DOI:
10.1039/C4PY00967C
(Paper)
Polym. Chem., 2015,
6, 143-149
Injectable and cross-linkable polyphosphazene hydrogels for space-filling scaffolds†
Received
15th July 2014
, Accepted 11th September 2014
First published on 11th September 2014
Abstract
Low mechanical strength is a major disadvantage of injectable hydrogels. Although the mechanical properties of hydrogels can be improved by the introduction of robust materials, the use of conventional reinforcing agents limits the medical application of injectable hydrogels due to their non-biodegradable property. We synthesized and self-assembled an injectable hydrogel from biodegradable mixed-substituted polyphosphazene and the natural organic molecule α-cyclodextrin (αCD) by host–guest inclusion. Photo-crosslinking was then employed for the hydrogel to transform it into the solid state with the desired shape. By this approach, the mechanical properties and anti-water solubility of the polyphosphazene hydrogel can meet the requirements of potential applications. In addition, the surface of the cured hydrogel can be readily tuned from cell-philic to cell-phobic by changing the ratio of two side chains of substituted polyphosphazene.
1. Introduction
Developments in medical techniques have led to a large demand for a variety of injectable space-filling scaffolds, as they are easy to manipulate and their use effectively avoids trauma.1–3 In this regard, hydrogels are ideal materials because they are not only injectable but also solidify under specific conditions.4,5 Injectable hydrogels have been regarded as new and emerging biomedical materials,6,7 and prepared by the use of physical and/or chemical cross-linking,8,9 for example, chitosan,10,11 hyaluronic acid,12,13 gelatin,14 peptide hydrogels,15 and polyphosphazene hydrogels.16–18 Polyphosphazene polymers are inorganic–organic hybrid polymers that consist of an inorganic –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P– backbone and two organic units attached to each phosphorus atom. These polymers are easily designable and exhibit various characteristics arising from their substituents which greatly influence their properties.19 Their major advantage in medical applications is their excellent biocompatibility and tunable biodegradability, especially seen for amino acid ester substituted polyphosphazenes.20–27 Furthermore, their degradation products are nontoxic.28–31 Therefore, the use of polyphosphazene hydrogels in biomedical materials has received increasing attention in the last decade.32–34 For example, Lee et al. reported a thermosensitive poly(organophosphazene) with functional groups that forms a hydrogel via phase transition at a temperature above the lower critical solution temperature of the aqueous polymer solution.35,36 Tian et al. employed noncovalent interactions between polyethylene and α-cyclodextrin (αCD), adamantine, or β-cyclodextrin to prepare polyphosphazene hydrogels.37,38 However, the poor anti-water solubility and poor mechanical properties of these hydrogels limit their practical application. Potta et al. attempted to introduce chemical cross-linking groups into a polyphosphazene hydrogel to prevent it from dissolving in water,39–41 but the mechanical performance of the hydrogel still did not allow its use in biomaterial applications. In order to enhance their mechanical strength, nanoparticles,42 carbon fibers,43 graphene,44,45 and montmorillonite46 are usually added as cross-linked sites to hydrogels. Nevertheless, these reinforcing agents are not biodegradable, thus limiting the application of their hydrogels in medical scaffolds. We observed that inclusion of polyethylene glycol (PEG) and αCD into hydrogels could improve the mechanical strength of the hydrogels by increasing the steric hindrance of polymer chains (Scheme 1). This strategy may have potential use in developing space-filling hydrogels. Another problem with hydrogels is their poor resistance to water, resulting in their dissolution in water after a certain period of contact with the solvent. Therefore, cross-linking of injectable hydrogels under specific conditions in real applications must also be taken into account.
P– backbone and two organic units attached to each phosphorus atom. These polymers are easily designable and exhibit various characteristics arising from their substituents which greatly influence their properties.19 Their major advantage in medical applications is their excellent biocompatibility and tunable biodegradability, especially seen for amino acid ester substituted polyphosphazenes.20–27 Furthermore, their degradation products are nontoxic.28–31 Therefore, the use of polyphosphazene hydrogels in biomedical materials has received increasing attention in the last decade.32–34 For example, Lee et al. reported a thermosensitive poly(organophosphazene) with functional groups that forms a hydrogel via phase transition at a temperature above the lower critical solution temperature of the aqueous polymer solution.35,36 Tian et al. employed noncovalent interactions between polyethylene and α-cyclodextrin (αCD), adamantine, or β-cyclodextrin to prepare polyphosphazene hydrogels.37,38 However, the poor anti-water solubility and poor mechanical properties of these hydrogels limit their practical application. Potta et al. attempted to introduce chemical cross-linking groups into a polyphosphazene hydrogel to prevent it from dissolving in water,39–41 but the mechanical performance of the hydrogel still did not allow its use in biomaterial applications. In order to enhance their mechanical strength, nanoparticles,42 carbon fibers,43 graphene,44,45 and montmorillonite46 are usually added as cross-linked sites to hydrogels. Nevertheless, these reinforcing agents are not biodegradable, thus limiting the application of their hydrogels in medical scaffolds. We observed that inclusion of polyethylene glycol (PEG) and αCD into hydrogels could improve the mechanical strength of the hydrogels by increasing the steric hindrance of polymer chains (Scheme 1). This strategy may have potential use in developing space-filling hydrogels. Another problem with hydrogels is their poor resistance to water, resulting in their dissolution in water after a certain period of contact with the solvent. Therefore, cross-linking of injectable hydrogels under specific conditions in real applications must also be taken into account.
 |
| | Scheme 1 The mechanism of host–guest inclusion and its rigid enhancing process. | |
Herein, we synthesized a series of polyphosphazenes with two kinds of mixed substituents, the glycine ethyl ester group (GlyEE) and the monoacrylic-terminated PEG group (PEGac). The former was used to impart good cell affinity and biocompatibility, and the latter was used to enable self-assembly in the presence of αCD and to impart anti-biological adhesion. The pendant polyphosphazene could self-assemble in the presence of αCD in water to form an injectable supermolecular hydrogel cream. After irradiation with ultraviolet (UV) light, the cream could form a cross-linked polyphosphazene hydrogel with good mechanical strength and high stability in water. The effects of the ratio of GlyEE to PEGac on the mechanical strength of the hydrogel and on cell adhesion were investigated.
2. Experimental
2.1. Materials
Hexachlorocyclotriphosphazene (HCCP) was purchased from Adamas-beta (Shanghai, China) and then purified twice by heating it at 50 °C under vacuum (about −0.1 MPa). Glycine ethyl ester hydrochloride, αCD, NaH (60% in paraffin), and PEG with an average molecular weight (Mn) of 600–1000 g mol−1 were obtained from Adamas-beta, acrylic anhydride was purchased from Xiya Reagent, and triethylamine (TEA) was purchased from Sigma-Aldrich. These were used without further purification. Tetrahydrofuran (THF; Shanghai Chemical Reagent Corporation) was dried by refluxing over sodium metal and then distilled under an argon atmosphere. All glassware were dried overnight under vacuum at 110 °C before use. Bovine serum albumin (66 kDa, >98% purity), fetal bovine serum (FBS), and Dulbecco's modified Eagle medium (DMEM) were purchased from Gibco BRL. HeLa cells were obtained from the Cell Resource Center of Shanghai Biological Sciences Institutes. Water used in the experiments was purified to a resistivity higher than 18.2 MΩ cm using a Hitech system.
2.2. Equipment
1H NMR and 31P NMR spectra were recorded on a Varian Mercury-400 spectrometer, 1H NMR spectra were referenced to solvent signals, and 31P NMR spectra were referenced to signals obtained with 85% phosphoric acid (external reference). Fourier transform infrared spectra (FTIR) were scanned on a Paragon 1000 (Perkin-Elmer) spectrometer. The molecular weight and polydispersity were estimated using a Perkin-Elmer Series 200 gel permeation chromatograph (GPC) at 30 °C equipped with two linear mixed-B columns (Polymer Lab Corporation; pore size: 10 μm, column size: 300 × 7.5 mm) and a refractive index detector. DMF (0.01 mol L−1 LiBr) and polystyrene were used as the eluent (elution rate: 1.0 mL min−1) and the calibration standard, respectively. Wide-angle X-ray diffraction (WAXD) patterns of powdered samples were obtained on a Shimadzu XRD-6000 X-ray diffractometer with a Cu Kα radiation source (1.54 Å wavelength), and the supplied voltage and current were set to 40 kV and 30 mA, respectively. Diffraction patterns were collected from 0.5° to 40° at a speed of 5° min−1. Transmission electron microscopy (TEM) images were taken using a JEM-2010/INCA Oxford TEM (JEOL/Oxford) operated at 200 kV accelerating voltage. Samples were prepared on the surface of 300-mesh Formvar–carbon film-coated copper grids. Visual images were captured using a Canon IXUS 800IS digital camera (Canon, Japan). Rheology behaviors of the hydrogel samples at room temperature were determined with a TA-ARG2 rheometer using a 40 mm parallel-plate geometry. Compressive stress–strain tests were conducted on water-swollen gels using a tensile-compressive tester (Zwick/Roell Z020). A cylindrical gel sample with 15 mm diameter and 10 mm thickness was set on the lower plate and then compressed with the upper plate at a strain rate of 10% min−1. The strain under compression is defined as the change in the thickness relative to the freestanding thickness of the sample.
2.3. Synthesis of the crosslinkable PEG precursor PEG600ac
Poly(ethylene glycol) 600 (PEG600; 6.0 g, 10 mmol) was dissolved in 300 mL of anhydrous THF under an argon atmosphere. Subsequently, 1.514 g of acrylic anhydride (12.0 mmol) was added dropwise to this solution at 0 °C. The resulting mixture was stirred for 2 h and then heated at 55 °C overnight. The reaction mixture was precipitated in diethyl ether, filtered, washed with a large amount of diethyl ether, and then dried under vacuum to obtain PEG600ac. Yield: 73%. 1H NMR (D2O): 6.45 (d, 1H, –CHCH2), 6.24 (t, 1H, –CHCH2), 6.03 (s, 1H, –CHCH2), 4.36 (d, 2H, –OCH2CH2–), 3.88–3.53 (br, 50H, PEG600ac, –OCH2CH2–) (Fig. S1†).
2.4. Synthesis of the crosslinkable PEG precursor PEG1000ac
Preparation of monoacrylic polyethylene glycol 1000 was similar to that of PEG600ac. We utilized 10.0 g (10 mmol) of polyethylene glycol 1000 (PEG1000) and 1.640 g of acrylic anhydride (13.0 mmol). Yield: 81%. 1H NMR (D2O): 6.45 (d, 1H, –CHCH2), 6.24 (t, 1H, –CHCH2), 6.03 (s, 1H, –CHCH2), 4.36 (d, 2H, –OCH2CH2–), 3.88–3.53 (br, 86H, PEG1000ac, –OCH2CH2–) (Fig. S2†).
2.5. Synthesis of the guest polyphosphazene polymer
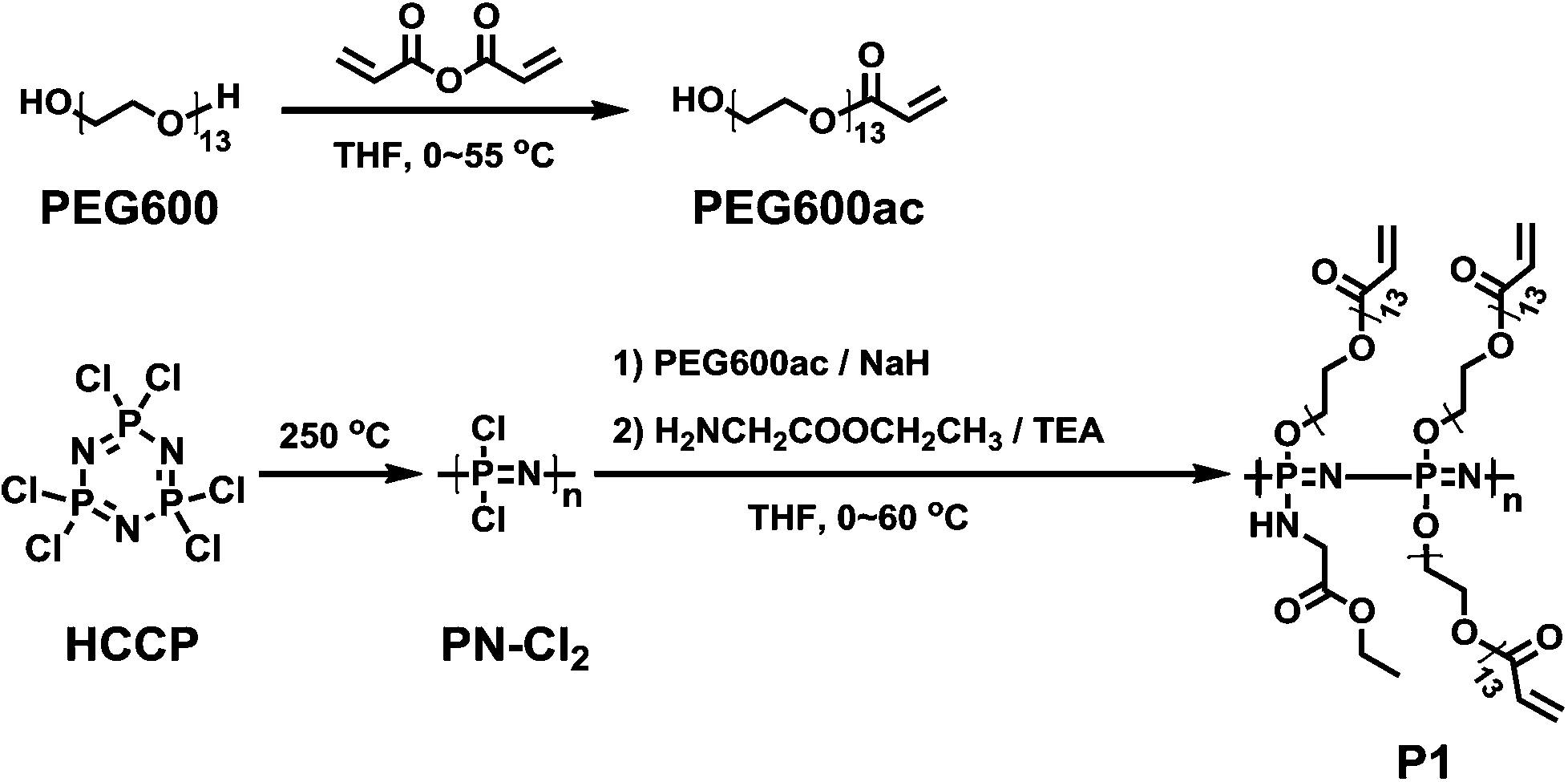
The synthetic route of the cross-linkable polyphosphazene polymer is shown in Scheme 2. Poly(dichlorophosphazene) (PN-Cl2) was firstly prepared by thermal ring-opening polymerization of HCCP at 250 °C in evacuated Pyrex tubes.47
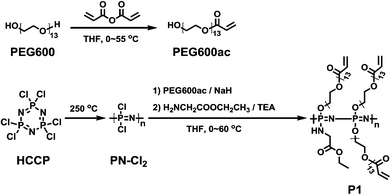 |
| | Scheme 2 Synthesis route of crosslinkable guest polyphosphazene. | |
2.5.1. PN-PEG600ac-70%gly (P1).
PEG600ac (3.38 g, 5.17 mmol) and NaH (0.207 g, 5.17 mmol) were mixed in anhydrous THF (60 mL) at 0 °C to form a clear yellowish suspension, which was afterwards added dropwise to poly(dichlorophosphazene) (1 g, 8.62 mmol) dissolved in anhydrous THF (50 mL) at 0 °C for 1 hour and then kept at 40 °C for 1 day to give a partially substituted polymer. Meanwhile, in another separate flask, glycine ethyl ester hydrochloride (3.36 g, 24.0 mmol) was added to anhydrous THF (20 mL) containing triethylamine (4.85 g, 48.0 mmol), and the suspension obtained was stirred at 60 °C for 1 day and then filtered. The obtained aqueous solution was subsequently added to the polymer solution dropwise at 0 °C for 1 hour and then at 60 °C overnight to complete the substitution reaction. Finally, the by-product was removed by centrifugation, and the solution was concentrated and precipitated twice in n-hexane. The crude product was further dialyzed versus methanol, methanol–water (90/10), and again methanol, each for 1 day at room temperature. The final product was dried under vacuum to obtain a gel-like polymer. Yield: 63%. 31P NMR (CDCl3): δ 1.88 (br), δ −2.81 (br), δ −5.35 (br). 1H NMR (CDCl3): δ 6.23 (d, 1H, PEG600ac, –CHCH2), δ 5.97 (s, 1H, PEG600ac, –CHCH2), δ 5.71 (s, 1H, PEG600ac, –CHCH2), δ 4.44 (br, 2H, PEG600ac, –CH2COO–), 4.20 (s, 2H, GlyEE, –OCH2CH3), δ 4.03 (d, 2H, –OCH2CH2–), δ 3.81 (s, 2H, GlyEE, –CH2COO–), δ 3.64–3.49 (br, 44–52H, PEG600ac, –OCH2CH2–), δ 1.97 (br, H, GlyEE, –NH–), δ 1.23 (s, 3H, GlyEE, –CH2CH3).
2.5.2. PN-PEG600ac-30%gly (P2).
PEG600ac (3.96 g, 6.05 mmol), NaH (0.242 g, 6.05 mmol), poly(dichlorophosphazene) (0.5 g, 4.31 mmol), glycine ethyl ester hydrochloride (0.72 g, 5.15 mmol), containing triethylamine (1.04 g, 10.3 mmol). 31P NMR (CDCl3): δ 1.88 (br), δ −2.81 (br), δ −5.35 (br). 1H NMR (CDCl3): δ 6.22 (s, 1H, PEG600ac, –CHCH2), δ 5.97 (s, 1H, PEG600ac, –CHCH2), δ 5.68 (s, 1H, PEG600ac, –CHCH2), δ 4.44 (br, 2H, PEG600ac, –CH2COO–), 4.20 (s, 2H, GlyEE, –OCH2CH3), δ 4.03 (d, 2H, –OCH2CH2–), δ 3.81 (s, 2H, GlyEE, –CH2COO–), δ 3.64–3.49 (br, 44–52H, PEG600ac, –OCH2CH2–), δ 1.97 (br, H, GlyEE, –NH–), δ 1.23 (s, 3H, GlyEE, –CH2CH3).
2.5.3. PN-PEG1000ac-30%gly (P3).
PEG1000ac (6.38 g, 6.05 mmol), NaH (0.242 g, 6.05 mmol), poly(dichlorophosphazene) (0.5 g, 4.31 mmol), glycine ethyl ester hydrochloride (0.72 g, 5.15 mmol), containing triethylamine (1.04 g, 10.3 mmol). 31P NMR (CDCl3): δ 1.88 (br), δ −2.81 (br), δ −5.35 (br). 1H NMR (CDCl3): δ 6.22 (s, 1H, PEG1000ac, –CHCH2), δ 5.97 (s, 1H, PEG1000ac, –CHCH2), δ 5.68 (s, 1H, PEG1000ac, –CHCH2), δ 4.44 (br, 2H, PEG1000ac, –CH2COO–), 4.20 (s, 2H, GlyEE, –OCH2CH3), δ 4.03 (d, 2H, –OCH2CH2–), δ 3.81 (s, 2H, GlyEE, –CH2COO–), δ 3.64–3.49 (br, 80–88H, PEG1000ac, –OCH2CH2–), δ 1.97 (br, H, GlyEE, –NH–), δ 1.23 (s, 3H, GlyEE, –CH2CH3).
2.6. Supermolecular inclusion hydrogel
A representative procedure for preparing the supermolecular inclusion hydrogel is described as follows. Two kinds of precursors, P2 (9 wt%) and αCD (9 wt%) water solutions, were mixed at room temperature. The clear mixture turned into a white and opaque gel within several minutes under magnetic stirring.
2.7. Rheological measurements
A TA-ARG2 rheometer was used to investigate the gelation kinetics, and time sweep tests were performed at 5% strain at a constant oscillatory frequency of 1 rad s−1. The samples were loaded on parallel plates with 25 mm diameter. The gap distance between the two plates was fixed at 0.3 mm.
2.8. Anti-dissolving ability of the hydrogel in water
The anti-dissolving ability of the hydrogel cream before and after UV cross-linking was compared using a 10 mm Petri dish filled with water. The dissolution process was recorded using a digital camera.
2.9. Load-bearing test
Samples tested were the cross-linked P3 (marked as P3U), P3 included in αCD (P3C), UV irradiated P3 included in αCD (P3CU), and P1C, P1U, P1CU and P2C, P2U and P2CU (the assembly systems of P1 and P2 were marked according to the same principle as P3). The first three were analyzed to determine the contribution of inclusion and cross-linking to the mechanical strength, and the latter two were analyzed to investigate the influence of PEG length on hydrogel strength.
2.10. Cell adhesion study
The hydrogel samples P1CU, P2CU, and P3CU, cut into 8 mm × 8 mm squares, were selected for cell adhesion study. The samples were sterilized and then added to a 24-well plate seeded with HeLa cells with a density of 1 × 105 cells per well. The cells were then cultured for 24 h in a medium consisting of DMEM, 10% FBS, and 1% penicillin–streptomycin solution at 37 °C and 5% CO2. All samples were visualized under a Nikon-C1 laser scanning confocal microscope.
3. Results and discussion
3.1. Characterization of mixed-substituent polyphosphazene
In order to avoid chain entanglement and ensure smooth progress of the following substitution reaction of GlyEE, we used PEG600 and PEG1000, which have relatively low molecular weights. This low-molecular-weight PEG also facilitates elimination by kidney in vivo after degradation of the polyphosphazene hydrogel, thus enabling practical clinical applications of the hydrogel.
Herein, polyphosphazenes with various molar ratios of monoacrylic PEG groups to GlyEE groups were prepared. The 31P NMR spectrum of P1 (Fig. S3†) shows signals at 1.88, −2.81, and −5.35 ppm, which are due to vibrations of [N–P–N], [N–P–O], and [O–P–O], respectively. The signal at −17.05 ppm corresponding to [Cl–P–Cl] disappeared, indicating that complete substitution had taken place. The amounts of PEG600ac and GlyEE were calculated according to the integral ratio of the peaks at 6.22 and 1.23 ppm in the 1H NMR spectrum (Fig. S4†). The percentage of PEG600ac obtained by calculations was very close to the feed ratio (Table 1), proving the synthesis of the mixed-substituent polyphosphazene.
Table 1 Number-average molecular weight (Mn), and weight-average molecular weights (Mw) and molecular weight distribution (Mw/Mn) of mixed-substituent polyphosphazene and the proportions of each substitute in the corresponding polymers
| Polymer |
PEGac |
GlyEE |
M
n
|
M
w
|
M
w/Mn |
| P1 (PEG600) |
29.7% |
70.3% |
14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 907 907 |
46763 |
3.137 |
| P2 (PEG600) |
68.9% |
31.1% |
35196 |
82640 |
2.348 |
| P3 (PEG1000) |
71.4% |
28.6% |
46814 |
122465 |
2.616 |
3.2. Preparation of the supermolecular inclusion hydrogel cream
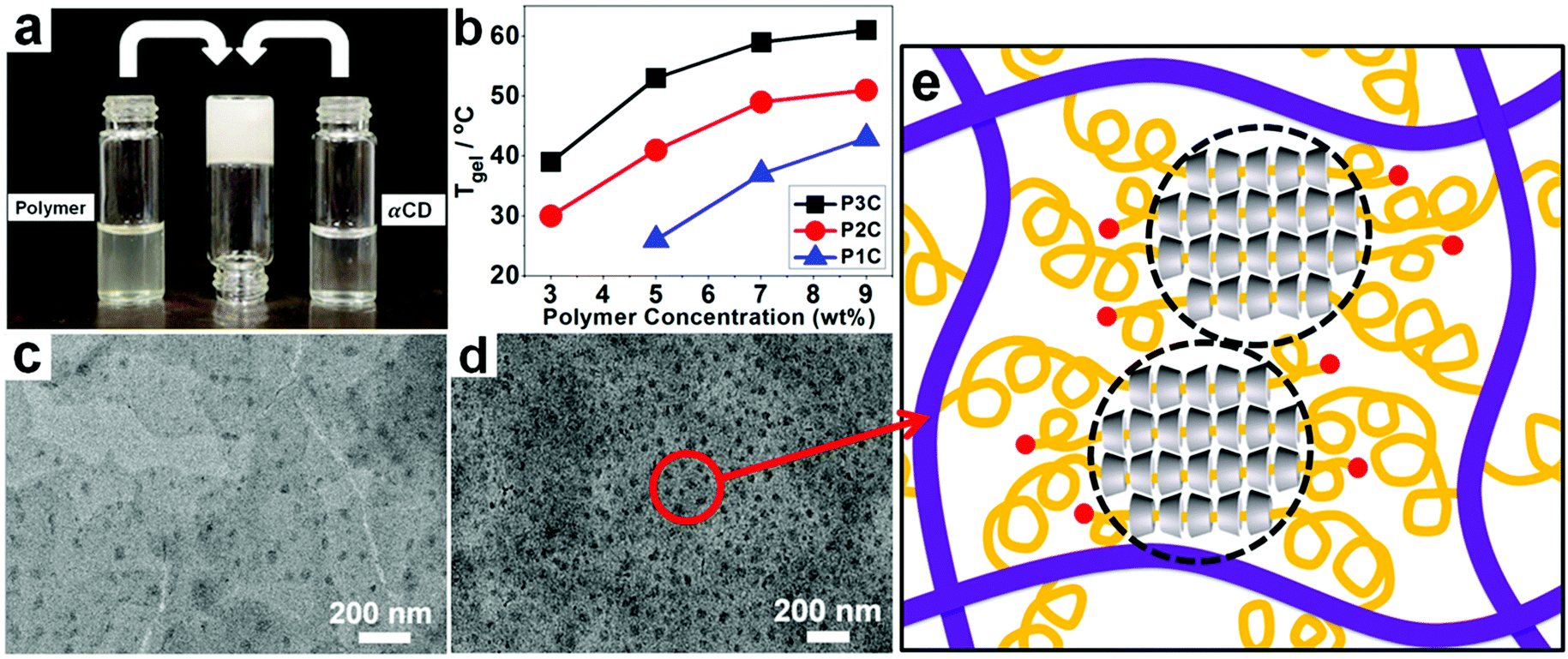
The supermolecular inclusion hydrogel creams were prepared by mixing an aqueous solution of the polymer and αCD (eq. concentration). The gelation time was dependent on the concentrations of the two precursors; that is, higher concentrations resulted in shorter gelation time. For example, when aqueous solutions of P2 (9 wt%) and αCD (9 wt%) were mixed at 25 °C, an opaque white gel was formed within 30 s, whereas 1.5 min was needed for gelation with 5 wt% P2 and αCD (Fig. 1a). This difference is due to the more opportunities to form inclusions at higher concentrations of polymer and αCD. In addition, Tgel was closely related to the fraction and length of side-chain PEGac groups of polyphosphazene. As shown in Fig. 1b, increasing the molecular weight and the amount of PEG segments resulted in higher Tgel, as more self-assembly sites formed. It has been proved that αCDs can form nanocrystallites with PEG.48–52 TEM observations revealed the presence of nanocrystallite domains and the formation of nanocrystallites at higher concentrations of polyphosphazene and αCD (Fig. 1c and d). Abundant hydrogen bonds outside the threaded αCDs provide the physical cohesion force that induces αCD crystallization.
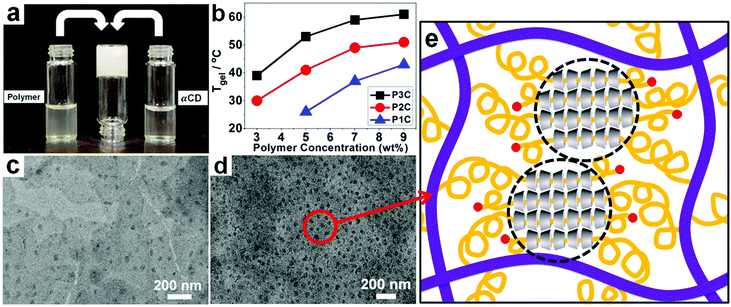 |
| | Fig. 1 Supermolecular hydrogel consisting of P3C inclusions. (a) Formation of the hydrogel; (b) the relationship between gelation temperature (Tgel) and polymer concentration; (c) & (d) TEM images of P2C with 3 wt% and 9 wt% P2, respectively. (e) Illustration of the self-assembly structure. | |
A plausible structure of the assembly between αCD and the polyphosphazene with PEGac groups is illustrated in Fig. 1e.
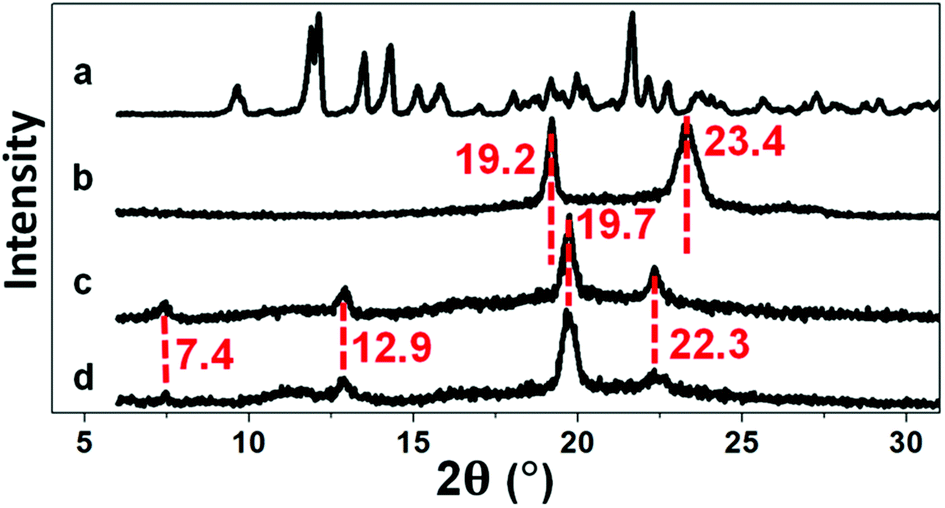
XRD is an effective tool for determining the inclusion structure of the host–guest inclusion hydrogels. Herein, XRD was employed to confirm whether the inclusion structure remained in the hydrogel after irradiation. Diffraction peaks at 7.4°, 12.9°, 19.7°, and 22.3° (Fig. 2) imply the formation of the supermolecular inclusion structure.53–63 After curing of the hydrogel under UV light, the intensity of the diffraction peaks remained the same, indicating retention of the inclusion structure.
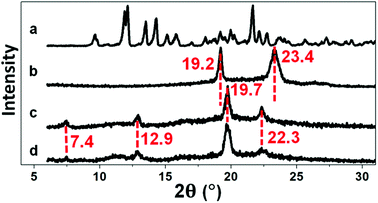 |
| | Fig. 2 XRD spectra of (a) αCD, (b) P2, (c) P2C, and (d) P2CU. | |
3.3. Light curing of the supermolecular inclusion hydrogel and its anti-water solubility
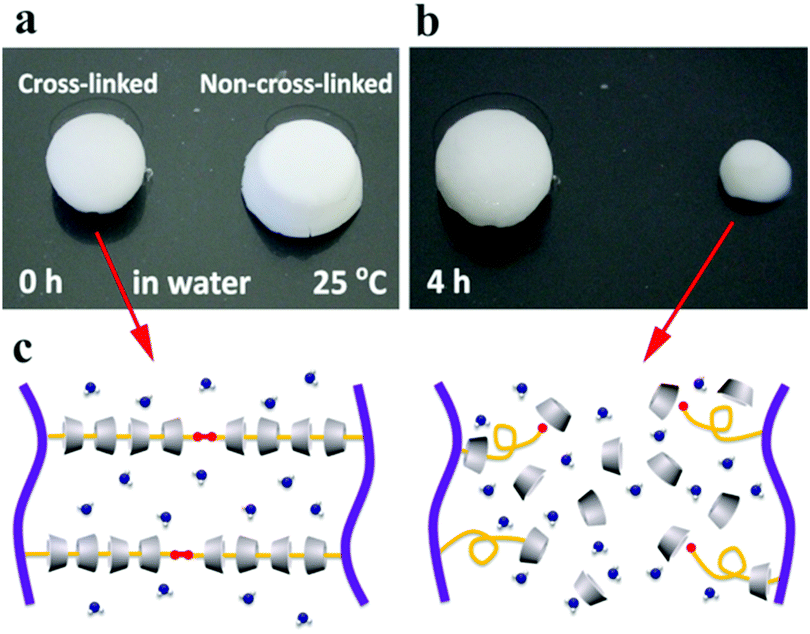
The anti-water solubility of the supermolecular inclusion hydrogel cream before and after UV irradiation is compared in Fig. 3. The cross-linked hydrogel showed high anti-water-solubility, whereas that without cross-linking partially dissolved in water within 4 h. Light curing induces cross-linking of acryloyl groups at the terminal of PEGac, producing abundant chemical cross-linking bonds and preventing the dethreading of αCD from the PEG segment.
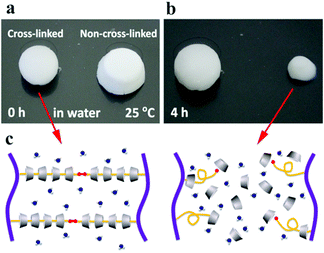 |
| | Fig. 3 Anti-water solubility test of the P2C hydrogel cream before (a) and after (b) UV irradiation and the proposed mechanisms of dissolution (c). | |
3.4. Mechanical performance of the cross-linked supermolecular hydrogels
3.4.1. Rheological study.
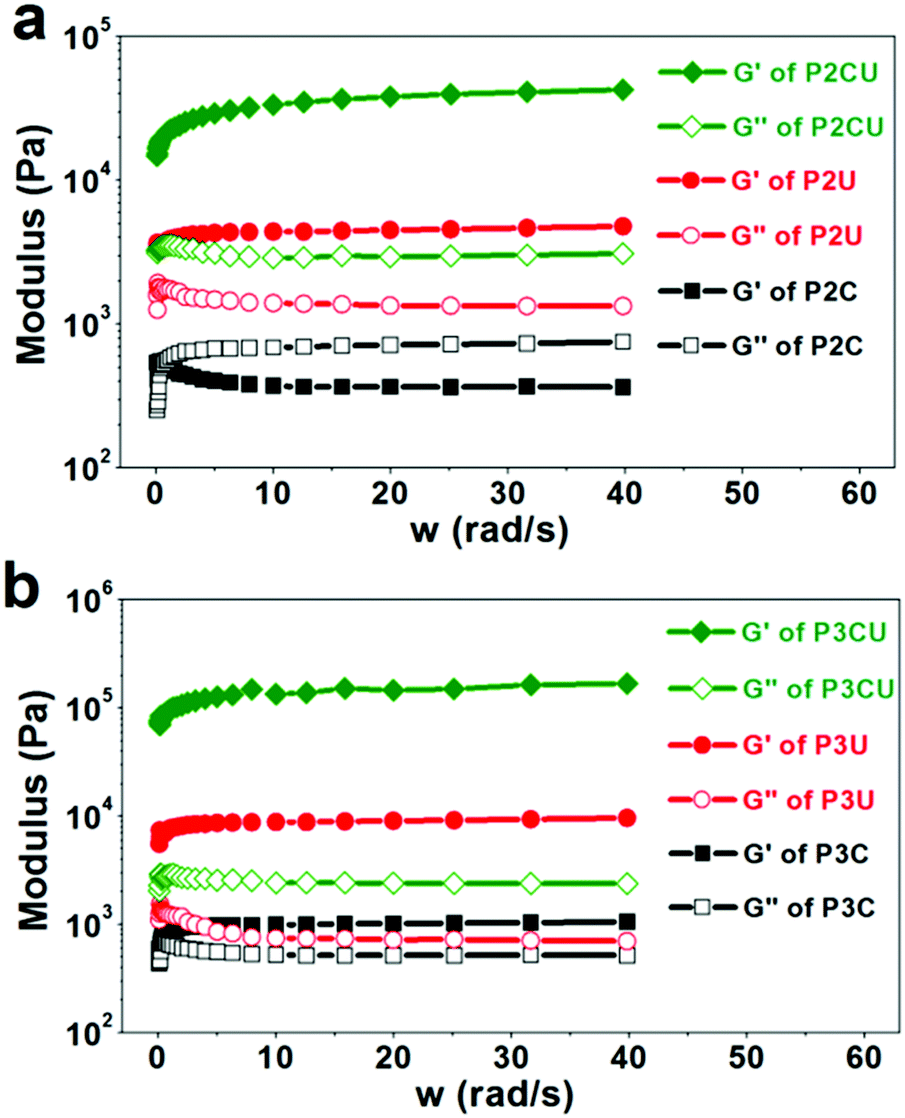
By definition, cream cannot return to its initial shape upon deformation. According to Fig. 4a, the sample P2C was a typical cream, as its storage modulus (220 Pa) was lower than its loss modulus (∼1100 Pa). UV curing could induce the cream to turn into a hydrogel (P2CU) whose storage modulus is higher than its loss modulus. In addition, the storage modulus of P2CU (threaded with αCD) was approximately eight times higher than that of P2U without αCD, proving that the host–guest inclusion approach effectively enhanced the mechanical strength of the hydrogels. When a longer PEG segment was used, the rheological performance was enhanced (Fig. 4b). For instance, the storage modulus of P3CU reached 130 kPa. This enhancement is due to the greater number of rigid inclusion structures in the hydrogel cream, which increases the mechanical strength. Even the storage modulus of the non-cross-linked P3C was higher than its loss modulus. However, its storage modulus is still too low (<1000 kPa); it was a white viscous liquid that could easily be extruded from a narrow needle (diameter: d = 0.9 mm). This property, however, allows its application as an injectable hydrogel.
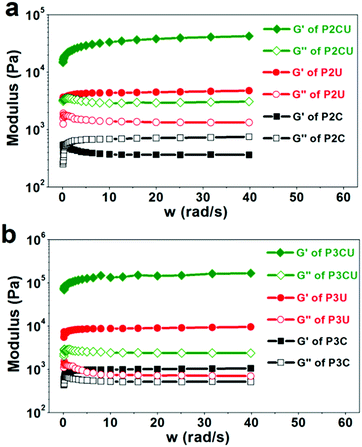 |
| | Fig. 4 Rheology curves of the supermolecular inclusion hydrogel cream with (a) P2 and (b) P3. | |
3.4.2. Load-bearing study.
A space-filling hydrogel must not only be anti-water-soluble and injectable, but also have good load-bearing properties. The rigid structure formed via the host–guest inclusion process improves the mechanical strength of the hydrogel from 178 kPa (P3U) to 1036 kPa (P3CU), as shown in Fig. 5. In addition, P3CU could bear more weight than P2CU (566 kPa), consistent with the result of the rheological study.
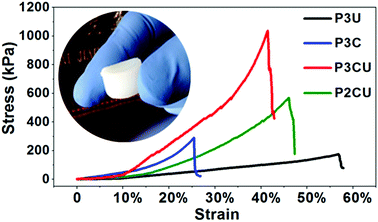 |
| | Fig. 5 Load-bearing test of the supermolecular inclusion hydrogel cream P3C and of the hydrogels P3U, P3CU, and P2CU. | |
3.5. Cell adhesion study
In general, a space-filling hydrogel requires a specific cell adhesion property. It may have a cell-philic surface or a cell-phobic (antifouling) surface, depending on the surgical purpose.64 Herein we tried to tune the cell adhesion properties of the hydrogel surface by adjusting the proportion of GlyEE to PEGac on the polyphosphazene backbone. Fig. 6 shows the cell adhesion behavior on the cross-linked polyphosphazene hydrogels. P1CU provided a cell-philic surface as it contained a high proportion of GlyEE pendants (Fig. 6a), whereas P3CU, which had abundant PEG pendants, exhibited cell-phobic properties (Fig. 6c). The hydrophilic PEG segments and the outer layer of the threaded αCD formed a hydration layer on the surface of P3CU, thereby preventing cells from attaching to the surface (Fig. 6d). We could also shorten the PEG segment to obtain a surface for moderate cell adhesion (Fig. 6b). Evidently, the cell adhesion properties of the polyphosphazene hydrogel could be tuned by changing the ratio of PEG to GlyEE. Both cell-philic and cell-phobic (antifouling) surfaces are important to the surface design of biological materials.
 |
| | Fig. 6 Cellular adhesion properties of (a) P1CU, (b) P2CU, and (c) P3CU. (d) An illustration of the cell-phobic mechanism. | |
4. Conclusions
A series of polyphosphazenes with various ratios of GlyEE and PEGac were prepared. Host–guest inclusion using αCD and photo-cross-linking could markedly enhance the mechanical strength (up to seven times) and anti-water-solubility of their hydrogels. Through this approach, we prepared scaffold material from polyphosphazene hydrogels. In particular, the high-strength solid, P3CU, which was obtained by curing the injectable hydrogel cream P3C under UV light, was found to satisfy the requirements for medical space-filling scaffolds. Furthermore, the surface of the hydrogel could be converted from cell-philic to cell-phobic by tuning the ratio of GlyEE to PEGac. Our synthetic procedure provides a new approach for the surface design of biomedical scaffold materials.
Acknowledgements
This work was supported by the Major Project of Chinese National Programs for Fundamental Research and Development (973 Project: 2009CB930400 and 2012CB933803), the National Science Foundation for Distinguished Young Scholars (50925310), and the National Science Foundation of China (20874059 and 21174087).
Notes and references
- J. D. Kretlow, S. Young, L. Klouda, M. Wong and A. G. Mikos, Adv. Mater., 2009, 21, 3368–3393 CrossRef CAS PubMed.
- J. D. Kretlow, L. Klouda and A. G. Mikos, Adv. Drug Delivery Rev., 2007, 59, 263–273 CrossRef CAS PubMed.
- A. T. Hillel, S. Unterman, Z. Nahas, B. Reid, J. M. Coburn, J. Axelman, J. J. Chae, Q. Guo, R. Trow, A. Thomas, Z. Hou, S. Lichtsteiner, D. Sutton, C. Matheson, P. Walker, N. David, S. Mori, J. M. Taube and J. H. Elisseeff, Sci. Transl. Med., 2011, 3, 93ra67 CAS.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS PubMed.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS.
-
J. Huang, J. Hao, D. P. Anderson and P. R. Chang, in Advanced Healthcare Materials, John Wiley & Sons, Inc., 2014, pp. 405–438 Search PubMed.
-
L. S. Nair, C. T. Laurencin and M. Tandon, in Advanced Biomaterials, John Wiley & Sons, Inc., 2010, pp. 179–203 Search PubMed.
- D. J. Overstreet, D. Dutta, S. E. Stabenfeldt and B. L. Vernon, J. Biomed. Mater. Res., Part B, 2012, 50, 881–903 CAS.
- Y. Li, J. Rodrigues and H. Tomas, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- L. S. Nair, T. Starnes, J.-W. K. Ko and C. T. Laurencin, Biomacromolecules, 2007, 8, 3779–3785 CrossRef CAS PubMed.
- C. Chen, L. Wang, L. Deng, R. Hu and A. Dong, J. Biomed. Mater. Res., Part A, 2013, 101A, 684–693 CrossRef CAS PubMed.
- Y. Luo, J. B. Kobler, J. T. Heaton, X. Jia, S. M. Zeitels and R. Langer, J. Biomed. Mater. Res., Part B, 2010, 93B, 386–393 CrossRef CAS PubMed.
- F. Yu, X. Cao, Y. Li, L. Zeng, B. Yuan and X. Chen, Polym. Chem., 2014, 5, 1082–1090 RSC.
- C. Liu, Z. Zhang, X. Liu, X. Ni and J. Li, RSC Adv., 2013, 3, 25041–25049 RSC.
- A. Dasgupta, J. H. Mondal and D. Das, RSC Adv., 2013, 3, 9117–9149 RSC.
- H. R. Allcock, Soft Matter, 2012, 8, 7521–7532 RSC.
- J.-K. Cho, J. W. Park and S.-C. Song, J. Pharm. Sci., 2012, 101, 2382–2391 CrossRef CAS PubMed.
- T. Potta, C. Chun and S.-C. Song, Macromol. Biosci., 2011, 11, 689–699 CrossRef CAS PubMed.
- H. R. Allcock and N. L. Morozowich, Polym. Chem., 2012, 3, 578–590 RSC.
- M. Heyde, M. Claeyssens and E. H. Schacht, Biomacromolecules, 2008, 9, 672–677 CrossRef CAS PubMed.
- Y. Yang, Z. Zhang, L. Chen, W. Gu and Y. Li, Biomacromolecules, 2010, 11, 927–933 CrossRef CAS PubMed.
- A. b. Cuetos, M. a. L. Valenzuela, I. n. Lavandera, V. Gotor and G. A. Carriedo, Biomacromolecules, 2010, 11, 1291–1297 CrossRef CAS PubMed.
- M. Deng, L. S. Nair, S. P. Nukavarapu, S. G. Kumbar, J. L. Brown, N. R. Krogman, A. L. Weikel, H. R. Allcock and C. T. Laurencin, J. Biomed. Mater. Res., Part A, 2010, 92A, 114–125 CrossRef CAS PubMed.
- M. Deng, S. G. Kumbar, L. S. Nair, A. L. Weikel, H. R. Allcock and C. T. Laurencin, Adv. Funct. Mater., 2011, 21, 2641–2651 CrossRef CAS.
- M. Deng, L. S. Nair, S. P. Nukavarapu, T. Jiang, W. A. Kanner, X. Li, S. G. Kumbar, A. L. Weikel, N. R. Krogman, H. R. Allcock and C. T. Laurencin, Biomaterials, 2010, 31, 4898–4908 CrossRef CAS PubMed.
- A. Marin, D. P. DeCollibus and A. K. Andrianov, Biomacromolecules, 2010, 11, 2268–2273 CrossRef CAS PubMed.
- S. P. Nukavarapu, S. G. Kumbar, J. L. Brown, N. R. Krogman, A. L. Weikel, M. D. Hindenlang, L. S. Nair, H. R. Allcock and C. T. Laurencin, Biomacromolecules, 2008, 9, 1818–1825 CrossRef CAS PubMed.
- A. K. Andrianov, A. Marin and B. E. Roberts, Biomacromolecules, 2005, 6, 1375–1379 CrossRef CAS PubMed.
- A. K. Andrianov, A. Marin and J. Chen, Biomacromolecules, 2005, 7, 394–399 CrossRef PubMed.
- A. K. Andrianov and A. Marin, Biomacromolecules, 2006, 7, 1581–1586 CrossRef CAS PubMed.
- D. P. DeCollibus, A. Marin and A. K. Andrianov, Biomacromolecules, 2010, 11, 2033–2038 CrossRef CAS PubMed.
-
A. K. Andrianov and R. Langer, in Polyphosphazenes for Biomedical Applications, John Wiley & Sons, Inc., 2008, pp. 1–13 Search PubMed.
- S. Doppalapudi, A. Jain, W. Khan and A. J. Domb, Polym. Adv. Technol., 2014, 25, 427–435 CrossRef CAS.
- M. Deng, S. G. Kumbar, Y. Wan, U. S. Toti, H. R. Allcock and C. T. Laurencin, Soft Matter, 2010, 6, 3119–3132 RSC.
- B. H. Lee, Y. M. Lee, Y. S. Sohn and S.-C. Song, Macromolecules, 2002, 35, 3876–3879 CrossRef CAS.
- B. H. Lee and S.-C. Song, Macromolecules, 2004, 37, 4533–4537 CrossRef CAS.
- Z. Tian, C. Chen and H. R. Allcock, Macromolecules, 2013, 46, 2715–2724 CrossRef CAS.
- Z. Tian, C. Chen and H. R. Allcock, Macromolecules, 2014, 47, 1065–1072 CrossRef CAS.
- T. Potta, C. Chun and S.-C. Song, Biomaterials, 2009, 30, 6178–6192 CrossRef CAS PubMed.
- T. Potta, C. Chun and S.-C. Song, Biomacromolecules, 2010, 11, 1741–1753 CrossRef CAS PubMed.
- T. Potta, C. Chun and S.-C. Song, Macromol. Rapid Commun., 2010, 31, 2133–2139 CrossRef CAS PubMed.
- M. K. Shin, G. M. Spinks, S. R. Shin, S. I. Kim and S. J. Kim, Adv. Mater., 2009, 21, 1712–1715 CrossRef CAS.
- S. Bhattacharyya, S. Guillot, H. Dabboue, J.-F. Tranchant and J.-P. Salvetat, Biomacromolecules, 2008, 9, 505–509 CrossRef CAS PubMed.
- C. Li and G. Shi, Adv. Mater., 2014, 26, 3992–4012 CrossRef CAS PubMed.
- H. Fan, L. Wang, K. Zhao, N. Li, Z. Shi, Z. Ge and Z. Jin, Biomacromolecules, 2010, 11, 2345–2351 CrossRef CAS PubMed.
- J. Aalaie, E. Vasheghani-Farahani, A. Rahmatpour and M. A. Semsarzadeh, Eur. Polym. J., 2008, 44, 2024–2031 CrossRef CAS PubMed.
- H. R. Allcock, R. L. Kugel and K. J. Valan, Inorg. Chem., 1966, 5, 1709–1715 CrossRef CAS.
- R. Bleta, S. Menuel, B. Leger, A. Da Costa, E. Monflier and A. Ponchel, RSC Adv., 2014, 4, 8200–8208 RSC.
- C. Travelet, P. Hébraud, C. Perry, C. Brochon, G. Hadziioannou, A. Lapp and G. Schlatter, Macromolecules, 2010, 43, 1915–1921 CrossRef CAS.
- C. Travelet, G. Schlatter, P. Hébraud, C. Brochon, A. Lapp and G. Hadziioannou, Langmuir, 2009, 25, 8723–8734 CrossRef CAS PubMed.
- A. Harada, T. Nishiyama, Y. Kawaguchi, M. Okada and M. Kamachi, Macromolecules, 1997, 30, 7115–7118 CrossRef CAS.
- H. Okumura, Y. Kawaguchi and A. Harada, Macromolecules, 2001, 34, 6338–6343 CrossRef CAS.
- A. Harada, J. Li and M. Kamachi, Nature, 1992, 356, 325–327 CrossRef CAS.
- A. Harada, J. Li and M. Kamachi, Nature, 1993, 364, 516–518 CrossRef CAS.
- A. Harada, J. Li and M. Kamachi, Nature, 1994, 370, 126–128 CrossRef CAS.
- J. Li, NPG Asia Mater, 2010, 2, 112–118 CrossRef.
- X. Li, J. Li and K. W. Leong, Macromolecules, 2003, 36, 1209–1214 CrossRef CAS.
- J. Li, X. Ni, Z. Zhou and K. W. Leong, J. Am. Chem. Soc., 2003, 125, 1788–1795 CrossRef CAS PubMed.
- J. Li, X. Li, X. Ni, X. Wang, H. Li and K. W. Leong, Biomaterials, 2006, 27, 4132–4140 CrossRef CAS PubMed.
- C. Yang, X. Wang, H. Li, S. H. Goh and J. Li, Biomacromolecules, 2007, 8, 3365–3374 CrossRef CAS PubMed.
- J. Li and X. J. Loh, Adv. Drug Delivery Rev., 2008, 60, 1000–1017 CrossRef CAS PubMed.
- Z. Li, H. Yin, Z. Zhang, K. L. Liu and J. Li, Biomacromolecules, 2012, 13, 3162–3172 CrossRef CAS PubMed.
- Y. Chen, X.-H. Pang and C.-M. Dong, Adv. Funct. Mater., 2010, 20, 579–586 CrossRef CAS.
- A. Leal-Egaña, A. Díaz-Cuenca and A. R. Boccaccini, Adv. Mater., 2013, 25, 4049–4057 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed information on the preparation method, water contact angle, protein adsorption, anticoagulant properties of the membranes, platelet adhesion, cell culture, and cell morphology on the membranes. See DOI: 10.1039/c4py00967c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.