
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical chain termination resolves the timing of ketoreduction in a partially reducing iterative type I polyketide synthase†
Hirokazu
Kage
a,
Elena
Riva
b,
James S.
Parascandolo
b,
Martin F.
Kreutzer
a,
Manuela
Tosin
*b and
Markus
Nett
*a
aLeibniz Institute for Natural Product Research and Infection Biology, Hans-Knöll-Institute, Adolf-Reichwein-Str. 23, D-07745 Jena, Germany. E-mail: markus.nett@hki-jena.de; Fax: +49 3641 5320811; Tel: +49 3641 5321297
bDepartment of Chemistry, University of Warwick, Library Road, Coventry CV4 7AC, UK. E-mail: m.tosin@warwick.ac.uk; Tel: +44 (0)2476572878
First published on 28th October 2015
Abstract
Synthetic chain terminators were used to capture the biosynthetic intermediates from a partially reducing iterative type I polyketide synthase, which is integrated into a multimodular biosynthesis enzyme. The off-loaded metabolites clarified the timing of ketoreduction and aromatization in the assembly of the antibiotic micacocidin.
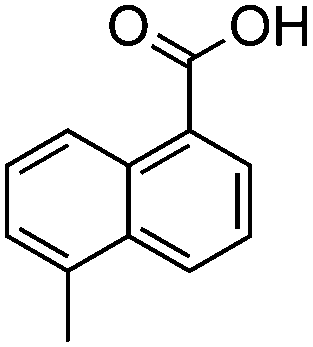
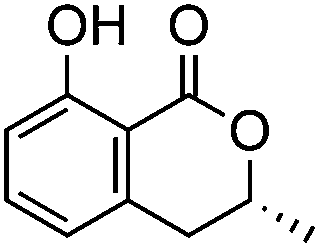
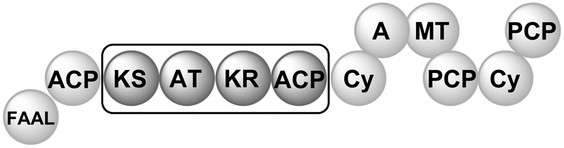
Polyketides represent an important class of secondary metabolites that are found in bacteria, fungi, plants and marine organisms alike.1 Their structural diversity arises from the action of large multifunctional proteins. The polyketide synthases (PKSs) select the acyl-CoA building blocks for incorporation and they control the number of chain extensions, the reductive processing and cyclization pattern.2 Depending on their mode of action, that is, whether or not they catalyse multiple elongation steps, PKSs are categorized as iterative or noniterative.3 While the programming of noniterative PKSs is often predictable,4 the mechanistic rules of iterative PKSs remain to be fully understood.5 This is particularly true for partially reducing (PR)-PKSs, which introduce hydroxyl groups at specific sites of the growing polyketide chain and, thereby, set the structural basis for subsequent cyclization reactions. In general, PR-PKSs share a highly conserved domain architecture, which distinguishes them from non-reducing (NR) and highly reducing (HR) PKSs. They typically possess a ketoreductase (KR) domain, but they lack dehydratase (DH) or enoylreductase (ER) domains for further reductive processing. PR-PKSs are mostly encountered as discrete proteins, catalysing the production of small aromatic molecules (Table 1). Examples include 6-methylsalicylic acid synthase (6-MSAS), which is quite common in fungi6 but can also be found in several bacteria,7 NcsB and AziB, which provide the biosynthetic building blocks 2-hydroxyl-5-methyl-naphthoic acid and 5-methyl-naphthoic acid,8,9 as well as the mellein synthase SACE5532.10
| Protein | Domain architecture | PKS product |
|---|---|---|
| 6-MSAS |

|

|
| NcsB |
|

|
| AziB |
|
|
| SACE5532 |

|
|
| MicC |
|

|
Recently, we identified a PR-PKS as a constituent of the multimodular enzyme MicC in the biosynthetic pathway to the antibiotic micacocidin.11In vitro studies confirmed that the PR-PKS is loaded with hexanoic acid via intraprotein transfer from an N-terminal acyl carrier protein (ACP) domain prior to iterative chain elongation.12,13 Although the described priming mechanism appears to be quite rare for PR-PKSs, it is commonly observed in fungal NR-PKSs.14 Another deviation from the prototypical PR-PKS is the lack of a thioester hydrolase (TH) domain in MicC. In 6-MSAS biosynthesis, the TH domain was shown to mediate the final product release,15 and the same function is also conceivable for NcsB, AziB and SACE5532. In micacocidin biosynthesis, however, the assumed product of the PR-PKS reaction (i.e., 6-pentylsalicylic acid) is immediately transferred to a non-ribosomal peptide synthetase (NRPS) module. A premature offload by a TH domain is thus not required. Functional interrogation of the micacocidin PR-PKS in a heterologous host already confirmed that the integrated KR domain is indispensable for the correct assembly of 6-pentylsalicylic acid.12 Due to the peculiarities of MicC in comparison with other PR-PKSs, several questions concerning its function have remained unsolved. In particular, the timing of the regioselective ketoreduction as well as the cyclization and aromatization of the linear tetraketide precursor were not clear. To achieve a better understanding of the programming of a PR-PKS, which is embedded in a multimodular enzyme, we sought to elucidate the sequence of the aforementioned reactions.
Initially, the active site of the KR domain in MicC was inactivated by site-directed mutagenesis. The mutation was introduced into the chromosome of the native micacocidin producer Ralstonia solanacearum GMI1000 by homologous recombination. The resulting mutant strain RS36 was incapable of synthesizing the antibiotic (1), but accumulated a metabolite (2) that was only present at low levels in the wild type strain (Fig. 1). After purifying this compound by HPLC, 2 could be identified as 4-methoxy-6-pentyl-2H-pyran-2-one through comparison of its NMR and mass spectroscopic data with literature values (Fig. S1 and S2†).16 Biosynthetically, 2 is proposed to originate from hexanoic acid, which would be extended by two decarboxylative Claisen condensations with malonyl-CoA and, eventually, undergo a lactonization reaction. A disruption of the ketosynthase (KS) domain in MicC abolished the production of 2 and, thereby, confirmed the proposed biosynthetic relationship (Fig. 1A, profile c). Despite thorough analysis of the extract from strain RS36 by LC/MS, only traces of putative tetraketide products were detected. This suggests that the ketoreduction of the intact micacocidin PR-PKS takes place at the stage of the triketide intermediate. If this step is omitted, e.g., due to an impaired KR domain, the assembly line-based biosynthesis would likely be aborted and lead to the occurrence of 2.
 | ||
| Fig. 1 Metabolic profiles of crude culture extracts from the R. solanacearum strains RS36 (profile a, MicC-KR mutant), GMI1000 (profile b, wild type), and RS31 (profile c, ΔmicC mutant) (A). Chemical structures of the metabolites 1 and 2 (B). | ||
To get further insights into the sequence of biosynthetic reactions and to verify the proposed timing of ketoreduction, we decided to intercept the enzymatic assembly of 6-pentylsalicylic acid. Chemical probes, such as malonyl carba(dethia) pantetheines and N-acetylcysteamines have been shown to compete with acyl carrier protein (ACP)-bound substrates for polyketide chain elongation and to induce a premature release of truncated intermediates. Since the resulting products cannot be reloaded onto the assembly line, they become available for LC/MS characterization.17 It should be noted that the aforementioned probes have been successfully utilised both for in vitro17 and in vivo18–21 investigations of modular PKSs.
In the present study, R. solanacearum GMI1000 was initially grown in the presence of malonyl carba(dethia)-N-acetyl cysteamine methyl ester 3a, which hydrolyses in vivo to generate the active probe 6a.18–20 Following a 3-day cultivation, the bacterial cultures were extracted with ethyl acetate to recover the off-loaded intermediates. LC/HRMS analysis revealed the presence of molecular species, which were not present in control samples and could be assigned to putative diketide and tetraketide intermediates (Fig. 2A and S3–S7†). The effective occurrence of these molecules was verified in parallel experiments in which R. solanacearum cultures were supplemented with the deuterated probe 3b in order to introduce a distinctive mass shift in the corresponding species. Feeding experiments with the N-pentynoyl probe 4 led to analogous results and the corresponding diketide and tetraketide intermediates were also subjected to mass fragmentation studies, which support their putative structure (Fig. S8–S12†). Although feeding experiments with probes 3a, 3b and 4 were repeated several times with varying concentrations of the synthetic chain terminator, no masses corresponding to triketide species were detected and the precise timing of the ketoreduction remained unclear. Assuming that cellular uptake and/or in vivo hydrolysis of the fed methyl esters might be limiting factors for the detection of all biosynthetic intermediates,18,19 we resorted to the use of malonyl carba(dethia)-N-decanoyl cysteamine methyl ester (5), which has been found to be more efficiently hydrolysed in vivo to its corresponding carboxylate 8.21 LC/MS analyses of R. solanacearum extracts grown in the presence of 5 revealed the capture of the same diketide and tetraketide species observed in the previous feeding experiments. More importantly, the masses of putative triketide species were also detected (Fig. 2B and ESI†). MS/MS analysis of the putative reduced triketide yielded diagnostic peaks at m/z 226 and 208, which correspond to the formation of a protonated cyclic imine following loss of the decanoyl chain during fragmentation and subsequent dehydration.21 This confirmed the identity of the hydroxylated triketide species and established the timing of the ketoreduction during the MicC-mediated assembly of 6-pentylsalicylic acid (Fig. 3).
 | ||
| Fig. 2 Chain termination probes 6–8 utilised for the capture of polyketide intermediates in micacocidin assembly (A). Extracted ion chromatograms for putative tetraketides (sodiated addicts) captured from R. solanacearum GMI1000 cultures grown in the presence of the methyl esters 3a and 3b, which hydrolyse to 6a and 6b (B).18–21 Extracted ion chromatogram and HR-MS2 analyses for the protonated adduct of a putative reduced triketide identified from the growth of R. solanacearum GMI1000 in the presence of the N-decanoyl probe 8 (C).21 | ||
 | ||
| Fig. 3 Model for the assembly of enzyme-bound 6-pentylsalicylate by the PR-PKS from micacocidin biosynthesis. | ||
The selective β-ketoreduction would hence take place immediately after the second Claisen condensation with malonyl-CoA. Because no distinct masses of unsaturated triketides were observed during the different chain termination experiments, we assume that the intermediary hydroxyl group is not eliminated before the final chain elongation. Subsequently, an elongated 5-hydroxy tetraketide must undergo an intramolecular aldol condensation and a likely spontaneous dehydration, with enolisation eventually giving rise to enzyme-bound 6-pentylsalicylate. These experiments also confirm that MicC encodes for the minimum set of proteins necessary to generate 6-alkyl salicylic acid derivatives. The mechanism of the 6-pentylsalicylate moiety transfer to the MicC downstream domains and others is currently under investigation.
Conclusions
In the present study, chemical probes were used to elucidate the programming of a PR-PKS which, unlike other related enzymes, is integrated into a multimodular assembly line. Notwithstanding this specific feature, our results show that the investigated PR-PKS follows the same logic as the previously characterized 6-MSAS,15,22 thereby establishing a unified paradigm for the biosynthetic mechanism of these proteins. Moreover, we demonstrate that chemical chain termination is a useful tool for the functional interrogation of iterative PKSs under in vivo conditions. The possibility to trap an entire range of biosynthetic intermediates in simple feeding experiments with nonhydrolysable malonyl ACP analogues complements existing methods for the analysis of these enzymes. Since selective domain inactivations can abort the biosynthesis at a specific step, the functional study of iterative PKSs usually involves domain swaps,22,23 heterologous reconstitution,10a,24 or the synthesis of highly reactive substrates for in vitro tests.10b When compared to these methods, the advantage of chemical chain termination clearly lies in its convenience.Acknowledgements
The idea for this project was born on occasion of the 3rd Transatlantic Frontiers of Chemistry Symposium, which was organised by the American Chemical Society (ACS), the Gesellschaft Deutscher Chemiker (GDCh), and the Royal Society of Chemistry (RSC). The authors gratefully acknowledge Andrea Perner (Hans-Knöll-Institute Jena) and Dr Lijiang Song (Warwick Chemistry) for initial LC/HRMS measurements. Financial support was provided by the Leibniz Association, BBSRC (project grant BB/J007250/1 to M. T.), the Institute of Advanced Studies (IAS) at Warwick (Postdoctoral Fellowship to E. R.) and EPSRC (DTA studentship to J. S. P.)Notes and references
- (a) D. O'Hagan, The Polyketide Metabolites, 1999, Ellis Horwood, Chichester Search PubMed; (b) K. J. Weissman, Methods Enzymol., 2009, 459, 3 CAS.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688 CrossRef CAS PubMed.
- R. J. Cox, Org. Biomol. Chem., 2007, 5, 2010 CAS.
- M. Nett, Prog. Chem. Org. Nat. Prod., 2014, 99, 199 Search PubMed.
- K. M. Fisch, RSC Adv., 2013, 3, 18228 RSC.
- (a) J. Beck, S. Ripka, A. Siegner, E. Schiltz and E. Schweizer, Eur. J. Biochem., 1990, 192, 487–498 CrossRef CAS PubMed; (b) I. Fujii, Y. Ono, H. Tada, K. Gomi, Y. Ebizuka and U. Sankawa, Mol. Gen. Genet., 1996, 253, 1 CrossRef CAS PubMed; (c) P. Lu, A. Zhang, L. M. Dennis, A. M. Dahl-Roshak, Y.-Q. Xia, B. Arison, Z. An and J. S. Tkacz, Mol. Genet. Genomics, 2005, 273, 207 CrossRef CAS PubMed.
- (a) S. Gaisser, A. Trefzer, S. Stockert, A. Kirschning and A. Bechthold, J. Bacteriol., 1997, 179, 6271 CAS; (b) W. Ding, C. Lei, Q. He, Q. Zhang, Y. Bi and W. Liu, Chem. Biol., 2010, 17, 495 CrossRef CAS PubMed; (c) S. G. Van Lanen, T. J. Oh, W. Liu, E. Wendt-Pienkowski and B. Shen, J. Am. Chem. Soc., 2007, 129, 13082 CrossRef CAS PubMed; (d) W. L. Lu, N. Roongsawang and T. Mahmud, Chem. Biol., 2011, 18, 425 CrossRef CAS PubMed.
- B. Sthapit, T. J. Oh, R. Lamichhane, K. Liou, H. C. Lee, C. G. Kim and J. K. Sohng, FEBS Lett., 2004, 566, 201 CrossRef CAS PubMed.
- Q. F. Zhao, Q. L. He, W. Ding, M. C. Tang, Q. J. Kang, Y. Yu, W. Deng, Q. Zhang, J. Fang, G. L. Tang and W. Liu, Chem. Biol., 2008, 15, 693 CrossRef CAS PubMed.
- (a) H. Sun, C. L. Ho, F. Ding, I. Soehano, X.-W. Liu and Z.-X. Liang, J. Am. Chem. Soc., 2012, 134, 11924 CrossRef CAS PubMed; (b) I. Soehano, L. Yang, F. Ding, H. Sun, Z. J. Low, X. Liu and Z.-X. Liang, Org. Biomol. Chem., 2014, 12, 8542 RSC.
- M. F. Kreutzer, H. Kage, P. Gebhardt, B. Wackler, H. P. Saluz, D. Hoffmeister and M. Nett, Appl. Environ. Microbiol., 2011, 77, 6117 CrossRef CAS PubMed.
- H. Kage, M. F. Kreutzer, B. Wackler, D. Hoffmeister and M. Nett, Chem. Biol., 2013, 20, 764 CrossRef CAS PubMed.
- M. F. Kreutzer, H. Kage, J. Herrmann, J. Pauly, R. Hermenau, R. Müller, D. Hoffmeister and M. Nett, Org. Biomol. Chem., 2014, 12, 113 CAS.
- (a) J. M. Crawford, B. C. R. Dancy, E. A. Hill, D. W. Udwary and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16728 CrossRef CAS PubMed; (b) H. Zhou, J. Zhan, K. Watanabe, X. Xie and Y. Tang, Proc. Natl. Acad. Sci. U. S. A., 2006, 105, 6249 CrossRef PubMed; (c) A. Al Fahad, A. Abood, K. M. Fisch, A. Osipow, J. Davison, M. Avramović, C. P. Butts, J. Piel, T. J. Simpson and R. J. Cox, Chem. Sci., 2014, 5, 523 RSC.
- T. Moriguchi, Y. Kezuka, T. Nonaka, Y. Ebizuka and I. Fujii, J. Biol. Chem., 2010, 285, 15637 CrossRef CAS PubMed.
- A. Evidente, M. C. Zonno, A. Andolfi, C. Troise, A. Cimmino and M. Vurro, J. Antibiot., 2012, 65, 203 CrossRef CAS PubMed.
- M. Tosin, L. Betancor, E. Stephens, W. M. A. Li, J. B. Spencer and P. F. Leadlay, ChemBioChem, 2010, 11, 539 CrossRef CAS PubMed.
- M. Tosin, Y. Demydchuk, J. S. Parascandolo, C. Blasco Per, F. J. Leeper and P. F. Leadlay, Chem. Commun., 2011, 47, 3460 RSC.
- M. Tosin, L. Smith and P. F. Leadlay, Angew. Chem., Int. Ed., 2011, 50, 11930 CrossRef CAS PubMed.
- J. Franke, K. Ishida and C. Hertweck, Angew. Chem., Int. Ed., 2012, 51, 11611 CrossRef CAS PubMed.
- E. Riva, I. Wilkening, S. Gazzola, W. M. A. Li, L. Smith, P. F. Leadlay and M. Tosin, Angew. Chem., Int. Ed., 2014, 53, 11944 CrossRef CAS PubMed.
- T. Moriguchi, Y. Ebizuka and I. Fujii, ChemBioChem, 2008, 9, 1207 CrossRef CAS PubMed.
- K. M. Fisch, W. Bakeer, A. A. Yakasai, Z. Song, J. Pedrick, Z. Wasil, A. M. Bailey, C. M. Lazarus, T. J. Simpson and R. J. Cox, J. Am. Chem. Soc., 2011, 133, 16635 CrossRef CAS PubMed.
- (a) J. Antosch, F. Schaefers and T. A. M. Gulder, Angew. Chem., Int. Ed., 2014, 53, 3011 CrossRef CAS PubMed; (b) Y. Li, H. Chen, Y. Xie, H. Wang, R. L. Cerny, Y. Shen and L. Du, Angew. Chem., Int. Ed., 2014, 53, 7524 CrossRef CAS PubMed; (c) B. Busch, N. Ueberschaar, S. Behnken, Y. Sugimoto, M. Werneburg, N. Traitcheva, J. He and C. Hertweck, Angew. Chem., Int. Ed., 2013, 52, 5285 CrossRef CAS PubMed; (d) B. Busch, N. Ueberschaar, Y. Sugimoto and C. Hertweck, J. Am. Chem. Soc., 2012, 134, 12382 CrossRef CAS PubMed; (e) J. M. Winter, M. Sato, S. Sugimoto, G. Chiou, N. K. Garg, Y. Tang and K. Watanabe, J. Am. Chem. Soc., 2012, 134, 17900 CrossRef CAS PubMed; (f) A. O. Zabala, Y.-H. Chooi, M. S. Choi, H.-C. Lin and Y. Tang, ACS Chem. Biol., 2014, 9, 1576 CrossRef CAS PubMed; (g) Y.-H. Chooi, C. Krill, R. A. Barrow, S. Chen, R. Trengove, R. P. Oliver and P. S. Solomon, Appl. Environ. Microbiol., 2015, 81, 177 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob02009c |
| This journal is © The Royal Society of Chemistry 2015 |