
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of RO3201195 and a pyrazolyl ketone P38 MAPK inhibitor library on the proliferation of Werner syndrome cells†
Mark C.
Bagley
*a,
Jessica E.
Dwyer
a,
Mohammed
Baashen‡
a,
Matthew C.
Dix
b,
Paola G. S.
Murziani
b,
Michal J.
Rokicki
c,
David
Kipling
c and
Terence
Davis
*c
aDepartment of Chemistry, School of Life Sciences, University of Sussex, Falmer, Brighton, East Sussex, BN1 9QJ, UK. E-mail: m.c.bagley@sussex.ac.uk
bSchool of Chemistry, Main Building, Cardiff University, Park Place, Cardiff, CF10 3AT, UK
cInstitute of Cancer and Genetics, Cardiff University School of Medicine, Heath Park, Cardiff, CF14 4XN, UK
First published on 12th November 2015
Abstract
Microwave-assisted synthesis of the pyrazolyl ketone p38 MAPK inhibitor RO3201195 in 7 steps and 15% overall yield, and the comparison of its effect upon the proliferation of Werner Syndrome cells with a library of pyrazolyl ketones, strengthens the evidence that p38 MAPK inhibition plays a critical role in modulating premature cellular senescence in this progeroid syndrome and the reversal of accelerated ageing observed in vitro on treatment with SB203580.
Introduction
P38α is one isoform of the mitogen-activated protein kinase (MAPK) intracellular enzymes which are central to the regulation of cytokine biosynthesis and inflammatory cell signalling.1,2 The reduction of pro-inflammatory cytokine levels offers a means for the treatment of inflammatory disorders such as rheumatoid arthritis and atherosclerosis.3,4 In addition to inflammatory disorders, p38α is known to play a role in the resistance of various cancers to chemotherapy, cancer proliferation and metastasis.5,6 This makes p38α a compelling therapeutic target for the design of safe and efficacious inhibitors suitable for clinical investigation.2,7 Following the discovery that pyridinylimidazole p38α inhibitors such as SB203580 mediate multiple cellular responses,1 including the production of inflammatory cytokines, a wide variety of structurally-distinct chemotypes8,9 have been discovered to inhibit this enzyme with notable differences in binding motif.10 Many of these have been used in stage II or III clinical trials.11Werner syndrome (WS) is a rare autosomal recessive disorder.12 The mutated gene (WRN) encodes for a RecQ helicase involved in DNA replication, recombination and repair. Individuals living with the syndrome display the premature onset of many of the clinical features of old age, show early susceptibility to a number of major age-related diseases and have an abbreviated median life expectancy (47 years). Consequently, WS is widely used as a model disease to investigate the mechanisms underlying normal human ageing. Associated with these aged features, fibroblasts from WS individuals have a much-reduced replicative capacity when compared with fibroblasts from normal individuals, and have a stressed, aged, morphology and high levels of activated p38α.13–15 This accelerated cellular senescence is thought to underlie whole body ageing in this syndrome. As part of our interest in mechanisms of premature cell senescence, we showed that administering the p38α inhibitor SB203580 to WS cells rescues all of the features of accelerated replicative decline.13 This observation suggested that the abbreviated life span of WS cells is due to stress-induced growth arrest mediated by p38α MAPK, which we speculate is transduced from the frequently stalled DNA replication forks observed in these cells.16 It also offers a means to clinically regulate this process and, by therapeutic means, intervene in a premature ageing syndrome.16
However, it has been shown that SB203580 is not specific for p38α, but also inhibits the related N-terminal c-Jun kinases (JNKs) that may play a role in cellular senescence.17–19 Thus more work is needed using inhibitors that do not target the JNKs to verify the role played by p38α in WS cells. In addition, SB203580 is not suitable for possible in vivo use.11
This manuscript describes a rapid route to one such inhibitor, RO3201195 (1),20 the study of this inhibitor in WS cells, and its comparison with the canonical p38α inhibitor SB203580 to investigate the role of p38α MAPK signal transduction in replicative senescence. This pyrazolyl ketone p38α MAPK inhibitor progressed to clinical trials, is orally bioavailable, exhibits high kinase selectivity, and inhibited the production of the cytokines IL-6 and TNFα upon lipopolysaccharide (LPS) challenge in rats.20 Thus, RO3201195 could be a valuable chemical tool in dissecting the accelerated ageing pathophysiology seen in WS cells and its rescue in vitro using small molecule inhibitors of this stress signalling pathway.13,21 Our rapid approach to the inhibitor employs microwave irradiation to accelerate a number of steps, preparing the central pyrazole motif through cyclocondensation of hydrazine 3 and a suitably protected benzoylnitrile 2, derivatized by homologation prior to heterocycle formation (Fig. 1). This approach had been successful for the synthesis of a pyrazolyl ketone library (4)22 and so should be suitable for rapid production of the target inhibitor under microwave-assisted conditions.
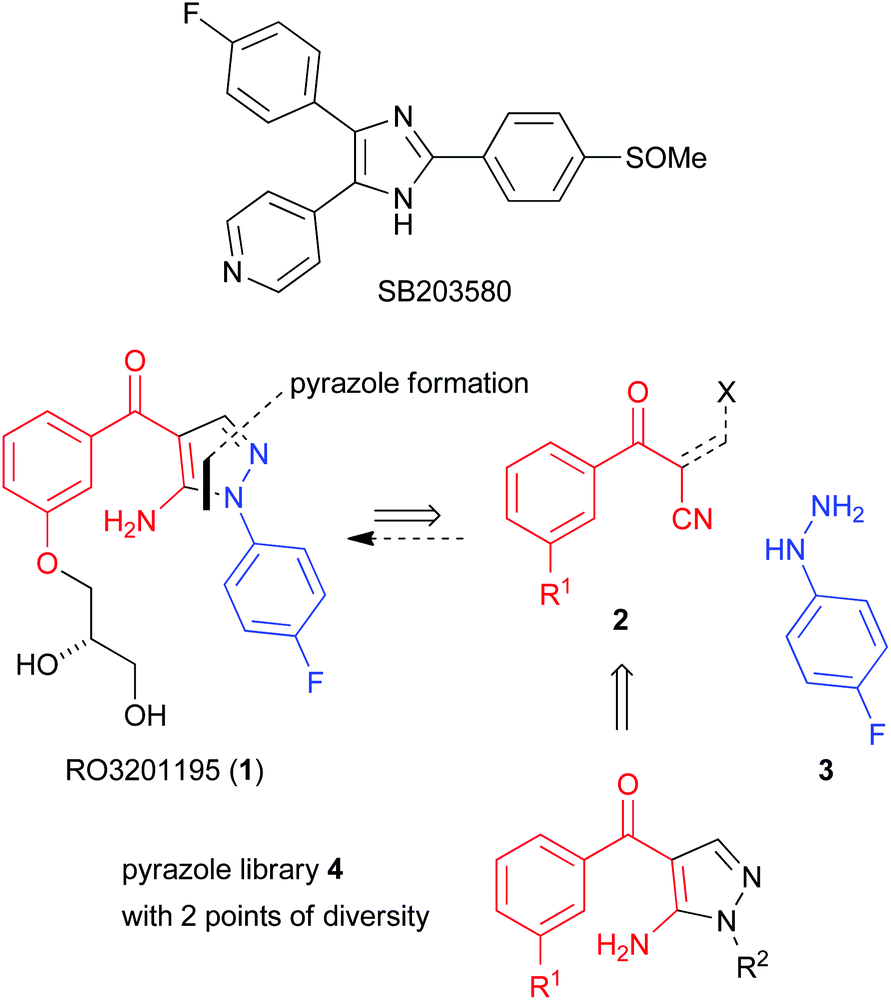 | ||
| Fig. 1 P38 MAPK inhibitors SB203580 and RO3201195 (1), a pyrazolyl ketone library 4 and disconnective approach to 1. | ||
Results and discussion
The synthesis of a suitably functionalized β-ketonitrile derivative 2, as a precursor for pyrazole formation, was first investigated using the Pd-catalyzed Heck reaction23,24 of 3-iodoanisole and 3-methoxyacrylonitrile (5). This approach had the advantage that, potentially, it could access 3-methoxybenzoylacetonitrile (6) directly from commercially-available materials, upon acid hydrolysis of the coupled product, methyl vinyl ether 7 (Scheme 1, top), providing a very rapid route into the pyrazole skeleton. Heck reactions of α- or β-substituted enol ethers are known and have been shown to provide efficient access to 1-arylpropanone derivatives.25 However, although there are numerous examples of Heck reactions of α,β-disubstituted alkenes such as methylacrylates,26 3-ethoxyacrylates,27 cinnamates,28 crotonates29 and alkoxypropenes,30 there are very few examples of β-alkoxyacrylonitriles in these and related processes. Enol ethers are less reactive than electron-poor alkenes in Heck transformations and so these slow reactions often require high catalyst loadings, more reactive aryl iodides as substrates and the use of certain additives, particularly when performed under solid–liquid phase transfer conditions.31 Furthermore, α,β-disubstituted alkenes may suffer from steric constraints and so, although direct, this approach for the synthesis of benzoylacetonitriles was untested. Ortar et al.32 did describe the arylation of an α-methoxyacrylate back in 1987 using aryl iodides, palladium acetate (3 mol%), NaHCO3 base and tetrabutylammonium chloride as additive. Using Jeffery's conditions for Heck-type processes as a starting point,33 methods related to the conditions for arylation of acrylonitrile with aryl iodides34 were investigated in the Heck reaction of 3-iodoanisole and 3-methoxyacrylonitrile (5). The use of Pd(OAc)2 (10 mol%) as catalyst in MeCN–H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Table 1) under microwave-assisted heating at 130 °C for 1.5 h (entry 4) with K2CO3 as base and tetrabutylammonium bromide (TBAB) as additive gave, following aqueous work up, the α-aryl enol ether 7 as a mixture of (E)- and (Z)-diastereoisomers, in a 2:1 ratio, as determined by 1H NMR spectroscopic analysis. The diastereoisomers could be separated by column chromatography, albeit in variable and poor yield. Low conversions and poor diastereocontrol has been observed before in Heck reactions of β-alkoxyacrylonitriles25 but, in our case, separation would not be required if the mixture could be converted directly to the target benzoylacetonitrile 6.
:1) (Table 1) under microwave-assisted heating at 130 °C for 1.5 h (entry 4) with K2CO3 as base and tetrabutylammonium bromide (TBAB) as additive gave, following aqueous work up, the α-aryl enol ether 7 as a mixture of (E)- and (Z)-diastereoisomers, in a 2:1 ratio, as determined by 1H NMR spectroscopic analysis. The diastereoisomers could be separated by column chromatography, albeit in variable and poor yield. Low conversions and poor diastereocontrol has been observed before in Heck reactions of β-alkoxyacrylonitriles25 but, in our case, separation would not be required if the mixture could be converted directly to the target benzoylacetonitrile 6.
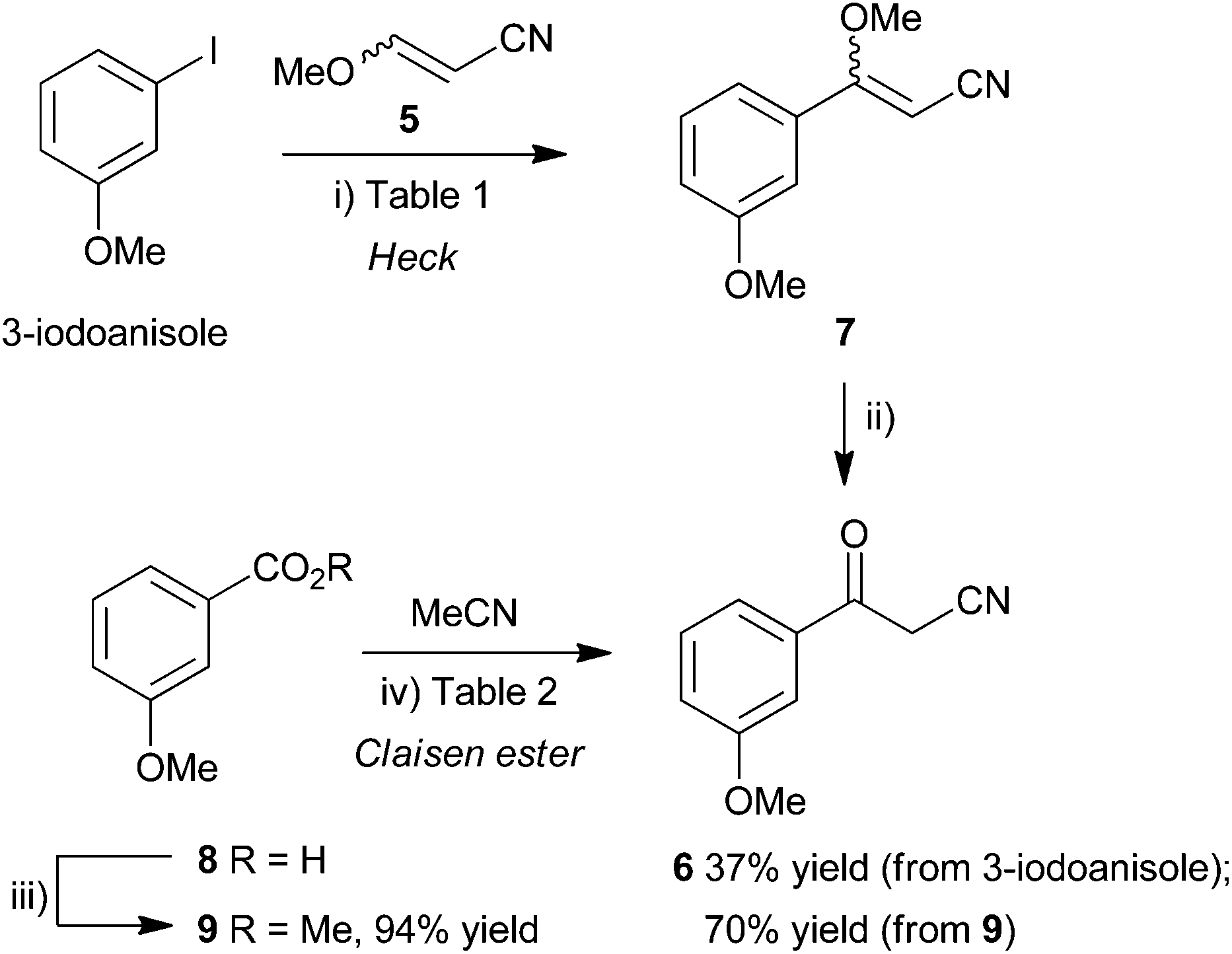 | ||
| Scheme 1 Synthesis of 3-methoxybenzoylacetonitrile (6) by Heck coupling (top) or Claisen ester condensation (bottom). Reagents and conditions: (i) see Table 1; (ii) cHCl–MeOH (1:4), microwaves, 110 °C, 30 min; (iii) MeOH, H2SO4, reflux, 2.5 h (90%); or MeOH, H2SO4, microwaves, 110 °C, 10 min (94%); (iv) see Table 2; HCl (aq). | ||
| Entry | Base | Additive | Conditionsa | Yieldb (%) |
|---|---|---|---|---|
|
a Isolated yield of benzoylacetonitrile 6.
b Reaction of 3-iodoanisole (1 equiv.), 3-methoxyacrylonitrile (5) (4 equiv.), Pd(OAc)2 (10 mol%), base (2 equiv.) and additive (10 mol%) in MeCN–H2O (3:1) under the given conditions, followed by aqueous work up, microwave irradiation in cHCl–MeOH (1:4) at 110 °C (hold time) for 30 min (by moderation of initial power, 150 W) and aqueous work up, followed by purification by column chromatography on silica.
c Microwave irradiation in a sealed tube at the given temperature, as measured by the instrument's in-built IR sensor, for the given time (hold time) by moderation of the initial power (150 W).
|
||||
| 1 | K2CO3 | TBAB | Conductive heating, 90 °C, 24 h | 36 |
| 2 | NaHCO3 | TBAB | Conductive heating, 90 °C, 24 h | 34 |
| 3 | K2CO3 | TBAB | Microwaves,c 130 °C, 1 h | 26 |
| 4 | K2CO3 | TBAB | Microwaves,c 130 °C, 1.5 h | 20 |
| 5 | NaHCO3 | TBAB | Microwaves,c 130 °C, 1.5 h | 37 |
| 6 | NaHCO3 | TBAB | Microwaves,c 140 °C, 1 h | 19 |
| 7 | K2CO3 | DPPP | Microwaves,c 150 °C, 1.5 h | 31 |
Surprisingly the hydrolysis of the enol ether diastereoisomeric mixture 7 required very forcing conditions. Simple treatment of the Heck reaction mixture with acid under ambient conditions failed to provide β-ketonitrile 6. The hydrolysis of related alkoxyacrylonitriles were notable, by their absence, from the study of Doucet and Santelli following Heck reaction of 3-ethoxyacrylonitrile, catalyzed by a tetraphosphane/palladium complex.25 However, by resorting to more forcing conditions, gratifyingly, microwave irradiation of the isolated diastereoisomeric mixture 7 in cHCl–MeOH (1:4) at 110 °C for 30 min finally provided benzoylacetonitrile 6 after purification by column chromatography but only in modest isolated yield over the two steps (Table 1, entry 1, 36%).
Efforts to improve the overall yield of this process by changing the base from K2CO3 to NaHCO3, in accordance with Heck methods for the synthesis of cinnamonitriles,34 carrying out the hydrolysis in cHCl–MeOH (1:4) under microwave irradiation as before (Table 1, entry 2), gave no improvement in yield (34%). Similarly, carrying out the Heck reaction in a sealed tube under microwave irradiation using K2CO3 at 130 °C for 1 h (entry 3) or 1.5 h (entry 4) was poorly efficient. The microwave-assisted reaction using NaHCO3 at 130 °C for 1.5 h (entry 5) did give a modest yield of product (37%), similar to the longer conductive heating processes, but this was not improved further by raising the temperature of reaction to 140 °C (entry 6) or to 150 °C in the presence of an alternative additive DPPP (10 mol%) (entry 7). Although the route had been successful for the preparation of ketonitrile 6, the need to isolate enol ether 7, in order to carry out the hydrolysis under high-temperature conditions, and the modest yield for the overall process (entry 5 gave the best yield, at 37%) meant that an alternative route had to be sought.
Considering the original approach to RO3201195 (1) by Roche,20 efforts were made to access ketonitrile 6 by a more traditional Claisen ester approach. Fischer esterification of 3-methoxybenzoic acid (8) in methanol using sulfuric acid as catalyst at reflux for 3 h gave methyl ester 9 in good yield (90%). This was improved further by carrying out the process in a sealed tube under microwave irradiation at 110 °C for 10 min to give methyl ester 9 in 94% yield (Scheme 1, bottom). A well-established method35 for Claisen condensation using sodium ethoxide or sodium methoxide as base was frustrated by the almost immediate formation of a homogenous gelatinous mass and so a range of conditions were investigated (Table 2). Heating this mixture in acetonitrile at reflux for 24 h gave only a very low yield (9% in both cases) of the benzoylacetonitrile 6 (entries 1 and 2) and significant return of unreacted ester and so alternative methods were employed. Carrying out the reaction using NaH in THF at reflux for 1.5 h gave an immediate improvement in efficiency and provided the condensation product 6 in 15% yield (entry 3), 35% yield if heated at 150 °C for 1 h in a sealed tube under microwave irradiation (entry 4). Changing the solvent to toluene and heating the mixture at 90 °C for 16 h gave a similar yield (entry 5, 34%). However, the use of freshly-prepared LDA as base at −50 °C in THF (entry 7) gave the Claisen condensation product 6 in good yield (70%), whereas at lower temperatures was inefficient (entry 6). Given the high yield and speed of the microwave-assisted esterification, this route now represented the quickest and most efficient means to access this key intermediate.
| Entry | Base | Conditions | Yielda (%) |
|---|---|---|---|
| a Isolated yield of benzoylacetonitrile 6 from methyl benzoate 9 after purification by column chromatography on silica. b Microwave irradiation in a sealed tube at the given temperature, as measured by the instrument's in-built IR sensor, for the given time (hold time) by moderation of the initial power. | |||
| 1 | NaOEt | MeCN, RT; reflux, 24 h | 9 |
| 2 | NaOMe | MeCN, RT; reflux, 24 h | 9 |
| 3 | NaH | THF, reflux, 1 h | 15 |
| 4b | NaH | THF, microwaves, 150 °C, 1 h | 35 |
| 5 | NaH | PhMe, 90 °C, 16 h | 34 |
| 6 | LDA | THF, −78 °C, 1.5 h | 18 |
| 7 | LDA | THF, −50 °C, 3 h | 70 |
Considering our previous success in the use of microwave irradiation in cyclocondensation reactions for the preparation of pyrazoles,17,36,37 the aminopyrazole core motif of BIRB 796,38 and pyrazolyl ketone libraries,22,39 benzoylacetonitrile 6 was homologated in a microwave-assisted Knoevenagel condensation using N,N′-diphenylformamidine (10) in xylenes at 180 °C for 20 min to give enaminone 11 in good yield (74%) (Scheme 2). Cyclocondensation by microwave irradiation with the hydrochloride salt of 4-fluorophenylhydrazine (3) in the presence of Et3N in EtOH at 140 °C for 1 h gave 5-aminopyrazole 12 in superior yield (86%) to a range of other methods,39 after purification by column chromatography. Protodemethylation using BBr3 in CH2Cl2 (1 M) at RT overnight gave the corresponding phenol 13 in quantitative yield, which was reacted in a SN2 displacement reaction with ketal-protected tosyl glycerate 14 under basic conditions. The reaction in DMF in the presence of K2CO3 at 80 °C for 24 h gave only a very low yield of product 15 (9%)39 but switching to DMSO and carrying out the SN2 displacement at a slightly higher temperature (100 °C) for prolonged period (40 h) gave phenyl ether 15 in improved yield (42%) after purification by column chromatography, followed by recrystallization. Submitting the product of tosylate displacement 15 to ketal deprotection directly, prior to recrystallization, under acid-catalyzed aqueous conditions at 50 °C for 18 h gave RO3201195 (1) in 37% yield over the two steps, after purification by column chromatography and recrystallization. The target inhibitor exhibited spectroscopic data that matched literature values.22,39 It was prepared in 15% overall yield in 7 separate steps from commercially-available material, three of them employing microwave irradiation, to provide rapid access to material suitable for examination in WS cells.
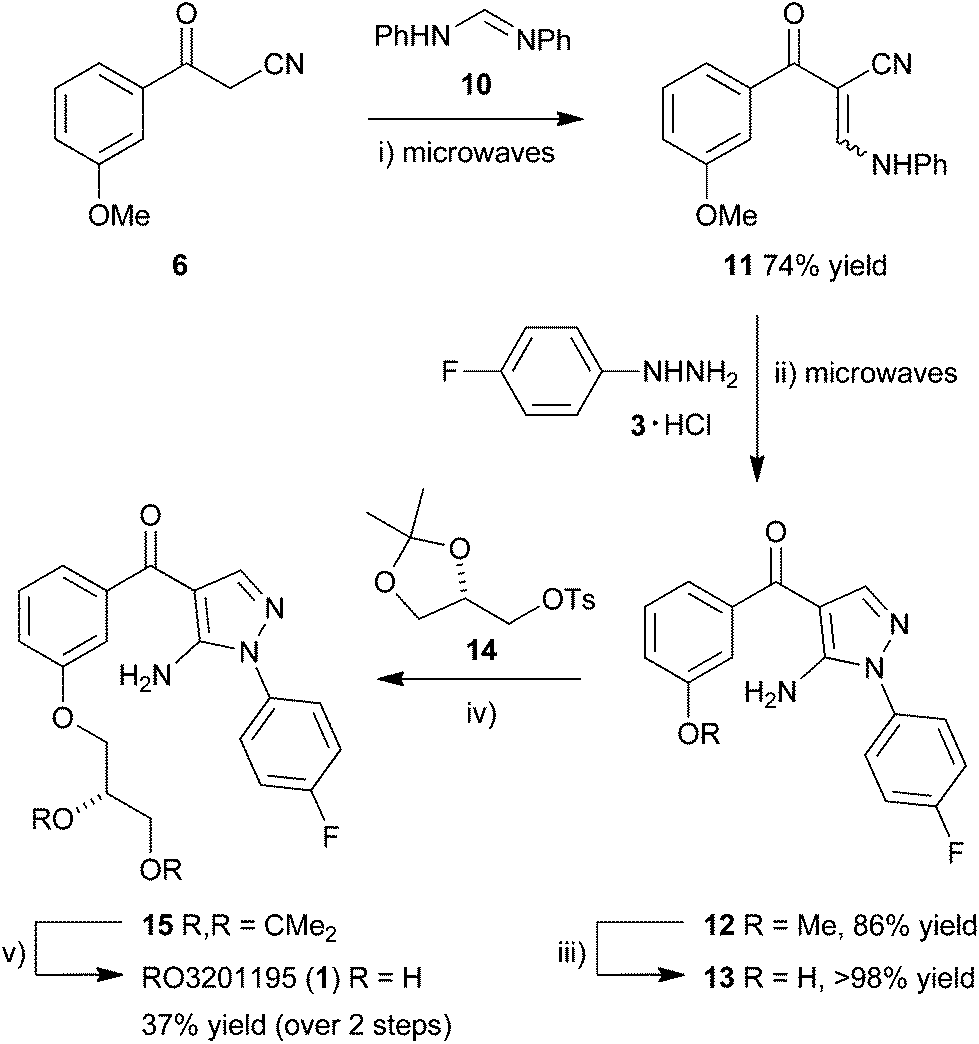 | ||
| Scheme 2 Microwave-assisted synthesis of RO3201195. Reagents and conditions: (i) xylenes, microwaves, 180 °C, 20 min; (ii) Et3N, EtOH, 140 °C, 1 h; (iii) BBr3in CH2Cl2 (1 M), 0 °C-RT, 18 h; (iv) K2CO3, DMSO, 100 °C, 40 h; (v) TsOH, MeOH–H2O, 50 °C, 18 h. | ||
Due to limited supplies of primary WS fibroblasts, initial experiments to determine the effects of novel kinase inhibitors on cellular proliferation were performed using WStert cells (see materials and methods for a description of these cells). Despite being immortal, these telomerized cells retain the slow proliferation rate typical of primary WS cells and show a similar response to SB203580 treatment.14 Thus, these cells can be used to test the effects of modulators of the p38-signalling pathway prior to their use on primary cells.
WStert cells were grown in standard medium supplemented with SB203580 at a final concentration ranging from 10 nM to 50 μM (Fig. 2a). SB203580 inhibited p38α even at the lowest doses used of 10 nM with maximum inhibition seen at 1.0 μM and above (Fig. 2c), and has an IC50 for p38α of approximately 30 nM as measured by us, and others.40,41 SB203580 treatment resulted in an increased proliferation rate of the WStert cells compared to DMSO-treated control cells even at the lowest concentration used, with the effect on proliferation rate increasing steadily with increasing SB203580 concentration and reaching a maximum between 2.5 μM and 10 μM. When the level of p38α activity was plotted against the effects upon proliferation rate a sigmoid curve was found (Fig. 2e). At 2.5 μM SB203580 resulted in an increase in proliferation rate of approximately 22% compared to DMSO controls (Fig. 2a and e).
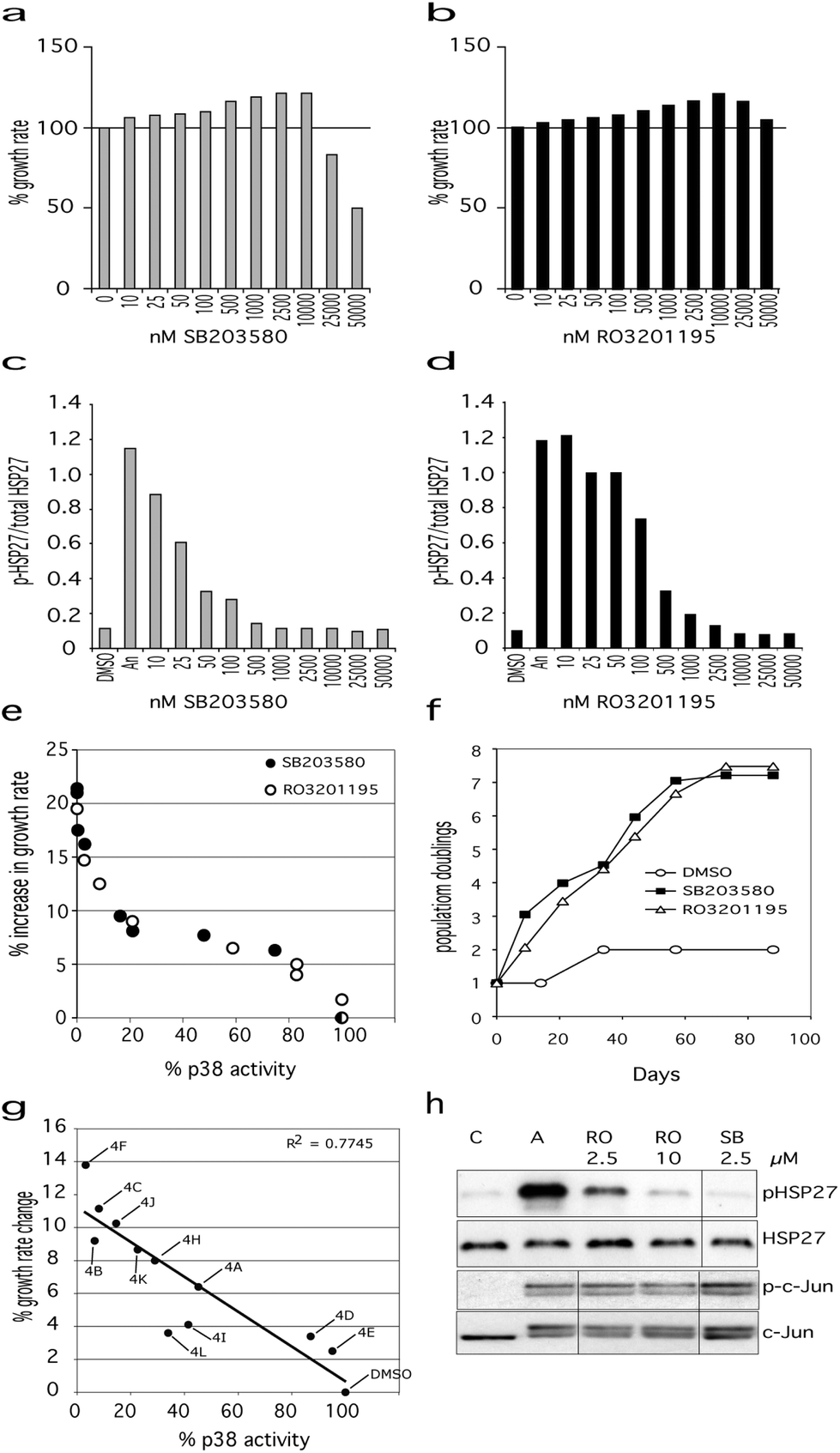 | ||
| Fig. 2 Effects of p38α inhibitors on Werner syndrome cells. Effects of SB203580 (a) and RO3201195 (b) on the proliferation rate of WStert cells, with 100% representing the DMSO control. Titration curves for inhibition of p38α activity for SB203580 (c) and RO3201195 (d) measured as the ratio of phosphorylated HSP27 to total HSP27 (measured by ELISA). For (a) to (d) DMSO are control cells, ‘An’ are cells treated with anisomycin to activate p38α, 10 to 50000 are cells treated with p38 inhibitors at the indicated concentration. (e) Plot of % p38α activity versus % increase in WStert cell proliferation rate for both SB203580 and RO3201195. (f) Proliferation of Werner syndrome AG05229 primary fibroblasts in the presence of SB203580 (2.5 μM) or RO3201195 (10 μM) measured as population doublings versus days. (g) Plot of % increase in proliferation rate of WStert cells against % p38α activity using pyrazolyl ketone library 4. See Table 3 for compound identities. (h) Immunoblot showing that neither SB203580 nor RO3201195 inhibit JNK at the optimal concentrations for their effects on cellular proliferation rates. C = DMSO control, A = cells treated with anisomycin to activate p38α and JNK, and the other three lanes show the effect of the given inhibitor concentrations in anisomycin-treated cells. | ||
RO3201195 has an IC50 of approximately 190 nM with maximal inhibition between 2.5 and 10 μM.39 In this work, RO3201195 inhibited p38α at a minimum concentration of 25 nM, with the maximal effect seen at 10.0 μM (Fig. 2d). RO3201195 had an increasing effect on the proliferation rate of WStert cells up to 10.0 μM concentration, thereafter becoming inhibitory (Fig. 2b). As seen for SB203580, when the level of p38α activity was plotted against the effects of RO3201195 upon proliferation rate a sigmoid curve was found (Fig. 2e): indeed the curve was essentially identical with the curve obtained using SB203580. The maximal increase seen in the proliferation rate of WStert cells was 19% at 10.0 μM (Fig. 2b and e).
The above data show that the optimal concentration of these inhibitors for the maximal effects on WStert cells are 2.5 μM for SB203580 and 10.0 μM for RO3201195. The inhibitors completely inhibited p38α but did not inhibit total JNK activity in WStert cells at these concentrations as measured by analyzing the phosphorylation of their downstream targets HSP27 and c-Jun by immunoblot (Fig. 2h). WStert cells are stimulated with anisomycin that activates p38α and JNKs as shown by the increased presence of the band in the A lane of the p-HSP27 panel, and the apperance of the doublet in the p-c-Jun and c-Jun panels (Fig. 2h). RO3201195 at 10.0 μM and SB203580 at 2.5 μM concentration reduced the p-HSP27 band to the level seen in the unstimulated control (C lane), but had no effect on the doublet seen in p-c-Jun and c-Jun.
We next used the p38α inhibitors on the primary Werner syndrome fibroblast strain WAG05229 that was close to replicative senescence to determine their effects on cellular senescence. For SB203580 a concentration of 2.5 μM was used, whereas for RO3201195 10.0 μM concentration was employed, as these were the lowest concentrations that resulted in maximal proliferation rate and 100% p38α inhibition (see Fig. 2). The cells were re-fed with DMEM containing the inhibitors as described previously.15 DMSO-treated fibroblasts proliferated very slowly and only achieved a single population doubling in the 88 days of the experiment (Fig. 2f). Inhibitor treated cells, however, had a much elevated proliferation rate, and managed approximately 7.5 population doublings prior to growth arrest (Fig. 2f). There was essentially no difference whichever inhibitor was used. Thus, as the effects of these inhibitors on WS cells are essentially identical and yet they show very different kinase specificity profiles at the concentrations used (the only common denominator being p38), the conclusion is that the effects on proliferation seen in both WStert cells and primary WS fibroblasts result from the inhibition of p38α.
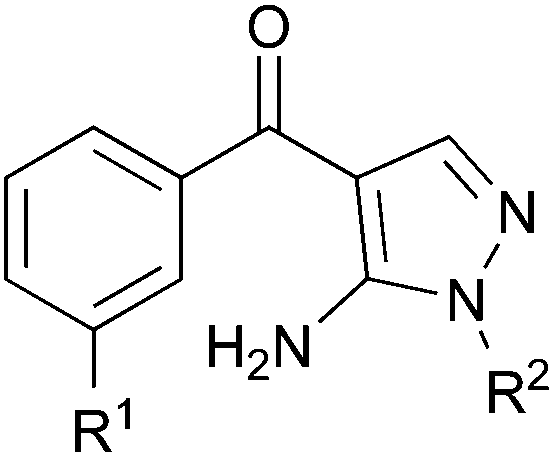
Finally, we used a pyrazolyl ketone library 4 (Table 3) of compounds related to RO320119522 for their effects on the proliferation of WStert cells (Fig. 2g). These were used at a concentration of 1.5 μM that resulted in a varied degree of p38α inhibition from approximately 5% to greater than 95% (Fig. 2g and ref. 22). When the effect on the proliferation rate of WStert cells by this pyrazolyl ketone library were plotted against the % p38α activity (as assessed by ELISA22), a linear response was seen (Fig. 2g), providing further support that these effects on cellular proliferation are due to p38α inhibition.
The data presented in this paper showing that RO3201195 has essentially identical effects on the proliferation of WS cells as SB203580, and that the proliferation rate is dependent on p38 activity, provide very strong support that the prevention of premature senescence seen in WS fibroblasts by SB203580 reported previously13 is via p38α signalling. If premature cellular senescence does underlie the accelerated ageing seen in WS individuals, these data also provide further support that the use of p38 inhibitors in vivo may alleviate this accelerated ageing.16 Finally, RO3201195 is shown to be a useful tool for the dissection of signalling pathways in human cells, as it is efficacious, relatively potent and is more kinase selective than SB203580.20 In addition, it has shown therapeutic efficacy in vivo in rats in regulating cytokine production upon LPS challenge,20 so may prove useful for future in vivo use.
Experimental
Materials and methods
Commercially available reagents were used without further purification; solvents were dried by standard procedures. Light petroleum refers to the fraction with bp 40–60 °C and ether refers to diethyl ether. Unless otherwise stated, reactions were performed under an atmosphere of air. Flash chromatography was carried out using Merck Kieselgel 60 H silica or Matrex silica 60. Analytical thin layer chromatography was carried out using aluminium-backed plates coated with Merck Kieselgel 60 GF254 that were visualised under UV light (at 254 and/or 360 nm). Microwave irradiation experiments were performed in a sealed Pyrex tube using a self-tunable CEM Discover or CEM Explorer focused monomodal microwave synthesizer at the given temperature using the instrument's in-built IR temperature measuring device, by varying the irradiation power (initial power given in parentheses).Fully characterized compounds were chromatographically homogeneous. Melting points were determined on a Kofler hot stage apparatus or Stanford Research Systems Optimelt and are uncorrected. Specific rotations were measured at the indicated temperature (in °C) using a ADP440 polarimeter (Bellingham + Stanley) at the sodium D line and are given in deg cm3 g−1 dm−1 with concentration c in 10−2 g cm−3. Infra-red spectra were recorded in the range 4000–600 cm−1 on a Perkin-Elmer 1600 series FTIR spectrometer using an ATR probe or using KBr disks or as a nujol mull for solid samples and thin films between NaCl plates for liquid samples and are reported in cm−1. NMR spectra were recorded using a Varian VNMRS instrument operating at 400 or 500 MHz or a Bruker Avance III 400 MHz for 1H spectra and 101 or 126 MHz for 13C spectra; J values were recorded in Hz and multiplicities were expressed by the usual conventions. Low resolution mass spectra were determined using a Waters Q-TOF Ultima using electrospray positive ionization, A Waters LCT premier XE using atmospheric pressure chemical ionization (APcI), an Agilent 6130 single quadrupole with an APcI/electrospray dual source, a Fisons Instrument VG Autospec using electron ionization at 70 eV (low resolution) or a ThermoQuest Finnigan LCQ DUO electrospray, unless otherwise stated. TOFMS refers to time-of-flight mass spectrometry, ES refers to electrospray ionization, CI refers to chemical ionization (ammonia), FTMS refers to Fourier transform mass spectrometry, NSI refers to nano-electrospray ionization and EI refers to electron ionization. A number of high resolution mass spectra were obtained courtesy of the EPSRC Mass Spectrometry Service at University College of Wales, Swansea, UK using the ionization methods specified.
Synthetic procedures
:3), gave the title compound (0.82 g, 70%) as a yellow solid, mp 86.3–87.4 °C (MeOH) (lit.,42 mp 87–88 °C) (Found [ES]: 198.0523. C10H9NO2Na [MNa] requires 198.0525); IR (neat) νmax/cm−1 2948 (CH str.), 2252 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N str.), 1694 (C
N str.), 1694 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O str.), 1580 (CC), 1451 (CH), 1258 (C–O), 1012 (C–O); 1H NMR (500 MHz, CDCl3) δH/ppm 7.49–7.41 (3H, 2-, 5- and 6-CH), 7.21 (1H, d, J = 8, 4-CH), 4.07 (2H, s, CH2), 3.88 (3H, s, Me); 13C NMR (126 MHz, CDCl3) δC/ppm 186.9 (C), 160.2 (3-C), 135.7 (1-C), 130.1 (5-CH), 121.2 (4-CH), 120.9 (6-CH), 113.6 (CN), 112.8 (2-CH), 55.6 (Me), 29.4 (CH2); m/z (EI) 175 (M˙+, 36%), 135 ([M − CH2CN]+, 100).
:3), and recrystallization (EtOAc–hexanes) gave the title compound (0.36 g, 34%) as a colourless solid, mp 87.3–88.2 °C (EtOAc) (lit.,42 mp 87–88 °C), with identical spectroscopic properties.
:1) (3 mL) under Ar and the mixture was heated at 90 °C for 20 h (entries 1 and 2) or irradiated at 130 °C or 140 °C for 1–1.5 h in a pressure-rated glass vial (10 mL) using a CEM Discover microwave synthesizer (entries 3–6) by moderating the initial power (150 W). After cooling, the solution was partitioned between water and EtOAc and the aqueous layer was further extracted with EtOAc (2 × 10 mL). The organic extracts were combined, washed with brine (20 mL), dried (Na2SO4) and evaporated in vacuo. The crude residue was dissolved in MeOH (2 mL) in air and concentrated HCl was added (10.2 M; 0.5 mL). The mixture was irradiated at 110 °C for 1 h in a pressure-rated glass vial (10 mL) using a CEM Discover microwave synthesiser by moderating the initial power (150 W). After cooling in a flow of compressed air, the solution was partitioned between water and EtOAc, and the aqueous layer was further extracted with EtOAc (2 × 10 mL). The organic extracts were combined, washed with brine (20 mL), dried and evaporated in vacuo. Purification by column chromatography on silica gel (dry load), gradient eluting with hexanes to EtOAc–hexanes (2:3), gave the title compound as a yellow solid, mp 87.0–87.6 °C (MeOH) (lit.,42 mp 87–88 °C), with identical spectroscopic properties.
:1) (12 mL) under Ar and the solution was stirred at 90 °C for 24 h. After cooling, the solution was partitioned between water and EtOAc and the aqueous layer was further extracted with EtOAc (2 × 20 mL). The organic extracts were combined, washed with brine (20 mL), dried (Na2SO4) and evaporated in vacuo. Purification by column chromatography on silica gel (dry load), gradient eluting with hexanes to EtOAc–hexanes (15:85), gave the title compounds (0.26 g, 68%; Z:E (1:2)) as yellow oils [Rf 0.5 (Z)-7 and 0.3 (E)-7 in EtOAc–light petroleum (1:4)] (Found [ES]: 212.0679. C11H11NO2Na [MNa] requires 212.0682); IR (neat) νmax/cm−1 3069 (C–H str.), 2950 (C–H str.), 2198 (CN str.), 1605 (CC str.), 1579 (C–C str.), 1437 (C–H bend), 1215 (C–O str.), 1117 (C–O str.), 1047 (C–O str.); 1H NMR (500 MHz, CDCl3) δH/ppm [(Z)-7] 7.31 (1H, t, J = 8, 5′-CH), 7.11 (1H, d, J = 8, 6′-CH), 7.06 (1H, s, 2′-CH), 7.00 (1H, d, J = 8, 3′-CH), 4.96 (1H, s, 2-CH), 4.24 (3H, s, 3-OMe), 3.84 (3H, s, 3′-OMe); δH/ppm [(E)-7] 7.40–7.33 (2H, 5′,6′-CH), 7.30 (1H, s, 2′-CH), 7.03 (1H, d, J = 7, 4′-CH), 4.65 (1H, s, 2-CH), 3.85 (6H, app s, 3- and 3′-OMe); 13C NMR (126 MHz, CDCl3) δC/ppm [(Z)-7] 170.9 (3-C), 159.7 (3′-C), 135.3 (1′-C), 129.7 (5′-CH), 118.9 (6′-CH), 117.3 (CN), 116.7 (4′-CH), 112.2 (2′-CH), 71.2 (2-CH), 59.6 (3-OMe), 55.4 (3′-OMe); δC/ppm [(E)-7] 173.0 (3-C), 159.4 (3′-C), 134.1 (1′-C), 129.5 (5′-CH), 120.3 (6′-CH), 118.4 (CN), 117.4 (4′-CH), 112.9 (2′-CH), 69.9 (2-CH), 56.8 (3-OMe), 55.4 (3′-OMe); m/z (EI) 189 (M˙+, 100%), 132 (29), 69 (39).
O str.), 1601 (CC str.), 1276 (C–O str.), 1221 (C–O str.); 1H NMR (500 MHz, CDCl3) δH/ppm 7.64 (1H, d, J = 8, 6-CH), 7.57 (1H, br. s, 2-CH), 7.35 (1H, t, J = 8, 5-CH), 7.11 (1H, dd, J = 8, 2, 4-CH), 3.92 (3H, s, CO2Me), 3.86 (3H, s, OMe); 13C NMR (126 MHz, CDCl3) δC/ppm 166.9 (C), 159.6 (3-C), 131.5 (1-C), 129.3 (5-CH), 122.0 (6-CH), 199.5 (4-CH), 144.0 (2-CH), 55.4 (OMe), 52.1 (CO2Me); m/z (EI): 166 (M˙+, 14%), 135 ([M − OMe]+, 39), 93 (100).
N str.), 1634 (CO str.), 1596 (N–H bend), 1571 (C–C str.), 1392 (C–C str.), 1313 (C–N str.), 1225 (C–O str.), 1044 (C–O str.), 990 (C–H bend), 867 (N–H wag); 1H NMR (500 MHz, CDCl3) δH/ppm 12.76 (1H, d, J = 12, NH), 8.06 (1H, d, J = 12, 3-CH), 7.57 (1H, d, J = 8, 6′-CH), 7.46 (3H, m, 2′-, 3′′, and 5′′-CH), 7.40 (1H, t, J = 8, 5′-CH), 7.30 (1H, m, 4′′-CH), 7.23 (2H, d, J = 8, 2′′- and 6′′-CH), 7.11 (1H, d, J = 8, 6′-CH), 3.89 (3H, s, Me); 13C NMR (126 MHz, CDCl3) δC/ppm 192.1 (C), 159.6 (3′-C), 154.1 (3-CH), 139.2 (1′-C), 138.1 (1′′-C), 130.2 (3′′, 5′′-CH), 129.4 (5′-CH), 126.6 (4′′-CH), 120.5 (6′-CH), 120.3 (1-CN), 118.9 (4′-CH), 117.9 (2′′, 6′′-CH), 112.7 (2′-CH), 83.5 (2-C), 55.5 (Me); m/z (EI): 278 (M˙+, 49%), 277 ([M − H]+, 100), 135 (52).
:7), gave the title compound (0.11 g, 86%) as a pink solid, mp 130.1–132.0 °C (Found [ES]: 312.1138. C17H15FN3O2 [MH] requires 312.1143); IR (neat) νmax/cm−1 3318 (N–H str.), 2925 (C–H str.), 1610 (CO str.), 1595 (N–H bend), 1539 (C–C str.), 1286 (C–N str.), 1223 (C–O str.), 1051 (C–O str.), 838 (N–H wag); 1H NMR (400 MHz, CDCl3) δH/ppm 7.81 (1H, s, 3-CH), 7.56 (2H, m, 2′, 6′-CH), 7.42 (2H, m, 5′′, 6′′-CH), 7.35 (1H, s, 2′′-CH), 7.25 (2H, m, 3′, 5′-CH), 7.11 (1H, m, 4′′-CH), 6.04 (2H, bs, exch. D2O, NH2), 3.89 (3H, s, Me); 13C NMR (101 MHz, CDCl3) δC/ppm 189.4 (C), 162.2 (d, 1JCF = 250, 4′-CF), 159.8 (3′′-C), 150.6 (5-C), 142.0 (3-CH), 141.1 (1′′-C), 133.3 (d, 4JCF = 4, 1′-C), 129.5 (5′′-CH), 126.1 (d, 3JCF = 9, 2′, 6′-CH), 120.7 (6′′-CH), 117.8 (4′′-CH), 116.9 (d, 2JCF = 23, 3′, 5′-CH), 113.0 (2′′-CH), 104.8 (4-C), 55.4 (OMe); m/z (APcI) 312 (MH+, 100%).
O str.), 1592 (N–H bend), 1538 (C–C str.), 1310 (C–N str.), 1126 (C–O str.), 841 (N–H wag); 1H NMR (500 MHz, CD3OD) δH/ppm 7.78 (1H, s, 3-CH), 7.59 (2H, m, 2′, 6′-CH), 7.37–7.29 (3H, 3′, 5′- and 5′′-CH), 7.23 (1H, d, J = 7, 6′′-CH), 7.18 (1H, s, 2′′-CH), 7.00 (1H, dd, J = 7, 1, 4′′-CH); 13C NMR (100 MHz, CD3OD) δC/ppm 191.6 (C), 164.1 (d, 1JCF = 247, 4′-CF), 159.2 (3′′-C), 153.4 (5-C), 143.6 (3-CH), 142.7 (1′′-C), 135.0 (d, 4JCF = 3, 1′-C), 131.1 (5′′-CH), 128.5 (d, 3JCF = 9, 2′, 6′-CH), 120.6 (6′′-CH), 120.1 (4′′-CH), 118.0 (d, 3JCF = 24, 3′, 5′-CH), 116.0 (2′′-CH), 105.7 (4-C); m/z (EI) 297 (M˙+, 91%), 296 ([M − H]+, 100), 204 (31).
:3), gave [5-amino-1-(4-fluorophenyl)-1H-pyrazol-4-yl] {3-[(R)-2,2-dimethyl-1,3-dioxolan-4-yl)methoxy]phenyl} ketone (15), which was dissolved in MeOH–water (4:1) (4 mL). p-Toluene sulfonic acid monohydrate (40 mg, 0.21 mmol) was added and the solution was heated at 50 °C for 18 h. After cooling, the solvent was evaporated in vacuo. The residue was dissolved in EtOAc, washed with saturated aqueous NaHCO3 solution, dried (MgSO4) and evaporated in vacuo. Purification by column chromatography on silica gel (dry load), gradient eluting with light petroleum to EtOAc, followed by recrystallization (EtOAc–hexanes) gave the title compound (0.22 g, 37%) as a colourless solid, mp 154.1–154.6 °C (EtOAc) (Found [ES]: 372.1353. C19H19FN3O4 [MH] requires 372.1354); [α]21D −28 (c 0.2, EtOAc); [α]22D + 6.5 (c 0.6, MeOH); IR (neat) νmax/cm−1 3440 (N–H str.), 3330 (O–H str.), 3229 (O–H str.), 2896 (C–H str.), 1633 (CO str.), 1597 (NH2 str.), 1540 (CN str.), 1498 (C–C str.), 1292 (C–O str.), 1222 (C–F str.), 1053 (C–O str.), 839 (NH2 wag.); 1H NMR (500 MHz, d6-DMSO) δH/ppm 7.80 (1H, s, 3H), 7.61 (2H, m, 2′, 6′-CH), 7.47–7.37 (3H, 3′, 4′- and 5′′-CH), 7.33 (1H, d, J = 7, 6′′-CH), 7.25 (1H, m, 2′′-CH), 7.14 (3H, 4′′-CH and NH2), 4.95 (1H, bs, CHOH), 4.65 (1H, bs, CH2OH), 4.09 (1H, m, OCHH), 3.95 (1H, m, OCHH), 3.82 (1H, m, CHOH), 3.47 (2H, d, J = 5, CH2OH); 13C NMR (126 MHz, d6-DMSO) δC/ppm 187.5 (C), 161.1 (d, 1JCF = 244, 4′-C), 158.7 (3′′-C), 151.2 (5-C), 141.4 (3-CH), 140.9 (1′′-C), 133.7 (d, 4JCF = 3, 1′-C), 129.6 (5′′-CH), 126.4 (d, 3JCF = 9, 2′, 6′-CH), 120.1 (6′′-CH), 117.8 (4′′-CH), 116.3 (d, 2JCF = 23, 3′, 5′-CH), 113.3 (2′′-CH), 103.5 (4-C), 69.9 (CHOH), 69.8 (CH2O), 62.6 (CH2OH); m/z (EI) 371 (M˙+, 10%), 325 (33), 160 (100).
O str.), 1580 (CC), 1451 (CH), 1258 (C–O), 1012 (C–O); 1H NMR (500 MHz, CDCl3) δH/ppm 7.49–7.41 (3H, 2-, 5- and 6-CH), 7.21 (1H, d, J = 8, 4-CH), 4.07 (2H, s, CH2), 3.88 (3H, s, Me); 13C NMR (126 MHz, CDCl3) δC/ppm 186.9 (C), 160.2 (3-C), 135.7 (1-C), 130.1 (5-CH), 121.2 (4-CH), 120.9 (6-CH), 113.6 (CN), 112.8 (2-CH), 55.6 (Me), 29.4 (CH2); m/z (EI) 175 (M˙+, 36%), 135 ([M − CH2CN]+, 100).
:3), and recrystallization (EtOAc–hexanes) gave the title compound (0.36 g, 34%) as a colourless solid, mp 87.3–88.2 °C (EtOAc) (lit.,42 mp 87–88 °C), with identical spectroscopic properties.
:1) (3 mL) under Ar and the mixture was heated at 90 °C for 20 h (entries 1 and 2) or irradiated at 130 °C or 140 °C for 1–1.5 h in a pressure-rated glass vial (10 mL) using a CEM Discover microwave synthesizer (entries 3–6) by moderating the initial power (150 W). After cooling, the solution was partitioned between water and EtOAc and the aqueous layer was further extracted with EtOAc (2 × 10 mL). The organic extracts were combined, washed with brine (20 mL), dried (Na2SO4) and evaporated in vacuo. The crude residue was dissolved in MeOH (2 mL) in air and concentrated HCl was added (10.2 M; 0.5 mL). The mixture was irradiated at 110 °C for 1 h in a pressure-rated glass vial (10 mL) using a CEM Discover microwave synthesiser by moderating the initial power (150 W). After cooling in a flow of compressed air, the solution was partitioned between water and EtOAc, and the aqueous layer was further extracted with EtOAc (2 × 10 mL). The organic extracts were combined, washed with brine (20 mL), dried and evaporated in vacuo. Purification by column chromatography on silica gel (dry load), gradient eluting with hexanes to EtOAc–hexanes (2:3), gave the title compound as a yellow solid, mp 87.0–87.6 °C (MeOH) (lit.,42 mp 87–88 °C), with identical spectroscopic properties.
:1) (12 mL) under Ar and the solution was stirred at 90 °C for 24 h. After cooling, the solution was partitioned between water and EtOAc and the aqueous layer was further extracted with EtOAc (2 × 20 mL). The organic extracts were combined, washed with brine (20 mL), dried (Na2SO4) and evaporated in vacuo. Purification by column chromatography on silica gel (dry load), gradient eluting with hexanes to EtOAc–hexanes (15:85), gave the title compounds (0.26 g, 68%; Z:E (1:2)) as yellow oils [Rf 0.5 (Z)-7 and 0.3 (E)-7 in EtOAc–light petroleum (1:4)] (Found [ES]: 212.0679. C11H11NO2Na [MNa] requires 212.0682); IR (neat) νmax/cm−1 3069 (C–H str.), 2950 (C–H str.), 2198 (CN str.), 1605 (CC str.), 1579 (C–C str.), 1437 (C–H bend), 1215 (C–O str.), 1117 (C–O str.), 1047 (C–O str.); 1H NMR (500 MHz, CDCl3) δH/ppm [(Z)-7] 7.31 (1H, t, J = 8, 5′-CH), 7.11 (1H, d, J = 8, 6′-CH), 7.06 (1H, s, 2′-CH), 7.00 (1H, d, J = 8, 3′-CH), 4.96 (1H, s, 2-CH), 4.24 (3H, s, 3-OMe), 3.84 (3H, s, 3′-OMe); δH/ppm [(E)-7] 7.40–7.33 (2H, 5′,6′-CH), 7.30 (1H, s, 2′-CH), 7.03 (1H, d, J = 7, 4′-CH), 4.65 (1H, s, 2-CH), 3.85 (6H, app s, 3- and 3′-OMe); 13C NMR (126 MHz, CDCl3) δC/ppm [(Z)-7] 170.9 (3-C), 159.7 (3′-C), 135.3 (1′-C), 129.7 (5′-CH), 118.9 (6′-CH), 117.3 (CN), 116.7 (4′-CH), 112.2 (2′-CH), 71.2 (2-CH), 59.6 (3-OMe), 55.4 (3′-OMe); δC/ppm [(E)-7] 173.0 (3-C), 159.4 (3′-C), 134.1 (1′-C), 129.5 (5′-CH), 120.3 (6′-CH), 118.4 (CN), 117.4 (4′-CH), 112.9 (2′-CH), 69.9 (2-CH), 56.8 (3-OMe), 55.4 (3′-OMe); m/z (EI) 189 (M˙+, 100%), 132 (29), 69 (39).
O str.), 1601 (CC str.), 1276 (C–O str.), 1221 (C–O str.); 1H NMR (500 MHz, CDCl3) δH/ppm 7.64 (1H, d, J = 8, 6-CH), 7.57 (1H, br. s, 2-CH), 7.35 (1H, t, J = 8, 5-CH), 7.11 (1H, dd, J = 8, 2, 4-CH), 3.92 (3H, s, CO2Me), 3.86 (3H, s, OMe); 13C NMR (126 MHz, CDCl3) δC/ppm 166.9 (C), 159.6 (3-C), 131.5 (1-C), 129.3 (5-CH), 122.0 (6-CH), 199.5 (4-CH), 144.0 (2-CH), 55.4 (OMe), 52.1 (CO2Me); m/z (EI): 166 (M˙+, 14%), 135 ([M − OMe]+, 39), 93 (100).
N str.), 1634 (CO str.), 1596 (N–H bend), 1571 (C–C str.), 1392 (C–C str.), 1313 (C–N str.), 1225 (C–O str.), 1044 (C–O str.), 990 (C–H bend), 867 (N–H wag); 1H NMR (500 MHz, CDCl3) δH/ppm 12.76 (1H, d, J = 12, NH), 8.06 (1H, d, J = 12, 3-CH), 7.57 (1H, d, J = 8, 6′-CH), 7.46 (3H, m, 2′-, 3′′, and 5′′-CH), 7.40 (1H, t, J = 8, 5′-CH), 7.30 (1H, m, 4′′-CH), 7.23 (2H, d, J = 8, 2′′- and 6′′-CH), 7.11 (1H, d, J = 8, 6′-CH), 3.89 (3H, s, Me); 13C NMR (126 MHz, CDCl3) δC/ppm 192.1 (C), 159.6 (3′-C), 154.1 (3-CH), 139.2 (1′-C), 138.1 (1′′-C), 130.2 (3′′, 5′′-CH), 129.4 (5′-CH), 126.6 (4′′-CH), 120.5 (6′-CH), 120.3 (1-CN), 118.9 (4′-CH), 117.9 (2′′, 6′′-CH), 112.7 (2′-CH), 83.5 (2-C), 55.5 (Me); m/z (EI): 278 (M˙+, 49%), 277 ([M − H]+, 100), 135 (52).
:7), gave the title compound (0.11 g, 86%) as a pink solid, mp 130.1–132.0 °C (Found [ES]: 312.1138. C17H15FN3O2 [MH] requires 312.1143); IR (neat) νmax/cm−1 3318 (N–H str.), 2925 (C–H str.), 1610 (CO str.), 1595 (N–H bend), 1539 (C–C str.), 1286 (C–N str.), 1223 (C–O str.), 1051 (C–O str.), 838 (N–H wag); 1H NMR (400 MHz, CDCl3) δH/ppm 7.81 (1H, s, 3-CH), 7.56 (2H, m, 2′, 6′-CH), 7.42 (2H, m, 5′′, 6′′-CH), 7.35 (1H, s, 2′′-CH), 7.25 (2H, m, 3′, 5′-CH), 7.11 (1H, m, 4′′-CH), 6.04 (2H, bs, exch. D2O, NH2), 3.89 (3H, s, Me); 13C NMR (101 MHz, CDCl3) δC/ppm 189.4 (C), 162.2 (d, 1JCF = 250, 4′-CF), 159.8 (3′′-C), 150.6 (5-C), 142.0 (3-CH), 141.1 (1′′-C), 133.3 (d, 4JCF = 4, 1′-C), 129.5 (5′′-CH), 126.1 (d, 3JCF = 9, 2′, 6′-CH), 120.7 (6′′-CH), 117.8 (4′′-CH), 116.9 (d, 2JCF = 23, 3′, 5′-CH), 113.0 (2′′-CH), 104.8 (4-C), 55.4 (OMe); m/z (APcI) 312 (MH+, 100%).
O str.), 1592 (N–H bend), 1538 (C–C str.), 1310 (C–N str.), 1126 (C–O str.), 841 (N–H wag); 1H NMR (500 MHz, CD3OD) δH/ppm 7.78 (1H, s, 3-CH), 7.59 (2H, m, 2′, 6′-CH), 7.37–7.29 (3H, 3′, 5′- and 5′′-CH), 7.23 (1H, d, J = 7, 6′′-CH), 7.18 (1H, s, 2′′-CH), 7.00 (1H, dd, J = 7, 1, 4′′-CH); 13C NMR (100 MHz, CD3OD) δC/ppm 191.6 (C), 164.1 (d, 1JCF = 247, 4′-CF), 159.2 (3′′-C), 153.4 (5-C), 143.6 (3-CH), 142.7 (1′′-C), 135.0 (d, 4JCF = 3, 1′-C), 131.1 (5′′-CH), 128.5 (d, 3JCF = 9, 2′, 6′-CH), 120.6 (6′′-CH), 120.1 (4′′-CH), 118.0 (d, 3JCF = 24, 3′, 5′-CH), 116.0 (2′′-CH), 105.7 (4-C); m/z (EI) 297 (M˙+, 91%), 296 ([M − H]+, 100), 204 (31).
:3), gave [5-amino-1-(4-fluorophenyl)-1H-pyrazol-4-yl] {3-[(R)-2,2-dimethyl-1,3-dioxolan-4-yl)methoxy]phenyl} ketone (15), which was dissolved in MeOH–water (4:1) (4 mL). p-Toluene sulfonic acid monohydrate (40 mg, 0.21 mmol) was added and the solution was heated at 50 °C for 18 h. After cooling, the solvent was evaporated in vacuo. The residue was dissolved in EtOAc, washed with saturated aqueous NaHCO3 solution, dried (MgSO4) and evaporated in vacuo. Purification by column chromatography on silica gel (dry load), gradient eluting with light petroleum to EtOAc, followed by recrystallization (EtOAc–hexanes) gave the title compound (0.22 g, 37%) as a colourless solid, mp 154.1–154.6 °C (EtOAc) (Found [ES]: 372.1353. C19H19FN3O4 [MH] requires 372.1354); [α]21D −28 (c 0.2, EtOAc); [α]22D + 6.5 (c 0.6, MeOH); IR (neat) νmax/cm−1 3440 (N–H str.), 3330 (O–H str.), 3229 (O–H str.), 2896 (C–H str.), 1633 (CO str.), 1597 (NH2 str.), 1540 (CN str.), 1498 (C–C str.), 1292 (C–O str.), 1222 (C–F str.), 1053 (C–O str.), 839 (NH2 wag.); 1H NMR (500 MHz, d6-DMSO) δH/ppm 7.80 (1H, s, 3H), 7.61 (2H, m, 2′, 6′-CH), 7.47–7.37 (3H, 3′, 4′- and 5′′-CH), 7.33 (1H, d, J = 7, 6′′-CH), 7.25 (1H, m, 2′′-CH), 7.14 (3H, 4′′-CH and NH2), 4.95 (1H, bs, CHOH), 4.65 (1H, bs, CH2OH), 4.09 (1H, m, OCHH), 3.95 (1H, m, OCHH), 3.82 (1H, m, CHOH), 3.47 (2H, d, J = 5, CH2OH); 13C NMR (126 MHz, d6-DMSO) δC/ppm 187.5 (C), 161.1 (d, 1JCF = 244, 4′-C), 158.7 (3′′-C), 151.2 (5-C), 141.4 (3-CH), 140.9 (1′′-C), 133.7 (d, 4JCF = 3, 1′-C), 129.6 (5′′-CH), 126.4 (d, 3JCF = 9, 2′, 6′-CH), 120.1 (6′′-CH), 117.8 (4′′-CH), 116.3 (d, 2JCF = 23, 3′, 5′-CH), 113.3 (2′′-CH), 103.5 (4-C), 69.9 (CHOH), 69.8 (CH2O), 62.6 (CH2OH); m/z (EI) 371 (M˙+, 10%), 325 (33), 160 (100).
Cells and cell growth
Human adult dermal primary Werner syndrome AG05229 and AG03141 fibroblasts were obtained from the Coriell Cell Repositories (Camden, NJ, USA). WStert cells are AG03141 fibroblasts that have been immortalised by the ectopic expression of human enzyme telomerase and have been described previously.14Cells were grown in DMEM growth medium as previously described.15 Cell proliferation rates for the WStert cells were measured as cumulated population doublings (CPDs) divided by the number of days of the experiment concerned and expressed as a percentage of the proliferation rate of control cells (with the control being 100%). The number of CPDs achieved at each passage of the cells was calculated according to the formula: PDs = log(Nt/No)/log2, where Nt is number of cells counted and No is number of cells seeded. For primary cells the CPDs were plotted versus days in culture. For assessing the effects of the various kinase inhibitors the culture medium was supplemented with the inhibitor dissolved in DMSO, with the medium being replaced daily. For controls an equivalent volume of the inhibitor solvent (DMSO) was added to the medium.
P38 inhibitor assay
The ability of SB203580 and RO3201195 to inhibit the p38α signalling pathway was tested in human immortalised HCA2 cells using an ELISA system (obtained from Cell Signalling, NEB, UK) as previously described.39 Kinase activity was detected using antibodies specific for HSP27 and its phosphorylated form, the degree of activation being measured as the ratio of phospho-protein/total protein (Fig. 2d and e). In this system activation of p38α by anisomycin activates the kinase MK2 that then phosphorylates the small heat shock protein HSP27. As MK2 is the major HSP27 kinase,43 the activity of p38α can be assessed by the phosphorylation status of HSP27.Immunoblot assays for stress kinase activity in WS cells
The ability of SB203580 and RO3201195 to inhibit the p38α and JNK signalling pathways in WStert cells was tested by immunoblot detection of the phosphorylated versions of their downstream targets HSP27 (pHSP27) and c-Jun (p-c-Jun) as previously described.15,44Conclusions
This manuscript describes the chemical synthesis and biological evaluation of a p38α MAPK inhibitor, RO3201195 (1), in WS cells. Two complementary routes for the chemical synthesis of a key intermediate, benzoylacetonitrile 6, are described, both of them involving the use of microwave dielectric heating for rapid reaction kinetics. Of these two routes, a classical approach based upon a Claisen ester condensation was found to be the most efficient. The benzoylacetonitrile intermediate 6 was transformed by Knoevenagel condensation, followed by cyclocondensation with a hydrazine, both under microwave irradiation, to give the core pyrazole motif. Phenoxide alkylation by SN2 displacement of tosylate, followed by ketal deprotection, gave the target inhibitor in a sequence of 7 steps and 15% overall yield.The effects of RO3201195 (1) on cellular proliferation were first examined using WStert cells, where it was found that the inhibitor elicited a very similar response to the prototypical p38 MAPK inhibitor SB203580. The maximal increase seen in the proliferation rate of WStert cells was 19% at 10.0 μM concentration of RO3201195 (1). At this concentration, the inhibitor completely inhibited p38α but did not inhibit total JNK activity in WStert cells, as determined by immunoblot assay. Following these findings, the cellular senescence of primary Werner syndrome fibroblast strain WAG05229 was investigated on treatment with SB203580 or RO3201195 (1). Despite the fact that these inhibitors showed very different kinase specificity profiles at the concentrations used, the effects on WS cells were essentially identical. Thus, we concluded that the effects on proliferation of treating WStert cells or primary WS fibroblasts with SB203580 or RO3201195 (1) resulted from inhibition of p38α. This was further supported by studying the effects of a pyrazolyl ketone library on WStert cells at a given concentration, which showed a linear relationship between the proliferation rate and % p38α activity. Thus, we conclude RO3201195 (1) is a useful chemical tool for dissecting signalling pathways in human cells in order to understand the effect of p38 MAPK signalling upon cell proliferation in this human progeroid syndrome.
Acknowledgements
We thank the EPSRC-BBSRC-MRC sponsored network SMS-Drug (EP/I037229/1; award to MCB), EPSRC (studentship award to JED), Saudi Cultural Bureau (support for MB), BBSRC (BB/D524140/1; award to DK, MCB and TD), SPARC (awards to MCB and TD), ESRC under the New Dynamics of Ageing Initiative (RES-356-25-0024; award to MCB, DK and TD) and the R M Phillips Trust (award to MCB) for support of this work and Dr Alaa Abdul-Sada at the University of Sussex and the EPSRC Mass Spectrometry Service at the University of Wales, Swansea UK for mass spectra.Notes and references
- J. C. Lee, J. T. Laydon, P. C. McDonnell, T. F. Gallagher, S. Kumar, D. Green, D. McNulty, M. J. Blumenthal, J. R. Heys and S. W. Landvatter, et al. , Nature, 1994, 372, 739 CrossRef CAS PubMed.
- G. L. Schieven, Curr. Top. Med. Chem., 2005, 5, 921 CrossRef CAS PubMed.
- E. Salgado, J. R. Maneiro, L. Carmona and J. J. Gomez-Reino, Ann. Rheum. Dis., 2014, 73, 871 CrossRef CAS PubMed.
- M. Back and G. K. Hansson, Nat. Rev. Cardiol., 2015, 12, 199–211 CrossRef PubMed.
- V. Grossi, A. Peserico, T. Tezil and C. Simone, World J. Gastroenterol., 2014, 20, 9744 CrossRef CAS PubMed.
- K. Yumoto, M. R. Eber, J. E. Berry, R. S. Taichman and Y. Shiozawa, Clin. Cancer Res., 2014, 20, 3384 CrossRef CAS PubMed.
- C. Dominguez, D. A. Powers and N. Tamayo, Curr. Opin. Drug Discovery Dev., 2005, 8, 421 CAS.
- D. J. Diller, T. H. Lin and A. Metzger, Curr. Top. Med. Chem., 2005, 5, 953 CrossRef CAS PubMed.
- D. M. Goldstein and T. Gabriel, Curr. Top. Med. Chem., 2005, 5, 1017 CrossRef CAS PubMed.
- S. T. Wrobleski and A. M. Doweyko, Curr. Top. Med. Chem., 2005, 5, 1005 CrossRef CAS PubMed.
- J. Saklatvala, Curr. Opin. Pharmacol., 2004, 4, 372 CrossRef CAS PubMed.
- D. Kipling, T. Davis, E. L. Ostler and R. G. Faragher, Science, 2004, 305, 1426 CrossRef CAS PubMed.
- T. Davis, D. M. Baird, M. F. Haughton, C. J. Jones and D. Kipling, J. Gerontol., Ser. A, 2005, 60, 1386 CrossRef.
- T. Davis, M. F. Haughton, C. J. Jones and D. Kipling, Ann. N. Y. Acad. Sci., 2006, 1067, 243 CrossRef CAS PubMed.
- T. Davis and D. Kipling, Biogerontology, 2009, 10, 253 CrossRef CAS PubMed.
- T. Davis and D. Kipling, Rejuvenation Res., 2006, 9, 402 CrossRef CAS PubMed.
- M. C. Bagley, T. Davis, M. J. Rokicki, C. S. Widdowson and D. Kipling, Future Med. Chem., 2010, 2, 193 CrossRef CAS PubMed.
- J. Bain, L. Plater, M. Elliott, N. Shpiro, C. J. Hastie, H. McLauchlan, I. Klevernic, J. S. Arthur, D. R. Alessi and P. Cohen, Biochem. J., 2007, 408, 297 CrossRef CAS PubMed.
- K. Godl and H. Daub, Cell Cycle, 2004, 3, 393 CrossRef CAS PubMed.
- D. M. Goldstein, T. Alfredson, J. Bertrand, M. F. Browner, K. Clifford, S. A. Dalrymple, J. Dunn, J. Freire-Moar, S. Harris, S. S. Labadie, J. La Fargue, J. M. Lapierre, S. Larrabee, F. Li, E. Papp, D. McWeeney, C. Ramesha, R. Roberts, D. Rotstein, B. San Pablo, E. B. Sjogren, O. Y. So, F. X. Talamas, W. Tao, A. Trejo, A. Villasenor, M. Welch, T. Welch, P. Weller, P. E. Whiteley, K. Young and S. Zipfel, J. Med. Chem., 2006, 49, 1562 CrossRef CAS PubMed.
- M. C. Bagley, T. Davis, P. G. S. Murziani, C. S. Widdowson and D. Kipling, Pharmaceuticals, 2010, 3, 1842 CrossRef CAS.
- M. C. Bagley, T. Davis, M. C. Dix, P. G. Murziani, M. J. Rokicki and D. Kipling, Future Med. Chem., 2010, 2, 203 CrossRef CAS PubMed.
- T. Mizoroki, K. Mori and A. Ozaki, Bull. Chem. Soc. Jpn., 1971, 44, 581 CrossRef CAS.
- R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320 CrossRef CAS.
- A. Battace, M. Feuerstein, M. Lemhadri, T. Zair, H. Doucet and M. Santelli, Eur. J. Org. Chem., 2007, 3122 CrossRef CAS.
- M. Beller and T. H. Riermeier, Tetrahedron Lett., 1996, 37, 6535 CrossRef CAS.
- T. Sakamoto, Y. Kondo, Y. Kashiwagi and H. Yamanaka, Heterocycles, 1988, 27, 257 CrossRef CAS.
- N. A. Cortese, C. B. Ziegler, B. J. Hrnjez and R. F. Heck, J. Org. Chem., 1978, 43, 2952 CrossRef CAS.
- A. F. Littke and G. C. Fu, J. Am. Chem. Soc., 2001, 123, 6989 CrossRef CAS PubMed.
- C. M. Andersson, A. Hallberg and G. D. Daves, J. Org. Chem., 1987, 52, 3529 CrossRef CAS.
- T. Jeffery, J. Chem. Soc., Chem. Commun., 1984, 1287 RSC.
- S. Cacchi, P. G. Ciattini, E. Morera and G. Ortar, Tetrahedron Lett., 1987, 28, 3039 CrossRef CAS.
- T. Jeffery, Tetrahedron, 1996, 52, 10113 CrossRef CAS.
- H. Zhao, M.-Z. Cai and C.-Y. Peng, Synth. Commun., 2002, 32, 3419 CrossRef CAS.
- J. B. Dorsch and S. M. McElvain, J. Am. Chem. Soc., 1932, 54, 2960 CrossRef CAS.
- M. C. Bagley, M. C. Lubinu and C. Mason, Synlett, 2007, 704 CrossRef CAS.
- M. C. Bagley, M. Baashen, V. L. Paddock, D. Kipling and T. Davis, Tetrahedron, 2013, 68, 8429 CrossRef.
- M. C. Bagley, T. Davis, M. C. Dix, C. S. Widdowson and D. Kipling, Org. Biomol. Chem., 2006, 4, 4158 CAS.
- M. C. Bagley, T. Davis, M. C. Dix, P. G. Murziani, M. J. Rokicki and D. Kipling, Bioorg. Med. Chem. Lett., 2008, 18, 3745 CrossRef CAS PubMed.
- S. P. Davies, H. Reddy, M. Caivano and P. Cohen, Biochem. J., 2000, 351, 95 CrossRef CAS PubMed.
- K. Godl, J. Wissing, A. Kurtenbach, P. Habenberger, S. Blencke, H. Gutbrod, K. Salassidis, M. Stein-Gerlach, A. Missio, M. Cotten and H. Daub, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15434 CrossRef CAS PubMed.
- C. Ota, S. Kumata and S. Kawaguchi, EP1544202A1, 2005, 56–57 Search PubMed.
- J. X. Shi, X. Su, J. Xu, W. Y. Zhang and Y. Shi, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2012, 302, L793 CrossRef CAS PubMed.
- T. Davis, M. C. Dix, M. J. Rokicki, A. J. C. Brook, C. S. Widdowson, D. Kipling and M. C. Bagley, Chem. Cent. J., 2011, 5, 83 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob02229k |
| ‡ Current address: Department of Chemistry, College of Science and Humanities, Shaqra University, Kingdom of Saudi Arabia. |
| This journal is © The Royal Society of Chemistry 2016 |