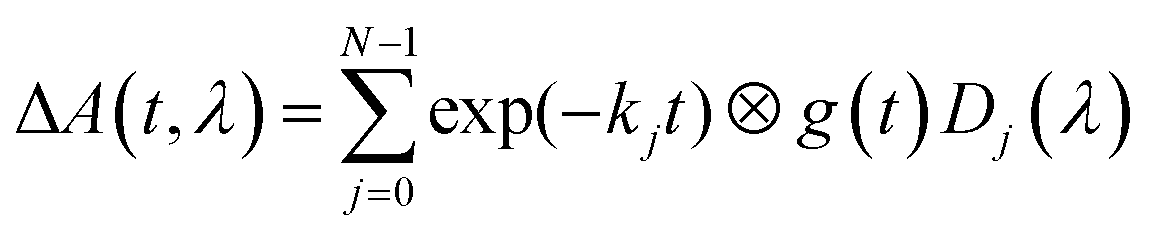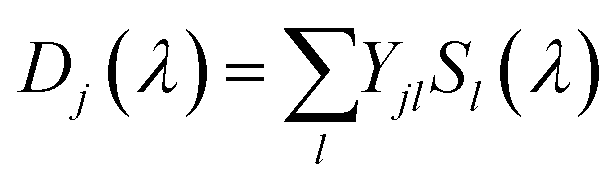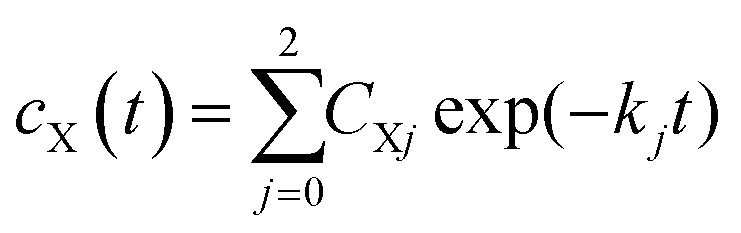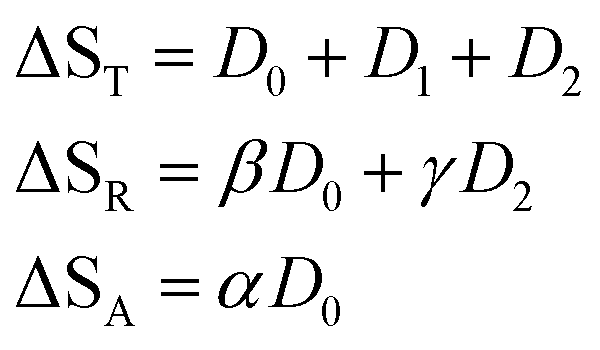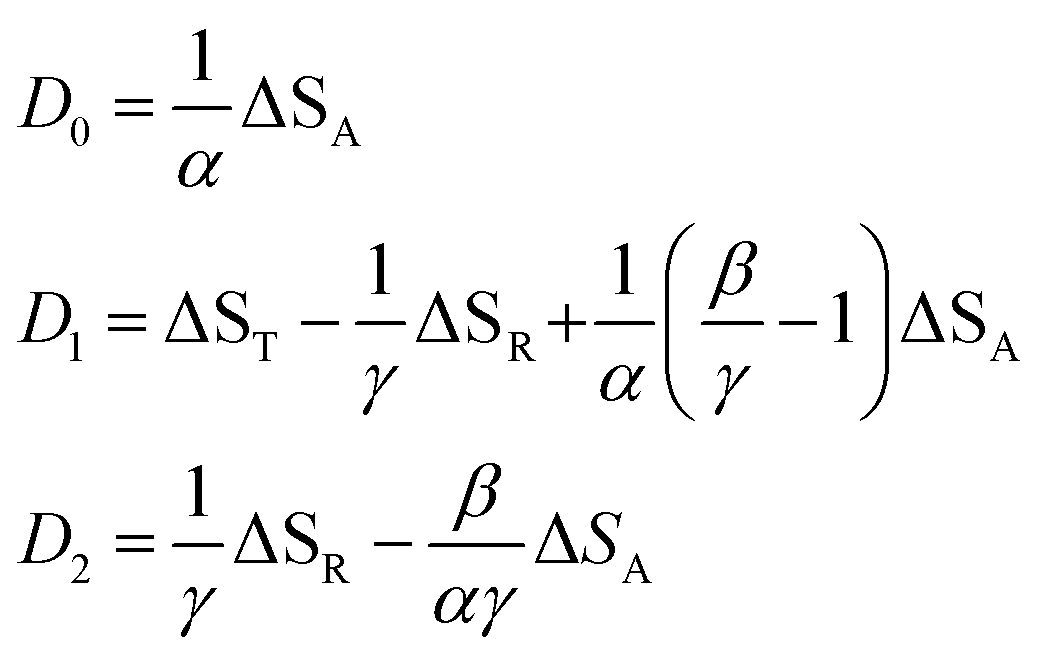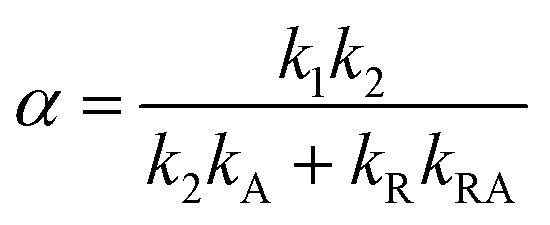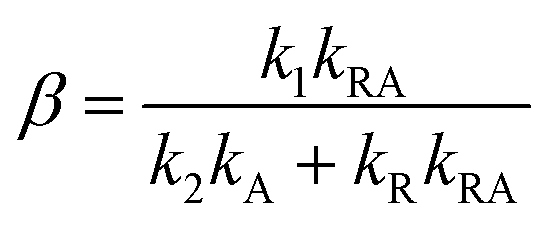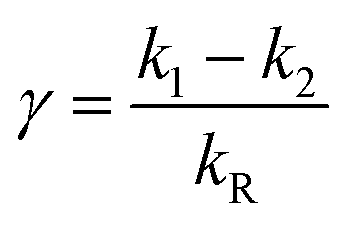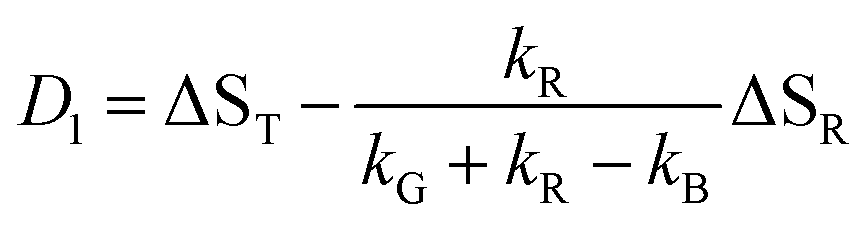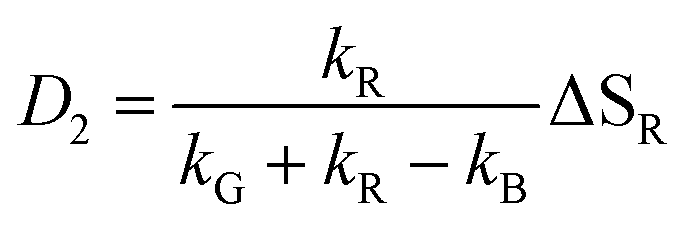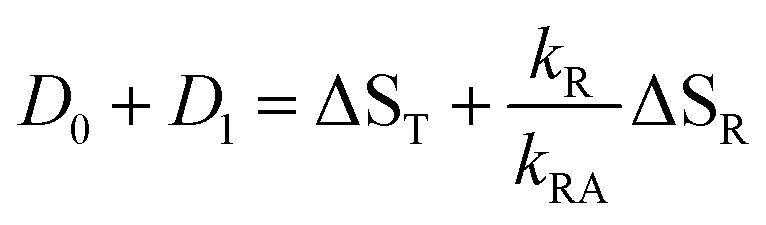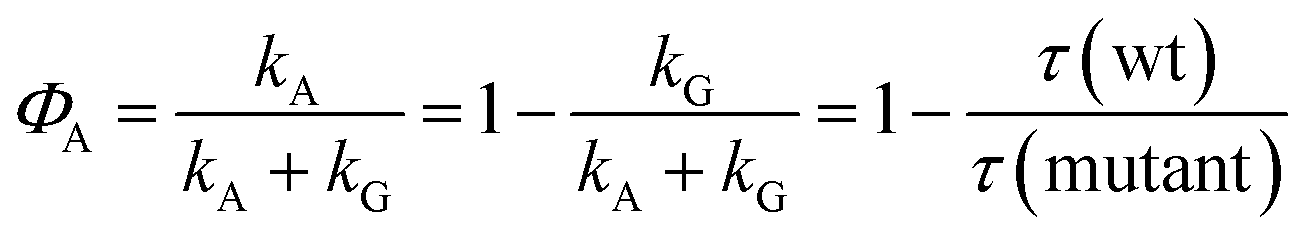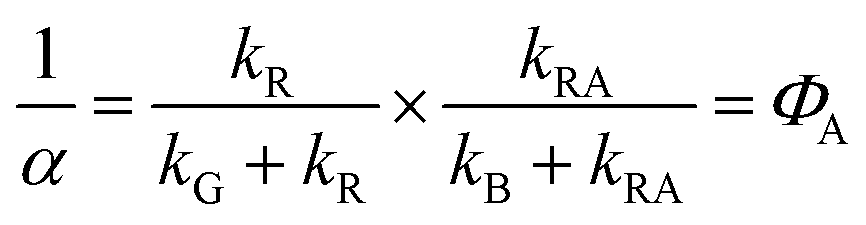A search for radical intermediates in the photocycle of LOV domains†
Received
29th April 2014
, Accepted 16th October 2014
First published on 17th October 2014
Abstract
LOV domains are the light sensitive parts of phototropins and many other light-activated enzymes that regulate the response to blue light in plants and algae as well as some fungi and bacteria. Unlike all other biological photoreceptors known so far, the photocycle of LOV domains involves the excited triplet state of the chromophore. This chromophore is flavin mononucleotide (FMN) which forms a covalent adduct with a cysteine residue in the signaling state. Since the formation of this adduct from the triplet state involves breaking and forming of two bonds as well as a change from the triplet to the singlet spin state, various intermediates have been proposed, e.g. a protonated triplet state 3FMNH+, the radical anion 2FMN˙−, or the neutral semiquinone radical 2FMNH˙. We performed an extensive search for these intermediates by two-dimensional transient absorption (2D-TA) with a streak camera. However, no transient with a rate constant between the decay of fluorescence and the decay of the triplet state could be detected. Analysis of the decay associated difference spectra results in quantum yields for the formation of the adduct from the triplet of ΦA(LOV1) ≈ 0.75 and ΦA(LOV2) ≈ 0.80. This is lower than the values ΦA(LOV1) ≈ 0.95 and ΦA(LOV2) ≈ 0.99 calculated from the rate constants, giving indirect evidence of an intermediate that reacts either to form the adduct or to decay back to the ground state. Since there is no measurable delay between the decay of the triplet and the formation of the adduct, we conclude that this intermediate reacts much faster than it is formed. The LOV1-C57S mutant shows a weak and slowly decaying (τ > 100 μs) transient whose decay associated spectrum has bands at 375 and 500 nm, with a shoulder at 400 nm. This transient is insensitive to the pH change in the range 6.5–10.0 but increases on addition of β-mercaptoethanol as the reducing agent. We assign this intermediate to the radical anion which is protected from protonation by the protein. We propose that the adduct is formed via the same intermediate by combination of the radical ion pair.
1. Introduction
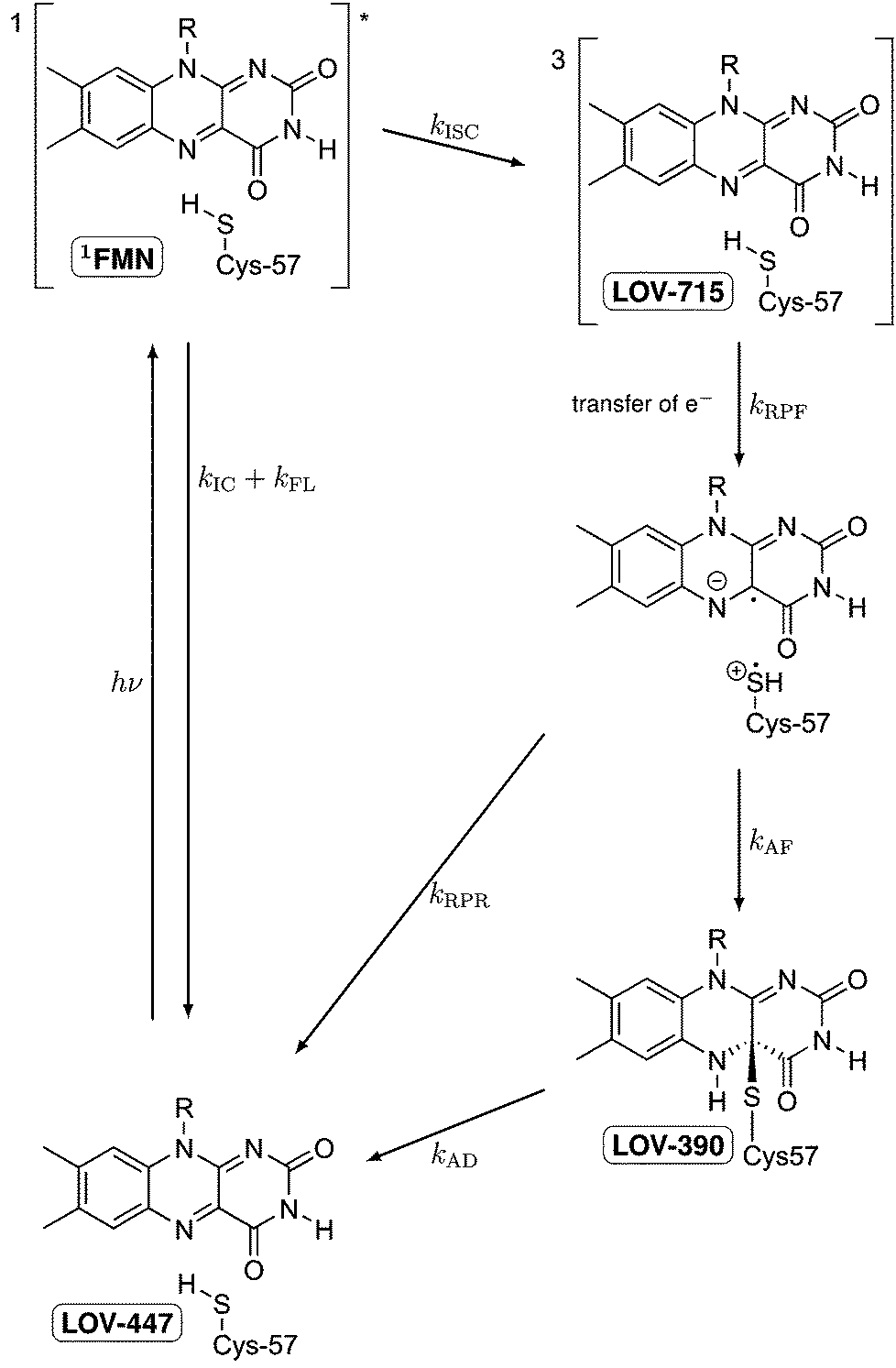
LOV domains are the light sensitive parts of many light-activated enzymes involved in regulating the response of plants and algae, as well as some fungi and bacteria, to blue light.1 Among these responses are chloroplast relocation,2,3 opening of stomata in leaves,4 phototropism,1,5,6 and the sexual life cycle of algae.7 Each LOV domain contains one flavin mononucleotide (FMN) as the chromophore with a characteristic absorption band in the visible spectral range of 420–470 nm. Immediately following the discovery of LOV domains several groups have studied their photocycles.8–12 Whereas in the dark form FMN is bound by hydrogen bonds,1 a covalent bond is formed between C4a of the flavin and the sulfur atom of a cysteine residue.8,13–15 The thermal reaction back to the dark state is rather slow, taking typically several minutes.9 In some organisms, the back reaction can take hours16 or is completely absent.17,18Fig. 1 shows the schematic of these studies, taking the first LOV domain of the PHOT protein from the green alga Chlamydomonas reinhardtii (C.r.) as an example. In this green alga the PHOT protein controls gametogenesis.7 Excitation of the dark form, LOV447, with blue light promotes the FMN chromophore to the excited singlet state 1FMN which decays either by fluorescence (τF = 2.9 ns)19 back to the ground state, or by intersystem crossing to the triplet excited state 3FMN. The latter is denoted as LOV715 according to the characteristic absorption band at 715 nm.19–21 The next species in the photocycle that is firmly established is the adduct state LOV390 with a single broad absorption band peaking at 390 nm.15,22
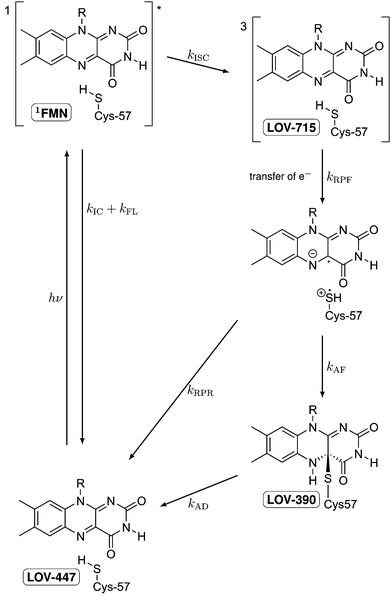 |
| | Fig. 1 Schematic of the photocycle of LOV domains. The established intermediates are the dark state LOV447, the excited singlet state 1FMN, the triplet state LOV715, and the signaling state LOV390. | |
LOV domains are the only biological photoreceptors for which a long lived triplet state has been identified as an intermediate in the photocycle.9 In all other photoreceptors for which the photocycle is known, e.g. rhodopsins,23–25 the primary photoreaction is completed within a few ps. Since the photoreaction in LOV domains is apparently a special case, the elucidation of the reaction mechanism is of particular interest. The formation of the adduct from the triplet state involves the breaking of two bonds, the formation of two new bonds, and the crossing from the triplet potential energy surface to that of the singlet. Although in principle a concerted reaction can be envisaged, a stepwise mechanism would appear more natural. Swartz et al. proposed a nucleophilic attack involving a protonated triplet state.9 However, this mechanism is ruled out by recent measurements of the transient IR spectra of triplet LOV26 and of protonated triplet flavin.27 TD-DFT calculations by Neiss and Saalfrank28 favored a mechanism involving the neutral flavin semiquinone radical and a thiyl radical. EPR data also led to the proposal of electron transfer as the primary event.29 This proposal was supported by QM/MM calculations on the triplet potential energy surface by Dittrich et al.30 and subsequently by MCSCF calculations on singlet and triplet states by Domratcheva et al.31 Corchnoy et al.32 observed a 5-fold decrease in the adduct formation when D2O was used as the solvent. They concluded that proton transfer is the rate limiting step, which implies an electron transfer preceding it. However, hydrogen transfer or concerted electron/proton transfer would also be in line with the experimental observation.
Previous experiments with mutated LOV domains in which the reactive cysteine is replaced by either serine or glycine have shown that triplet excited FMN easily forms the reduced semiquinone form FMNH˙ by electron transfer from a reducing agent (e.g. EDTA) followed by protonation.21,33–35 Hence electron transfer from the reactive cysteine to the triplet excited FMN is likely for the primary reaction step. When methyl mercaptan is used as the reducing agent together with the mutant in which the reactive cysteine had been replaced by glycine, also the adduct species is formed,34,35 whereas with bulkier mercaptans only the neutral radical FMNH˙ is observed. It must be concluded that in the wild type protein electron transfer from the reactive cysteine to FMN is possible. If this is followed by proton transfer, a neutral radical pair will result. As long as the distance between the two radicals is large enough, the total triplet and singlet spin states of this radical pair will be almost degenerate within the range of kBT at room temperature, and intersystem crossing should occur easily. Recombination of these radicals in the overall singlet state will lead to the adduct state. If this mechanism applies the radical intermediates FMN˙− and possibly also FMNH˙ should occur in the photocycle.
Recently, Bauer et al. have presented experimental data that support this mechanistic hypothesis.36 Snapshots of transient absorption spectra taken ca. 80 ns and 1 μs after excitation of the wild type domain differ by a small additional absorption in the region 550–620 nm where the absorption of the neutral radical is expected.38 The corresponding spectra recorded for the C57S mutant at 2 μs and 20 μs do not show this difference. It is concluded that the decay of the FMN triplet state yields the neutral radical as the next intermediate when the reactive cysteine is present. Since the radical pair combines faster to form the adduct than it is formed, the transient concentration of the radical is small. However, the same data could also be explained with the assumption that a certain small fraction of the LOV domains produces the radical in a side reaction, and that the main fraction forms the adduct with no detectable participation of the radical. In order to firmly establish a radical as an intermediate in the photocycle the kinetics of this species should be measured. However, the very long recovery time of the LOV domains makes the measurement of both the transient spectra and their kinetics rather difficult: laser excitation with high repetition rates cannot be used, and large quantities of the proteins for use in a flow cell are difficult to obtain.
For this purpose we have developed a novel apparatus based on a streak camera for obtaining transient absorption spectra.37 With this apparatus we can obtain the transient absorption spectra of a small sample amount (typically 300 μL with OD 0.3 at the excitation wavelength and 1 cm path length) with ca. 10–100 laser excitations. Here we report measurements of the photocycle of both LOV domains LOV1 and LOV2 from the PHOT protein of the green alga Chlamydomonas reinhardtii. The wild type domains LOV1-wt and LOV2-wt are compared with the mutants LOV1-C57S, LOV1-C57G, and LOV2-C250S in which the reactive cysteine has been replaced by either serine or glycine. In these mutants formation of the adduct should not be possible, i.e. we expect that their transient spectra are only due to the triplet species.
We could not detect any contribution of radical species to the photocycles of the wild type domains, i.e. no delay is observed between the decay of the triplet and the formation of the adduct. Analysis of our decay associated difference spectra (DADS) results in quantum yields for the formation of the adduct from the triplet of ΦA(LOV1) ≈ 0.75 and ΦA(LOV2) ≈ 0.80. These values are smaller than the values ΦA(LOV1) ≈ 0.95 and ΦA(LOV2) ≈ 0.99 estimated from the ratio of the triplet decay rates of wild types and inactive mutants. This can be explained by an intermediate that reacts faster than it is formed, leading either to decay back to the ground state or to formation of the adduct. The mutants show a long-lived transient which is assigned to the radical anion. We propose that this is also an intermediate in the photocycle of the wild-type domains.
2. Experiment and methods
All LOV constructs used in this study were expressed in E. coli and purified according to the standard protocols described previously.20,35 These were the two wild-type LOV domains LOV1-wt and LOV2-wt from the PHOT protein of the green alga Chlamydomonas reinhardtii as well as the mutants LOV1-C57S, LOV1-C57G, and LOV2-C250S in which the reactive cysteine had been replaced by either serine or glycine. All proteins were made with a His tag on the N-terminus. In the case of LOV2-C250S a sample fused with the maltose binding protein was also produced. All proteins were dialyzed four times against 10 mM phosphate buffer, pH 8, with 10 mM NaCl (all components purchased from Merck) in order to completely remove all FMN that are not bound to the LOV domains.
For the production of LOV2 with the His tag, the LOV2-encoding cDNA fragment (amino acids 199–336) was amplified by PCR from the cDNA. Replacement of the cysteine 250 by serine was done by site directed mutagenesis according to standard protocols. The cDNA fragments were digested with NdeI and SalI and cloned into the vector pET-28a(+) (Novagen) carrying a N-terminal His-tag. The LOV2 proteins were expressed and purified by the same protocol as LOV1.
Transient absorption measurements in the microsecond time range were performed with a streak camera. The setup and performance of this apparatus are described in detail in ref. 37. Briefly, the sample is placed in a fused silica flow cuvette of 2 mm optical path length for excitation and of 10 mm path length for probe light. Including the storage vessel and the peristaltic pump, the overall sample volume was about 5 mL. The control of the peristaltic pump was synchronized with the timing of the measuring process so that the sample was exchanged stepwise between each individual laser excitation.
The sample is excited through a cylindrical lens (f = 150 mm) along the short (2 mm) path with a laser flash (450 nm, 8 ns, 10 mJ) from an OPO pumped by a Nd:YAG laser (Surelite II, Continuum). The laser operates at a repetition rate of 10 Hz and a mechanical shutter selects every 20th pulse for sample excitation. This allows a 2 second delay between subsequent excitation cycles, enough time for the reference measurement and complete exchange of the excited sample volume by the peristaltic pump. White light from a 150 W pulsed Xe flashlamp of ca. 1 ms duration (MSP-05, Müller Elektronik-Optik) is passed along the long (10 mm) path through the sample cell and subsequently analyzed using an imaging spectrograph (Bruker 200is, grating 100 grooves per mm) coupled to a streak camera (C7700, Hamamatsu Photonics). The data were registered using a CCD camera (ORCA-CR, Hamamatsu Photonics) and stored in a computer. Global lifetime analysis was done with home-written software. The data are fitted to the expression
| | 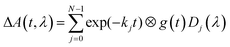 | (1) |
where
g(
t) is the instrument response function and ⊗ indicates convolution. The instrument response function is represented by a Gaussian. The results of this analysis are rate constants
kj and decay associated difference spectra (DADS)
Dj(
λ). We labelled the DADS according to increasing rate constants.
D0(
λ) contains the contribution that is quasi infinitely slow (
k0 ≈ 0) on the timescale of the measurement,
D1(
λ) is the next faster component, and so on.
In addition to the transmitted probe light, the streak camera also analyzes the fluorescence of flavin and some scattered light from the excitation laser. These events are faster than the time resolution of the streak camera in the fastest mode used here (i.e. ca. 10 ns in the 2 μs streak window). The fluorescence intensity is not small compared to that of the probe light, hence the detector is partially saturated and the time profile of the fluorescence is not well represented by the instrument response function. We could deal with this situation by excluding the part of the 2D image containing the fluorescence and the laser stray light from the fit using an algorithm described in ref. 37. However, this data area might also contain information on the radical intermediates. Therefore, we included the fluorescence in the fit and used a sum of up to four Gaussians for the time profile. The fitting can be performed directly to the 2D data matrices. However, since the noise at each individual pixel is rather large, the quality of the fit is difficult to judge from the residuals in this representation. Therefore, the fittings were performed to the leading components of singular value decomposition (SVD) of the data matrix. This allows easy visual inspection of the residuals. Fits and residuals can be found in the ESI.†
The result of the analysis of each dataset is a spectrum of the temporally unresolved component F(λ) which contains the fluorescence and scattered laser light, and a set of DADS each associated with a specific rate constant. These DADS are linear combinations of the species associated spectra (SAS) Sl(λ) of the various intermediates,
| | 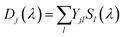 | (2) |
where the matrix of coefficients
Y depends on the mechanistic model. The test of different mechanistic models is done in two steps: first the unique DADS are determined by the global lifetime analysis described above, and then various kinetic schemes are projected onto these DADS according to
eqn (2). In contrast to fitting with model-specific time functions this procedure has the advantage that all interpretation is done with the same quality of the fit. This is due to the fact that the second step does not change the
χ2-value found in the first step.
3. Results and analysis
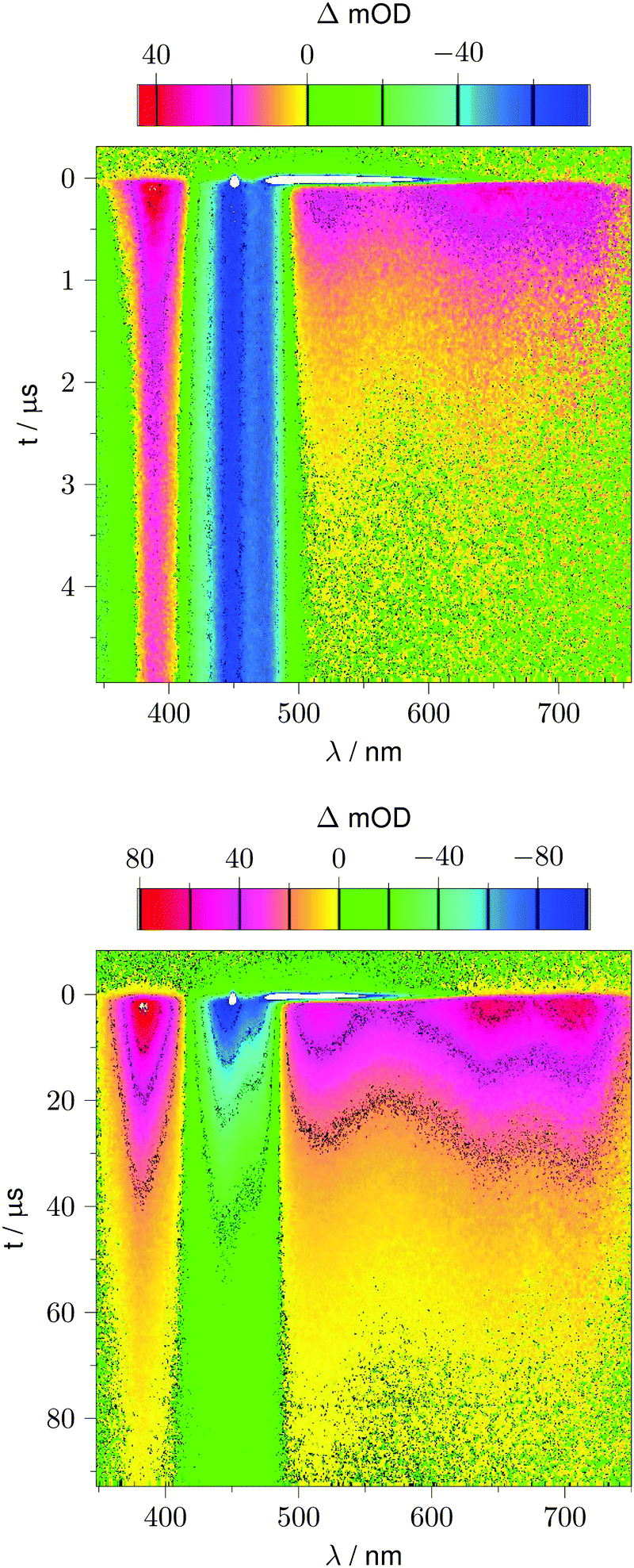
Fig. 2 shows 2D datasets, each obtained with 100 excitation cycles, for LOV1-wt and the LOV1-C57S mutant, respectively. In the false color representation, green and blue indicate the increasing negative absorption change, yellow and red correspond to increasing positive transient absorption. The bleaching of the ground state is clearly visible in both datasets as a blue stripe in the spectral range 420–480 nm. Positive TA appears in the ranges 350–410 nm and 500–740 nm. In the case of the LOV1-C57S mutant these transient features decay on a similar timescale and have almost vanished at the end of the streak window of 100 μs. In the case of the wild type domain the TA in the range 500–740 nm decays within the 5 μs streak window whereas the TA in the range 350–410 nm as well as the ground state bleach persists on this timescale. Apparently at least three different species are involved in the photocycles of these two domains. Before we proceed with the global lifetime analysis we note a small white spot at 450 nm and a white stripe in the spectral range 480–580 nm in a narrow time range near t = 0. These are strong negative signals outside the range of the false color scale, and are due to scattered laser light and fluorescence from the flavin. The dataset for the LOV2-wt domain looks similar to that for LOV1-wt, and datasets for the photoinactive mutants LOV1-C57G and LOV2-C250S look similar to that for LOV1-C57S.
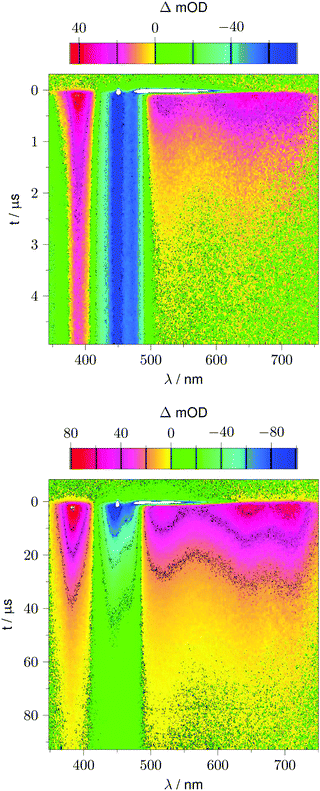 |
| | Fig. 2 False color representation of the 2D-TA data from 100 accumulated excitation cycles. Yellow and red indicate positive transient absorption, green and blue indicate negative transient absorption. Upper panel: LOV1-wt recorded with a streak window of 5 μs. Lower panel: LOV1-C57S recorded with a streak window of 100 μs. | |
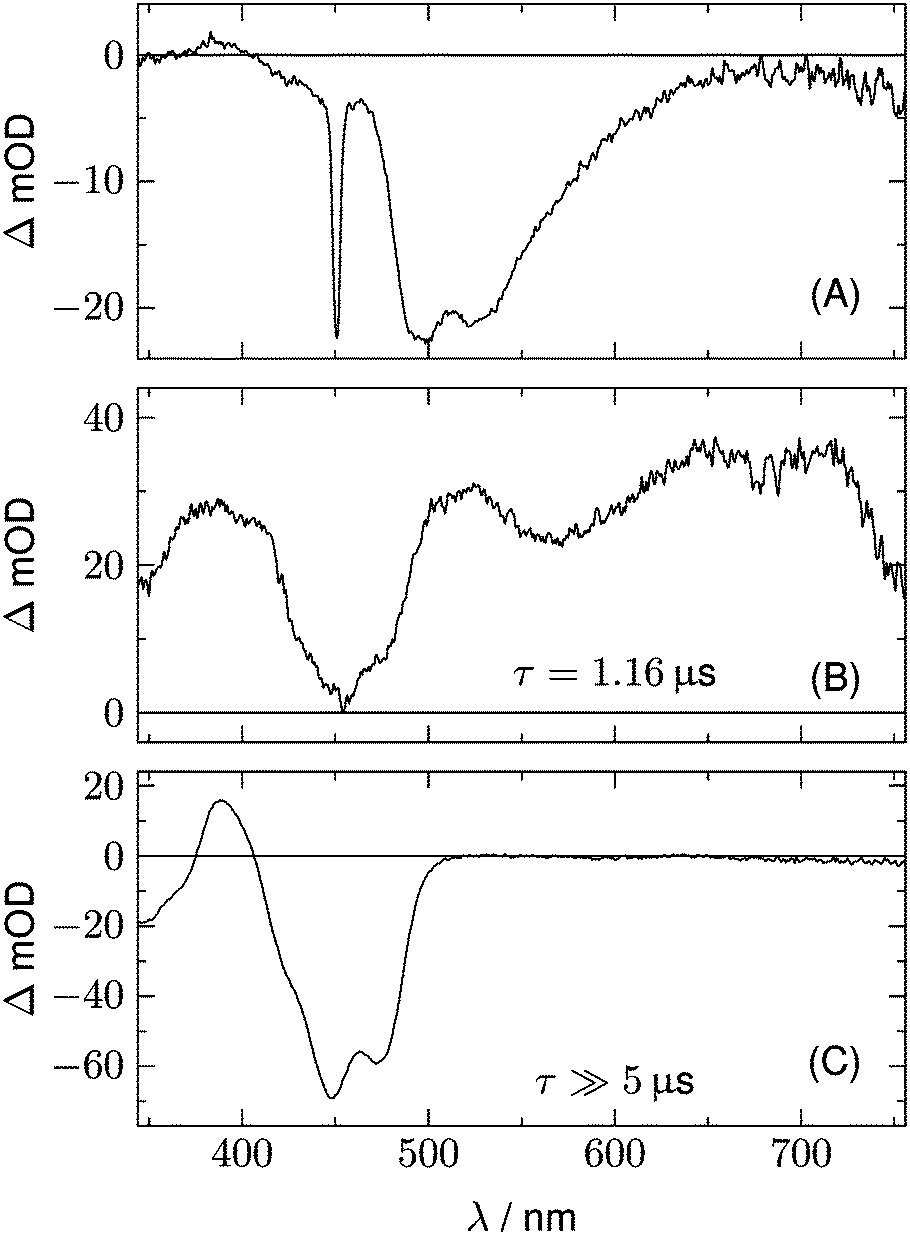
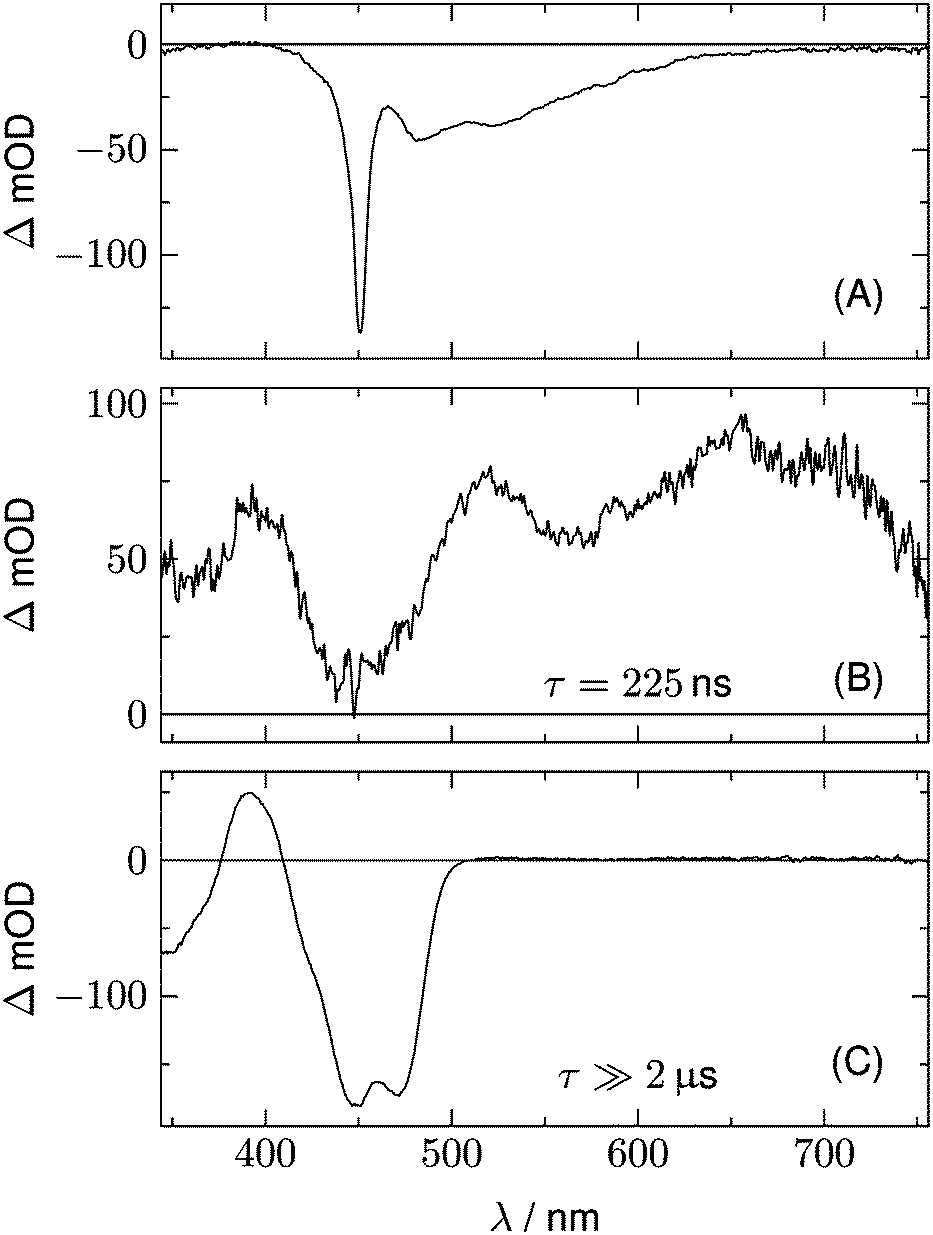
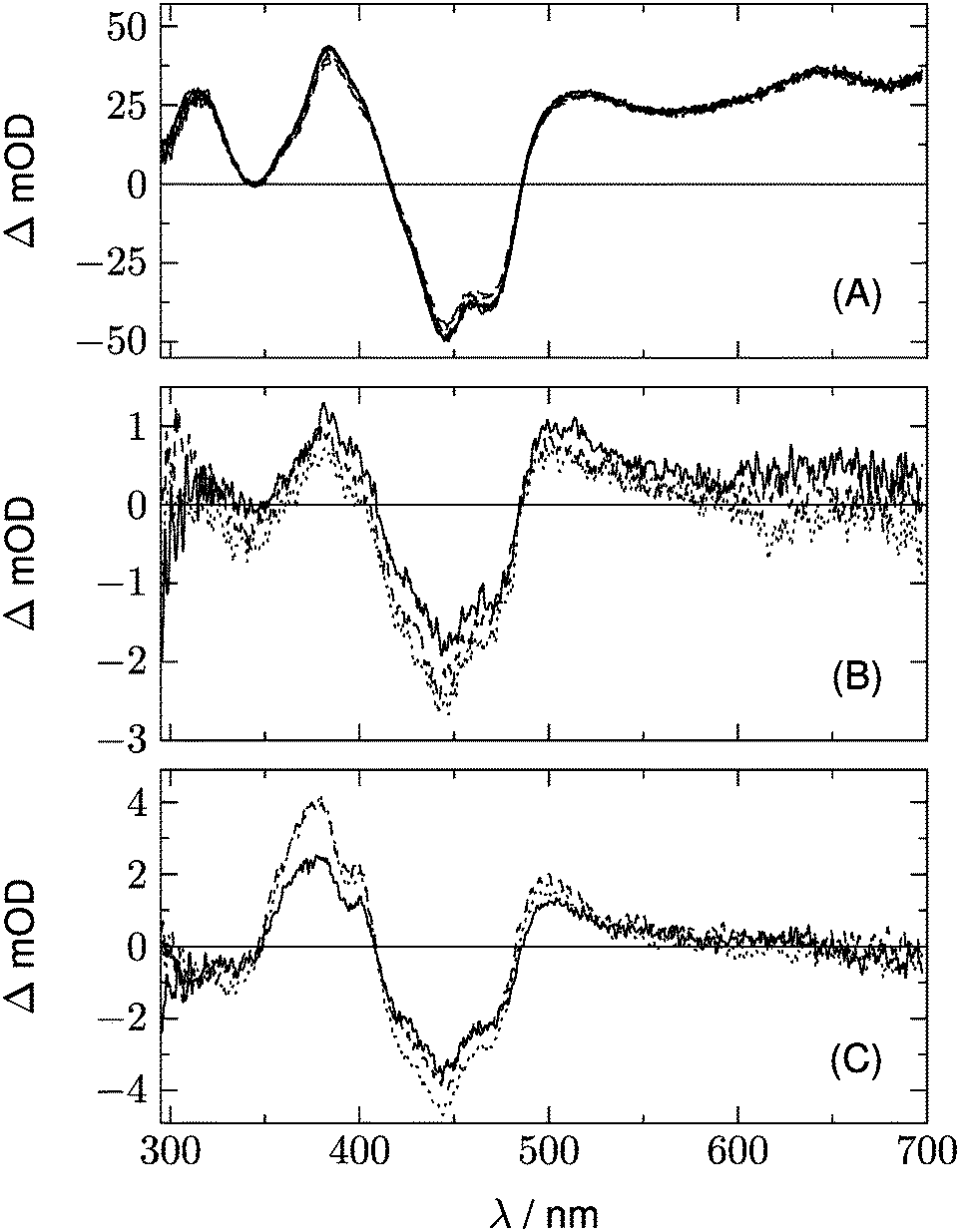
We begin with the global lifetime analysis of the wild type domains. The result is shown in Fig. 3 and 4. Each dataset can be fitted with three spectral components: panel A shows the spectrum of the temporally unresolved component, whose temporal structure is fitted by a sum of four Gaussians centered at t = 0 and widths below the time resolution of the streak camera. For the slit width of 30 μm, this time resolution is ca. 25 ns for the measurement of LOV1 (5 μs streak window) and ca. 10 ns for the measurement of LOV2 (2 μs streak window). The spectrum is easily recognized as the fluorescence of flavin with maxima at ca. 495 and 510 nm, and laser stray light at 450 nm. Since this light appears as an increase of the probe light, analysis in terms of transient absorption shows negative bands.
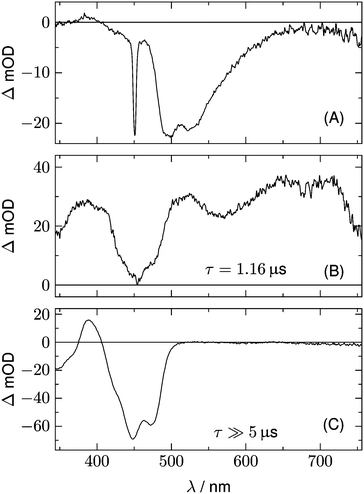 |
| | Fig. 3 Analysis of the 2D-TA data for LOV1-wt. (A) Spectrum of the temporally unresolved part, showing fluorescence and laser stray light. (B) and (C) show the DADS with decay times of 1.16 μs and ≫5 μs, respectively. | |
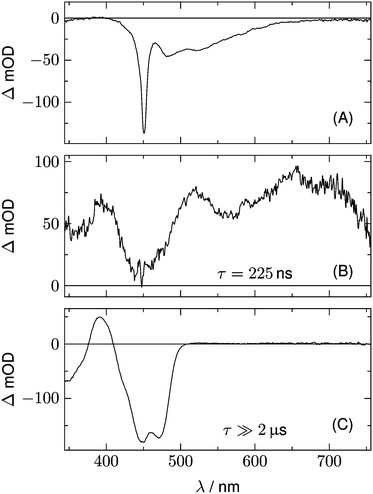 |
| | Fig. 4 Analysis of the 2D-TA data for LOV2-wt. (A) Spectrum of the temporally unresolved part, showing fluorescence and laser stray light. (B) and (C) show the DADS with decay times of 225 ns and ≫2 μs, respectively. | |
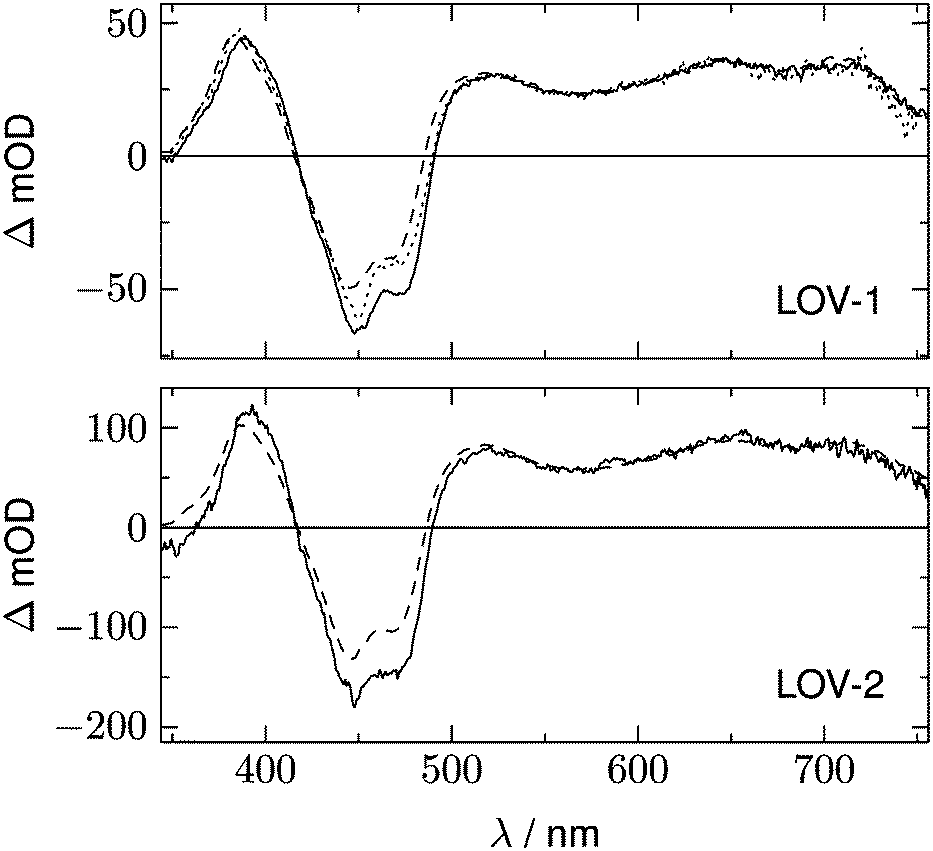
In addition to the fluorescence, two DADS are found in each case. One corresponds to a decay time within the streak window (1.16 μs for LOV1 and 225 ns for LOV2), shown in panel B. The second has a decay time much larger than the streak window and is shown in panel C. This DADS corresponds to the signaling state of LOV domains, i.e. it is the species associated difference spectrum (SADS) of the cysteine–flavin adduct, SA − SG. The adduct spectrum SA is obtained from stationary measurements and shows only one broad band with a maximum at 390 nm in the spectral range of our experiment. Within the spectral range shown here, the extinction coefficient of the adduct spectrum is larger than that of the ground state spectrum SG only in the small range between the isosbestic points at 373 and 404 nm. Note that both DADS in Fig. 3C and 4C are flat and coincident with the baseline at λ > 500 nm. This indicates that no radical species contribute to the photocycles of the wild-type domains on timescales greater than 5 μs.
The DADS in Fig. 3B and 4B correspond to the decay of the triplet state which occurs with time constants of 1.16 μs and 225 ns for LOV1 and LOV2, respectively. (In the first account of this decay in 2003, a double exponential fit with time constants of 0.8 μs and 4 μs was reported.20 As was shown later,19,21 samples of this preparation contained small amounts of free flavin. We measured a triplet decay time of 3.8 μs in the aerated buffer solution. In our present samples, free flavin was removed by dialysis. We observe always a single exponential decay in the wild-type domains.)
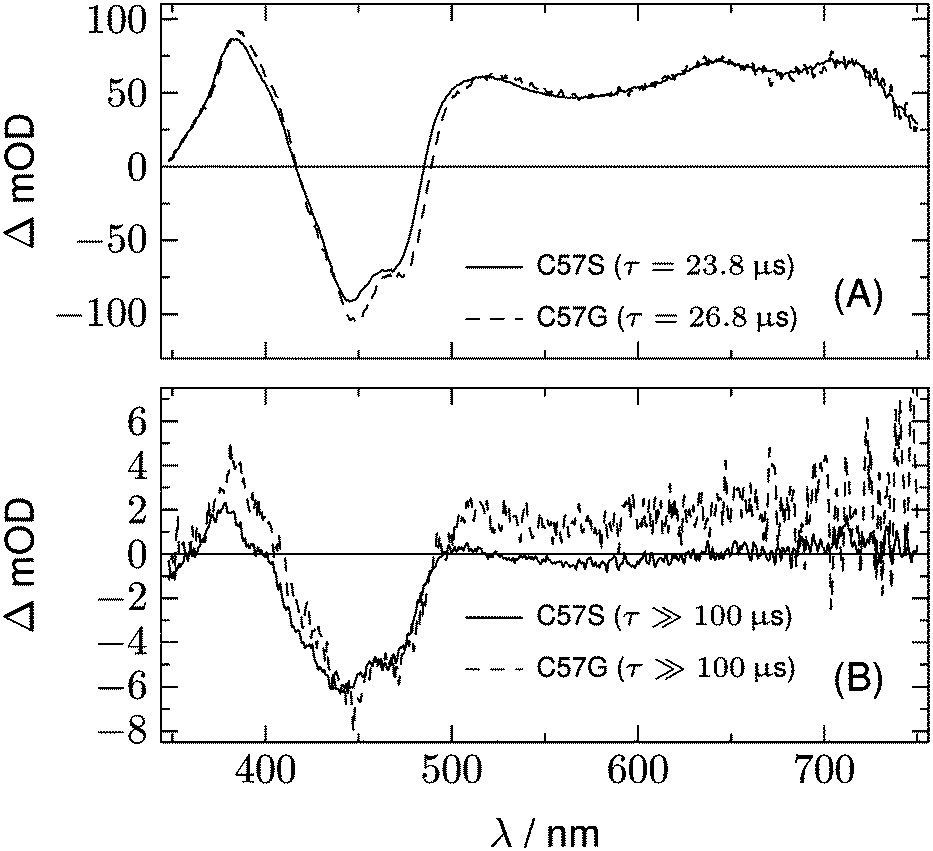
The datasets for the photoinactive mutants LOV1-C57S, LOV1-C57G, and LOV2-C250S can also be fitted with three components. The first is again the temporally unresolved fluorescence and laser stray light, and is not shown in the following figures. The second component is a DADS with a time constant of 23.8 μs for LOV1, 26.8 μs for LOV1-C57G, and 185 μs for LOV2. This is in good agreement with previously published triplet decay times for these species.21 The third component is weak and corresponds to a decay time beyond the streak window of 100 μs or 500 μs used for the LOV1- and LOV2-mutants, respectively.
Fig. 5 shows the two DADS for LOV1-C57S as the full lines, and the corresponding DADS of LOV1-C57G are also shown as dashed lines. The latter were scaled by a factor of 5.0 for better comparison. The first DADS is very similar for both mutants and corresponds to the decay of the triplet state of flavin. The second DADS of the C57G mutant looks also very similar to this triplet decay spectrum. Apparently, in this mutant the triplet state displays a biphasic decay with a slow component that has ca. 4% of the amplitude of the faster component. In degassed solution39 but also in the LOV2-C250S mutant, the flavin triplet decays with a time constant of ca. 200 μs, whereas the lifetime in LOV1-C57S is shorter by almost one order of magnitude. Apparently an additional quenching channel exists in LOV1 domains compared to LOV2 domains which could not be identified up to now. Calculations of the influence of the protein on the spin–orbit coupling have been reported,40–42 but these could not explain such a drastic effect. Hence the additional quenching channel has so far been unexplained. It is possible that the C57G mutant of LOV1 exists in a minor conformer in which this quenching channel is not active. Glycine requires less space than serine thus allowing for small molecules to enter the binding pocket next to the flavin. This has been shown for methylmercaptan35 and might also be true for one or two water molecules. Since the triplet decay is much slower in pure water, this might also cause a lengthening of the triplet decay time in the C57G mutant. The DADS with τ ≫ 100 μs of LOV1-C57S is, however, different from that of the C57G mutant (see Fig. 5B). The bleaching of the ground state is clearly visible, indicating that a small fraction of the triplets did not decay back to the ground state but to some other long-lived species. The DADS has no positive bands in the region 550–650 nm, hence this long-lived species cannot be the triplet or the neutral radical. The spectrum has some similarity to the second DADS of the wild-type domain. This could lead to the hypothesis that in the C57S mutant also an adduct is formed, albeit with rather low quantum yield and a lifetime in the range of a few ms only. The two spectra can indeed be overlaid nicely, however only with a shift of the DADS of the wild type by 8.5 nm to the blue. Since the DADS should be the difference of the species spectra of the unknown species minus that of the ground state, one should ask what other flavin species exist that might live for several 100 μs and have no absorption at λ > 550 nm.
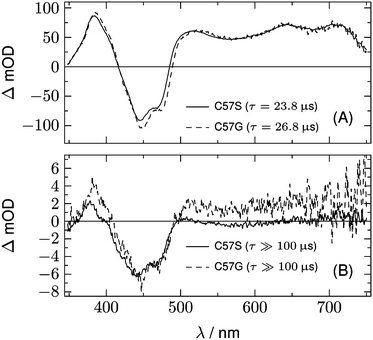 |
| | Fig. 5 DADS from the analysis of the 2D-TA data for LOV1-C57S (full lines) and LOV1-C57G (dashed lines). | |
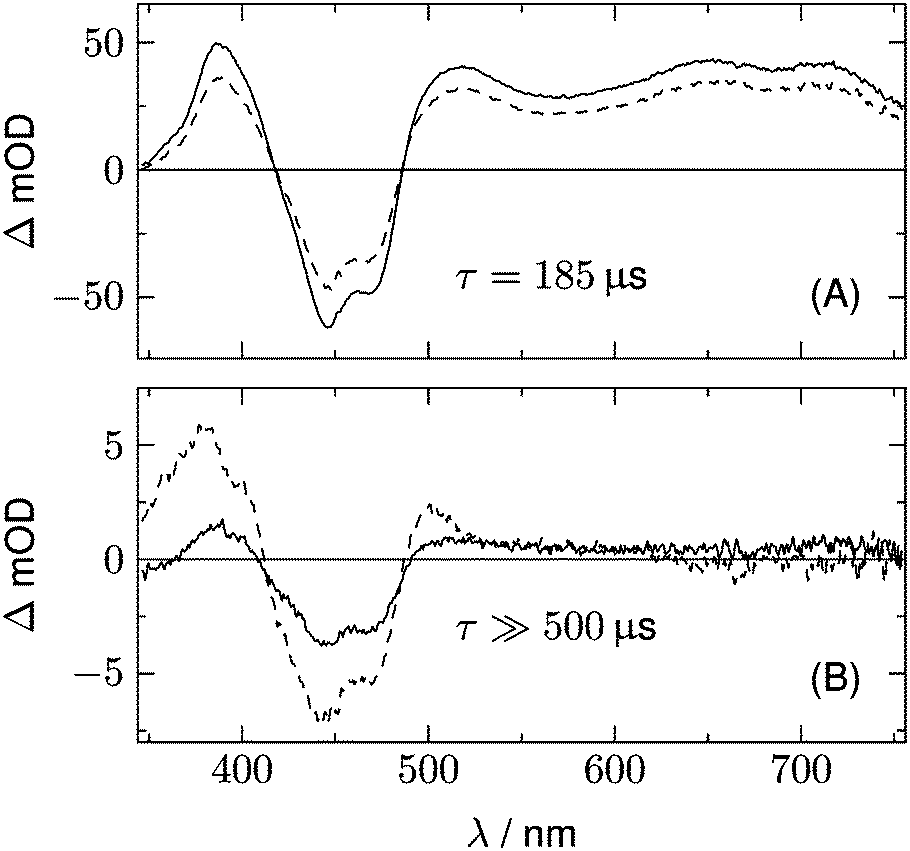
A further hint is found from the analysis of the data for LOV2-C250S. The two DADS of this domain are shown in Fig. 6. The first DADS corresponds to the well known triplet decay. The existence of a second DADS of LOV1-C57S as well as LOV2-C250S indicates that not all triplets decay to the ground state with the same time constant, but that a small fraction returns to the ground state on a longer timescale. In the second DADS of LOV2-C250S, a small positive amplitude is obvious in the range λ > 500 nm, which is only faintly seen in the case of LOV1-C57S. The corresponding species could be triplet states with a longer decay time, neutral radicals, or anion radicals. Formation of the radicals requires a reducing agent, which could be a tryptophane or a tyrosine within the protein. When the experiment is repeated with addition of 200 mM of β-mercaptoethanol (βME) to LOV2-C250S, the DADS shown as the dashed lines in Fig. 6 are found. Comparison of the two DADS in the lower panel shows that βME increased the yield of species absorbing at λ > 500 nm. The peak at 375 nm and the shoulder at 400 nm are characteristic of the flavin radical anion, in addition to a weaker band at 500 nm. The spectrum of the anion radical has been reported for a flavin in a cryptochrome under anaerobic conditions.43 There it shows a band at ca. 475 nm that extends to larger wavelengths than the first band of fully oxidized flavin, hence the difference spectrum shows a small positive maximum at 500 nm.
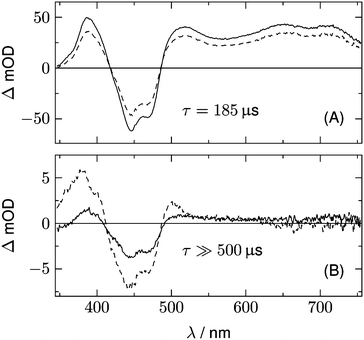 |
| | Fig. 6 DADS from the analysis of the 2D-TA data for LOV2-C250S (full lines). Dashed lines represent the corresponding spectra after addition of 200 mM of βME as the reducing agent. | |
It is surprising that this radical should be stable in a LOV domain under aerobic conditions on a timescale of 1 ms. Measurements in acetonitrile–water mixtures (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) have shown that the flavin radical anion is protonated with a time constant of 5.5 μs.44 In order to check protonation in a LOV domain we performed a series of measurements with the LOV1-C57S mutant at three pH values (6.5, 8.0, and 10.0) and in the presence and absence of βME. The results are collected in Fig. 7. In order to obtain more spectral information on the radical anion the spectral window was shifted by 50 nm to the blue. All datasets can be fitted well with two DADS, in addition to the fluorescence component. The first DADS corresponds to a lifetime of ca. 25 μs irrespective of the pH value and the presence or absence of βME. When scaled to the same value at 650 nm, all six spectra are identical within the noise level. The second DADS corresponds to lifetimes τ ≫ 100 μs. Even with the largest streak window of 500 μs no significant decay was observed. The three spectra for the different pH values in the absence of βME are rather similar (Fig. 7B), and the same is true for the three spectra in the presence of βME (Fig. 7C). However, the peaks at 375 and 500 nm as well as the shoulder at 400 nm increase significantly when βME is added. Apparently, the transient species is insensitive to pH, and the yield increases with addition of βME. We conclude that in both domains, LOV1 and LOV2, electron transfer from a mercaptan to flavin is possible, and that the majority of the resulting radical anions is not protonated for about 1 ms. Hence, if this electron transfer occurs from the cysteine as the electron donor in the wild-type domains, formation of the adduct could be the result of combination of the radical ion pair.
:1) have shown that the flavin radical anion is protonated with a time constant of 5.5 μs.44 In order to check protonation in a LOV domain we performed a series of measurements with the LOV1-C57S mutant at three pH values (6.5, 8.0, and 10.0) and in the presence and absence of βME. The results are collected in Fig. 7. In order to obtain more spectral information on the radical anion the spectral window was shifted by 50 nm to the blue. All datasets can be fitted well with two DADS, in addition to the fluorescence component. The first DADS corresponds to a lifetime of ca. 25 μs irrespective of the pH value and the presence or absence of βME. When scaled to the same value at 650 nm, all six spectra are identical within the noise level. The second DADS corresponds to lifetimes τ ≫ 100 μs. Even with the largest streak window of 500 μs no significant decay was observed. The three spectra for the different pH values in the absence of βME are rather similar (Fig. 7B), and the same is true for the three spectra in the presence of βME (Fig. 7C). However, the peaks at 375 and 500 nm as well as the shoulder at 400 nm increase significantly when βME is added. Apparently, the transient species is insensitive to pH, and the yield increases with addition of βME. We conclude that in both domains, LOV1 and LOV2, electron transfer from a mercaptan to flavin is possible, and that the majority of the resulting radical anions is not protonated for about 1 ms. Hence, if this electron transfer occurs from the cysteine as the electron donor in the wild-type domains, formation of the adduct could be the result of combination of the radical ion pair.
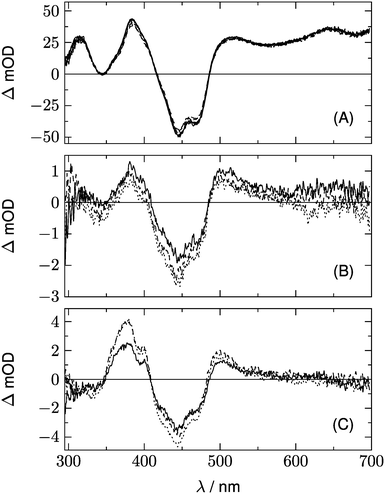 |
| | Fig. 7 Influence of pH and βME on the DADS of LOV1-C57S. (A) The first DADS, corresponding to a lifetime of ca. 25 μs, for pH = 6.5, 8.0, and 10.0. Full lines were measured in the absence of βME, dashed lines in the presence of βME. All curves were scaled to the same value at 650 nm. (B) The second DADS corresponding to quasi infinite lifetime, for pH = 6.5 (full line), 8.0 (dashed line), and 10.0 (dotted line) and in the absence of βME. (C) show the same spectra as (B) but with addition of βME (200 mM). | |
4. Discussion
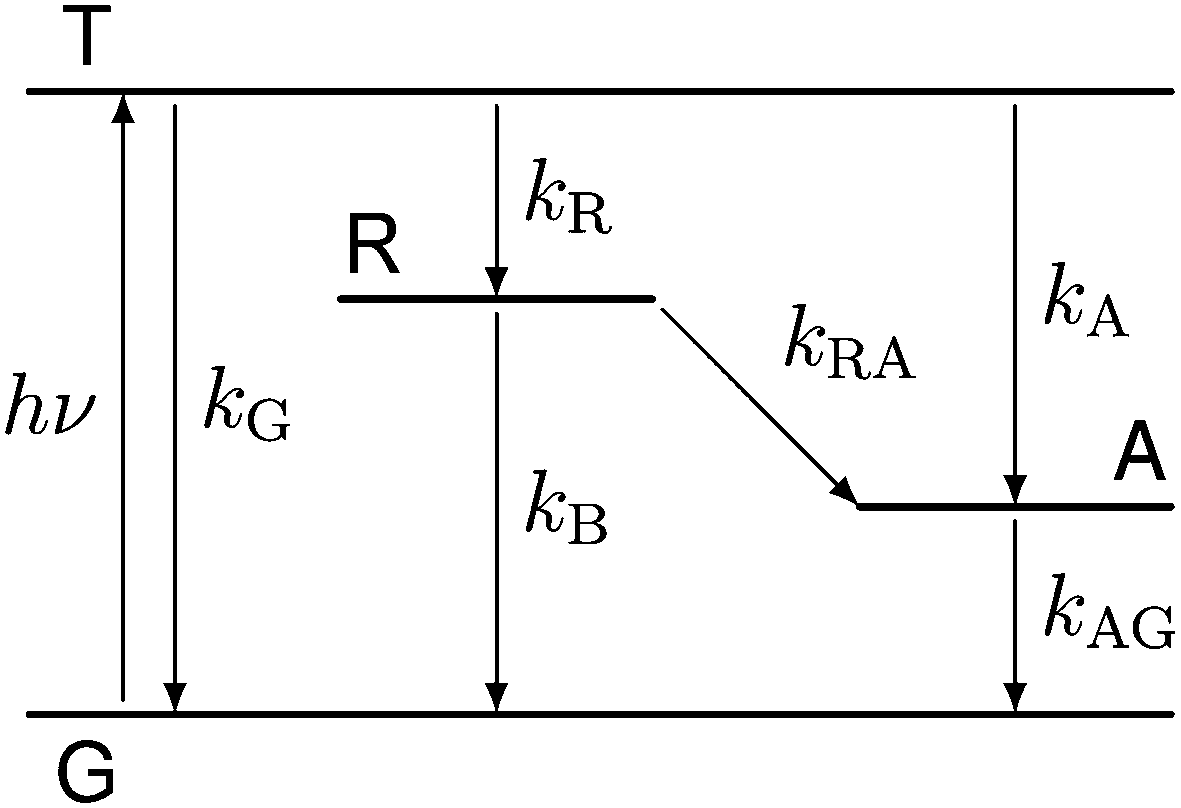
Our experiments show evidence for four transient species, namely the excited singlet state 1FMN, the triplet state 3FMN, the radical anion 2FMN˙−, and the adduct state between FMN and the active cysteine. In the following we shall denote these species as S, T, R, and A, respectively. The mechanism of the photocycle involves the ground state G of FMN as the fifth species. The decay of S occurs mostly to G by internal conversion and fluorescence, and to T by intersystem crossing. The rate constant of this decay, kS ≈ 3.4 × 108 s−1, is determined from measurements of the fluorescence decay.19,20 The decay of S is not resolved in our experiments. Hence we do not consider S in the kinetic scheme but assume that the initial state of our observation is the situation immediately after decay of the singlet. The basis of our discussion is thus the reaction scheme shown in Fig. 8. The rate constants kG, kR, and kA lead from the triplet state to the ground state, the radical, and the adduct, respectively. Two more rate constants kB and kRA account for the decay of the radical species back to the ground state or to the adduct. The reaction from the adduct back to the ground state is at least four orders of magnitude slower than all other reactions, i.e. on the millisecond timescale we can neglect the decay from A to G and set kAG = 0, whereas all other reactions are complete within this timescale. We assume that all these reactions are irreversible. This reaction scheme corresponds to a hierarchical ordering of the states T > R > A > G. As a consequence, the matrix of rate constants is lower triangular, and the set of differential equations has solutions for the concentrations cX(t) of each species X of the form| | 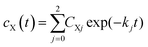 | (3) |
with
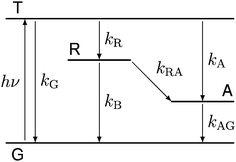 |
| | Fig. 8 Kinetic model used for data analysis, consisting of the ground state (G), the excited triplet state (T), the radical (R) and the adduct (A). | |
The transient absorption ΔE is given by
| | | ΔE = cT(t)ΔST + cR(t)ΔSR + cA(t)ΔSA = D0 + D1exp(−k1t) + D2exp(−k2t) | (7) |
In this expression, the ΔSX are the species associated difference spectra (SADS) SX − SG of the three species T, R, and A. The Dj are the decay associated difference spectra (DADS) for the three rate constants kj. The latter are the result of the global lifetime analysis. The species associated difference spectra are related to the DADS by
| | 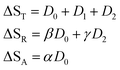 | (8) |
and the inverse relationship is
| | 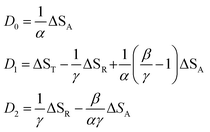 | (9) |
with
| | 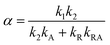 | (10) |
| | 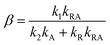 | (11) |
| | 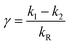 | (12) |
This model includes the photocycle of the mutant with a possible production of the radical in a side reaction (kA = kRA = 0). The hypothesis of R as an intermediate in the photocycle of the wild type is represented by kA = 0. If the radical in the wild type photocycle is not an intermediate but the result of a side reaction, this is accounted for by setting kRA = 0. According to eqn (8), the SADS of the triplet state is always given by the sum of all DADS, the SADS of the adduct state is always proportional to the DADS D0 with an infinite lifetime (i.e. much larger than the time window TW of the experiment), and the DADS D1 contributes only to the SADS of the triplet state. The models then only differ in the weights that are assigned to D0 and D2 in forming the SADS of the radical.
However, in all our experiments only two decay components (ka, kb) could be resolved. One corresponds to the decay of the triplet state (ka = k1), and the other is quasi constant (1/TW ≫ kb ≈ 0), where TW is the streak window. In the wild type domain the latter is obviously the SADS of the adduct. This means that k2 is either so small that it cannot be distinguished from k0, or so large that this component decays faster than the time resolution of our experiment (ca. 10 ns). Let us consider these two cases separately.
The observation made for the mutant is in favor of the first alternative: here we see a long-lived component with the signature of the radical, i.e. kB < 104 s−1. The DADS in this case are given by
| | 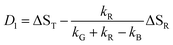 | (14) |
| |  | (15) |
Richter et al.45 have reported the formation of the radical anion in the C450A mutant of LOV2 from Avena sativa and assigned a tryptophan as the electron donor. LOV1 of C.r. has a tryptophan Trp98 at a distance of ca. 8.8 Å from the flavin that could act as the electron donor. Kay et al.46 also observed the anion radical in At-LOV2-C450A indirectly as a transient in nuclear polarization experiments, and the neutral radical in stationary UV/Vis spectra and EPR. Massey and Palmer47 have shown that the protein environment strongly influences the pKa of the neutral flavin radical. Hence, for a given pH, either the neutral radical or the anion radical can be the stationary stable form, whereas the other form is a transient. In Cr-LOV1 the neutral radical is in the stable form near pH = 8.21,33–35,38 Slow protonation of the radical anion has also been found for a photolyase,48 and even for a flavin outside a protein in an acetonitrile–water mixture.44 We observed that adding β-mercaptoethanol as an electron donor to the solution leads to a significant increase in the signature of the radical in the DADS of the long lived component. From measurements on riboflavin in solution we estimate49 that the radical anion has about twice the extinction coefficient at 500 nm as the triplet. Miura50 presents, apparently as a private communication, UV/Vis spectra of the neutral oxidized form as well as the neutral and anion radicals of d-amino acid oxidase that show a common isosbestic point at 500 nm with an extinction coefficient of ε = 4 mM−1 cm−1. Unfortunately, no experimental details are given. With these data we estimate the yield of the radical anion in LOV1-C57S of ca. 2% if no other species is produced that absorbs at 500 nm. If neutral tryptophan radicals are produced as the counter-radicals, this estimate reduces to ca. 1%.51 After addition of 200 mM of βME the yield increases to ca. 4%.
If the radical is not an intermediate in the photocycle of the wild type protein, then it will only be observed as a long-lived byproduct like in the mutants. In this case, k2 is much smaller than the inverse of the streak window, hence the observed DADS for k ≈ 0 is the sum of the model DADS D0 + D2. The latter is a superposition of the spectra of the adduct and radical,
| | | D0 + D2 = ΦAΔSA + ΦRΔSR | (16) |
where
ΦA and
ΦR are the quantum yields of formation of the adduct and long-lived radical from the excited triplet state. However, the corresponding DADS of LOV1-wt and LOV2-wt, shown in panel C of
Fig. 3 and
4, respectively, show no trace of any radical species, neither the neutral radical nor the anion. We conclude that the reaction leading to the adduct in the wild type domains is much faster than the reaction leading to long-lived radicals in the case of the mutants.
Let us now consider the second case, i.e. the radical is an essential intermediate in the formation of the adduct. Then kRA cannot be smaller than the inverse rise time of the adduct. Since we do not observe a second fast component in this lifetime range, we must conclude that k2 ≈ kRA is too large to be resolved by our experiment. The latter is limited by the duration of the excitation laser and the fluorescence decay. In this limit, the sum of all observed DADS is given by
| | 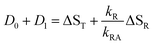 | (17) |
i.e., the signature of the radical should appear in the sum of the two observed DADS. In the case of the mutants, however, formation and further reactions of the radicals are slow and resolved in the observed DADS. Hence for the mutants the sum of all DADS corresponds to the transient spectrum extrapolated to
t = 0,
i.e. to ΔS
T. In
Fig. 9 these sums of all DADS are compared for the wild type and the mutants, where all spectra were scaled to the same intensity in the range 650–710 nm where the triplet is the only absorbing species. In the spectral range above 500 nm these curves are in agreement within the noise level,
i.e. no significant contribution of the radical spectrum can be detected. The SADS of the neutral radical should have a band in the region 500–620 nm with a similar intensity to the triplet spectrum. The differences of the spectra in
Fig. 9 are so small in this spectral region that we can estimate
kR/
kRA < 0.05 if the radical intermediate is the neutral radical. The SADS of the radical anion should show bands at 375 and 500 nm. This region overlaps already with the rising edge of the ground state absorption. The spectra in
Fig. 9 differ slightly in the region of the ground state bleach (410–480 nm). Apparently, not only the peak positions but also the oscillator strength of the ground state absorption (relative to that of the triplet bands) differs slightly for the various mutants. However, the intensity of the wild type spectra in
Fig. 9 is below that of the mutant spectra in the region around 500 nm, whereas the spectrum ΔS
R should be positive. Hence we conclude that also for the radical anion a contribution to the sum of all DADS is rather unlikely, and estimate
kR/
kRA < 0.05. Thus the transient spectra give no evidence for participation of the radical in the mechanism of the adduct formation. This would lead to the conclusion that the adduct is either formed by a concerted reaction, or the adduct formation from the radical is
ca. 20 times faster than the formation of the radical
via decay of the triplet by electron transfer, so that no measurable transient concentration of the radical can accumulate.
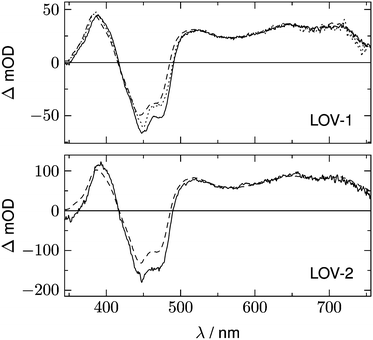 |
| | Fig. 9 Full lines represent the sum of all DADS for LOV1-wt (upper panel) and LOV2-wt (lower panel). The corresponding sum of DADS for the mutants LOV1-C57S (upper panel) and LOV2-C250S (lower panel) is shown as dashed lines, the sum of DADS for the LOV1-C57G mutants is shown with the dotted line. The spectra of the mutants are scaled to the same intensity as the wild types in the range 650–710 nm. For discussion see text. | |
There is, however, indirect evidence for the participation of an intermediate in adduct formation. This originates from a discussion of the quantum yield ΦA of the adduct formed from the triplet. If no intermediate is involved in that reaction, this quantum yield is given by the rate constants as
| | 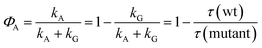 | (18) |
In the last step we have assumed that kG is the same for a wild type domain and the corresponding mutant. With the observed triplet decay times we obtain ΦA = 0.95 for LOV1 and ΦA > 0.99 for LOV2. The quantum yield of the adduct can also be obtained from the DADS. If we assume that the adduct is formed via an intermediate (kA = 0) and that this intermediate reacts much faster than it is formed (kRA ≫ kR) we obtain
| | 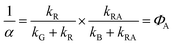 | (19) |
| |  | (20) |
with the consequence
| | | D1 = ST − ΦASA − ΦGSG | (22) |
In these expressions, SX is the species associated spectrum of species X, and ΦX is the quantum yield of species X from the triplet state. We now form a linear combination of these DADS
| | | Dη = D1 − ηD0 = ST − ΦASA(1 + η) − SG(ΦG − ηΦA) | (23) |
with
η =
ΦG/
ΦA the contribution of S
G vanishes from this linear combination. Considering that
ΦA +
ΦG = 1 we find in this case
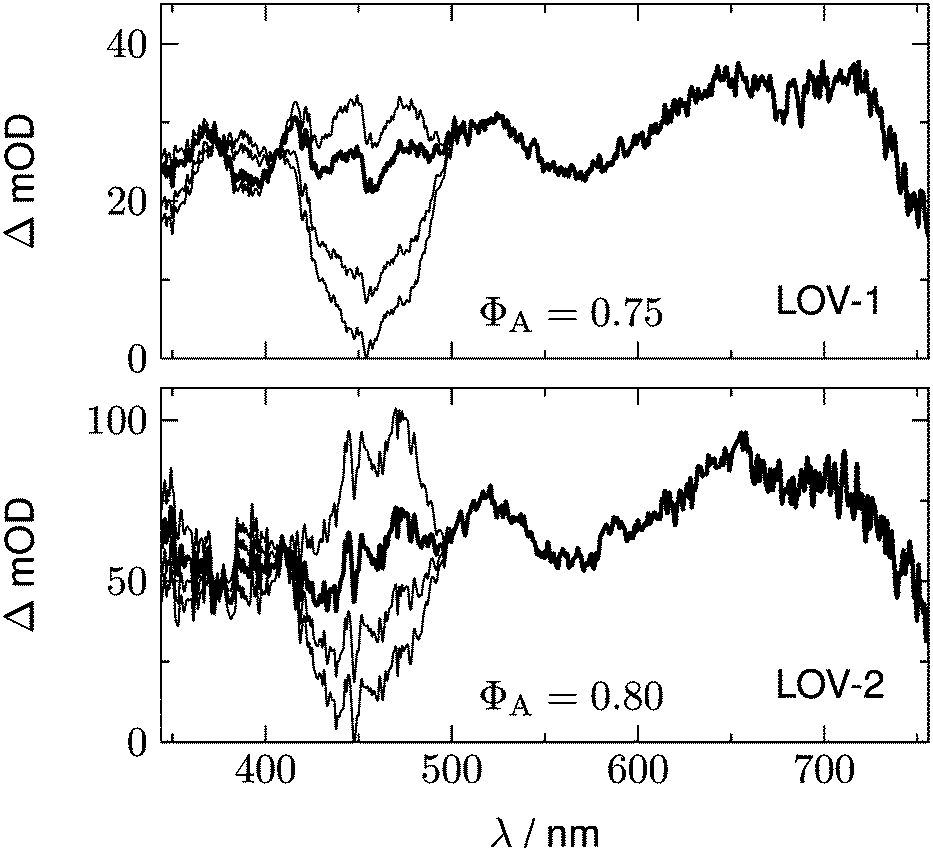
Fig. 10 shows a series of Dη for both LOV domains with ΦA varied in the range 0.7 < ΦA < 1.0. We expect only a very small contribution of the adduct spectrum in the range λ > 420 nm. According to published spectra of flavin triplets,52 the extinction coefficient of the triplet spectrum at 450 nm should be equal to or higher than that at the minimum at 550 nm. However, if we assume ΦA = 1.0 (lowest curves in Fig. 10), the triplet spectrum goes to zero at 450 nm. In order to fulfil the condition given above, we have to assume ΦA ≈ 0.75 for LOV1, and ΦA ≈ 0.80 for LOV2 (thick lines in Fig. 10). These quantum yields are smaller than those calculated from the ratio of the triplet decay rates of wild types and mutants. The discrepancy can be explained if the ratio of the triplet decay rates is assigned to the yield of the intermediate. This means that the radical anion is formed with a quantum yield of 0.95 (LOV1) and 0.99 (LOV2), respectively. The radical anion subsequently reacts to form the adduct or decay back to the ground state with a branching ratio of ca. 4:1.
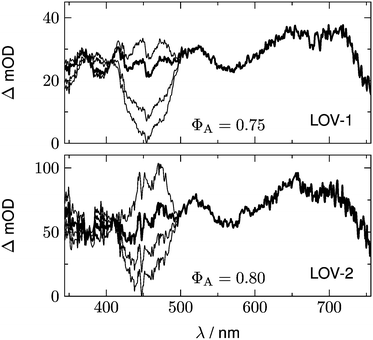 |
| | Fig. 10 Difference of the DADS, D1 − ηD0 for LOV1-wt (upper panel) and LOV2-wt (lower panel). In each panel, the scaling factor η corresponds to ΦA = 1.0 for the lowest curve, ΦA = 0.9 for the next curve, and ΦA = 0.7 for the highest curve. Best agreement with the published triplet spectrum of flavin is obtained with ΦA = 0.75 for LOV1 and ΦA = 0.80 for LOV2 (thicker curves). | |
5. Summary
The transient absorption data for all wild-type domains and mutants studied show, in addition to the unresolved fluorescence decay, one resolved exponentially decaying component and one component that is non-decaying within the time window of the streak camera measurement. The temporally resolved component always contains the signature of the flavin triplet state, decaying in 0.225, 1.16, 23.8, 26.8, and 185 μs for LOV2-wt, LOV1-wt, LOV1-C57S, LOV1-C57G, and LOV2-C250S, respectively.
The second DADS of the photoinactive mutants have rather small amplitude, but the bleaching of the ground state is clearly visible. This indicates that ca. 2–4% of the excited molecules did not return to the dark state within the time window of the experiment. A weak positive absorption near 500 nm hints to the formation of the flavin radical; in LOV1-C57G and LOV2-C250S a long-lived triplet component seems also to contribute. The second DADS of LOV1-C57S contains the signature of the flavin radical anion FMN˙−. The yield of this species increases when reducing agents are added to the solution, but is insensitive to pH in the range 6.5–10. The fact that the radical anion is observed on timescales of several 100 μs is surprising. Apparently, no acidic protons are available inside the binding pocket of the mutants, and the protein environment effectively shields the radical from the external water. Since the LOV mutants recover completely before the next laser excitation, all radical species are apparently oxidized back to the fully oxidized flavin by molecular oxygen on the ms timescale.
We do not find any evidence of radical species in the photocycles of the wild-type domains. Instead, there is no measurable delay between the decay of the triplet and the formation of the adduct. If electron transfer from cysteine to FMN initiates the reaction, it must be the rate limiting step, i.e. the following steps of protonation and/or radical combination must be much faster. Thus the mechanism appears to be concerted. However, whereas the ratio of the triplet decay rates for wild types and mutants indicates an efficiency of ΦA > 0.95 for the formation of an intermediate from the triplet in the wild-type domains, the transient spectra are more in agreement with a quantum yield of ΦA < 0.80 of the adduct. This can be explained with the assumption that the triplet decay leads to a radical species with a rate constant kR which subsequently reacts to form the adduct with a rate constant kRA > 20kR.
Acknowledgements
RJK acknowledges a postdoctoral fellowship by the German Science Foundation (Deutsche Forschungsgemeinschaft, DFG) as a fellow of the Research Training Group (GRK-1626) “Photocatalysis”. The streak camera used in this work was a grant by the DFG (project Di299/6 as part of FOR526).
References
- J. M. Christie, M. Salomon, K. Nozue, M. Wada and W. R. Briggs, LOV (light, oxygen, or voltage) domains of the blue-light photoreceptor phototropin (nph1): Binding sites for the chromophore flavin mononucleotide, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8779–8783 CrossRef CAS.
- J. A. Jarillo, H. Gabrys, J. Capel, J. M. Alonso, J. R. Ecker and A. R. Cashmore, Phototropin-related NPL1 controls chloroplast relocation induced by blue light, Nature, 2001, 410, 952–954 CrossRef CAS PubMed.
- T. Kagawa, T. Sakai, N. Suetsugu, K. Oikawa, S. Ishiguro, T. Kato, S. Tabata, K. Okada and M. Wada, Arabidopsis NPL1: A phototropin homolog controlling the chloroplast high-light avoidance response, Science, 2001, 291, 2138–2141 CrossRef CAS PubMed.
- T. Kinoshita, M. Doi, N. Suetsugu, T. Kagawa, M. Wada and K.-i. Shimazaki, phot1 and phot2 mediate blue light regulation of stomatal opening, Nature, 2001, 414, 656–660 CrossRef CAS PubMed.
- J. M. Christie, P. Reymond, G. K. Powell, P. Bernasconi, A. A. Raibekas, E. Liscum and W. R. Briggs, Arabidopsis NPH1: a flavoprotein with the properties of a photoreceptor for phototropism, Science, 1998, 282, 1698–1701 CrossRef CAS.
- E. Huala, P. W. Oeller, E. Liscum, I.-S. Han, E. Larsen and W. R. Briggs, Arabidopsis NPH1: a protein kinase with a putative redox-sensing domain, Science, 1997, 278, 2120–2123 CrossRef CAS.
- K. Huang and C. F. Beck, Phototropin is the blue-light receptor that controls multiple steps in the sexual life cycle of the green alga Chlamydomonas reinhardtii, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6269–6274 CrossRef CAS PubMed.
- M. Salomon, J. M. Christie, E. Knieb, U. Lempert and W. R. Briggs, Photochemical and Mutational Analysis of the FMN-Binding Domains of the Plant Blue Light Receptor, Phototropin, Biochemistry, 2000, 39, 9401–9410 CrossRef CAS PubMed.
- T. E. Swartz, S. B. Corchnoy, J. M. Christie, J. W. Lewis, I. Szundi, W. R. Briggs and R. A. Bogomolni, The photocycle of a flavin-binding domain of the blue light photoreceptor phototropin, J. Biol. Chem., 2001, 276, 36493–36500 CrossRef CAS PubMed.
- J. T. M. Kennis, S. Crosson, M. Gauden, I. H. M. van Stokkum, K. Moffat and R. van Grondelle, Primary Reactions of the LOV2 Domain of Phototropin, a Plant Blue-Light Photoreceptor, Biochemistry, 2003, 42, 3385–3392 CrossRef CAS PubMed.
- A. Losi, Flavin-based blue-light photosensors: a photobiophysics update, Photochem. Photobiol., 2007, 83, 1283–1300 CrossRef CAS PubMed.
- S.-H. Song, D. Madsen, J. B. van der Steen, R. Pullman, L. H. Freer, K. J. Hellingwerf and D. S. Larsen, Primary Photochemistry of the Dark- and Light-Adapted States of the YtvA Protein from Bacillus subtilis, Biochemistry, 2013, 52, 7951–7963 CrossRef CAS PubMed.
- M. Salomon, W. Eisenreich, H. Dürr, E. Schleicher, E. Knieb, V. Massey, W. Rüdiger, F. Müller, A. Bacher and G. Richter, An optomechanical transducer in the blue light receptor phototropin from Avena sativa, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12357–12361 CrossRef CAS PubMed.
- S. Crosson and K. Moffat, Photoexcited Structure of a Plant Photoreceptor Domain Reveals a Light-Driven Molecular Switch, Plant Cell, 2002, 14, 1067–1075 CrossRef CAS.
- R. Fedorov, I. Schlichting, E. Hartmann, T. Domratcheva, M. Fuhrmann and P. Hegemann, Crystal structures and molecular mechanism of a light-induced signaling switch: The Phot-LOV1 domain from Chlamydomonas reinhardtii, Biophys. J., 2003, 84, 2474–2482 CrossRef CAS.
- A. Losi, B. Quest and W. Gaertner, Listening to the blue: the time-resolved thermodynamics of the bacterial blue-light receptor YtvA and its isolated LOV domain, Photochem. Photobiol. Sci., 2003, 2, 759–766 CAS.
- S. Kikuchi, M. Unno, K. Zikihara, S. Tokutomi and S. Yamauchi, Vibrational Assignment of the Flavin-Cysteinyl Adduct in a Signaling State of the LOV Domain in FKF1, J. Phys. Chem. B, 2009, 113, 2913–2921 CrossRef CAS PubMed.
- Y. Nakasone, K. Zikihara, S. Tokutomi and M. Terazima, Kinetics of Conformational Changes of the FKF1-LOV Domain upon Photoexcitation., Biophys. J., 2010, 99, 3831–3839 CrossRef CAS PubMed.
- W. Holzer, A. Penzkofer, M. Fuhrmann and P. Hegemann, Spectroscopic characterization of flavin mononucleotide bound to the LOV1 domain of Phot1 from Chlamydomonas reinhardtii, Photochem. Photobiol., 2002, 75, 479–487 CrossRef CAS.
- T. Kottke, J. Heberle, D. Hehn, B. Dick and P. Hegemann, Phot-LOV1: Photocycle of a blue-light receptor domain from the green alga Chlamydomonas reinhardtii, Biophys. J., 2003, 84, 1192–1201 CrossRef CAS.
- H. Guo, T. Kottke, P. Hegemann and B. Dick, The phot LOV2 domain and its interaction with LOV1, Biophys. J., 2005, 89, 402–412 CrossRef CAS PubMed.
- T. Bednarz, A. Losi, W. Gaertner, P. Hegemann and J. Heberle, Functional variations among LOV domains as revealed by FT-IR difference spectroscopy, Photochem. Photobiol. Sci., 2004, 3, 575–579 CAS.
- M. A. van der Horst and K. J. Hellingwerf, Photoreceptor proteins, “star actors of modern times”: a review of the functional dynamics in the structure of representative members of six different photoreceptor families, Acc. Chem. Res., 2004, 37, 13–20 CrossRef CAS PubMed.
- A. Moeglich, X. Yang, R. A. Ayers and K. Moffat, Structure and function of plant photoreceptors, Annu. Rev. Plant Biol., 2010, 61, 21–47 CrossRef PubMed.
- O. P. Ernst, D. T. Lodowski, M. Elstner, P. Hegemann, L. S. Brown and H. Kandori, Microbial and animal rhodopsins: Structures, functions, and molecular mechanisms, Chem. Rev., 2014, 114, 126–163 CrossRef CAS PubMed.
- M. T. A. Alexandre, T. Domratcheva, C. Bonetti, L. J. G. W. van Wilderen, R. van Grondelle, M.-L. Groot, K. J. Hellingwerf and J. T. M. Kennis, Primary reactions of the LOV2 domain of phototropin studied with ultrafast mid-infrared spectroscopy and quantum chemistry, Biophys. J., 2009, 97, 227–237 CrossRef CAS PubMed.
- C. Thoeing, A. Pfeifer, S. Kakorin and T. Kottke, Protonated triplet-excited flavin resolved by step-scan FTIR spectroscopy: implications for photosensory LOV domains, Phys. Chem. Chem. Phys., 2013, 15, 5916–5926 RSC.
- C. Neiss and P. Saalfrank, Ab initio quantum chemical investigation of the first steps of the photocycle of phototropin: a model study, Photochem. Photobiol., 2003, 77, 101–109 CrossRef CAS.
- E. Schleicher, R. M. Kowalczyk, C. W. M. Kay, P. Hegemann, A. Bacher, M. Fischer, R. Bittl, G. Richter and S. Weber, On the Reaction Mechanism of Adduct Formation in LOV Domains of the Plant Blue-Light Receptor Phototropin, J. Am. Chem. Soc., 2004, 126, 11067–11076 CrossRef CAS PubMed.
- M. Dittrich, P. L. Freddolino and K. Schulten, When light falls in LOV: A quantum mechanical/molecular mechanical study of photoexcitation in Phot-LOV1 of Chlamydomonas reinhardtii, J. Phys. Chem. B, 2005, 109, 13006–13013 CrossRef CAS PubMed.
- T. Domratcheva, R. Fedorov and I. Schlichting, Analysis of the Primary Photocycle Reactions Occurring in the Light, Oxygen, and Voltage Blue-Light Receptor by Multiconfigurational Quantum-Chemical Methods, J. Chem. Theory Comput., 2006, 2, 1565–1574 CrossRef CAS.
- S. B. Corchnoy, T. E. Swartz, J. W. Lewis, I. Szundi, W. R. Briggs and R. A. Bogomolni, Intramolecular Proton Transfers and Structural Changes during the Photocycle of the LOV2 Domain of Phototropin 1, J. Biol. Chem., 2003, 278, 724–731 CrossRef CAS PubMed.
- G. Noell, G. Hauska, P. Hegemann, K. Lanzl, T. Noell, M. von Sanden-Flohe and B. Dick, Redox properties of LOV domains: chemical versus photochemical reduction, and influence on the photocycle, ChemBioChem, 2007, 8, 2256–2264 CrossRef CAS PubMed.
- K. Lanzl, G. Noell and B. Dick, LOV1 protein from Chlamydomonas reinhardtii is a template for the photoadduct formation of FMN and methylmercaptane, ChemBioChem, 2008, 9, 861–864 CrossRef CAS PubMed.
- K. Lanzl, M. v. Sanden-Flohe, R.-J. Kutta and B. Dick, Photoreaction of mutated LOV photoreceptor domains from Chlamydomonas reinhardtii with aliphatic mercaptans: Implications for the mechanism of wild-type LOV, Phys. Chem. Chem. Phys., 2010, 12, 6594–6604 RSC.
- C. Bauer, C.-R. Rabi, J. Heberle and T. Kottke, Indication for a radical intermediate preceding the signaling state in the LOV domain photocycle, Photochem. Photobiol., 2011, 87, 548–553 CrossRef CAS PubMed.
- R.-J. Kutta, T. Langenbacher, U. Kensy and B. Dick, Setup and performance of a streak camera apparatus for transient absorption measurements in the ns to ms range, Appl. Phys. B: Lasers Opt., 2013, 111, 203–216 CrossRef CAS.
- T. Kottke, B. Dick, R. Fedorov, I. Schlichting, R. Deutzmann and P. Hegemann, Irreversible Photoreduction of Flavin in a Mutated Phot-LOV1 Domain, Biochemistry, 2003, 42, 9854–9862 CrossRef CAS PubMed.
- B. J. Fritz, K. Matsui, S. Kasai and A. Yoshimura, Triplet lifetimes of some flavins, Photochem. Photobiol., 1987, 45, 539–541 CrossRef CAS PubMed.
- K. Zenichowski, M. Gothe and P. Saalfrank, Exciting flavins: Absorption spectra and spin–orbit coupling in light-oxygen-voltage (LOV) domains, J. Photochem. Photobiol., A, 2007, 190, 290–300 CrossRef CAS PubMed.
- S. Salzmann, V. Martinez-Junza, B. Zorn, S. E. Braslavsky, M. Mansurova, C. M. Marian and W. Gaertner, Photophysical Properties of Structurally and Electronically Modified Flavin Derivatives Determined by Spectroscopy and Theoretical Calculations, J. Phys. Chem. A, 2009, 113, 9365–9375 CrossRef CAS PubMed.
- S. Salzmann and C. M. Marian, The photophysics of alloxazine: a quantum chemical investigation in vacuum and solution., Photochem. Photobiol. Sci., 2009, 8, 1655–1666 CAS.
- B. Liu, H. Liu, D. Zhong and C. Lin, Searching for a photocycle of the cryptochrome photoreceptors, Curr. Opin. Plant Biol., 2010, 13, 578–586 CrossRef CAS PubMed.
- U. Megerle, M. Wenninger, R.-J. Kutta, R. Lechner, B. König, B. Dick and E. Riedle, Unraveling the flavin-catalyzed photooxidation of benzylic alcohol with transient absorption spectroscopy from sub-pico- to microseconds., Phys. Chem. Chem. Phys., 2011, 13, 8869–8880 RSC.
- G. Richter, S. Weber, W. Roemisch, A. Bacher, M. Fischer and W. Eisenreich, Photochemically Induced Dynamic Nuclear Polarization in a C450A Mutant of the LOV2 Domain of the Avena sativa Blue-Light Receptor Phototropin, J. Am. Chem. Soc., 2005, 127, 17245–17252 CrossRef CAS PubMed.
- C. W. M. Kay, E. Schleicher, A. Kuppig, H. Hofner, W. Ruediger, M. Schleicher, M. Fischer, A. Bacher, S. Weber and G. Richter, Blue light perception in plants., J. Biol. Chem., 2003, 278, 10973–10982 CrossRef CAS PubMed.
- V. Massey and G. Palmer, The existence of spectrally distinct classes of flavoprotein semiquinones. A new method for the quantitative production of flavoprotein semiquinones, Biochemistry, 1966, 5, 3181–3189 CrossRef CAS.
- K. B. Henbest, K. Maeda, P. J. Hore, M. Joshi, A. Bacher, R. Bittl, S. Weber, C. R. Timmel and E. Schleicher, Magnetic-field effect on the photoactivation reaction of Escherichia coli DNA photolyase, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14395–14399 CrossRef CAS PubMed.
-
R. J. Kutta, U. Kensy and B. Dick, Measuring photochemical intermediates with a streak camera – a new technique for transient absorption, in Chemical Photocatalysis, ed. B. König De Gruyter, 2013 Search PubMed.
- R. Miura, Versatility and specificity in flavoenzymes: control mechanisms of flavin reactivity, Chem. Rec., 2001, 1, 183–194 CrossRef CAS PubMed.
- T. Langenbacher, D. Immeln, B. Dick and T. Kottke, Microsecond Light-induced Proton Transfer to Flavin in the Blue Light Sensor Plant Cryptochrome, J. Am. Chem. Soc., 2009, 131, 14274–14280 CrossRef CAS PubMed.
- M. Sakai and H. Takahashi, One-electron photoreduction of flavin mononucleotide: time-resolved resonance Raman and absorption study, J. Mol. Struct., 1996, 379, 9–18 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4pp00155a |
|
| This journal is © The Royal Society of Chemistry and Owner Societies 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a