
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetic isotope effect of proton-coupled electron transfer in a hydrogen bonded phenol–pyrrolidino[60]fullerene†
Janneke
Ravensbergen
a,
Chelsea L.
Brown
b,
Gary F.
Moore
b,
Raoul N.
Frese
a,
Rienk
van Grondelle
a,
Devens
Gust
b,
Thomas A.
Moore
b,
Ana L.
Moore
*b and
John T. M.
Kennis
*a
aDepartment of Physics and Astronomy, Faculty of Sciences, VU University, De Boelelaan 1081, 1081 HV, Amsterdam, The Netherland. E-mail: j.t.m.kennis@vu.nl
bCenter for Bioenergy and Photosynthesis, Department of Chemistry and Biochemistry, Arizona State University, Tempe, AZ 85287-1604, USA. E-mail: Ana.Moore@asu.edu
First published on 27th October 2015
Abstract
Proton-coupled electron transfer (PCET) plays a central role in photosynthesis and potentially in solar-to-fuel systems. We report a spectroscopy study on a phenol–pyrrolidino[60]fullerene. Quenching of the singlet excited state from 1 ns to 250 ps is assigned to PCET. A H/D exchange study reveals a kinetic isotope effect (KIE) of 3.0, consistent with a concerted PCET mechanism.
Proton-coupled electron transfer (PCET) is of key importance in photosynthesis and various other biological processes.1 In photosystem II, after the initial charge separation the oxidized chlorophyll is reduced in a PCET step by a tyrosine (TyrZ). The tyrosine phenolic proton is donated to a nearby histidine residue (His 190).2,3 When the tyrosine is reduced by the water oxidation complex it regains a proton. By the coupling of proton movement to these redox reactions photosynthesis avoids high energy intermediates and stabilizes the charge separated state.4–6
In artificial photosynthesis one aim is to use design principles from nature to develop a solar fuel producing system. PCET is one of these principles that can play a crucial role in bridging the timescale of short-lived, reactive intermediates formed by photoinduced charge separation to that of the relative slow process of multi-electron catalysis. Various studies have been performed on artificial PCET systems with various degrees of molecular complexity.1,5,7–16 In search of a minimal biomimetic construct for photoinduced PCET, Moore et al., have previously reported a phenol–pyrrolidino[60]fullerene in which the fullerene fluorescence lifetime in benzonitrile is reduced from 1.3 ns to 260 ps. This reduction of the fluorescence lifetime was not found in acidified solvent, strongly suggesting a PCET mechanism for the quenching of excited fullerene.17 This could be either a ‘proton first’, ‘electron first’ or concerted transfer process. We will refer to both the step-wise and concerted mechanisms as PCET.
Here we report a transient absorption spectroscopy study of this putative PCET in the phenol–pyrrolidino[60]fullerene 1 depicted in Fig. 1. In this isomer the phenol hydroxyl group is ortho to the pyrrolidine moiety and designed to hydrogen bond to the lone pair electrons of the nitrogen on the pyrrolidine. This internal hydrogen bond provides a well-defined structural framework for the PCET process. Reference compound 2 has a hydroxyl group at the para position where the internal hydrogen bond cannot be formed. Reference compound 3 lacks the hydroxyl group.
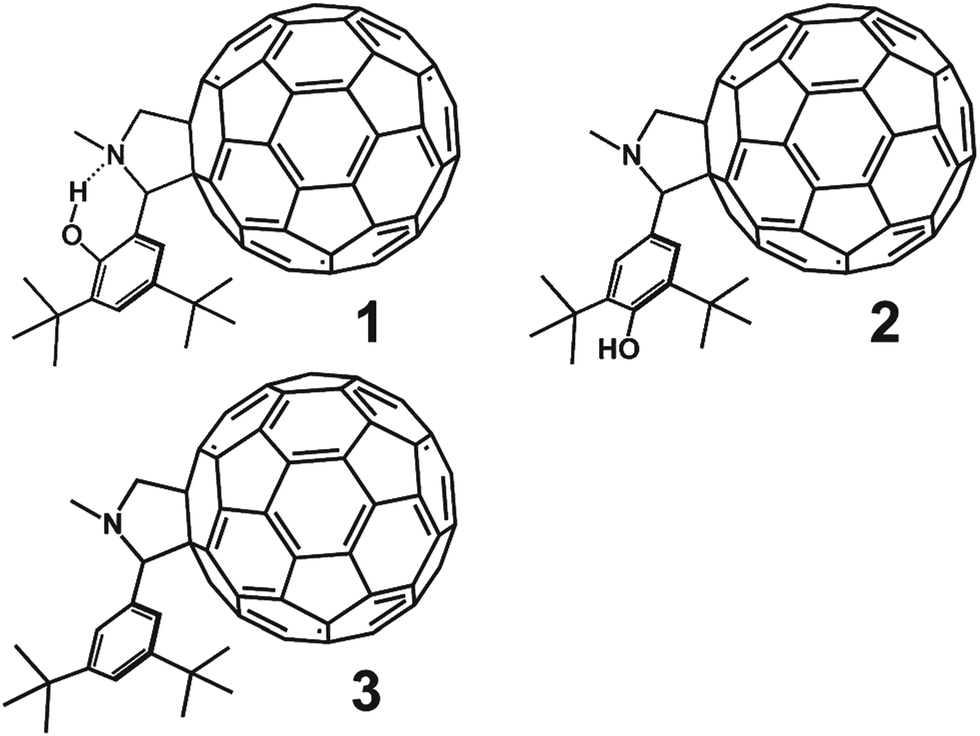 | ||
| Fig. 1 Phenol–pyrrolidino[60]fullerene compounds investigated in the present work. | ||
Previously reported electrochemical measurements revealed that the phenol moiety of compound 1 is thermodynamically capable of reducing singlet excited C60. The phenol moiety in compound 2 and the phenyl group in 3 are thermodynamically incapable of reducing singlet excited C60. This also holds for compound 1 in acidified solvent, where the internal hydrogen bond is disrupted.17
The steady-state absorption spectra in Fig. 2 are dominated by features from fullerene.18 The extinction coefficient of the compounds is largest in the UV. A small amplitude tail spans the visible part of the spectrum, where maxima are found at 433 nm and 705 nm.
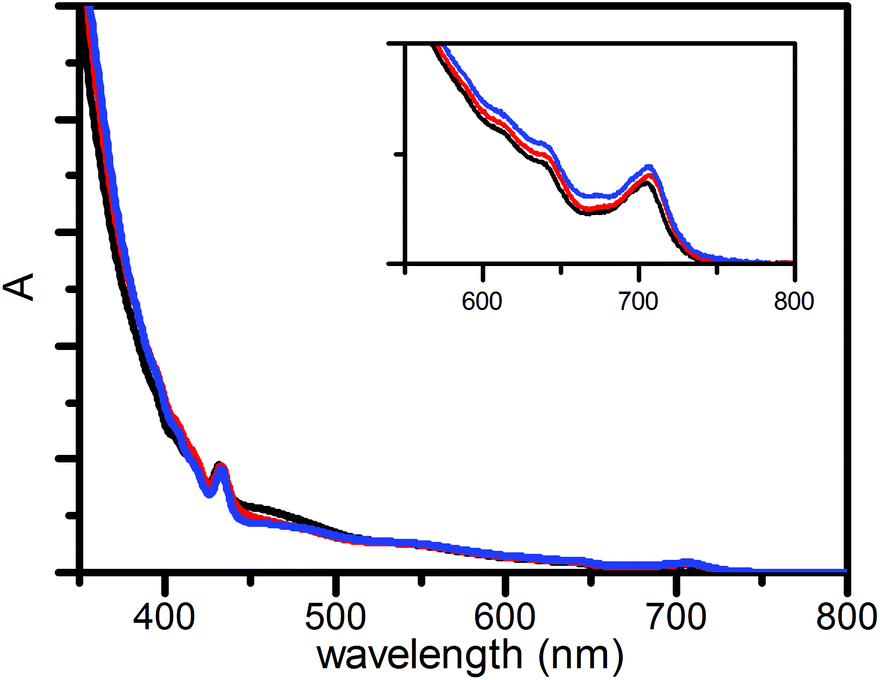 | ||
| Fig. 2 Normalized absorption spectra of 1 (black), 2 (red) and 3 (blue) in benzonitrile, normalized at 433 nm. | ||
For transient absorption spectroscopy the compounds were excited at 705 nm in the lowest-energy absorption band of fullerene; the instrument response time was 100 fs. Global analysis yields the Evolution Associated Difference Spectra (EADS) in Fig. 3. These are the interconverting spectra that follow from analysis with a sequential model. Further details of the measurements and analysis are given in the Materials and Methods section in the ESI.† Raw transient absorption data is presented in Fig. S2.†
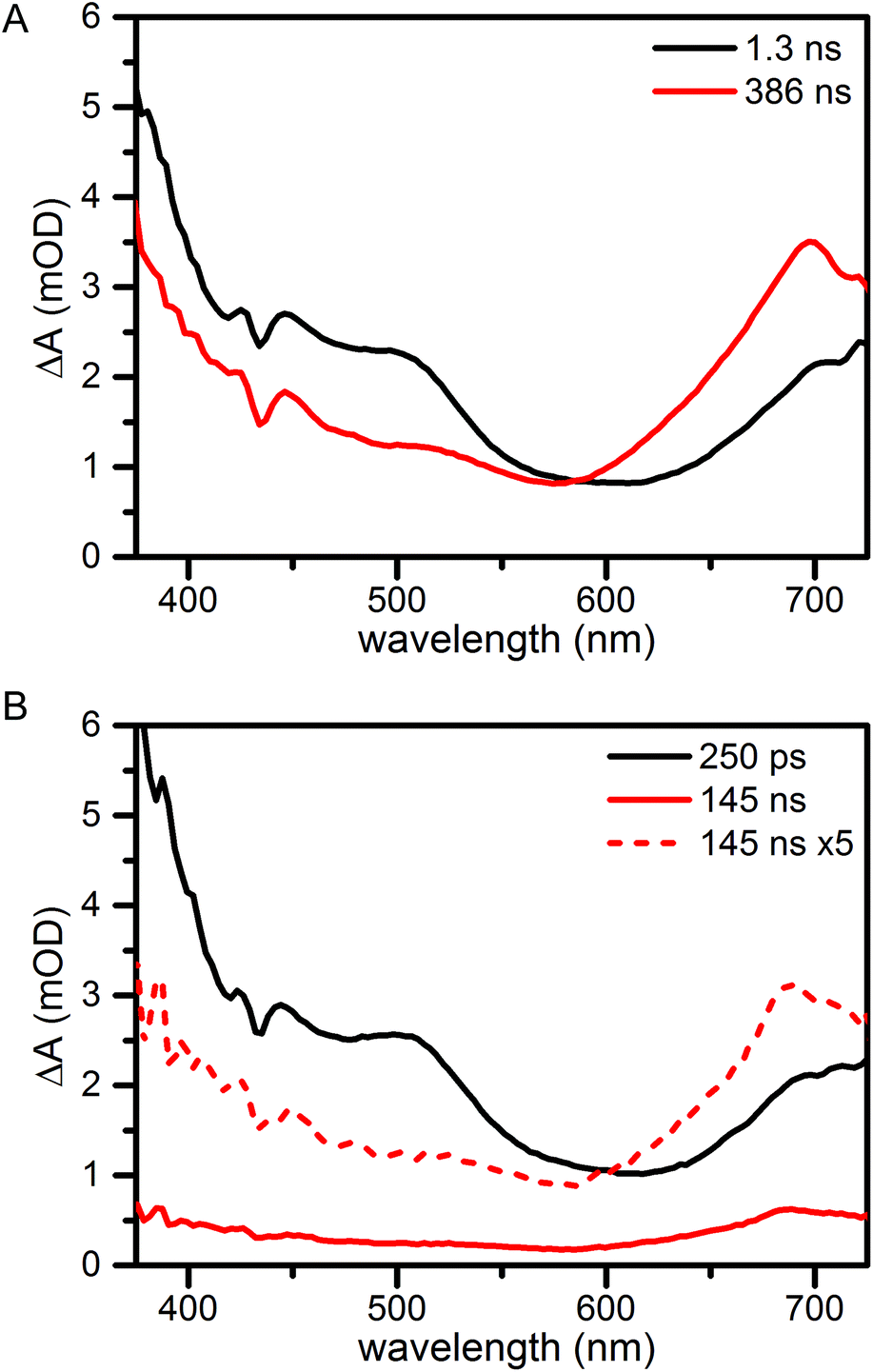 | ||
| Fig. 3 Evolution Associated Difference Spectra (EADS) of compounds 2 (A) and 1 (B) in aerated benzonitrile upon 705 nm excitation. The lifetimes of the interconverting spectra are given in the legend. | ||
For compound 2 (Fig. 3A) in aerated benzonitrile two lifetime components are needed for a sufficient fit of the data: 1.3 ns and 386 ns. The spectra are assigned to the singlet and triplet excited state of fullerene and are very similar to those reported for methylfulleropyrrolidine.18 The first EADS (black) assigned to the singlet state shows excited state absorption in the full probed window. Overlapping, but with smaller amplitude we find ground state bleach. This has a negative contribution to the ΔA signal that can be recognized by the minima at 433 nm and 705 nm. The singlet excited state has a lifetime of 1.3 ns. The second EADS, assigned to the fullerene triplet, has lower amplitude at wavelengths shorter than 585 nm and higher amplitude at longer wavelengths. A maximum is found around 700 nm. The triplet decays to the ground state in 386 ns. Very similar results are found for 3 (Fig. S1†).
The singlet excited state spectrum of 1 (Fig. 3B, black) closely resembles the singlet spectra of 2 and 3. The lifetime of the singlet state is found to be 250 ps, in agreement with the reported fluorescence lifetime.17 As a result of the shorter singlet lifetime, less triplet is formed, and the triplet spectrum (which is similar to that of 2 and 3), is smaller in amplitude (Fig. 3B, red and red dash). The reduced singlet lifetime has been assigned to a proton-coupled electron transfer process forming a neutral phenoxyl radical and a zwitterionic pyrrolidino[60]fullerene group (PhO˙–PyrH+–C60˙−). The zwitterionic state could not be detected. Most probably, the rate constant for recombination is larger than that for formation of the zwitterionic state. In such ‘inverted kinetics’, the zwitterionic state signal would rise with the time constant of its lifetime, and its transient concentration would be low. The detection of such recombination time constants on fast timescales is further complicated by a large coherent and cross-phase modulation artefact around zero time delay.
To confirm the involvement of proton migration in the photophysical pathway we performed transient absorption spectroscopy of compound 1 in aerated benzonitrile with 2% v/v H2O or D2O. The protons or deuterium from H2O or D2O will exchange with the phenolic proton, the only exchangeable proton of compound 1. The H/D exchange was confirmed by 1H-NMR. The 1H-NMR spectrum of the compound in equal parts CDCl3/CS2 with 2% H2O was compared to the 1H-NMR spectrum in equal parts CDCl3/CS2 with 2% D2O. The disappearance of the peak for the phenolic proton was observed after addition of D2O (Fig. S3 and S4†). We refer to the deuterated compound as 1-D (1-H for H2O). Fig. 4 shows the time trace for the absorption of both compounds at 538 nm. For 1-H we find a singlet lifetime of 250 ps, similar to the measurement in benzonitrile. In contrast, the singlet lifetime of 1-D is 546 ps. Based on the lifetime for decay of 2 of 1.3 ns, we estimate the rate of PCET at 3.2 × 109 s−1 and 1.1 × 109 s−1 for 1-H and 1-D respectively.‡ Thus, excited state quenching by PCET is associated with a H/D kinetic isotope effect (KIE) of 3.0. For compounds 2 and 3 in benzonitrile with 2% v/v H2O or D2O no kinetic isotope effect was found.
 | ||
| Fig. 4 Transient absorption spectroscopy time trace of 1 at 538 nm in aerated benzonitrile with 2% H2O (1-H, black) and 2% D2O (1-D, red). The sharp peak at 0 ps is due to a coherent and cross-phase modulation artefact. | ||
This significant kinetic isotope effect for 1 confirms the involvement of proton migration in the quenching of the singlet state. We propose that this PCET process occurs by a concerted mechanism. The size of the KIE shows that the proton is involved in the rate determining step and excludes the ‘electron first’ mechanism for this non-equilibrium PCET process.7–11,19,20 The ‘proton first’ mechanism is excluded by the large difference in acidity. The pyrrolidine is about 10 pKa units more acidic than the phenol, based on values in acetonitrile.21,22 Prato et al. have shown that pyrrolidine is even more acidic when attached to fullerene.23 The pKa of pyrrolidine is not expected to change much upon excitation of fullerene, because the HOMO and LUMO do not include the atoms around the H-bond.24 Due to the large difference in acidity, proton transfer from phenol to pyrrolidine prior to electron migration would be energetically steeply uphill. Concerted PCET avoids this high energy intermediate.
Costentin et al. reported a phenol–pyrrolidine system with an electrochemically observed KIE of 1.8.25 A possible explanation for the larger KIE in our experiments is the lower pKa of pyrrolidinofullerene, as compared to pyrrolidine, which leads to a weaker hydrogen bond. Hammes-Schiffer and co-workers discussed the KIE dependence on hydrogen bond strength in PCET processes.26 The trend of increasing KIE with weaker hydrogen bond is explained by the more localized wavefunction in case of deuterium, leading to a faster decay of overlap with increasing donor–acceptor distance. However the opposite trend has been observed in some systems and explained by the role of vibronic states.26
To summarize, our transient absorption spectroscopy results for compound 1 show a quenching of the singlet excited fullerene, assigned to a proton coupled electron transfer process. We observe a H/D kinetic isotope effect of 3.0 consistent with a concerted PCET mechanism.
Notes and references
- B. A. Barry, Reaction dynamics and proton coupled electron transfer: Studies of tyrosine-based charge transfer in natural and biomimetic systems, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 46–54 CrossRef CAS PubMed.
- S. Nakamura, R. Nagao, R. Takahashi and T. Noguchi, Fourier Transform Infrared Detection of a Polarizable Proton Trapped between Photooxidized Tyrosine YZ and a Coupled Histidine in Photosystem II: Relevance to the Proton Transfer Mechanism of Water Oxidation, Biochemistry, 2014, 53, 3131–3144 CrossRef CAS PubMed.
- T. Noguchi, Fourier transform infrared difference and time-resolved infrared detection of the electron and proton transfer dynamics in photosynthetic water oxidation, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 35–45 CrossRef CAS PubMed.
- M. Hambourger, G. F. Moore, D. M. Kramer, D. Gust, A. L. Moore and T. A. Moore, Biology and technology for photochemical fuel production, Chem. Soc. Rev., 2009, 38, 25–35 RSC.
- L. Hammarström and S. Styring, Proton-coupled electron transfer of tyrosines in Photosystem II and model systems for artificial photosynthesis: the role of a redox-active link between catalyst and photosensitizer, Energy Environ. Sci., 2011, 4, 2379 Search PubMed.
- M. H. V. Huynh and T. J. Meyer, Proton-Coupled Electron Transfer, Chem. Rev., 2007, 107, 5004–5064 CrossRef CAS PubMed.
- I. J. Rhile, T. F. Markle, H. Nagao, A. G. DiPasquale, O. P. Lam, M. A. Lockwood, K. Rotter and J. M. Mayer, Concerted Proton−Electron Transfer in the Oxidation of Hydrogen-Bonded Phenols, J. Am. Chem. Soc., 2006, 128, 6075–6088 CrossRef CAS PubMed.
- J. M. Mayer, I. J. Rhile, F. B. Larsen, E. A. Mader, T. F. Markle and A. G. DiPasquale, Models for Proton-coupled Electron Transfer in Photosystem II, Photosynth. Res., 2006, 87, 3–20 CrossRef CAS PubMed.
- J. J. Concepcion, M. K. Brennaman, J. R. Deyton, N. V. Lebedeva, M. D. E. Forbes, J. M. Papanikolas and T. J. Meyer, Excited-State Quenching by Proton-Coupled Electron Transfer, J. Am. Chem. Soc., 2007, 129, 6968–6969 CrossRef CAS PubMed.
- T. Irebo, O. Johansson and L. Hammarström, The Rate Ladder of Proton-Coupled Tyrosine Oxidation in Water: A Systematic Dependence on Hydrogen Bonds and Protonation State, J. Am. Chem. Soc., 2008, 130, 9194–9195 CrossRef CAS PubMed.
- C. Bronner and O. S. Wenger, Kinetic Isotope Effects in Reductive Excited-State Quenching of Ru(2,2′-bipyrazine)32+ by Phenols, J. Phys. Chem. Lett., 2011, 3, 70–74 CrossRef.
- F. Lachaud, A. Quaranta, Y. Pellegrin, P. Dorlet, M.-F. Charlot, S. Un, W. Leibl and A. Aukauloo, A Biomimetic Model of the Electron Transfer between P680 and the TyrZ–His190 Pair of PSII, Angew. Chem., Int. Ed., 2005, 44, 1536–1540 CrossRef PubMed.
- S. Y. Reece, J. M. Hodgkiss, J. Stubbe and D. G. Nocera, Proton-coupled electron transfer: the mechanistic underpinning for radical transport and catalysis in biology, Philos. Trans. R. Soc., B, 2006, 361, 1351–1364 CrossRef CAS PubMed.
- J. D. Megiatto Jr., D. D. Méndez-Hernández, M. E. Tejeda-Ferrari, A.-L. Teillout, M. J. Llansola-Portolés, G. Kodis, O. G. Poluektov, T. Rajh, V. Mujica, T. L. Groy, D. Gust, T. A. Moore and A. L. Moore, A bioinspired redox relay that mimics radical interactions of the Tyr–His pairs of photosystem II, Nat. Chem., 2014, 6, 423–428 CrossRef PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Proton-Coupled Electron Transfer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- J. Chen, M. Kuss-Petermann and O. S. Wenger, Distance Dependence of Bidirectional Concerted Proton–Electron Transfer in Phenol-Ru(2,2′-bipyridine)32+ Dyads, Chem. – Eur. J., 2014, 20, 4098–4104 CrossRef CAS PubMed.
- G. F. Moore, J. D. Megiatto, M. Hambourger, M. Gervaldo, G. Kodis, T. A. Moore, D. Gust and A. L. Moore, Optical and electrochemical properties of hydrogen-bonded phenol-pyrrolidino[60]fullerenes, Photochem. Photobiol. Sci., 2012, 11, 1018 CAS.
- D. M. Guldi and M. Prato, Excited-State Properties of C60 Fullerene Derivatives, Acc. Chem. Res., 2000, 33, 695–703 CrossRef CAS PubMed.
- S. Hammes-Schiffer and A. A. Stuchebrukhov, Theory of Coupled Electron and Proton Transfer Reactions, Chem. Rev., 2010, 110, 6939–6960 CrossRef CAS PubMed.
- A. Hazra, A. V. Soudackov and S. Hammes-Schiffer, Isotope Effects on the Nonequilibrium Dynamics of Ultrafast Photoinduced Proton-Coupled Electron Transfer Reactions in Solution, J. Phys. Chem. Lett., 2011, 2, 36–40 CrossRef CAS PubMed.
- A. Kütt, V. Movchun, T. Rodima, T. Dansauer, E. B. Rusanov, I. Leito, I. Kaljurand, J. Koppel, V. Pihl, I. Koppel, G. Ovsjannikov, L. Toom, M. Mishima, M. Medebielle, E. Lork, G.-V. Röschenthaler, I. A. Koppel and A. A. Kolomeitsev, Pentakis(trifluoromethyl)phenyl, a Sterically Crowded and Electron-withdrawing Group: Synthesis and Acidity of Pentakis(trifluoromethyl)benzene, -toluene, -phenol, and -aniline, J. Org. Chem., 2008, 73, 2607–2620 CrossRef PubMed.
- I. Kaljurand, A. Kütt, L. Sooväli, T. Rodima, V. Mäemets, I. Leito and I. A. Koppel, Extension of the Self-Consistent Spectrophotometric Basicity Scale in Acetonitrile to a Full Span of 28 pKa Units: Unification of Different Basicity Scales, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS PubMed.
- M. Prato and M. Maggini, Fulleropyrrolidines: A Family of Full-Fledged Fullerene Derivatives, Acc. Chem. Res., 1998, 31, 519–526 CrossRef CAS.
- I. D. Petsalakis, N. Tagmatarchis and G. Theodorakopoulos, Theoretical Study of Fulleropyrrolidines by Density Functional and Time-Dependent Density Functional Theory, J. Phys. Chem. C, 2007, 111, 14139–14149 CAS.
- C. Costentin, M. Robert and J.-M. Savéant, Electrochemical and Homogeneous Proton-Coupled Electron Transfers: Concerted Pathways in the One-Electron Oxidation of a Phenol Coupled with an Intramolecular Amine-Driven Proton Transfer, J. Am. Chem. Soc., 2006, 128, 4552–4553 CrossRef CAS PubMed.
- S. J. Edwards, A. V. Soudackov and S. Hammes-Schiffer, Analysis of Kinetic Isotope Effects for Proton-Coupled Electron Transfer Reactions, J. Phys. Chem. A, 2009, 113, 2117–2126 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5pp00259a |
| ‡ From the measured 1.3 ns singlet lifetime of reference compound 2 it follows that the rate of singlet decay is kref = 1/(1.3 ns) = 7.7 × 108 s−1. This rate includes intersystem crossing and internal conversion in compound 2 and is used as estimation for these processes in 1-H and 1-D. For 1-H and 1-D the singlet decay rate is the sum of kref and the PCET rate as ksum = kref + kPCET. Here, ksum corresponds to the observed singlet decay rate, which is (1/250 ps) for 1-H and (1/546 ps) for 1-D. From this follows that kPCET is 3.2 × 109 s−1 and 1.1 × 109 s−1 for 1-H and 1-D respectively. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2015 |