
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComplementary isonitrile-based multicomponent reactions for the synthesis of diversified cytotoxic hemiasterlin analogues†
Giordano
Lesma
a,
Ivan
Bassanini
a,
Roberta
Bortolozzi
b,
Chiara
Colletto
a,
Ruoli
Bai
c,
Ernest
Hamel
c,
Fiorella
Meneghetti
d,
Giulia
Rainoldi
a,
Mattia
Stucchi
a,
Alessandro
Sacchetti
e,
Alessandra
Silvani
*a and
Giampietro
Viola
*b
aUniversità di Milano, Dipartimento di Chimica, via Golgi 19, Milano, 20133, Italy. E-mail: alessandra.silvani@unimi.it
bUniversità degli Studi di Padova, Dipartimento di Salute della Donna e del Bambino, via Giustiniani 2, Padova, 35128, Italy. E-mail: giampietro.viola.1@unipd.it
cScreening Technologies Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute, Frederick National Laboratory for Cancer Research, National Institutes of Health, Frederick, Maryland 21702, USA
dDipartimento di Scienze Farmaceutiche, Università degli Studi di Milano, via L. Mangiagalli 25, 20133 Milano, Italy
ePolitecnico di Milano, Dipartimento di Chimica, Materiali ed Ing. Chimica ‘Giulio Natta’, Piazza Leonardo da Vinci 32, Milano, 20133, Italy
First published on 8th October 2015
Abstract
A small family of structural analogues of the antimitotic tripeptides, hemiasterlins, have been designed and synthesized as potential inhibitors of tubulin polymerization. The effectiveness of a multicomponent approach was fully demonstrated by applying complementary versions of the isocyanide-based Ugi reaction. Compounds strictly related to the lead natural products, as well as more extensively modified analogues, have been synthesized in a concise and convergent manner. In some cases, biological evaluation provided evidence for strong cytotoxic activity (six human tumor cell lines) and for potent inhibition of tubulin polymerization.
Introduction
Multicomponent reactions (MCRs) are convergent chemical processes that involve the one pot condensation of more than two reactants to form a product that incorporates most of each reagent, containing ideally all atoms. In addition to generating structural complexity with greater atom economy, they usually also offer the advantage of simplicity and synthetic efficiency over conventional chemical reactions.1 In particular, isonitrile-based MCRs (IMCRs) are widely applied in diversity-oriented synthetic strategies, due to the considerable ability of isocyanides to undergo α-addition with electrophiles and nucleophiles and due to the various possibilities to exploit the different secondary reactions of the obtained α-adducts. Among IMCRs, the Ugi reaction has undergone developments over the years, and various modifications of the classic protocol have been used successfully. As a consequence, more than linear, peptide-like adducts can be obtained by the introduction of unusual building blocks, by transformation of the MCR products using post-condensation reactions or by performing intramolecular IMCRs with bifunctional inputs.2Nevertheless, with regard to the target-oriented synthesis of natural products or their derivatives, the rational design of practical and versatile approaches employing MCRs, and in particular the Ugi reaction and its modifications, remained, until recently, a largely unexplored area of chemical research.3
As a result of our interest in the MCR-based approach to conformationally constrained peptidomimetics,4 in this work we show the use of complementary Ugi-type reactions for the synthesis of a small family of cytotoxic hemiasterlin analogues.
Hemiasterlins are a family of natural tripeptides, discovered and isolated from the South African marine sponge Hemiastrella minor some years ago.5 The most active members of the family show cytotoxicity in the nanomolar range and are highly potent inhibitors of microtubule polymerization, binding in the vinca domain of tubulin.6 Relative to other known antimitotic agents, hemiasterlins possess an attractive combination of structural simplicity and potent antimitotic activity, which makes them ideal targets for synthetic modification.7
Recently, synthetic analogues of hemiasterlin 1 (Fig. 1), namely taltobulin (HTI-286) 2 and the closely related 3,8,9 wherein aryl groups replace the indol-3-yl substituent, and the piperidine-based E7974 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 advanced into clinical trials, due to their more potent in vivo cytotoxicity and antimitotic activity. Moreover, unlike taxanes and vincas, such synthetic derivatives are poor substrates for P-glycoprotein drug transporters and maintain toxicity towards cell lines with high expression of multidrug resistant (MDR) efflux pumps. Further, since 4 binds predominantly to the α-subunit of the tubulin, with minor binding to the β-subunit, it offers significant promise of activity in taxane-resistant tumor types, regardless of whether the mechanism driving resistance is based on P-glycoprotein or tubulin mutations.11
10 advanced into clinical trials, due to their more potent in vivo cytotoxicity and antimitotic activity. Moreover, unlike taxanes and vincas, such synthetic derivatives are poor substrates for P-glycoprotein drug transporters and maintain toxicity towards cell lines with high expression of multidrug resistant (MDR) efflux pumps. Further, since 4 binds predominantly to the α-subunit of the tubulin, with minor binding to the β-subunit, it offers significant promise of activity in taxane-resistant tumor types, regardless of whether the mechanism driving resistance is based on P-glycoprotein or tubulin mutations.11
 | ||
| Fig. 1 Tubulin polymerization inhibitors: natural hemiasterlins and synthetic analogues. | ||
Hemiasterlins and their derivatives contain three highly modified amino acids (A, B and C segments, see Fig. 1) and their successful synthesis has always relied on amide bond synthesis in a sterically challenged environment.12 This approach has prevented more extensive structural modifications, for instance at the central (L)-valine or (L)-tert-leucine amino acid residue.
Since the Ugi reaction and its modifications are less sensitive to steric hindrance than peptide coupling, we envisioned that a multicomponent strategy could be suitable for the generation of a wide range of hemiasterlin derivatives, also including non-peptidic analogues. By means of a Ugi four-component reaction (U-4CR), we achieved the synthesis of 5 (Fig. 2), a compound closely related to taltobulin, in which we employed (L)-valine in the place of (L)-tert-leucine, as it represents a variation that could allow substantial bioequivalence. By the same approach, we achieved also the unprecedented indole-based analogue 6. Applying a Ugi-like three-component reaction (U-like-3CR), oxazole-based compounds 7–9 could be easily obtained. To the best of our knowledge, these compounds represent the first example of hemiasterlin analogues with major modifications of the central B core. Lastly, a Ugi–Joullié three-component reaction (U-J-3CR) allowed us to prove the applicability of the multicomponent approach for the synthesis of piperidine-based compounds, such as 10–12, closely related to E7974.
 | ||
| Fig. 2 Structures of hemiasterlin analogues 5–12. | ||
Results and discussion
The aldehyde components 13–16, which were necessary in the U-4CR and U-like-3CR strategies, were prepared as described in Scheme 1. The syntheses relied on an allylpalladium-catalyzed α-arylation of isobutyraldehyde with an appropriate aryl or heteroaryl bromide, in the presence of catalytic Q-phos,13 cleanly affording the desired aldehydes in yields up to 75%. Alternative palladium-catalyzed protocols, based on palladium diacetate as the catalyst,14 or involving vinyl acetates as coupling components,15 proved to be less effective. | ||
| Scheme 1 Synthesis of aldehyde components 13–16. Reagents and conditions: (a) isobutyraldehyde, [Pd(η3-allyl)Cl]2, Q-phos, Cs2CO3, THF, reflux (13: 75%; 14: 57%; 15: 50%; 16: 46%). | ||
Many synthetic procedures are reported for the preparation of isocyanides from α-amino acid ester hydrochlorides. In order to achieve the enantiomerically pure α-isocyanoacetate component 17 (Scheme 2), we selected a two-step sequence, involving formylation of the precursor by reaction with trimethyl orthoformate under neat conditions, followed by dehydration of the obtained α-N-formylamino acid methyl ester, using triphosgene as a mild dehydrating agent and N-methylmorpholine as the base.16 Trifluoroacetic acid and methylamine were chosen as the suitable carboxylic acid and amine for the U-4CR process.
 | ||
| Scheme 2 First multicomponent approach: the 4C-Ugi reaction. Reagents and conditions: (a) MeOH, MgSO4, rt (18a: 32%; 18b: 31%; 19a: 37%; 19b: 38%). | ||
To preserve the optical purity of the isocyanoacetate, the Ugi reactions employing aldehydes 13 or 14 as carbonyl components were conducted after a precondensation time of 2 h between the aldehyde and methylamine, in the presence of MgSO4 used as the dehydrating promoter.17 Ugi compounds 18 and 19 were obtained in good overall yields (63% for 18, 75% for 19), both as 1:1 diastereoisomeric mixtures, which could be easily separated by flash chromatography (FC).
Relying on a valuable literature suggestion,18 the stereochemistry of both compounds 18 and 19 was postulated by NMR, and in particular performing the NOESY experiment on the separated a and b diastereoisomers. Besides, with the aim to unambiguously confirm the stereochemistry of these intermediates, we performed X-ray diffraction analysis on compound 18b, for which good diffracting single crystals were isolated from a methanol solution. The crystallographic structure of 18b disclosed an (R,S)-configuration (Fig. 3), leading us to select diastereoisomers 18a and 19a for continuing the synthesis, as the stereochemistry reported for potent taltobulin derivatives is (S,S,S).
 | ||
| Fig. 3 ORTEP19 view of compound 18b, anti (R,S), and the relative atom-numbering scheme (thermal ellipsoids at 40% probability). | ||
To complete the synthesis, methyl esters 18a and 19a were carefully converted into the corresponding acids under mild basic conditions, with the preservation of the trifluoroacetamide functional group, and then condensed with the known amino ester fragment 20,20 in acceptable yields using HTBU and DIPEA. From intermediates 21 and 22, the final compounds 5 and 6 were eventually recovered as amino acids by basic hydrolysis of both the ethyl ester and the trifluoroacetamide group (Scheme 3).
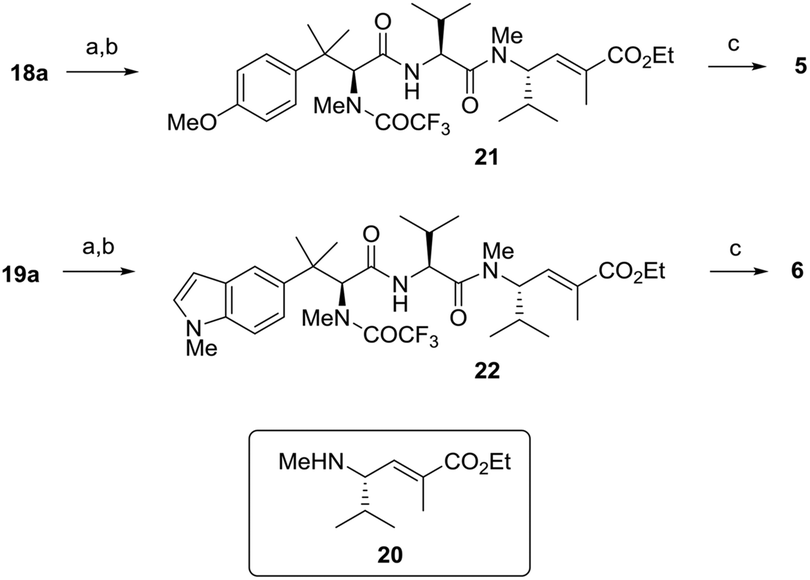 | ||
| Scheme 3 Synthesis of analogues 5 and 6. Reagents and conditions: (a) LiOH, 50% aq. MeOH, rt; then (b) compound 20, HBTU, DIPEA, CH2Cl2, rt (21: overall 58%; 22: overall 52%). (c) LiOH, 50% aq. MeOH, 60 °C (5: 76%; 6: 65%). | ||
With the aim of evaluating more extensively modified analogues, even compounds lacking amide bonds, we looked at a U-like-3CR and pursued the synthesis of oxazole-based compounds 7–9, as depicted in Scheme 4. In this case, the key intermediate is the α-isocyanoacetamide 23. Compared with α-isocyanoacetates, α-isocyanoacetamides are much more configurationally stable. They show a higher Lewis basicity of the amide oxygen compared with that of the corresponding esters, and this should kinetically favor the cyclization step with the irreversible formation of the oxazole ring.21 Isocyanopeptide 23 was efficiently prepared starting from amine 24,22 through intermediate formation of formamide 25 and subsequent dehydration using diphosgene at −30 °C,23 as depicted in Scheme 5. By stirring compound 23 with aldehydes 13, 15 or 16 in the presence of methylamine and MgSO4, we easily obtained the final compounds 7–9, in satisfactory yields as an inseparable 1:1 to 1.5:1 mixture of diastereoisomers. Since for such extensively modified scaffolds the preliminary indication of activity can be considered the main goal, we performed the biological evaluation on the diastereoisomeric mixture (see below).
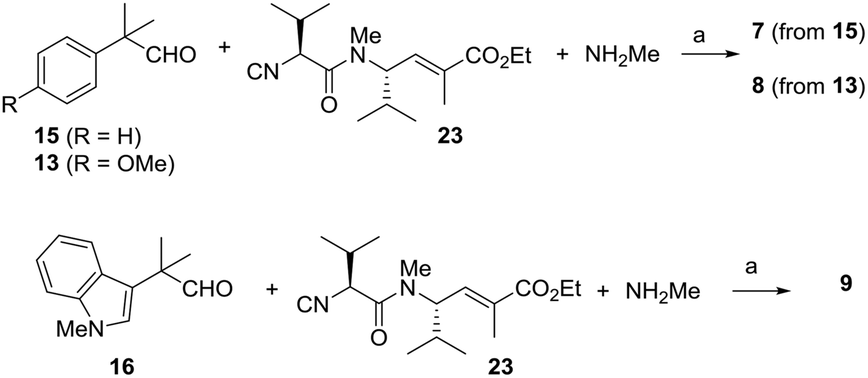 | ||
| Scheme 4 Second multicomponent approach: the 3C-Ugi-like reaction. Synthesis of analogues 7–9. Reagents and conditions: (a) MeOH, MgSO4, rt (7: 51%; 8: 68%; 9: 64%). | ||
 | ||
| Scheme 5 Synthesis of the isocyanopeptide 23. Reagents and conditions: (a) acetic formic anhydride, CH2Cl2, 0 °C to rt (25: quant. yield). (b) N-methylmorpholine, diphosgene, THF, −30 °C to 0 °C (23: 80%). | ||
In order to exploit the multicomponent strategy for the synthesis of piperidine-based E7974 analogues, we relied on the U-J-3CR, a modification of the Ugi protocol involving the use of cyclic imines and resulting in the synthesis of α-substituted nitrogen heterocycles. Being aware of the reported risk of isocyanoacetate epimerization related to the manner in which the cyclic imine was prepared, we followed the protocol of inducing a reversible trimerization of Δ1-piperideine, yielding crystalline and easily isolable tripiperideine 26, as the starting component. Carrying out the multicomponent reaction of tripiperideine, isocyanoacetate 17 and 5-pentenoic acid as the acid component, we obtained the expected peptide 27 in good yield, as a 1:1 inseparable diastereoisomeric mixture. Unfortunately, this mixture could not be resolved at any stage of the synthesis of the final compounds 11 and 12. In our approach, the 5-pentenoic acid was chosen because the pentenoyl moiety can be selectively removed by iodolactonization24 after the multicomponent reaction and the resulting secondary amine could be functionalized in various ways. Once the NH piperidine derivative 28 was synthesized, we looked at the reductive amination as a route to install selected lipophilic moieties on the piperidine ring. Therefore, after temporary Boc protection of the piperidine secondary nitrogen to give 29 and subsequent methylester hydrolysis and amide coupling with fragment 20, we easily synthesized compound 10. From 10, Boc deprotection gave the key intermediate 30. Reductive amination with acetone or cyclohexenone, using sodium triacetoxyborohydride and acetic acid, afforded, respectively, the final compounds 11 and 12 (Scheme 6).
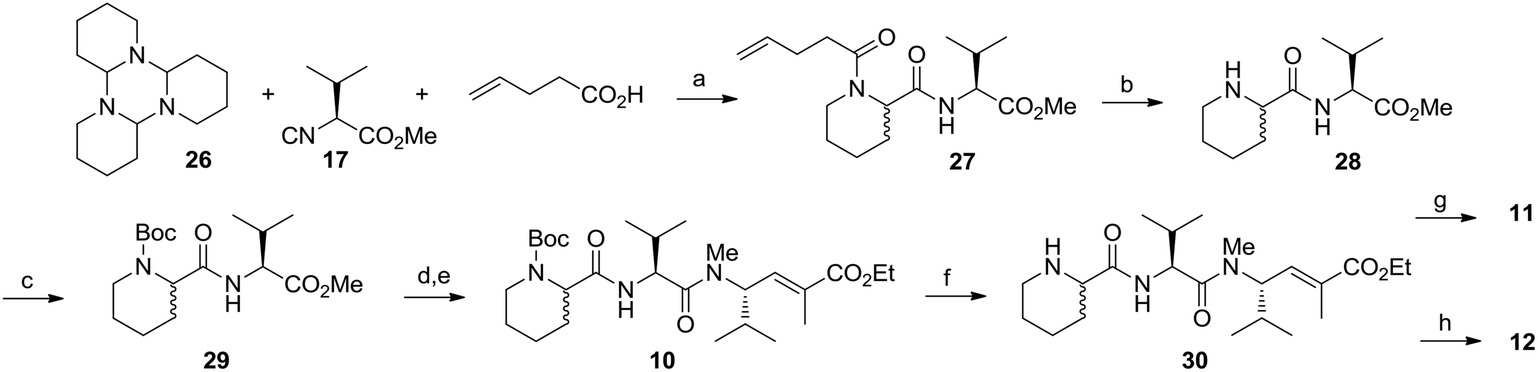 | ||
| Scheme 6 Third multicomponent approach: the 3C-Ugi–Joullié reaction. Synthesis of analogues 10–12. Reagents and conditions: (a) MeOH, rt (27: 46%). (b) Iodine, aq. Na2S2O3, THF/H2O, rt (28: 85%). (c) (Boc)2O, CH2Cl2, rt (29: 92%). (d) LiOH, 50% aq. MeOH, rt; then (e) compound 20, HBTU, DIPEA, CH2Cl2, rt (10: overall 47%). (f) 50% TFA in CH2Cl2, rt (30: quant. yield). (g) Acetone, Na(OAc)3BH, AcOH, CH2Cl2, rt (11: quant. yield). (h) Cyclohexenone, Na(OAc)3BH, AcOH, CH2Cl2, rt (12: quant. yield). | ||
Compounds 5–12 were evaluated in vitro for their cytotoxic activity against a panel of six human tumor cell lines, and the results are summarized in Table 1. Two of the analogues synthesized during this work, namely compounds 6 and 11, possessed cytotoxicity against all lines, though being 10-fold less active compared to the model compound HTI-286. The other compounds showed modest (compound 5) activity or were practically devoid of any significant activity, having GI50 values in the micromolar range. The two highly active compounds 6 and 11 were also examined for their effects on tubulin polymerization and as inhibitors of the binding of [3H]vinblastine, [3H]dolastatin 10, and [3H]halichondrin B to tubulin (Table 2). In these studies, they were found to be active as tubulin inhibitors, although less active than HTI-286 (compound 2). Their reduced activity in the tubulin assays is in agreement with their reduced cytotoxicity as compared with 2 (compare data in Tables 1 and 2). We think it is most likely that their interactions with tubulin are similar to those of hemiasterlin (1) and HTI-286 (2). Compound 6 retains a high structural similarity to the natural product hemiasterlin 1, highlighting the possibility that further modifications of the aromatic moiety in the first (A) amino acid segment will yield interesting and active agents. With regard to compound 11, closely related structurally to E7974 (4), its potent activity suggests a marginal role of the piperidine ring stereogenic centre configuration, opening the way to more reliable and straightforward synthetic approaches. Lastly, the poor activity found with the oxazole-based derivatives 7–9 discourages further extensive modifications on the central (B) amino acid segment. In particular, the consistent structural modification brought by the presence of the oxazole ring caused a remarkable conformational bending, presumably forcing the molecule into a less favorable conformation with respect to bioactive compounds.
| Compd | GI50a (nM) |
|||||
|---|---|---|---|---|---|---|
| HT-29 | HeLa | MCF-7 | Jurkat | HL-60 | RS4;11 | |
| a GI50 = compound concentration required to inhibit tumor cell growth by 50%. Data are presented as the mean ± SE (Standard Error) from the dose–response curves of at least three independent experiments. | ||||||
| HTI-286 (2) | 0.4 ± 0.05 | 0.3 ± 0.06 | 2.0 ± 0.6 | 0.2 ± 0.08 | 0.4 ± 0.1 | 0.3 ± 0.1 |
| 5 | 3000 ± 356 | 700 ± 259 | 3750 ± 943 | 176.7 ± 28.5 | 34.3 ± 5.6 | 430 ± 224 |
| 6 | 8.0 ± 2.4 | 11.2 ± 0.5 | 7.3 ± 1.7 | 0.8 ± 0.1 | 1.1 ± 0.1 | 2.3 ± 0.3 |
| 7 | 12580 ± 738 |
21300 ± 2979 |
16800 ± 4217 |
2333 ± 120 | 3067 ± 120 | 2967 ± 418 |
| 8 | 23500 ± 512 |
10580 ± 5203 |
22300 ± 1250 |
2441 ± 203 | 923 ± 79.3 | 2000 ± 600 |
| 9 | 4700 ± 711 | 8533 ± 654 | 8300 ± 1525 | 2433 ± 296 | 3800 ± 833 | 6833 ± 917 |
| 10 | 36433 ± 2882 |
13333 ± 4826 |
13956 ± 6233 |
4400 ± 458 | 10166 ± 1524 |
405 ± 45 |
| 11 | 4.2 ± 1.1 | 0.9 ± 0.3 | 25.3 ± 5.1 | 0.9 ± 0.2 | 0.8 ± 0.4 | 0.9 ± 0.4 |
| 12 | 18780 ± 7486 |
22760 ± 1311 |
17160 ± 1513 |
223.3 ± 18.6 | 320 ± 35.1 | 125.3 ± 33 |
| Inhibition of bindingb of | |||||||
|---|---|---|---|---|---|---|---|
| Inhibition of tubulin assemblyIC50 (μM) ± SDa | [3H]vinblastine | [3H]dolastatin 10 | [3H]halichondrin B | ||||
| % inhibition ± SDa | |||||||
| 5 μM | 20 μM | 5 μM | 20 μM | 5 μM | 20 μM | ||
| inhibitor | inhibitor | inhibitor | |||||
| a SD = standard deviation. b Ligand binding studies were performed in 0.1 M 4-morpholinethanesulfonate (pH 6.9 in 1 M stock solution adjusted with NaOH)–0.5 mM MgCl2 containing 10 μM tubulin (1.0 mg ml−1), 10 μM radiolabeled ligand, and inhibitors as indicated. The reaction volume was 0.3 mL and the incubation time was 15 min at RT (around 20 °C). Ligands were mixed prior to tubulin addition. Duplicate aliquots of each reaction mixture were applied to syringe columns of Sephadex G-50 (superfine) swollen in 0.1 M Mes–0.5 mM MgCl2. At least two experiments performed for each condition. | |||||||
| HTI-286 (2) | 0.94 ± 0.01 | 41 ± 10 | 62 ± 20 | 2 ± 1 | 22 ± 3 | 21 ± 4 | 62 ± 10 |
| 6 | 10 ± 0.6 | 3 ± 1 | 22 ± 7 | 2 ± 1 | 27 ± 4 | 1 ± 1 | 11 ± 4 |
| 11 | 15 ± 2 | 4 ± 2 | 23 ± 8 | 0 | 21 | 0 | 0 |
To demonstrate the presumptive antimitotic activity of 6 and 11, based on their antitubulin activities, we analyzed their effects on cell cycle progression in HeLa cells. As shown in Fig. 4, the two compounds caused a significant G2/M arrest in a concentration-dependent manner. In particular, compound 11 was very active, inducing cell cycle arrest at 5 nM, similar to the activity of HTI-286 (2). Compound 6 was less active, inducing a G2/M block only at 50 nM. The increase in the proportion of cells in the G2/M phase was accompanied by a sharp decrease in the proportion of cells in the other phases of the cell cycle.
 | ||
| Fig. 4 Percentage of cells in each phase of the cell cycle in HeLa cells treated with HTI-286 (2) (panel A), 6 (panel B) and 11 (panel C) at the indicated concentrations for 24 h. Cells were fixed and labeled with propidium iodide and analyzed by flow cytometry as described in the Experimental section. | ||
Conclusions
In summary, the preparation of new hemiasterlin derivatives was achieved, in which either the A or the B fragment was alternatively replaced. The procedures exploited multicomponent approaches, applied in three complementary isonitrile-based versions, and were highly valuable for the rapid and convergent synthesis of a small family of analogues. Our multicomponent approach was not previously used in preparing hemiasterlin analogues and allowed us to prepare compounds with unconventional modifications, such as compounds 7–9. Biological evaluation confirmed that we had prepared two cytotoxic molecules, for which tubulin assembly inhibition and ligand binding studies were also performed, with the activity for the two analogues obtained in these assays. The two analogues also caused a G2/M arrest in HeLa cells. We plan to continue our target-oriented synthesis programs, using addition strategies relying on MCRs. Our goal is to replace the multistep generation of sterically hindered amide functions with more reliable multicomponent assembly reactions.Experimental section
General information
All commercial materials (Aldrich, Fluka) were used without further purification. All solvents were of reagent grade or HPLC grade. All reactions were carried out under a nitrogen atmosphere. All reactions were monitored by thin layer chromatography (TLC) on precoated silica gel 60 F254; spots were visualized with UV light or by treatment with a 1% aqueous KMnO4 solution. Products were purified by flash chromatography (FC) on silica gel 60 (230–400 mesh). 1H NMR spectra and 13C NMR spectra were recorded on 300 and 400 MHz spectrometers. Chemical shifts are reported in parts per million relative to the residual solvent. 13C NMR spectra have been recorded using the APT pulse sequence. Multiplicities in 1H NMR are reported as follows: s = singlet, d = doublet, t = triplet, m = multiplet, br s = broad singlet. High-resolution MS spectra were recorded with an FT-ICR (Fourier Transform Ion Cyclotron Resonance) instrument, equipped with an ESI source.:3, n-hexane/DCM); 75% yield; yellow oil; Rf 0.27 (7:3, n-hexane/dichloromethane); 1H NMR (300 MHz, CDCl3) and 13C NMR (75 MHz, CDCl3) in accordance with the literature. HRMS (ESI) calcd for C11H15O2+ [MH]+ 179.1067, found 179.1075.
:3, n-hexane/DCM); 57% yield; oil; Rf 0.2 (1.5:1, n-hexane/dichloromethane); 1H NMR (300 MHz, CDCl3) δ 9.50 (s, 1H), 7.56 (s, 1H), 7.26 (d, J = 8.5 Hz, 1H), 7.18–7.03 (m, 2H), 6.47 (d, J = 2.9 Hz, 1H), 3.75 (s, 3H), 1.53 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 202.7, 135.8, 131.8, 129.5, 128.8, 120.6, 118.8, 109.6, 101.1, 51.0, 33.6, 23.2 (2C); HRMS (ESI) calcd for C13H15NNaO+ [MNa]+ 224.1046, found 224.1054.
:3, n-hexane/DCM); 50% yield; yellow oil; Rf 0.2 (4:1, n-hexane/dichloromethane); 1H NMR (300 MHz, CDCl3) and 13C NMR (75 MHz, CDCl3) in accordance with the literature. HRMS (ESI) calcd for C10H12NaO+ [MNa]+ 171.0780, found 171.0792.
:3, n-hexane/DCM); 46%; oil; Rf 0.2 (1.5:1, n-hexane/dichloromethane); 1H NMR (300 MHz, CDCl3) δ 9.48 (s, br, 1H), 7.55 (d, br, J = 7.7 Hz, 1H), 7.32 (d, J = 7.8 Hz, 1H), 7.24 (t, br, J = 7.8 Hz, 1H), 7.10 (t, J = 7.7 Hz, 1H), 6.96 (s, br, 1H), 3.79 (s, br, 3H), 1.56 (m, br, 6H); 13C NMR (75 MHz, CDCl3) δ 202.3, 137.7, 130.9, 126.2, 121.9, 120.3, 119.4, 115.1, 109.6, 46.5, 32.8, 22.0 (2C); HRMS (ESI) calcd for C13H16NO+ [MH]+ 202.1226, found 202.1234.
:1, n-hexane/ethyl acetate) to give 18a (200 mg, 32%) and 18b (194 mg, 31%). 18a: white amorphous solid; Rf (9:1 n-hexane/ethyl acetate) 0.17; [α]21D = +46.4 (c = 0.1, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.42 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 8.7 Hz, 2H), 5.68 (d, br, J = 7.8 Hz, 1H), 5.47 (s, 1H), 4.30 (dd, J = 8.7, 4.9 Hz, 1H), 3.78 (s, 3 H), 3.69 (s, 3H), 3.26 (s, br, 3H), 1.97 (m, 1H), 1.61 (s, 3H), 1.41 (s, 3H), 0.74 (d, J = 6.8 Hz, 3H), 0.64 (d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 171.6, 168.0, 158.9 (q, J = 34.9 Hz), 158.5, 137.8, 127.5 (2C), 116.5 (q, J = 287.7 Hz), 113.8 (2C), 65.0, 57.1, 55.2, 42.0, 41.7, 33.7, 30.5, 27.5, 25.5, 18.8, 17.4; HRMS (ESI) calcd for C21H29F3N2O5+ [MNa]+ 469.1921, found 469.1919. 18b: white amorphous solid; Rf (9:1 n-hexane/ethyl acetate) 0.18; [α]21D = +26.3 (c = 0.1, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.39 (d, J = 8.7 Hz, 2H), 6.86 (d, J = 8.7 Hz, 2H), 5.82 (d, J = 8.1 Hz, 1H), 5.45 (s, 1H), 4.30 (dd, J = 8.4, 4.7 Hz, 1H), 3.77 (s, 3H), 3.65 (s, 3H), 3.22 (s, 3H) 1.96 (m, 1H), 1.61 (s, 3H), 1.41 (s, 3H), 0.70 (d, J = 6.8 Hz, 3H), 0.69 (d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 171.5, 167.7, 159.4 (q, J = 34.9 Hz), 158.4, 137.5, 127.7 (2C), 116.4 (q, J = 287.7 Hz), 113.8 (2C), 64.8, 57.2, 55.3, 52.2, 41.8, 33.5, 30.7, 27.2, 25.5, 18.7, 17.7; HRMS (ESI) calcd for C21H29F3N2O5+ [MNa]+ 469.1921, found 469.1931.
:1, n-hexane/ethyl acetate) to give 19a (215 mg, 37%) and 19b (221 mg, 38%). 19a: white amorphous solid; Rf (5.7:1 n-hexane/ethyl acetate) 0.17; [α]21D = +53.0 (c = 0.1, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.77 (s, br, 1H), 7.43 (dd, J = 8.8 and 2.0 Hz, 1H), 7.33 (d, br, J = 8.8 Hz, 1H), 7.04 (d, J = 2.9 Hz, 1H), 6.46 (d, br, J = 2.9 Hz, 1H), 5.75 (s, 1H), 5.65 (d, br, J = 7.8 Hz, 1 H), 4.22 (dd, J = 7.8 and 4.9 Hz, 1H), 3.77 (s, 3H), 3.60 (s, 3H), 3.31 (s, br, 3H), 1.83 (m, 1H), 1.72 (s, 3H), 1.45 (s, 3H), 0.59 (d, J = 6.8 Hz, 3H), 0.37 (d, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 171.6, 168.4, 158.4 (q, J = 34.9 Hz), 136.9, 135.5, 129.5, 128.7, 120.0, 119.6, 116.5 (q, J = 287.7 Hz), 109.8, 101.0, 65.6, 57.2, 51.8, 42.2, 34.1, 32.8, 30.2, 28.3, 25.2, 18.7, 17.0; HRMS (ESI) calcd for C23H30F3N3NaO4+ [MNa]+ 492.2081, found 492.2071. 19b: white amorphous solid; Rf (5.7:1 n-hexane/ethyl acetate) 0.18; [α]D21 = +30.2 (c = 0.1, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.69 (s, br, 1H), 7.40 (dd, J = 8.7 and 2.0 Hz, 1H), 7.31 (d, br, J = 8.7 Hz, 1H), 7.03 (d, J = 2.9 Hz, 1H), 6.43 (d, J = 2.9 Hz, 1H), 5.67 (d, br, J = 7.8 Hz, 1H), 5.58 (s, 1H), 4.25 (dd, J = 7.8 and 4.9 Hz, 1H), 3.77 (s, 3H), 3.56 (s, 3H), 3.31 (s, 3H), 1.78 (m, 1H), 1.71 (s, 3H), 1.48 (s, 3H), 0.53 (d, J = 6.8 Hz, 3H), 0.51 (d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 171.3, 167.9, 158.4 (q, J = 34.9 Hz), 136.5, 135.6, 129.4, 128.6, 120.1, 118.4, 116.5 (q, J = 287.7 Hz), 109.4, 101.2, 65.2, 57.2, 51.9, 42.3, 33.6, 32.8, 30.6, 27.9, 25.5, 18.3, 17.5; HRMS (ESI) calcd for C23H30F3N3NaO4+ [MNa]+ 492.2081, found 492.2066.
:1, n-hexane/ethyl acetate) to give 21 (48 mg, 58%). Pale yellow oil; Rf (4:1, n-hexane/ethyl acetate) 0.25; [α]23D = −57.4 (c = 0.12, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 8.8 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 6.61 (dq, br, J = 9.2 and 1.5 Hz, 1H), 6.09 (d, br, J = 8.6 Hz, 1H), 5.44 (s, 1H), 5.01 (dd, J = 10.5 and 9.2 Hz, 1H), 4.52 (dd, J = 8.6 and 6.8 Hz, 1H), 4.18 (q, J = 7.0 Hz, 2H), 3.77 (s, 3H), 3.15 (q, br, J = 1.7 Hz, 3H), 2.88 (s, 3H), 1.93–1.75 (m, br, 2H), 1.85 (d, J = 1.5 Hz, 3H), 1.54 (s, 3H), 1.40 (s, 3H), 1.28 (t, J = 7.0 Hz, 3 H), 0.88 (d, J = 6.6 Hz, 3H), 0.81 (d, J = 6.6 Hz, 3H), 0.78 (d, J = 6.8 Hz, 3H), 0.65 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.3, 167.9, 167.7, 158.4 (q, J = 34.9 Hz), 158.0, 138.2, 137.6, 132.9, 127.5 (2C), 116.6 (q, J = 287.7 Hz), 114.0 (2C), 65.0, 60.9, 56.4, 55.3, 54.0, 41.6, 33.5, 30.8, 30.3, 30.0, 27.3, 26.4, 19.4 (2C), 18.8, 17.3, 14.2, 13.7; HRMS (ESI) calcd for C31H46F3N3NaO6+ [MNa]+ 636.3231, found 636.32423.
:1, n-hexane/ethyl acetate) to give 22 (58 mg, 52%). Pale yellow foam; Rf (3:1, n-hexane/ethyl acetate) 0.28; [α]21D = −78.1 (c = 0.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.80 (s, br, 1H), 7.41 (d, br, J = 8.7 Hz, 1H), 7.34 (d, J = 8.7 Hz, 1H), 7.06 (d, J = 2.9 Hz, 1H), 6.64 (d, br, J = 8.9 Hz, 1H), 6.49 (d, J = 2.9 Hz, 1H), 6.14 (d, J = 8.2 Hz, 1H), 5.71 (s, 1 H), 5.04 (t, J = 9.9 Hz, 1H), 4.46 (t, J = 7.6 Hz, 1H), 4.21 (q, J = 6.7 Hz, 2H), 3.80 (s, 3H), 3.21 (s, 3H), 2.89 (s, 3H), 1.88 (s, 3H), 1.93–1.78 (m, 1H), 1.78–1.64 (m, 1H), 1.69 (s, 3H), 1.50 (s, 3H), 1.82 (t, J = 6.7 Hz, 3H), 0.91 (d, J = 6.5 Hz, 3H), 0.80 (d, J = 6.5 Hz, 3H), 0.72 (d, J = 6.7 Hz, 3H), 0.47 (d, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.1, 168.9, 168.4, 158.9 (q, J = 35.3 Hz), 139.1, 137.4, 136.2, 133.5, 130.1, 129.3, 120.80, 119.1, 117.3 (q, J = 288.2 Hz), 110.3, 102.0, 66.3, 61.5, 57.0, 54.9, 42.7, 34.5, 33.5, 31.3, 30.9, 30.6, 30.6, 28.7, 27.0, 20.0, 19.9, 19.4, 17.9, 14.9; HRMS (ESI) calcd for C33H47F3N4NaO5+ [MNa]+ 659.3391, found 659.3384.
:1, n-hexane/ethyl acetate) to give 7 (73 mg, 51%) as a 1.5:1 inseparable mixture of two diastereoisomers. White foam; Rf 0.38 (1:1.5, n-hexane/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 7.56–7.18 (m, 5H), 6.67 (d, br, J = 9.8 Hz, 1H), 4.21 (q, J = 7.1 Hz, 2H), 3.76 (s, 0.6H), 3.73 (s, 0.4 H), 3.43 (m, 1H), 2.86 (m, 1H), 2.57 (s, 3H), 2.21 (s, 3H), 1.81 (s, 3H), 1.76 (m, 1H), 1.39 (s, 6H), 1.30 (t, J = 7.1 Hz, 3H), 1.22 (m, 6H), 0.91 (m, 3H), 0.84 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 168.3, 159.6, 149.9, 146.0, 140.1, 139.9, 135.0, 131.9, 129.3 (2C), 127.0 (2C), 126.5, 69.7, 67.3, 61.3, 42.6, 40.2, 35.8, 30.9, 26.5, 24.8, 24.3, 21.8 (2C), 19.9 (2C), 14.9; HRMS (ESI) calcd for C28H43N3NaO3+ [MNa]+ 492.3197, found 492.3209.
:3, n-hexane/ethyl acetate) to give 8 (66 mg, 68%) as a 1.5:1 inseparable mixture of two diastereoisomers. White foam; Rf 0.4 (1:1.5, n-hexane/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 7.24 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 6.67 (m, 1H), 4.21 (q, J = 7.1 Hz, 2H), 3.79 (s, 3H), 3.72 (s, 0.6H), 3.69 (s, 0.4H), 3.43 (m, 1H), 2.88 (m, 1H), 2.61 (s, 3H), 2.21 (s, 3H), 1.81 (s, br, 3H), 1.76 (m, 1H), 1.43 (s, 6H), 1.30 (t, J = 7.1 Hz, 3H), 1.19 (m, 6H), 0.97 (m, 3H), 0.85 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 167.6, 159.2, 157.9, 149.6, 139.4, 138.2, 134.3, 130.1, 127.5 (2C), 113.4 (2C), 69.2, 66.7, 60.6, 55.2, 41.4, 39.6, 35.1, 30.3, 26.5, 24.9, 24.3 (2C), 21.8 (2C), 19.9 (2C), 13.1; HRMS (ESI) calcd for C29H45N3NaO4+ [MNa]+ 522.3302, found 522.3317.
:1, n-hexane/ethyl acetate) to give 9 (66 mg, 64%) as a 1:1 inseparable mixture of two diastereoisomers. Thick oil; Rf 0.38 (1:1.5, n-hexane/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 8.2 Hz, 0.5H), 7.83 (d, J = 8.2 Hz, 0.5H), 7.30 (d, J = 8.1 Hz, 1H), 7.22 (t, br, J = 8.1 Hz, 1H), 7.09 (t, br, J = 7.9 Hz, 1H), 6.88 (s, 1H), 6.72 (d, br, J = 9.8 Hz, 0.5H), 6.69 (d, br, J = 9.8 Hz, 0.5H), 4.21 (q, J = 7.1 Hz, 2H), 4.13 (s, 0.5H), 4.11 (s, 0.5H), 3.75 (s, 3H), 3.47 (m, 1H), 2.86 (m, 1H), 2.59 (s, 1.5H), 2.57 (s, 1.5H), 2.14 (s, 3H), 1.92–1.81 (m, 4H), 1.50 (s, 3H), 1.41 (s, 3H), 1.30–1.21 (m, 10H), 0.95 (m, 3H), 0.84 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 168.4, 160.2, 150.0, 140.2, 140.0, 135.1, 134.9, 127.6 (2C), 126.7, 122.7, 121.9, 119.3, 110.8, 68.3, 67.3, 61.3, 40.1, 40.0, 36.0, 33.3, 31.0, 27.7, 27.5, 25.7, 24.5, 22.5, 21.8, 20.1, 20.5, 18.3; HRMS (ESI) calcd for C31H46N4NaO3+ [MNa]+ 545.3462, found 545.3455.
:1, n-hexane/ethyl acetate) to give 23 (0.67 g, 80%). Yellow oil; Rf 0.26 (4:1, n-hexane/ethyl acetate); [α]19D = −91.8 (c 1.1, CH3OH); 1H NMR (300 MHz, CD3OD) δ 6.70 (d, J = 9.2 Hz, 1H), 4.97 (dd, J = 10.0 and 9.2 Hz, 1H), 4.70 (d, J = 5.9 Hz, 1H), 4.20 (q, J = 7.1 Hz, 2H), 2.94 (s, 3H), 2.30 (m, 1H), 1.90 (m, 1H), 1.86 (s, 3H), 1.29 (t, J = 7.1 Hz, 3H), 1.09 (m, 6H), 0.85 (m, 6H); 13C NMR (75 MHz, CD3OD) δ 169.2, 168.1, 159.4, 139.2, 134.5, 62.4, 62.0, 59.3, 32.5, 31.8, 31.4, 19.4–19.3 (3C), 18.5, 14.5, 13.8; HRMS (ESI) calcd for C17H28N2NaO3+ [MNa]+ 331.1992, found 331.2008.
:3 to 1.5:1 gradient, n-hexane/ethyl acetate) to give 27 (843 mg, 46%) as an inseparable 1:1 mixture of diastereoisomers. Yellow oil; Rf 0.29 (7:3, n-hexane/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 6.69–6.59 (m, 1H), 5.96–5.83 (m, 1H), 5.32 (d, br, J = 5.4 Hz, 0.5H), 5.26 (d, br, J = 5.4 Hz, 0.5H), 5.10 (d, m, J = 17.1 Hz, 1H), 5.03 (d, br, J = 10.0 Hz, 1H), 4.50 (dd, J = 5.4 and 3.9 Hz, 0.5H), 4.48 (dd, J = 5.0 and 3.2 Hz, 0.5H), 3.85–3.75 (m, 1H), 3.74 (1.5 H, s), 3.73 (1.5 H, s), 3.17 (dt, J = 13.2 and 3.2 Hz, 0.5H), 3.14 (dt, J = 13.2 and 3.2 Hz, 0.5H), 2.58–2.50 (m, 2H), 2.50–2.41 (m, 2H), 2.33–2.14 (m, 2H), 1.78–1.65 (m, 3H), 1.60–1.42 (m, 2H), 0.96 (d, J = 6.8 Hz, 1.5H), 0.93 (d, J = 6.8 Hz, 1.5H), 0.88 (d, J = 6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.1 and 172.8 (1C), 172.5 and 172. 0 (1C), 171.3, 137.2, 115.4, 57.3, 52.1 and 52.0 (1C), 51.9 and 51.8 (1C), 43.8 and 43.7 (1C), 32.8 and 32.7 (1C), 31.0 and 30.7 (1C), 29.2, 25.5, 25.3 and 25.0 (1C), 20.4 and 20.3 (1C), 19.1, 17.7 and 17.6 (1C); HRMS (ESI) calcd for C17H28N2NaO4+ [MNa]+ 347.1941, found 347.1958.
:1 v/v) and extracted with EtOAc (3 × 20 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo to give a yellow residue that was taken in diethyl ether (10 mL) and washed with a 1 M aqueous solution of HCl (3 mL × 2). The aqueous phase was basified to pH 9 with a saturated aqueous solution of NaHCO3 and extracted with dichloromethane (3 × 10 mL). The combined organic phases were dried over Na2SO4 and concentrated in vacuo to give compound 28 (64 mg, 85%) as an inseparable 1:1 mixture of diastereoisomers. Yellow oil; 1H NMR (300 MHz, CDCl3) δ 7.40–7.30 (m, 1H), 4.51 (t, br, J = 5.8 Hz, 0.5H), 4.49 (t, br, J = 5.4 Hz, 0.5H), 3.70 (s, 3H), 3.40–3.28 (m, 1H), 3.14–3.00 (m, 1H), 2.79–2.65 (m, 1H), 2.65–2.48 (m, 1H), 2.17 (oct, J = 5.9 Hz, 1H), 2.13–1.88 (m, 1H), 1.85–1.69 (m, 1H), 1.63–1.37 (m, 4H), 0.95–0.87 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 173.7 and 173.5 (1C), 172.5 and 172.4 (1C), 60.1 and 60.0 (1C), 59.9 and 59.8 (1C), 56.8 and 56.7 (1C), 45.5, 33.8, 31.9 and 31.20 (1C), 25.5, 23.5 and 22.6 (1C), 19.2 and 19.1 (1C), 18.7 and 18.0 (1C); HRMS (ESI) calcd for C12H22N2NaO3+ [MNa]+ 265.1523, found 265.1510.
:1, n-hexane/ethyl acetate) to give compound 29 (39 mg, 92%) as an inseparable 1:1 mixture of diastereoisomers. Yellow oil; Rf 0.28 (9:1, n-hexane/ethyl acetate); 1H NMR (300 MHz, CDCl3, rotameric mixture of diastereoisomers) δ 6.60 (m, br, 0.5H), 6.47 (m, br, 0.5H), 4.78(m, br, 1H), 4.64 (d, br, J = 7.8 and 3.9 Hz, 1H), 4.28–3.89 (m, 1H), 3.72 (s, 3H), 2.85 (t, br, J = 12.7 Hz, 0.7H), 2.77 (m, br, 0.3H), 2.28 (m, br, 1H), 2.17 (m, 1H), 1.71– 1.32 (m, 5H), 1.48 (s, 3H), 1.47 (s, 6H), 0.93 (d, J = 6.8 Hz, 1.7H), 0.92 (d, J = 6.8 Hz, 1.3H), 0.87 (d, J = 6.8 Hz, 1.5H), 0.86 (d, J = 6.8 Hz, 1.5H); 13C NMR (75 MHz, CDCl3, rotameric mixture of diastereoisomers) δ 172.4 and 172.1 (1C), 171.3, 161.1, 80.6, 56.9, 52.1, 42.4 and 42.1 (1C), 31.30, 30.9, 28.4 (3C), 25.3, 24.9, 20.5, 19.0, 17.7 and 17.5 (1C); HRMS (ESI) calcd for C17H30N2NaO5+ [MNa]+ 365.2047, found 365.2038.
:3, n-hexane/ethyl acetate) to give 10 (12 mg, 62%) as an inseparable 1:1 mixture of diastereoisomers. White amorphous solid; Rf (7:3, n-hexane/ethyl acetate) 0.29; 1H NMR (400 MHz, CDCl3, rotameric mixture of diastereoisomers) δ 6.72–6.59 (m, 1H), 5.12–4.98 (m, 1H), 4.94–4.64 (m, 3H), 4.23 (q, J = 7.1 Hz, 1.2H), 4.22 (q, J = 7.1 Hz, 0.8H), 4.10–3.94 (m, 1H), 3.00 (s, 0.9H), 2.99 (s, 0.3H), 2.98 (s, 0.6H), 2.97 (s, 1.2H), 2.89–2.77 (m, 1H), 2.37–2.20 (m, 1H), 2.08–1.86 (2H), 1.91 (d, J = 1.4 Hz, 0.9H), 1.90 (d, J = 1.4 Hz, 1.3H), 1.89 (d, J = 1.4 Hz, 0.8H), 1.72–1.40 (m, 5H), 1.60 (s, br, 9H), 1.83 (t, J = 7.1 Hz, 1.8H), 1.82 (t, J = 7.1 Hz, 1.2H), 0.97–0.83 (m, 12H); 13C NMR (100 MHz, CDCl3, rotameric mixture of diastereoisomers) δ 172.0 and 171.5 (1C), 171.6 and 171.1 (1C), 167.7, 157.6 and 156.7 (1C), 138.3, 132.9, 80.5, 60.8, 56.9, 56.3, 53.9, 42.6 and 41.2 (1C), 31.2 and 31.1 (1C), 30.4, 30.0, 28.4 (3C), 25.8, 24.9, 20.6, 20.1–17.3 (4C), 14.3, 13.7 and 13.5 (1C); HRMS (ESI) calcd for C27H47N3NaO6+ [MNa]+ 532.3357, found 532.3366.
:1 mixture of diastereoisomers (102 mg, quantitative yield). Colorless oil; 1H NMR (400 MHz, CDCl3, rotameric mixture of diastereoisomers) δ 7.70 (d, br, J = 8.2 Hz, 0.25H), 7.48 (d, br, J = 8.4 Hz, 0.5H), 7.36 (d, J = 8.9 Hz, 0.25H), 6.69–6.71 (m, 1H), 5.15–4.92 (m, 1H), 4.86–4.63 (m, 1H), 4.23 (q, J = 7.1 Hz, 2H), 3.69 (m, br, 0.2H), 3.58 (m, br, 0.8H), 3.36–3.23 (m, 1H), 3.03 (s, 1H), 2.99 (s, 2H), 2.89 (m, 1H), 2.36–2.18 (m, 1H), 2.16–1.97 (m, 2H), 1.95–1.86 (m, 1H), 1.90 (s, br, 3H), 1.81 (m, br, 1H), 1.76–1.60 (m, 3H), 1.52 (m, 1H), 1.33 (t, J = 7.1 Hz, 3H), 1.05–0.82 (m, 12H); 13C NMR (100 MHz, CDCl3, rotameric mixture of diastereoisomers) δ 172.5 and 172.4 (1C), 172.2, 168.4, 138.9 and 138.7 (1C), 133.4, 61.6, 59.6 and 58.8 (1C), 57.8 and 57.5 (1C), 55.1 and 54.9 (1C), 45.3, 31.6 and 31.5 (1C), 31.1, 30.6, 29.4, 25.0 and 24.6 (1C), 23.4 and 23.1 (1C), 20.7–17.7 (4C), 14.9, 14.4; HRMS (ESI) calcd for C22H40N3O4+ [MH]+ 410.3013, found 410.3010.
:1 mixture of diastereoisomers (59 mg, quantitative yield). White amorphous solid; 1H NMR (400 MHz, CDCl3, rotameric mixture of diastereoisomers) δ 7.50–7.22 (m, br, 1H), 6.66 (d, br, J = 9.4 Hz, 0.7H), 6.62 (d, br, J = 9.4 Hz, 0.3H), 5.13–4.99 (m, 1H), 4.80–4.62 (m, 1H), 4.22 (q, J = 7.0 Hz, 1.4H), 4.21 (q, J = 7.0 Hz, 0.6H), 3.22–2.92 (m, 1.5H), 2.99 (s, 1H), 2.98 (s, 1H), 2.97 (s, 0.7H), 2.96 (s, 0.3H), 2.85 (m, br, 0.5H), 2.74 (m, br, 1H), 2.38–2.13 (m, br, 1H), 2.13–1.81 (m, br, 2H), 1.91 (d, br, J = 1.2 Hz, 1.5H), 1.90 (d, br, J = 1.2 Hz, 0.9H), 1.87 (d, br, J = 1.2 Hz, 0.6H), 1.76–1.37 (m, br, 4H), 1.82 (t, J = 7.0 Hz, 2.1H), 1.81 (t, J = 7.0 Hz, 0.9H), 1.24 (m, br, 2H), 1.04–0.79 (m, 18H); 13C NMR (100 MHz, CDCl3, rotameric mixture of diastereoisomers) δ 175.9 and 175.8 (1C), 172.5 and 172.3 (1C), 168.4, 139.1, 133.5 and 133.4 (1C), 65.6 and 64.8 (1C), 61.5, 57.4 and 56.9 (1C), 54.3 and 53.6 (1C), 51.9, 43.2, 31.6, 31.2 and 31.0 (1C), 30.6, 26.0 and 25.5 (1C), 24.2, 23.9, 20.6–18.4 (6C), 14.9, 14.4; HRMS (ESI) calcd for C25H45N3NaO4+ [MNa]+ 474.3302, found 474.3318.
:1 mixture of diastereoisomers (34 mg, quantitative yield). White amorphous solid; 1H NMR (400 MHz, CDCl3, rotameric mixture of diastereoisomers) δ 7.52 (m, br, 0.65H), 7.41 (m, br, 0.35H), 6.68–6.59 (m, 1H), 5.15–4.92 (m, br, 1H), 4.87–4.58 (m, 0.7H), 4.58–4.43 (m, 0.3H), 4.20 (q, J = 7.0 Hz, 1.3H), 4.19 (q, J = 7.0 Hz, 0.7H), 3.66–3.51 (m, 2H), 2.96 (s, 2H), 2.88 (s, 1H), 2.34 (t, J = 6.8 Hz, 2H), 2.07–1.49 (m, 15H), 1.37–1.10 (m, 15H), 0.98–0.76 (m, 6H); 13C NMR (100 MHz, CDCl3, mixture of diastereoisomers) δ 171.7 and 171.2 (1C), 167.8 and 167.2 (2C), 138.5, 132.8, 70.3, 60.8, 56.9 and 56.3 (1C), 53.6, 47.5, 42.0, 35.6 (2C), 31.4 and 30.3 (1C), 29.9, 29.8, 29.7, 27.0 (2C), 25.5, 25.0, 24.2, 19.5 and 19.4 (4C), 14.2, 13.8 and 13.7 (1C); HRMS (ESI) calcd for C28H49N3NaO4+ [MNa]+ 514.3615, found 514.3608.
Biological studies
Notes and references
- (a) H. Eckert, Molecules, 2012, 17, 1074 CrossRef CAS PubMed; (b) B. Ganem, Acc. Chem. Res., 2009, 42, 463 CrossRef CAS PubMed.
- (a) S. S. van Berkel, B. G. M. Bögels, M. A. Wijdeven, B. Westermann and F. P. J. T. Rutjes, Eur. J. Org. Chem., 2012, 3543 CrossRef CAS PubMed; (b) A. Shaabani, A. Maleki, A. Hossein Rezayan and A. Sarvary, Mol. Diversity, 2011, 15, 41 CrossRef CAS PubMed; (c) E. Soleimani and M. Zainali, J. Org. Chem., 2011, 76, 10206 CrossRef PubMed.
- O. Pando, S. Stark, A. Denkert, A. Porzel, R. Preusentanz and L. A. Wessjohann, J. Am. Chem. Soc., 2011, 133, 7692 CrossRef CAS PubMed.
- (a) M. Stucchi, S. Cairati, R. Cetin-Atalay, M. S. Christodoulou, G. Grazioso, G. Pescitelli, A. Silvani, D. C. Yildirim and G. Lesma, Org. Biomol. Chem., 2015, 13, 4993 RSC; (b) G. Lesma, F. Meneghetti, A. Sacchetti, M. Stucchi and A. Silvani, Beilstein J. Org. Chem., 2014, 10, 1383 CrossRef PubMed; (c) A. Silvani, G. Lesma, S. Crippa and V. Vece, Tetrahedron, 2014, 70, 3994 CrossRef CAS PubMed; (d) G. Lesma, R. Cecchi, S. Crippa, P. Giovanelli, F. Meneghetti, M. Musolino, A. Sacchetti and A. Silvani, Org. Biomol. Chem., 2012, 10, 9004 RSC.
- (a) W. R. Gamble, N. A. Durso, R. W. Fuller, C. K. Westergaard, T. R. Johnson, D. L. Sackett, E. Hamel, J. H. Cardellina II and M. R. Boyd, Bioorg. Med. Chem., 1999, 7, 1611 CrossRef CAS; (b) R. Talpir, Y. Benayahu, Y. Kashman, L. Pannell and M. Schleyer, Tetrahedron Lett., 1994, 35, 4453 CrossRef CAS.
- (a) H. J. Anderson, J. E. Coleman, R. J. Andersen and M. Roberge, Cancer Chemother. Pharmacol., 1997, 39, 223 CrossRef CAS; (b) R. Bai, N. A. Durso, D. L. Sackett and E. Hamel, Biochemistry, 1999, 38, 14302 CrossRef CAS PubMed.
- T. A. Dang Thi, C. P. The, Q. A. Ngo, T. H. Vu Thi, T. D. Nguyen, D. T. Doan, C. B. Thi, M. Jean, P. van de Weghe and T. N. Van, Bioorg. Med. Chem. Lett., 2014, 24, 5216 CrossRef PubMed.
- (a) A. Zask, J. Kaplan, S. Musto and F. Loganzo, J. Am. Chem. Soc., 2005, 127, 17667 CrossRef CAS PubMed; (b) A. Zask, G. Birnberg, K. Cheung, J. Kaplan, C. Niu, E. Norton, R. Suayan, A. Yamashita, D. Cole, Z. Tang, G. Krishnamurthy, R. Williamson, G. Khafizova, S. Musto, R. Hernandez, T. Annable, X. Yang, C. Discafani, C. Beyer, L. M. Greenberger, F. Loganzo and S. Ayral-Kaloustian, J. Med. Chem., 2004, 47, 4774 CrossRef CAS PubMed; (c) A. Zask, G. Birnberg, K. Cheung, J. Kaplan, C. Niu, E. Norton, A. Yamashita, C. Beyer, G. Krishnamurthy, L. M. Greenberger, F. Loganzo and S. Ayral-Kaloustian, Bioorg. Med. Chem. Lett., 2004, 14, 4353 CrossRef CAS PubMed.
- (a) B. A. Hadaschik, H. Adomat, L. Fazli, Y. Fradet, R. J. Andersen, M. E. Gleave and A. I. So, Clin. Cancer Res., 2008, 14, 1510 CrossRef CAS PubMed; (b) B. A. Hadaschik, S. Ettinger, R. Sowery, D. Richard, A. Zoubeidi, R. J. Andersen, M. Roberge and M. E. Gleave, Int. J. Cancer, 2008, 122, 2368 CrossRef CAS PubMed; (c) S. Ayral-Kaloustian and A. Zask, Drugs Future, 2005, 30, 254 CrossRef CAS; (d) F. Loganzo, M. Hari, T. Annable, X. Tan, D. B. Morilla, S. Musto, A. Zask, J. Kaplan, A. A. Minnick Jr., M. K. May, S. Ayral-Kaloustian, M. S. Poruchynsky, T. Fojo and L. M. Greenberger, Mol. Cancer Ther., 2004, 3, 1319 CAS; (e) F. Loganzo, C. M. Discafani, T. Annable, C. Beyer, S. Musto, M. Hari, X. Tan, C. Hardy, R. Hernandez, M. Baxter, T. Singanallore, G. Khafizova, M. S. Poruchynsky, T. Fojo, J. A. Nieman, S. Ayral-Kaloustian, A. Zask, R. J. Andersen and L. M. Greenberger, Cancer Res., 2003, 63, 1838 CAS.
- G. Kuznetsov, K. TenDyke, M. J. Towle, H. Cheng, J. Liu, J. P. Marsh, S. E. R. Schiller, M. R. Spyvee, H. Yang, B. M. Seletsky, C. J. Shaffer, V. Marceau, Y. Yao, E. M. Suh, S. Campagna, F. G. Fang, J. J. Kowalczyk and B. A. Littlefield, Mol. Cancer Ther., 2009, 8, 2852 CrossRef CAS PubMed.
- C. M. Rocha-Lima, S. Bayraktar, J. MacIntyre, L. Raez, A. M. Flores, A. Ferrell, E. H. Rubin, E. A. Poplin, A. R. Tan, A. Lucarelli and N. Zojwalla, Cancer, 2012, 4262 CrossRef CAS PubMed.
- (a) G. Lesma, A. Sacchetti, R. Bai, G. Basso, R. Bortolozzi, E. Hamel, A. Silvani, N. Vaiana and G. Viola, Mol. Diversity, 2014, 18, 357 CrossRef CAS PubMed; (b) J. A. Nieman, J. E. Coleman, D. J. Wallace, E. Piers, L. Y. Lim, M. Roberge and R. J. Andersen, J. Nat. Prod., 2003, 66, 183 CrossRef CAS PubMed; (c) R. J. Andersen, J. E. Coleman, E. Piers and D. J. Wallace, Tetrahedron Lett., 1997, 38, 317 CrossRef CAS.
- G. D. Vo and J. F. Hartwig, Angew. Chem., Int. Ed., 2008, 47, 2127 CrossRef CAS PubMed.
- (a) R. Martin and S. L. Buchwald, Org. Lett., 2008, 10, 4561 CrossRef CAS PubMed; (b) R. Martin and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 7236 CrossRef CAS PubMed.
- M. Jean, J. Renault and P. van de Weghe, Tetrahedron Lett., 2009, 50, 6546 CrossRef CAS PubMed.
- J. Zhu, X. Wu and S. J. Danishefsky, Tetrahedron Lett., 2009, 50, 577 CrossRef CAS PubMed.
- D. W. Carney, J. V. Truong and J. K. Sello, J. Org. Chem., 2011, 76, 10279 CrossRef CAS PubMed.
- C. Niu, D. M. Ho, A. Zask and S. Ayral-Kaloustian, Bioorg. Med. Chem. Lett., 2010, 20, 1535 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- E. Vedejs and C. Kongkittingam, J. Org. Chem., 2001, 66, 7355 CrossRef CAS PubMed.
- A. V. Gulevich, A. G. Zhdanko, R. V. A. Orru and V. G. Nenajdenko, Chem. Rev., 2010, 110, 5235 CrossRef CAS PubMed.
- D. Simoni, R. M. Lee, D. E. Durrant, N.-W. Chi, R. Baruchello, R. Rondanin, C. Rullo and P. Marchetti, Bioorg. Med. Chem. Lett., 2010, 20, 3431 CrossRef CAS PubMed.
- (a) N. Elders, R. F. Schmitz, F. J. J. de Kanter, E. Ruijter, M. B. Groen and R. V. A. Orru, J. Org. Chem., 2007, 72, 6135 CrossRef CAS PubMed; (b) E. Schwartz, H. J. Kitto, R. de Gelder, R. J. M. Nolte, A. E. Rowan and J. J. L. M. Cornelissen, J. Mater. Chem., 2007, 17, 1876 RSC.
- T. Wennekes, K. M. Bonger, K. Vogel, R. J. B. J. N. van den Berg, A. Strijland, W. E. Donker-Koopman, J. M. F. G. Aerts, G. A. van der Marel and H. S. Overkleeft, Eur. J. Org. Chem., 2012, 6420 CrossRef CAS PubMed.
- A. Porcheddu, G. Giacomelli and M. Salaris, J. Org. Chem., 2005, 70, 2361 CrossRef CAS PubMed.
- E. Hamel and C. M. Lin, Biochemistry, 1984, 23, 4173 CrossRef CAS.
- E. Hamel, Cell Biochem. Biophys., 2003, 38, 1 CrossRef CAS PubMed.
- R. Bai, T. L. Nguyen, J. C. Burnett, O. Atasoylu, M. H. G. Munro, G. R. Pettit, A. B. Smith III, R. Gussio and E. Hamel, J. Chem. Inf. Model., 2011, 51, 1393 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1062247. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob01882j |
| This journal is © The Royal Society of Chemistry 2015 |