (1, 30 mg, 0.14 mmol), PCBM (280 mg, 0.31 mmol) and DBTO (8 mg, 0.03 mmol) using the general method described above, and fractionated through a single silica gel column, eluting with toluene–methanol (9/1 v/v), to give PEG∼4(OPCB)2 (4, 25 mg, 9%) as a brown solid. Rf toluene–methanol (9:1 v/v) 0.43; δH (400 MHz, CDCl3) 2.20 (quin, J = 7.6 Hz, 4H), 2.57 (t, J = 7.3 Hz, 4H), 2.93 (t, J = 8.1 Hz, 4H), 3.60–3.77 (m, 17H), 4.25 (t, J = 4.3 Hz, 4H), 7.49 (t, J = 7.5 Hz, 2H), 7.56 (t, J = 7.6 Hz, 4H), 7.93 (d, J = 7.9 Hz, 4H) ppm; ν (FT–IR; ATR) 2917 (aliphatic C–H stretching), 2855 (aliphatic C–H stretching), 1732 (carbonyl stretching of PCBM ester), 1599, 1562, 1493, 1374, 1021 (C–O symmetric stretching of PEG), 797, 755, 695 cm−1; λ (UV-vis; toluene) 330, 433, 510 nm; m/z (MALDI-TOF) calc'd for [C152H42O8Na]+ 2017.23 m/z, found [M + Na]+ 2017.21 m/z.
Synthesis of mixture of PEG∼21(OPCB)3, 5a, and PEG∼21(OPCB)2(OH), 5b
Polydisperse tris-C60 3-arm branched PEG 5a was synthesized from PEG∼21(OH)3 (2, 50 mg, 0.05 mmol), PCBM (150 mg, 0.16 mmol) and DBTO (4 mg, 0.01 mmol) using the general method described above, and fractionated through silica gel columns (2×), followed by preparative TLC (3×), eluting with toluene–methanol (9:1 v/v), to give a mixture of PEG∼21(OPCB)3 (5a) and PEG∼21(OPCB)2(OH) (5b) (40 mg, 22%, 5a:5bca. 1:9 by 1H NMR integration) as a brown solid. Rf toluene–methanol (9:1 v/v) 0.30; δH (400 MHz, CDCl3) 2.08 (s, 5H), 2.20 (quin, J = 7.8 Hz, 4.4H), 2.57 (t, J = 7.5 Hz, 4.4H), 2.91 (t, J = 8.1 Hz, 4.4H), 3.47–3.85 (m, 84.6H), 4.25 (t, J = 4.3 Hz, 4.4H), 7.49 (t, J = 7.5 Hz, 2.2H), 7.57 (t, J = 7.6 Hz, 4.4H), 7.94 (d, J = 7.9 Hz, 4.4H) ppm; ν (FT-IR; ATR) 2912 (aliphatic C–H stretching), 2862 (aliphatic C–H stretching), 1731 (carbonyl stretching of PCBM ester), 1600, 1446, 1428 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) C stretching in fullerene), 1348, 1294, 1247, 1095 (C–O symmetric stretching of PEG), 944, 848, 755, 699 cm−1; λ (UV-vis; toluene) 330, 433, 510 nm; m/z (MALDI-TOF) calc'd for [C256H118O26Na]+ 3629.78 m/z, found [M + Na]+ 3627.87 m/z.
C stretching in fullerene), 1348, 1294, 1247, 1095 (C–O symmetric stretching of PEG), 944, 848, 755, 699 cm−1; λ (UV-vis; toluene) 330, 433, 510 nm; m/z (MALDI-TOF) calc'd for [C256H118O26Na]+ 3629.78 m/z, found [M + Na]+ 3627.87 m/z.
Synthesis of mixture of PEG∼15(OPCB)4, 6a, and PEG∼15(OPCB)3(OH), 6b
Polydisperse tetrakis-C60 4-arm branched PEG 6 was synthesized from PEG∼15(OH)4 (3, 40 mg, 0.05 mmol), PCBM (200 mg, 0.22 mmol) and DBTO (5 mg, 0.02 mmol) using the general method described above, and fractionated through silica gel columns (2×), followed by preparative TLC (3×), eluting with toluene–methanol (9:1 v/v), to give a mixture of PEG∼15(OPCB)4 (6a) and PEG∼15(OPCB)3(OH) (6b) (35 mg, 16%, 6a:6bca. 1:1 by 1H NMR integration) as a brown solid. Rf toluene–methanol (9:1 v/v) 0.32; δH (400 MHz, CDCl3) 2.21 (quin, J = 7.7 Hz, 8.1H), 2.58 (t, J = 7.5 Hz, 8.1H), 2.94 (t, J = 8.1 Hz, 8.1H), 3.45 (s, 5.4H), 3.53–3.67 (m, 61.2H), 4.26 (t, J = 4.3 Hz, 7.5H), 7.50 (t, J = 7.5 Hz, 4.1H), 7.58 (t, J = 7.6 Hz, 8.1H), 7.95 (d, J = 7.9 Hz, 8.1H); ν (FT-IR; ATR) 2924 (aliphatic C–H stretching), 2862 (aliphatic C–H stretching), 1731 (carbonyl stretching of PCBM ester), 1602, 1423 (CC stretching in fullerene), 1242, 1093 (C–O symmetric stretching of PEG), 952, 796, 755, 700 cm−1; λ (UV-vis; toluene) 330, 433, 510 nm; m/z (MALDI-TOF) calc'd for [C319H112O23Na]+ 4331.75 m/z, found [M + Na]+ 4332.85 m/z.
Synthesis of PEG24(OPCB)3, 8
Monodisperse tris-C60 3-arm branched PEG 8 was synthesized from PEG24(OH)3 (7, 25 mg, 0.02 mmol), PCBM (60 mg, 0.06 mmol) and DBTO (1.6 mg, 0.007 mmol) using the general method described above, and fractionated through silica gel columns (2×), followed by preparative TLC (3×), eluting with toluene–methanol (9:1 v/v), to give PEG24(OPCB)3 (8, 4 mg, 5%) as a brown solid. Rf toluene–methanol (9:1 v/v) 0.35; δH (400 MHz, CDCl3) 2.21 (quin, J = 8.1 Hz, 6H), 2.57 (t, J = 7.5 Hz, 6H), 2.93 (t, J = 8.1 Hz, 6H), 3.61–3.76 (m, 96H), 4.25 (t, J = 4.3 Hz, 6H), 4.56 (s, 6H), 7.25 (s, 3H), 7.51 (t, J = 7.5 Hz, 3H), 7.57 (t, J = 7.6 Hz, 6H), 7.94 (d, J = 7.9 Hz, 6H); ν (FT-IR; ATR) 2922 (aliphatic C–H stretching), 2852 (aliphatic C–H stretching), 1728 (carbonyl stretching of PCBM ester), 1462, 1378, 1265 (C–C stretching in fullerene), 1079 (C–O symmetric stretching of PEG), 1045, 889, 722 cm−1; m/z (MALDI-TOF) calc'd for [C270H138O30Na]+ 3881.92 m/z, found [M + Na]+ 3881.83 m/z.
Results and discussion
Multifullerene-end-capped PEGs, PEG∼4(OPCB)2 (4), PEG∼21(OPCB)3 (5a), PEG∼15(OPCB)4 (6a) and PEG24(OPCB)3 (8) were synthesized via a one-step transesterification of PCBM with the corresponding hydroxyl-terminated PEG derivatives [polydisperse PEG(OH)n1–3, and monodisperse PEG24(OH)37] in the presence of a catalytic amount of dibutyltinoxide (DBTO), at 140 °C in dry ortho-dichlorobenzene (o-DCB) (Scheme 1).29 Small aliquots from the crude reaction mixture were analysed after 2 h, and then every 24 h, up to 5 days by 1H NMR. 1H NMR analysis (see ESI,† Fig. S1) showed the appearance and growth of a new triplet signal at 4.25 ppm corresponding to the newly formed methylene ester protons (–CH2–O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).21 The signal intensity increased significantly during the first three days, after which point growth slowed, indicating that the transesterification reaction could proceed no further. As no pure mono-disperse PEGn(OH)3/4 (2 or 3) were commercially available, the study commenced with polydisperse PEG∼4(OH)2 (1, average molecular weight Mn ∼ 200 manufacturer value; Mn = 203 from NMR, see below), PEG∼21(OH)3 (2, average Mn ∼ 1000 manufacturer value; Mn = 1029 from NMR) and PEG∼15(OH)4 (3, average Mn ∼ 797 manufacturer value; Mn = 786 from NMR). After azeotropic drying with acetonitrile, these cores were used for the preparation of PEG∼4(OPCB)2 (4), PEG∼21(OPCB)3 (5a), and PEG∼15(OPCB)4 (6a), respectively, and the resulting compounds were purified by column and/or preparative thin layer chromatography (TLC). MALDI-TOF mass spectral analysis, supported by 1H NMR, FTIR and UV-vis techniques, were used for characterization of the phenyl-C61-butyryl (PCB)-functionalised PEGs.
O).21 The signal intensity increased significantly during the first three days, after which point growth slowed, indicating that the transesterification reaction could proceed no further. As no pure mono-disperse PEGn(OH)3/4 (2 or 3) were commercially available, the study commenced with polydisperse PEG∼4(OH)2 (1, average molecular weight Mn ∼ 200 manufacturer value; Mn = 203 from NMR, see below), PEG∼21(OH)3 (2, average Mn ∼ 1000 manufacturer value; Mn = 1029 from NMR) and PEG∼15(OH)4 (3, average Mn ∼ 797 manufacturer value; Mn = 786 from NMR). After azeotropic drying with acetonitrile, these cores were used for the preparation of PEG∼4(OPCB)2 (4), PEG∼21(OPCB)3 (5a), and PEG∼15(OPCB)4 (6a), respectively, and the resulting compounds were purified by column and/or preparative thin layer chromatography (TLC). MALDI-TOF mass spectral analysis, supported by 1H NMR, FTIR and UV-vis techniques, were used for characterization of the phenyl-C61-butyryl (PCB)-functionalised PEGs.
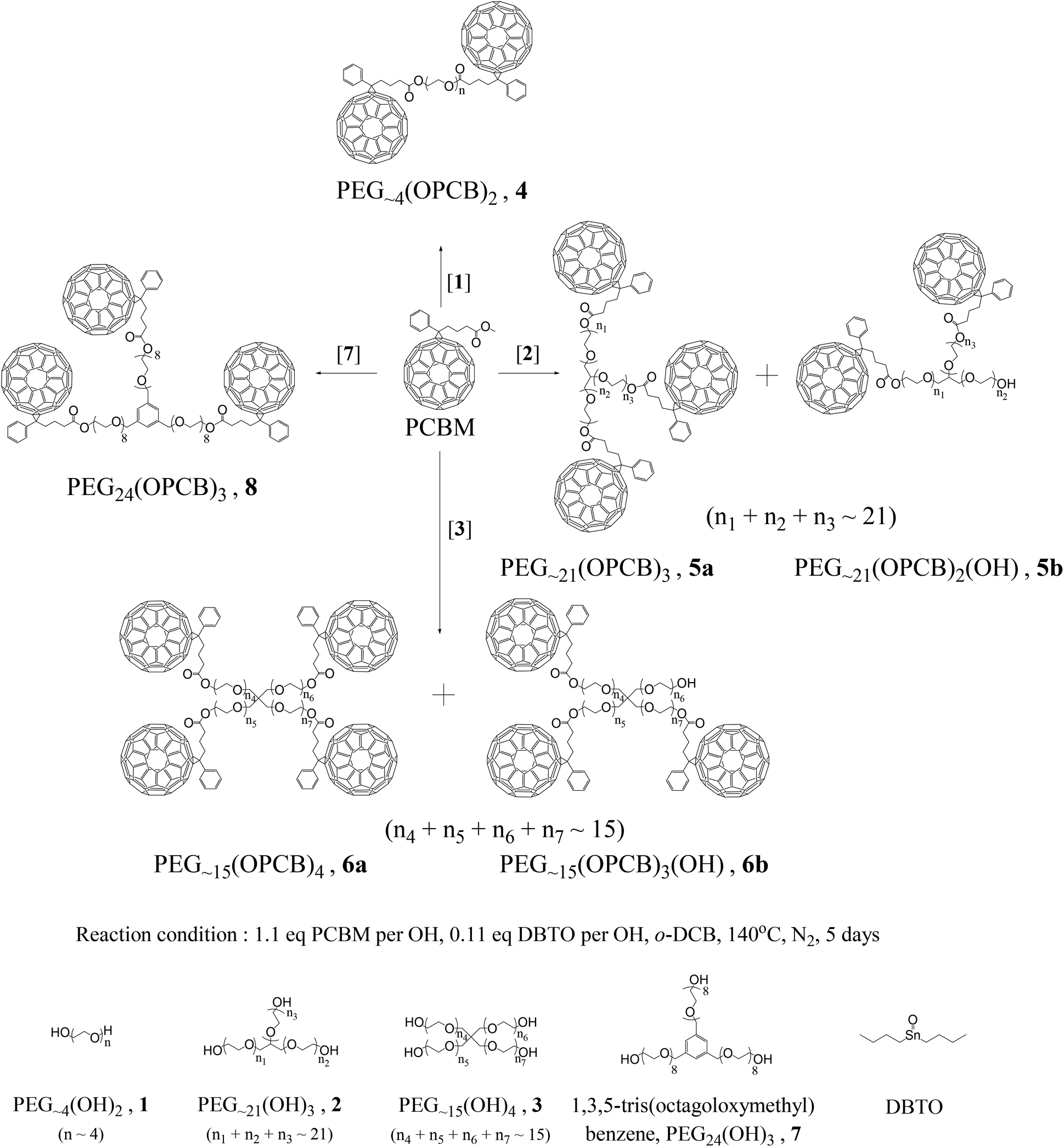 | ||
| Scheme 1 The transesterification of PCBM with PEGx(OH)n (1–3) and PEG24(OH)3 (7) to yield polydisperse PEGx(OPCB)n (4–6) and monodisperse PEG24(OPCB)3 (8). | ||
The extent of the transesterification reaction can be determined by the ratio of the chain terminal proton resonances to the polymeric backbone. However, the resonances at around 3.7 ppm corresponding to methylene (alpha proton, α) bearing the terminal hydroxyl groups overlap with the PEG backbone. To establish a suitable control and confirm the nature of starting materials, each starting PEGx(OH)n was treated with highly reactive trifluoroacetic anhydride (TFAA)–CDCl3 (1:19 v/v) in an NMR tube to give the corresponding per-acylated PEGx(OCOCF3)n derivative in situ.31 The electronegative trifluoroacetyl substituent deshields the adjacent acyloxymethylene protons (alpha proton, α), F3CCO2CH2CH2O, giving a signal at ca. 4.51 ppm well resolved from the PEG backbone. Thus, the average number of repeating units of each PEG compound was obtained from the integral ratio of the acyloxymethylene protons to the main PEG signals (see ESI,† Fig. S3–5), and found to be 4, 21 & 15 for bis-OH, tris-OH and tetrakis-OH terminated PEGs respectively. These 1H NMR findings are consistent with the number average of molecular weight distributions in the MALDI spectra of the pure PEG cores (ESI,† Fig. S8–9). The 1H NMR of PEG∼15(OCOCF3)4 (ESI,† Fig. S5) indicated that the tetrakis-OH terminated PEG contains a distinctive second variant; most likely a fraction of the mixture has a CH2-OH stub directly linked to the central sp3 carbon, rather than through an ether linkage, i.e. in 3, one of n4/5/6/7 = 0; (see ESI† for detailed characterisation). Since PCBM is a bulky molecule, a relatively longer side chain should assist the grafting reaction; steric effects may prevent fullerene attaching onto short side chains, particularly onto the CH2OH stub.
The MALDI-TOF mass spectrum of PEG∼4(OPCB)2 (4) (Fig. 1A) verified the formation of doubly transesterified PEG∼4(OH)2, showing four main clusters of positively charged [M + Na]+ ions ranging from m/z 1970 to 2160, where each peak region corresponds to the bis-fullerene end-capped PEG dumbbells with between 4 and 7 ethylene oxide repeat units (see ESI,† Fig. S10 for the full MALDI-TOF mass spectrum of 4; no mono-fullerene substituted species is visible). Considering the penta-ethylene oxide repeat unit cluster in detail (Fig. 1A*, upper), the experimental isotopic peak pattern of PEG∼4(OPCB)2 (4) was consistent with the simulated pattern (Fig. 1A*, lower) across all expected masses, [M + Na]+obs = 2017.23, [M + Na]+calc = 2017.21. In the IR spectrum of 4 (Fig. 2b), there is no OH stretch at around 3500 cm−1, indicating that there is very little, if any mono-fullerene substituted PEG∼4 in the product fraction. In the 1H NMR spectrum of PEG∼4(OPCB)2 (ESI,† Fig. S11), a new triplet at 4.25 ppm indicates the successful acylation of the linear PEG. The observed integral ratio between the newly formed acyloxymethylene resonance (alpha protons) and the main PEG signal was 1:4.25, whereas the ratio observed in fully trifluoroacetylated linear PEG was 1:3.25, Rather than incomplete reaction, the IR evidence suggests that this shift in ratio is more likely to arise due to the preferential double reaction of the longer PEG chains, because of steric constraints on the second fullerene addition. From the observed integral ratio, the as produced dumbbell derived under these conditions contains an average of 5 repeating units whereas the starting PEG∼4(OH)2 has an average of 4 repeating units.
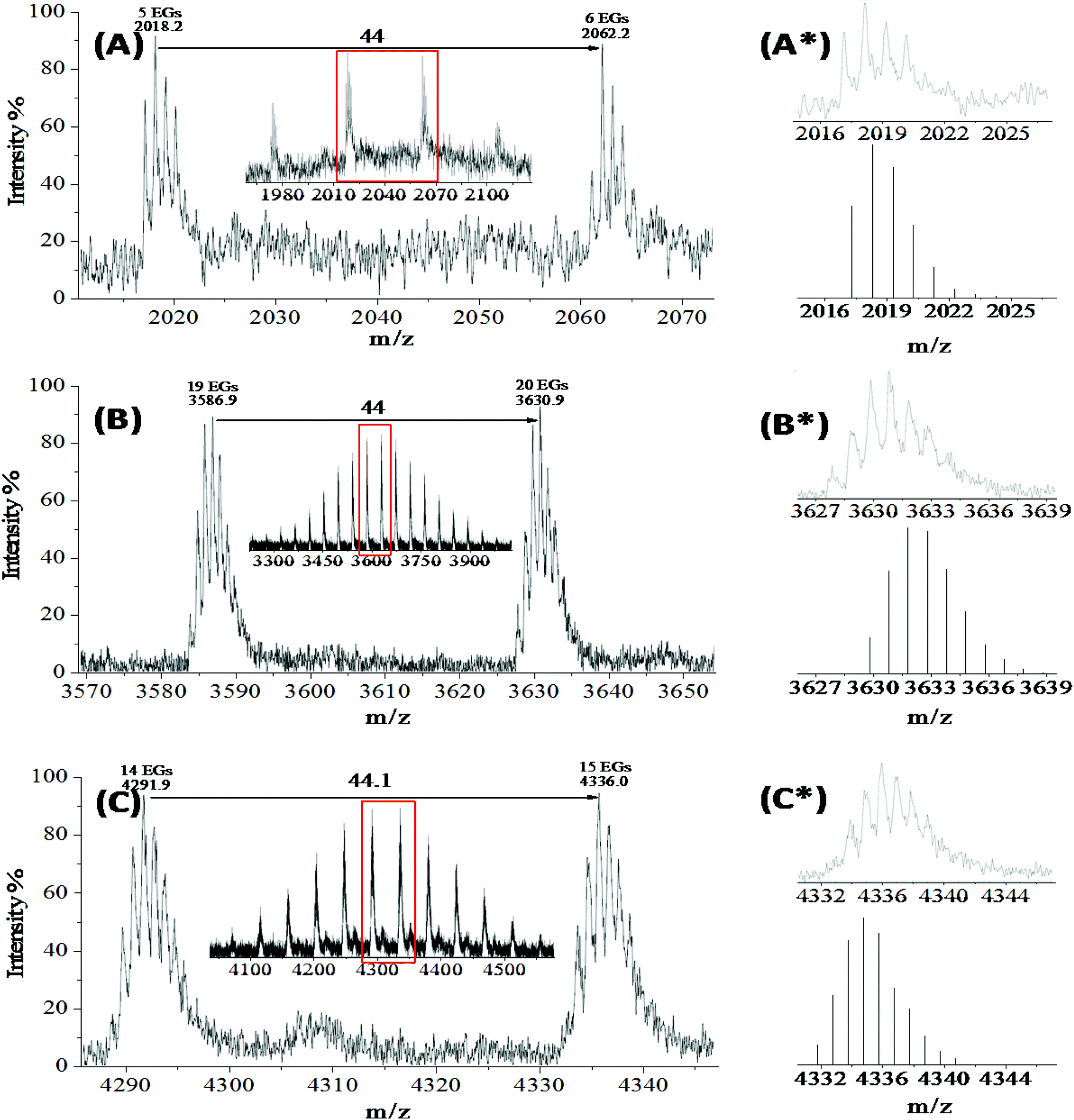 | ||
| Fig. 1 Experimental and simulated MALDI-TOF mass spectra of [PEG(OPCB)n+Na]+ (4–6) confirming successful transesterification of PCBM with polydisperse PEGs 1–3: (A) PEG∼4(OPCB)2 (4); (B) PEG∼21(OPCB)3 (5); (C) PEG∼15(OPCB)4 (6). The full spectra are given in the ESI.† For each of 4–6, the whole product cluster (inset) is expanded in the region of the red rectangle to show the PEG monomer spacing, and further expanded (A*, n = 5; B* n = 20; and C*, n = 15) to show the monoisotopic peak pattern (top), with comparison to the corresponding simulated peak pattern (below). | ||
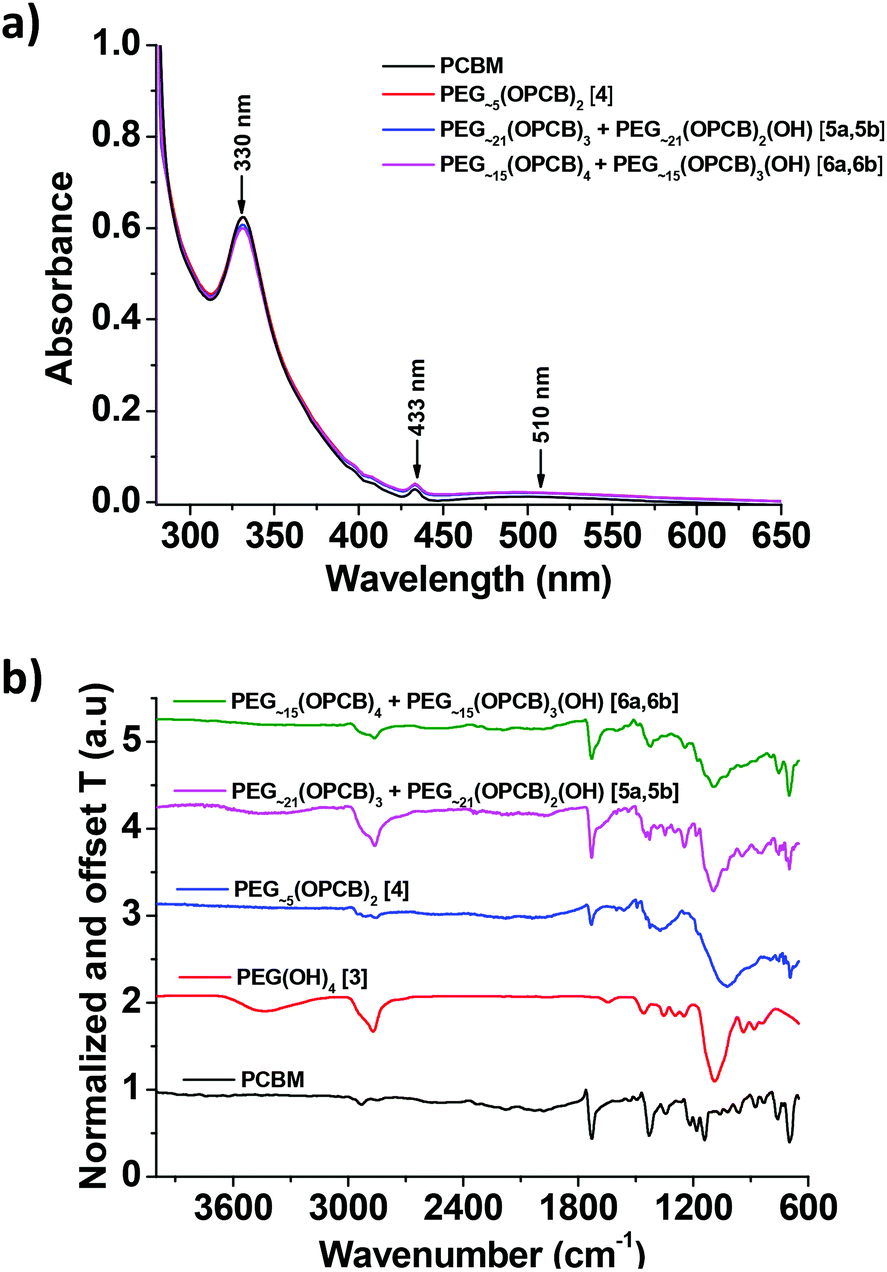 | ||
| Fig. 2 Photospectrometric data for PCB-capped PEG dendrimers: (a) UV-vis spectra of PCBM, PEG∼4(OPCB)2 (4), PEG∼21(OPCB)3 & PEG∼21(OPCB)2(OH) (5a and 5b), and PEG∼15(OPCB)4 & PEG∼15(OPCB)3(OH) (6a and 6b) recorded in toluene. Each concentration was adjusted to around Abs330 nm ∼ 0.6 for ease of comparison; (b) FTIR spectra of PCBM, PEG∼15(OH)4 (3), PEG∼4(OPCB)2 (4), PEG∼21(OPCB)3 (5) and PEG∼15(OPCB)4 (6). | ||
The purifications of all bis-, tris- and tetrakis-fullerene adducts were performed by multiple rounds of column chromatography and preparative TLC. Since PEGs bearing unreacted hydroxyl groups have a stronger affinity for silica gel, any remaining partially unreacted PEG is eluted in the later fractions. The polarity of the molecules decreases with increasing number of covalently grafted non-polar fullerenes. Hence, the most highly substituted adducts are collected in the first fraction to elute using Toluene–MeOH 9:1 v/v. All detailed analyses were carried out with these initially eluted chromatographic fractions.
Tris-fullerene substituted PEG
The MALDI spectrum of 5 (ESI,† Fig. S13) clearly confirms the successful formation of the tris-fullerene polymer (mean peak distribution at ∼3630 m/z). The observed monoisotopic mass of tris-fullerene substituted 3-arm PEG (with 20 repeating units) (Fig. 1B*) appeared to be shifted by 2 m/z to lower mass, most likely due to the limit of spectrometer sensitivity. The overall shape of the monoisotopic mass reassembled that of the calculated value for the tris-fullerene substituted 3-arm PEG. However, two other peak distributions (mean peak distribution at around 2750 and 1740 m/z) were also observed. Since the transesterification of fullerene and PEG hydroxyls is a sequential reaction, mono- and bis-fullerene substituted PEGs may be present along with the desired tris-fullerene adduct. In order to identify the origin of the peaks in the MALDI-TOF spectrum, each peak distribution corresponding to tris- and possible mono- and bis- fullerene PEG distributions were compared with that of the starting PEG (Fig. S14).† Both tris- and bis-substituted species had an average of 20 repeating units (mean peak distribution at around 3630 and 2750 m/z respectively), but the mono-substituted PEG peak had a mean peak distribution at around 1740 m/z, corresponding to an average of 17 repeating units. The expected mono-substituted 3-arm PEG would have an average of 20 repeating units with a mean peak distribution at around 1880 m/z; the difference of 3 repeating units between the observed mono-substituted species and the bis/tris substituted PEG star suggests that the mono-substituted distribution at 1740 m/z may result from fragmentation of the bis/tris PEG stars. An alternative explanation is that the shorter mono-fullerene substituted 3-arm PEG is less polar and eluted preferentially with the more substituted products; however, the change in length would be expected to be less significant than the change in the number of available hydroxyls.The high polydispersity of the fullerene end-capped PEG adducts PEG∼21(OPCB)3 (5a) made chromatographic purification of very difficult; the variations in PEG molecular weight and degree of fullerene substitution smeared the variation in polarity. Hence, further efforts were made to isolate PEG∼21(OPCB)3 (5a) from its mixture by repeated column chromatography, and multiple preparative TLC (up to 3 times). However, the purity did not improve significantly despite these lengthy steps. The signal intensity ratio of the bis- and tris- fullerene distribution in MALDI improved only to about 84:16 from 90:10 after initial work-up. As the MALDI signal intensity may not always reflect molecular abundance, the ratio was also determined from the 1H NMR integral (Fig. S12†). In the case of PEG∼21(OCOCF3)3, the ratio of chain terminal to backbone signals was 6:84 (confirming 21 ethylene oxide units per dendrimer molecule, as noted above). For the fullerene end-capped 3-arm dendrimer 5, the butyryl peaks of the PCB moiety and the acyloxymethylene of the chain termini are also well resolved from the polymeric PEG peak, and the corresponding ratio of fullerene ends to backbone signals was 4.3:84. Assuming that the product is a mixture of only PEG∼21(OPCB)2(OH) (5b) and PEG∼21(OPCB)3 (5a), (i.e. that the mono-fullerene substituted PEG star signal in MALDI-TOF spectrum is due to fragmentation) the best fit for the α-proton integration indicates a 90:10 ratio of PEG∼21(OPCB)2(OH) (5b) to PEG∼21(OPCB)3 (5a), a value comparable to the ratio derived from the MALDI peak intensities (84:16).
Tetrakis-fullerene substituted PEG
The MALDI-TOF spectra of PEG∼15(OPCB)4 (6a) and PEG∼15(OPCB)3(OH) (6b) exhibited similar distribution patterns to the tris-fullerene substituted PEG, but with different masses, having mean peak distributions around 4330 and 3410 m/z respectively (see ESI,† Fig. S16). The observed monoisotopic mass of the tetrakis-fullerene substituted 4-arm PEG (Fig. 1C*) appeared to be shifted by 2 m/z but resembled the shape of the calculated monoisotopic mass (with 15 repeating units). Within the limits of the spectrometer sensitivity, the result confirmed the formation the desired PEG∼15(OPCB)4 (6a). Similar to the synthesis of PEG∼21(OPCB)3 (5a), the incompletely substituted 4-arm PEG, PEG∼15(OPCB)3(OH) (6b) was also observed in the spectrum at around 3410 m/z. An additional mass distribution corresponding to the possible bis-fullerene substituted adduct was also observed at around 2240 m/z. However, detailed comparison (see ESI,† Fig. S17) indicates that only the tris- and tetrakis-fullerene derivatives are likely to be primary signals. The tris- and tetrakis-fullerene distribution both have an average of 15 repeating units, whereas the bis-fullerene distribution was found to have an average of 8 repeating units. MALDI-TOF spectra of the starting 4-arm PEG (Fig. S9†) showed no evidence of any 8 repeating unit fraction within the polymer. Therefore, it is clear that the peak distribution at around 2240 m/z in the tetrakis-fullerene substituted PEG spectra resulted from fragmentation of the higher mass species. Whilst the MALDI-TOF indicated a 50:50 ratio between the tris- and tetrakis-fullerene signal intensities (Fig. S16†), the NMR integral ratios provided a more reliable measure. The integral ratio of α-protons versus the main PEG backbone protons for fully acylated PEG∼15(OCOCF3)4 was 8:60 (see Fig. S5†). The observed ratio of the corresponding tris- and tetrakis-fullerene substituted PEG star in the 1H NMR of mixture of (6a, 6b) was 7.4:60 (see Fig. S15†). Assuming that, as shown in the MALDI-TOF analysis, the NMR integration solely results from the contribution of tris- and tetrakis-substituted PEG∼15 stars, the best fit to the α-proton integration is a 1:1 ratio of PEG∼15(OPCB)3(OH) (6b) to PEG∼15(OPCB)4 (6a). As in the case of PEG∼21(OPCB)3 (5a), extensive purification could not separate the species with differing degrees of fullerene substitution, presumably due to the level of PEG polydispersity.
Higher molecular mass species tend to be harder to ionise in MS and therefore peak intensity is not generally used in determination of natural abundance.32 However, considering the calculation based on the NMR peak integral, the values derived from both MALDI and NMR are very comparable. Although NMR integration is more reliable for determination of actual abundance, it seems that MALDI peak intensity could also be used as a good approximation in the current system.
The UV-vis spectra of PEG∼4(OPCB)2 (4), PEG∼21(OPCB)3 (5) and PEG∼15(OPCB)4 (6) products in toluene have three electronic absorption bands at 330, 433 and 510 nm (Fig. 2a), similar to PCBM, as expected. The retention of the optoelectronic properties should be helpful for PV applications. The starting PEG(OH)n (1–3) do not contribute to the absorbance (see ESI† Fig. S18 for UV-vis spectra of 1–3).23 However, in FTIR (Fig. 2b), the intensity of the –OH stretching bands at around 3400 cm−1 were significantly reduced in all products indicating successful transesterification. The –OH bands did not disappear completely in 3-arm 5 and 4-arm 6 as the substituted PEG still contained a small proportion of free terminal hydroxyl groups. As in previous reports,21,23 no spectral change was observed for the bands at 1732 cm−1 (carbonyl stretching of PEG-OPCB ester), 1180 cm−1 and 1430 cm−1 (C–C & CC stretching in fullerene) and 1095 cm−1 (C–O symmetric stretching of PEG) compared to the spectra of pure PCBM and PEG∼15(OH)4 (3) (Fig. 2b).
The MALDI-TOF results together with NMR, FTIR and UV-vis data confirmed the synthesis of multi-fullerene end-capped PEG dumbbells PEG∼4(OPCB)2 (4) and stars PEG∼21(OPCB)3 (5a) and PEG∼15(OPCB)4 (6a), through the transesterification of PCBM using polydisperse PEGs starting materials. However, the polydispersity, particularly of the tris and tetrakis fullerene products remained problematic. To synthesise monodisperse multi-fullerene end-capped PEG stars, a monodisperse PEG core was required. PEG24(OH)3 (7) was synthesized following a recent procedure;30 NMR and MALDI-TOF (see ESI,† Fig. S19, S20 and S21 for 1H, 13C NMR and MALDI-TOF spectra of PEG24(OH)3 (7)), confirmed the monodisperse nature of the starting material, with a mass of 1247.7 Da. This star-shaped PEG, PEG24(OH)3 (7), was then transesterified with PCBM using the same procedure described above for PEGx(OH)n (1–3). Due to the monodisperse character of PEG24(OH)3 (7), it was possible to separate the resulting monodisperse tris-fullerene end-capped PEG, PEG24(OPCB)3 (8) from the mixture of mono- and bis-fullerene end-capped PEG24(OPCB)x(OH)y employing two rounds of column chromatography, followed by three rounds of preparative thin layer chromatography. Since this same purification protocol had failed to isolate the pure materials from the earlier crude product mixtures, it is clear that the monodispersity of the PEG star is helpful in differentiating the products, although the separation is still lengthy.
Monodisperse PEG24(OPCB)3
The MALDI-TOF spectrum of isolated PEG24(OPCB)3 (8) (Fig. 3) showed the expected monoisotopic mass at 3881.82 Da, close to the calculated value of 3881.91 Da (Fig. 3D*). The mass spectrometric results were further supported by the 1H NMR spectrum which displayed a new triplet signal corresponding to the acyloxymethylene protons (–CH2–O–CO) adjacent to the newly formed ester carbonyl, at 4.25 ppm (see Fig. 4). The starting material PEG24(OH)3 (7) has three arms, each having eight ethylene oxide repeating units (96 protons in total). After transesterification, one would expect six of 96 protons to be converted into acyloxymethylene protons (d), resolving them from the large backbone resonance. The hydroxyl protons of PEG24(OH)3 (7) at 3.15 ppm (see ESI,† Fig. S19) disappeared after transesterification, and the integrals ratios of the acyloxymethylene (labelled as d in Fig. 4) to the PEG backbone (labelled as CH2CH2O in Fig. 4) protons was found to be 5.9:90 (consistent with the expected 6:90 for complete acylation). Both MALDI-TOF and 1H NMR analysis provide strong evidence for the successful formation and isolation of the desired monodispersed tris-fullerene substituted PEG24(OPCB)3 (8).
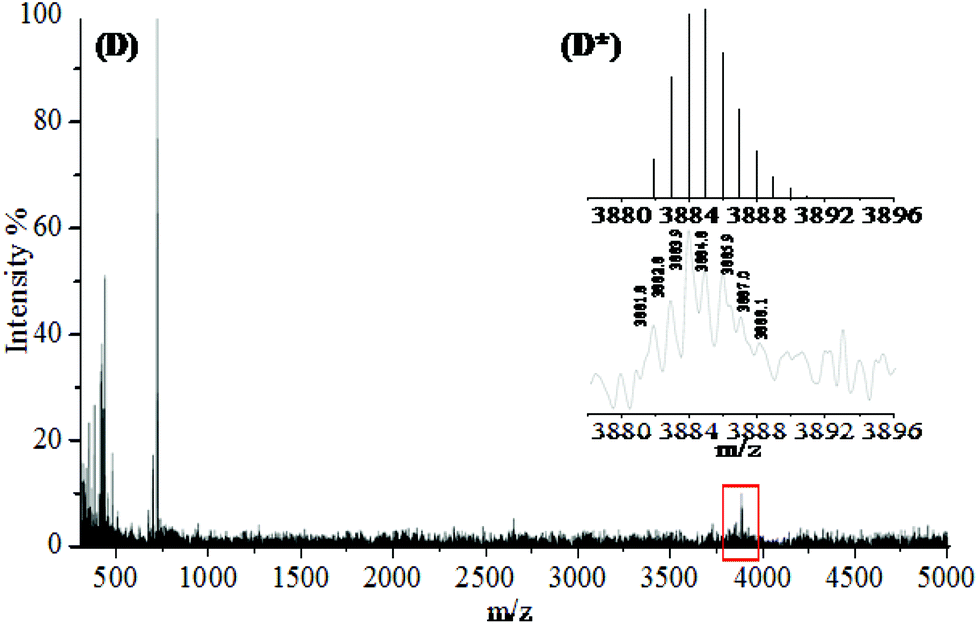 | ||
| Fig. 3 Experimental and theoretical MALDI-TOF spectra confirming successful transesterification of PCBM with PEG24(OH)3 (7). Full MALDI-TOF mass spectrum of PEG24(OPCB)3 (8) (D). Expanded region (represented by rectangle) shows the monoisotopic peak pattern of PEG24(OPCB)3 (8) (bottom D*). Theoretically simulated peak pattern (upper D*) for PEG24(OPCB)3 (8). The signal at 720 m/z was attributed to C60 and below 720 m/z features predominantly related to matrix DCTB. | ||
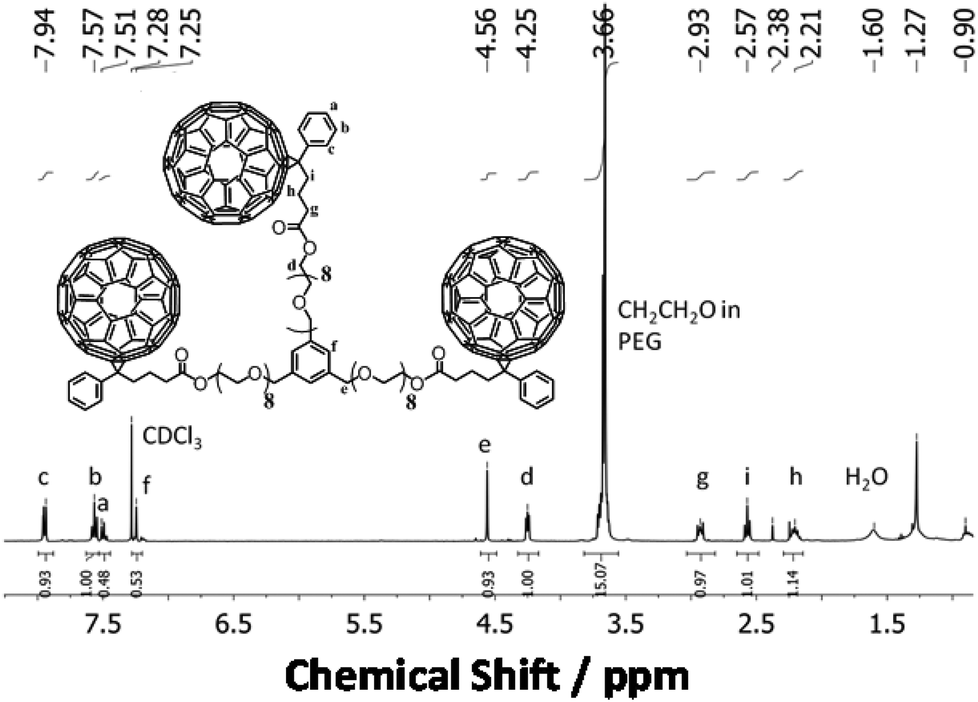 | ||
| Fig. 4 1H NMR spectrum of pure monodisperse PEG24(OPCB)3 (8). Integration of 1 unit = 6 H. Trace amounts of water (1.60 ppm) and vacuum grease (1.27 and 0.90 ppm) were observed. | ||
Due to the difficulty in purification, only the fractions containing the desired species were separated and studied closely. During column chromatography a large coloured fraction remained visible in the column. From the analysis of the primary fractions, the remaining materials in the column are likely to be species with lower degrees of substitution. Similar to the preparation of 3-arm adducts of oligothienylenevinylenes and C60,33 the yield of isolated PEG24(OPCB)3 (8) and all other adducts were low, even though the reaction medium was saturated with PCBM.29 The final yields of fully substituted products were 4% for PEG∼21(OPCB)3 (5), 8% for PEG∼15(OPCB)4 (6) and 5% for PEG24(OPCB)3 (8). In the case of PEG∼15(OPCB)4 (6), the presence of a minor 3-arm PEG impurity in the starting material may have affected the overall yield of the reaction. Due to the small yields, elemental analyses were not performed. An initial NMR integration study (Fig. S1†) of the α-protons (adjacent to newly formed ester) revealed that the reaction proceeded slowly (about 3 days), after which time the catalyst may have become deactivated. Furthermore, although the reaction was carried out at 140 °C, well above the boiling point of methanol, it is possible that the trace amount of methanol released by transesterification may have allowed the reverse reaction to occur to some extent. Finally, although the reaction was kept under nitrogen throughout, the work-up and column chromatography were performed in air. It is well documented that fullerene is a good photo-sensitizer which could easily convert molecular oxygen into singlet oxygen. Fullerene oxidises and/or polymerises in the presence of singlet oxygen, limiting its solubility and that of its adducts.34 Therefore, polymerisation of the initially produced fullerene end-capped PEGs during purification may have significantly limited the final purified yield.
Conclusions
Multiple fullerene end-capped PEGs were synthesized for the first time using single step transesterification of PCBM with PEGs. The monodisperse nature of PEG24(OH)3 (7) allowed isolation of the pure monodisperse fullerene end-capped PEG star PEG24(OPCB)3 (8) from the crude reaction mixture. Monodisperse PEGs with multiple fullerene arms are attractive for BHJ solar cells. It may be possible to tune the phase segregation and/or self-assembly of the fullerene-grafted PEGs by controlling the length of the starting PEG chains and the number of attached fullerene species, whilst retaining the optoelectronic character of PCBM. The synthetic procedure could be applied to the synthesis of other monodisperse multi-arm species with higher mass fullerenes (C70/C82) as experimental data suggest that higher PV efficiency may be achieved in this way.35 Such fullerene adducts are also not necessarily limited to a PEG core. Since the reaction relies on converting the hydroxyl into an ester, any cores with terminal OH groups could be used. The observed partial fullerene substitution of the 3/4-armed products is likely related to the low reaction yields, though steric effects related to short PEG arms may also have played a role. The reaction yield (and likely degree of substitution) might be increased by adjusting the catalyst system, or by using initial fullerene derivatives bearing a more reactive ester (e.g. pentafluorophenyl ester). Future applications of well-defined multiple fullerene stars are likely to emerge with wider availability of well-characterised monodisperse materials. Fullerene dyads have become a popular topic in PV applications. This synthetic procedure for well-defined fullerene derivatives may offer new opportunities for the future study of multiple fullerenes adducts, especially when a conjugated polymer is employed in the synthesis, in place of PEG.Acknowledgements
The authors would like to commemorate the late Dr Joachim H. G. Steinke (1964–2013) for his contribution to this work and to the polymer chemistry field. We acknowledge the EPSRC UK National Mass Spectrometry Service at Swansea and EPSRC research grant EP/G007314/1 and EP/J014974/1. MKB is particularly grateful to the Scientific and Technological Research Council of Turkey (TUBITAK) for post-doctoral research permission.References
- A. Dodabalapur, H. E. Katz, L. Torsi and R. C. Haddon, Science, 1995, 269, 1560–1562 CAS.
- R. C. Haddon, T. Siegrist, R. M. Fleming, P. M. Bridenbaugh and R. A. Laudise, J. Mater. Chem., 1995, 5, 1719–1724 RSC.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- L. S. Roman, M. R. Andersson, T. Yohannes and O. Inganas, Adv. Mater., 1997, 9, 1164–1168 CrossRef CAS.
- L. Garlaschelli, D. Pasini and F. Spiaggia, Eur. J. Org. Chem., 2005, 4322–4327 CrossRef CAS.
- Y. H. Zhu, S. Bahnmueller, C. B. Ching, K. Carpenter, N. S. Hosmane and J. A. Maguire, Tetrahedron Lett., 2003, 44, 5473–5476 CrossRef CAS.
- M. Carano, C. Corvaja, L. Garlaschelli, M. Maggini, M. Marcaccio, F. Paolucci, D. Pasini, P. P. Righetti, E. Sartori and A. Toffoletti, Eur. J. Org. Chem., 2003, 374–384 CrossRef CAS.
- M. Svensson, F. L. Zhang, S. C. Veenstra, W. J. H. Verhees, J. C. Hummelen, J. M. Kroon, O. Inganas and M. R. Andersson, Adv. Mater., 2003, 15, 988–991 CrossRef CAS.
- I. A. Howard, R. Mauer, M. Meister and F. Laquai, J. Am. Chem. Soc., 2010, 132, 14866–14876 CrossRef CAS PubMed.
- C.-H. Hsieh, Y.-J. Cheng, P.-J. Li, C.-H. Chen, M. Dubosc, R.-M. Liang and C.-S. Hsu, J. Am. Chem. Soc., 2010, 132, 4887–4893 CrossRef CAS PubMed.
- G. W. Wang, T. H. Zhang, E. H. Hao, L. J. Jiao, Y. Murata and K. Komatsu, Tetrahedron, 2003, 59, 55–60 CrossRef CAS.
- M. Reyes-Reyes, K. Kim and D. L. Carroll, Appl. Phys. Lett., 2005, 87, 083506 CrossRef PubMed.
- F. Lincker, P. Bourgun, H. Stoeckli-Evans, I. M. Saez, J. W. Goodby and R. Deschenaux, Chem. Commun., 2010, 46, 7522–7524 RSC.
- H. Xin, G. Ren, F. S. Kim and S. A. Jenekhe, Chem. Mater., 2008, 20, 6199–6207 CrossRef CAS.
- S. M. Tuladhar, M. Sims, S. A. Choulis, C. B. Nielsen, W. N. George, J. H. G. Steinke, D. D. C. Bradley and J. Nelson, Org. Electron., 2009, 10, 562–567 CrossRef CAS PubMed.
- C. M. Bjorstrom, K. O. Magnusson and E. Moons, Synth. Met., 2005, 152, 109–112 CrossRef CAS PubMed.
- P. W. M. Blom, V. D. Mihailetchi, L. J. A. Koster and D. E. Markov, Adv. Mater., 2007, 19, 1551–1566 CrossRef CAS.
- M. Campoy-Quiles, T. Ferenczi, T. Agostinelli, P. G. Etchegoin, Y. Kim, T. D. Anthopoulos, P. N. Stavrinou, D. D. C. Bradley and J. Nelson, Nat. Mater., 2008, 7, 158–164 CrossRef CAS PubMed.
- H. Hoppe and N. S. Sariciftci, J. Mater. Res., 2004, 19, 1924–1945 CrossRef CAS.
- C. Müller, T. A. M. Ferenczi, M. Campoy-Quiles, J. M. Frost, D. D. C. Bradley, P. Smith, N. Stingelin-Stutzmann and J. Nelson, Adv. Mater., 2008, 20, 3510–3515 CrossRef.
- J. W. Jung, J. W. Jo and W. H. Jo, Adv. Mater., 2011, 23, 1782–1787 CrossRef CAS PubMed.
- J.-Y. Jeng, M.-W. Lin, Y.-J. Hsu, T.-C. Wen and T.-F. Guo, Adv. Energy Mater., 2011, 1, 1192–1198 CrossRef CAS.
- Q. Tai, J. Li, Z. Liu, Z. Sun, X. Zhao and F. Yan, J. Mater. Chem., 2011, 21, 6848–6853 RSC.
- T. Song, S. H. Goh and S. Y. Lee, Polymer, 2003, 44, 2563–2567 CrossRef CAS.
- D. Taton, S. Angot, Y. Gnanou, E. Wolert, S. Setz and R. Duran, Macromolecules, 1998, 31, 6030–6033 CrossRef CAS.
- H. C. Yau, M. K. Bayazit, J. H. G. Steinke and M. S. P. Shaffer, Macromolecules, 2014, 47, 4870–4875 CrossRef CAS.
- M. Graetzel, R. A. J. Janssen, D. B. Mitzi and E. H. Sargent, Nature, 2012, 488, 304–312 CrossRef CAS PubMed.
- Q. Wei, T. Nishizawa, K. Tajima and K. Hashimoto, Adv. Mater., 2008, 20, 2211–2216 CrossRef CAS.
- J. C. Hummelen, F. B. Kooistra and D. F. Kronholm, USPATENT, US2005245606, 2005 Search PubMed.
- G. Szekely, M. Schaepertoens, P. R. J. Gaffney and A. G. Livingston, Polym. Chem., 2014, 5, 694–697 RSC.
- M. Tryznowski, K. Tomczyk, Z. Fras, J. Gregorowicz, G. Rokicki, E. Wawrzynska and P. G. Parzuchowski, Macromolecules, 2012, 45, 6819–6829 CrossRef CAS.
- G. Montaudo, F. Samperi and M. S. Montaudo, Prog. Polym. Sci., 2006, 31, 277–357 CrossRef CAS PubMed.
- C. Martineau, P. Blanchard, D. Rondeau, J. Delaunay and J. Roncali, Adv. Mater., 2002, 14, 283–287 CrossRef CAS.
- H. Watanabe, E. Matsui, Y. Ishiyama and M. Senna, Tetrahedron Lett., 2007, 48, 8132–8137 CrossRef CAS PubMed.
- X. Meng, W. Zhang, Z. a. Tan, Y. Li, Y. Ma, T. Wang, L. Jiang, C. Shu and C. Wang, Adv. Funct. Mater., 2012, 22, 2187–2193 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra, and MALDI-TOF spectra of the polydisperse multifullerene PEG adducts. See DOI: 10.1039/c4py01167h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |