
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A rapid and efficient one-pot method for the reduction of N-protected α-amino acids to chiral α-amino aldehydes using CDI/DIBAL-H†
Jakov
Ivkovic
,
Christian
Lembacher-Fadum
and
Rolf
Breinbauer
*
Institute of Organic Chemistry, Graz University of Technology, 8010 Graz, Austria. E-mail: breinbauer@tugraz.at
First published on 25th September 2015
Abstract
N-Protected amino acids can be easily converted into chiral α-amino aldehydes in a one-pot reaction by activation with CDI followed by reduction with DIBAL-H. This method delivers Boc-, Cbz- and Fmoc-protected amino aldehydes from proteinogenic amino acids in very good isolated yields and complete stereointegrity.
Introduction
Chiral N-protected α-amino aldehydes are tremendously valuable building blocks,1 which have seen numerous applications in the synthesis of biologically active molecules.2 Typically their synthesis starts from the chiral pool with readily accessible N-protected amino acids following two distinct routes (Scheme 1).3 In the first (Route A), the amino acid A is converted into an activated carboxylic acid derivative C, such as an ester4 or a Weinreb amide,5 and then directly reduced to the corresponding aldehyde B. The second (Route B) starts from the same N-protected amino acid A, which is first fully reduced to the corresponding amino alcohol D and selectively reoxidized to the desired α-amino aldehyde B.6 The major challenge for both routes is the intrinsic lability of the stereogenic centre of α-amino aldehydes, which is prone to epimerization especially in the presence of acid or base.7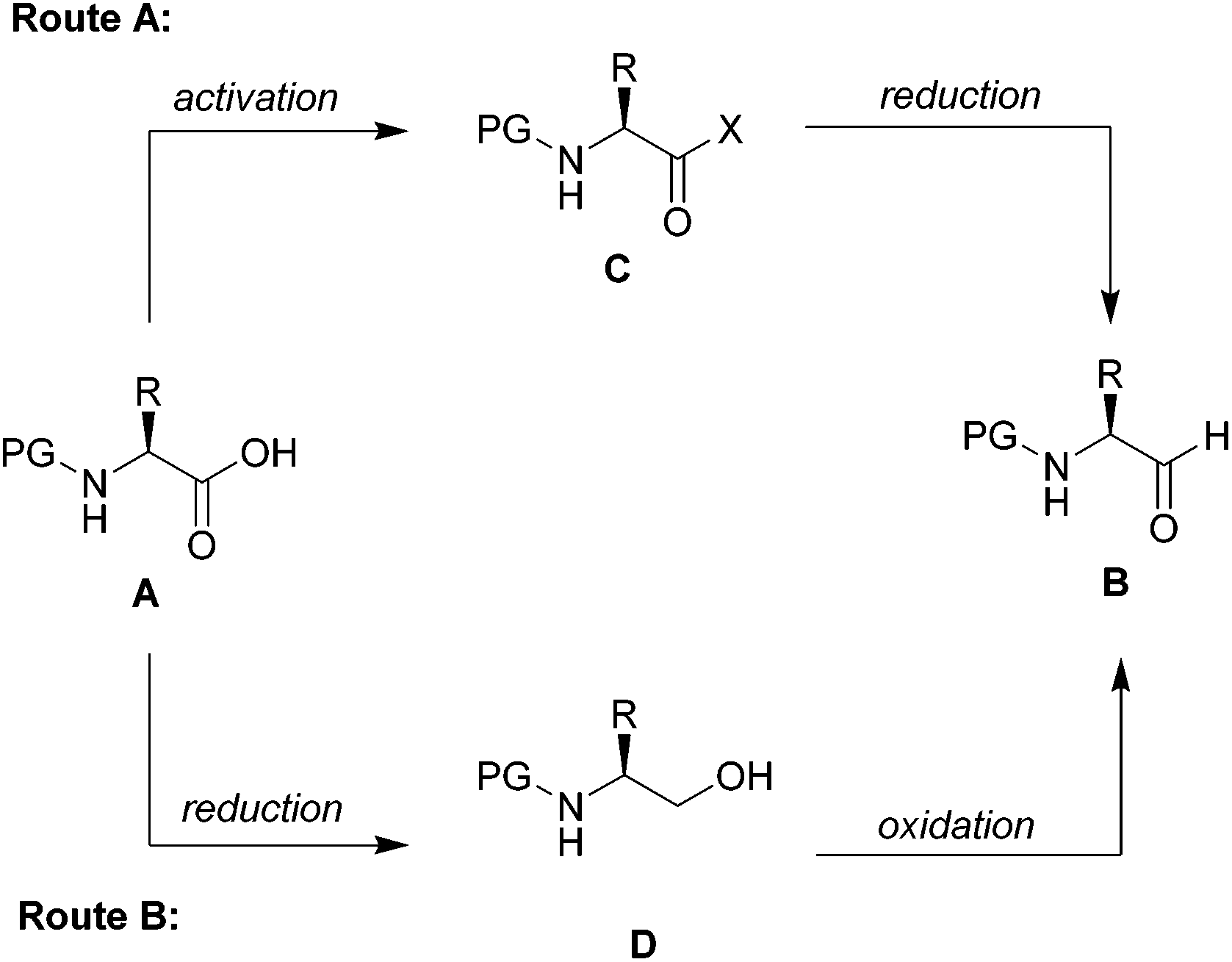 | ||
| Scheme 1 Two distinct routes for the synthesis of α-amino aldehydes and conversion of amino acids to amino aldehydes using CDI/DIBAL-H. | ||
In the course of an ongoing project in our group to design and synthesize transition state mimetics as inhibitors for a peptidase target protein, we required a scalable access to Boc-valinal. In our first attempt we synthesized the aldehyde following the procedure of Morwick by converting Boc-valine into the corresponding Weinreb amide using activation by Staab's reagent (CDI, 1,1′-carbonyldiimidazole).8 The Weinreb amide was isolated and converted to Boc-valinal by reduction with LiAlH4 in 85% yield under complete retention of stereoconfiguration. While this two-step method delivered the desired product, we regarded the necessity to isolate the intermediate Weinreb-amide as a time-consuming nuisance, and reasoned that this step could be avoided if the intermediate acyl imidazolide9 would serve as a substrate for the DIBAL-H reaction. Stammer et al. have already reported in 1979 such a method,10 which despite its apparent attractiveness has seen little application,11 very likely because in the original paper optical rotation studies, which have been pursued for only one product in detail (Cbz-leucinal), indicated that this product was produced in only 60% ee. Despite this caveat we were encouraged by this literature precedence and can now report that through optimization of reaction parameters the one-pot production and reduction of acyl imidazolides provides an attractive rapid and efficient access to chiral α-amino aldehydes.
Results and discussion
As a test substrate we chose Boc-valine to establish a suitable protocol for the CDI/DIBAL-H method. After considerable optimization in which we varied solvent, temperature, reaction time and workup conditions in comparison to the literature precedence of Stammer,10a we arrived at the following protocol in which a solution of N-protected amino acid in DCM was treated with 1.1 eq. of CDI at 0 °C for 60 min. Subsequently 2.1 eq. of DIBAL-H were added dropwise to the resulting solution at −78 °C (Scheme 2). In the course of our studies we recognized that the outcome of this process depended on the quality of the used CDI and we recommend the use of CDI recrystallized from dry THF,12 which can be stored in nitrogen or argon atmosphere at 4 °C for at least 8 weeks. Special attention had to be paid for the workup of the DIBAL-H reduction reaction. In order to avoid basic conditions and prolonged workup, which could cause loss of chiral integrity of the resulting amino aldehyde,7 we have devised a quick method of quenching and aluminium complexation using a solution of tartaric acid instead of the commonly used Rochelle-salt solution.13 This method provided slightly acidic quenching conditions, and dramatically shortened the dissolution times of aluminium salts to less than 20 min even on multigram scale reactions compared to 2 h when using Rochelle-salt. We were pleased to see that by this extractive workup we could isolate Boc-valinal (3a) already in pure form as judged by NMR and chiral GC, thereby avoiding purification via flash chromatography on silica.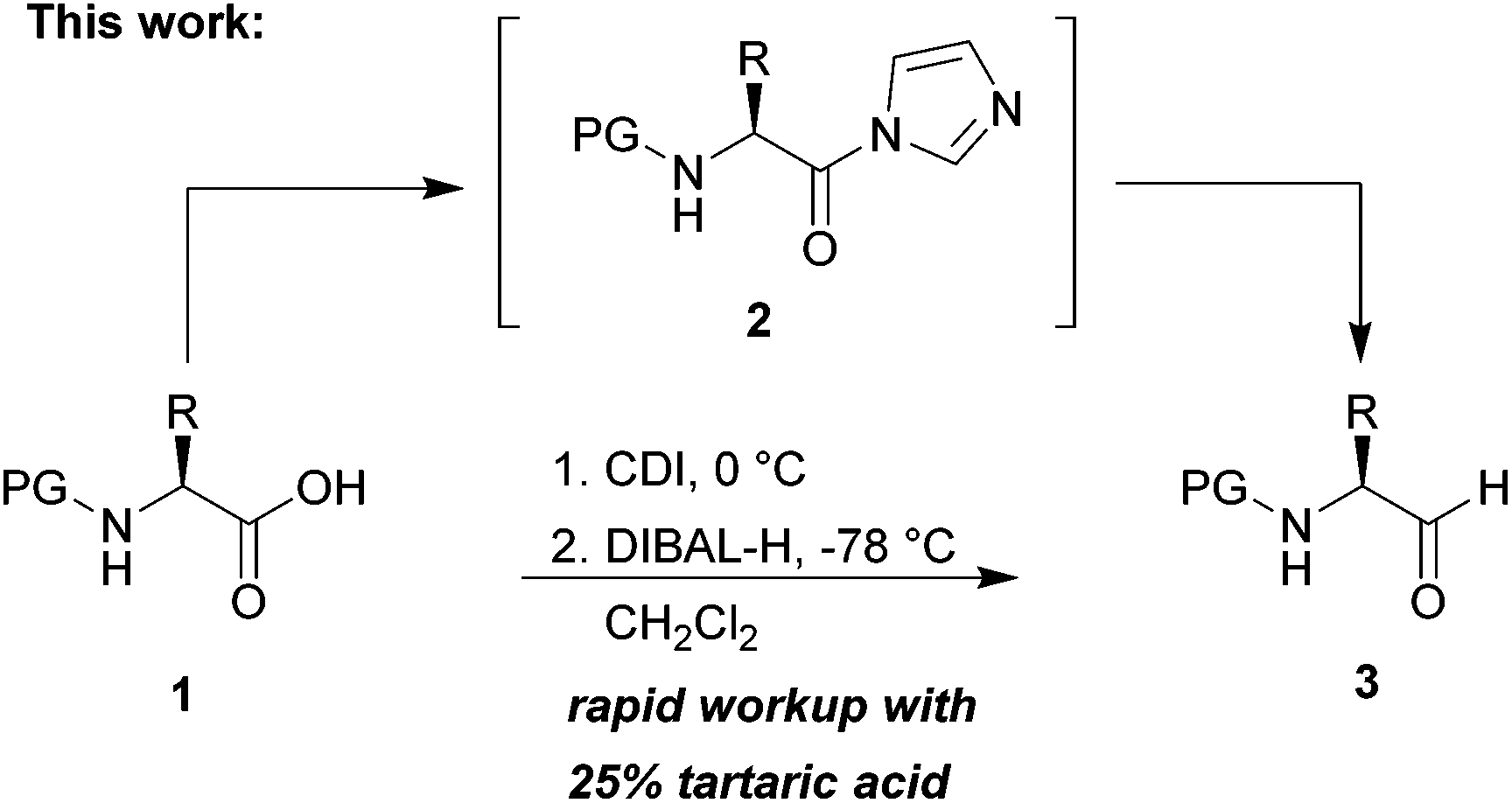 | ||
| Scheme 2 One-pot synthesis of α-amino aldehydes using CDI and DIBAL-H. | ||
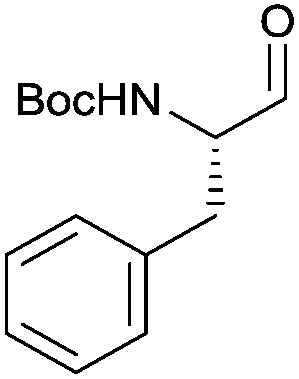
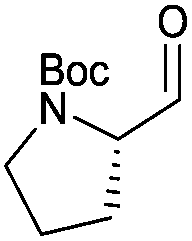
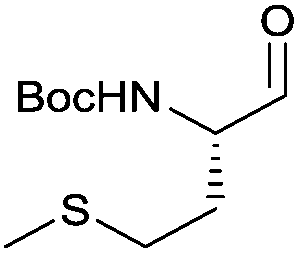
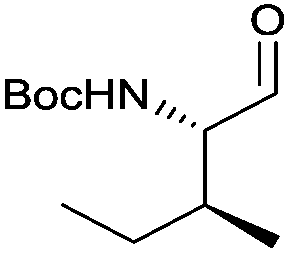
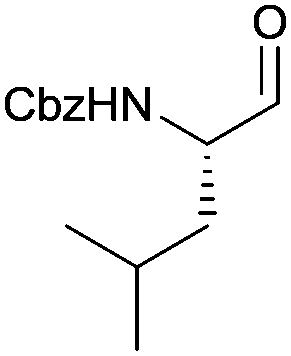
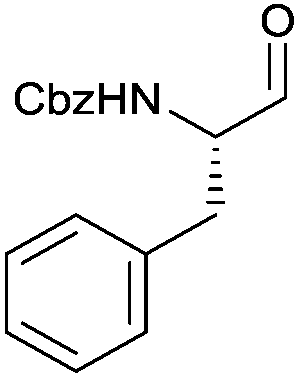
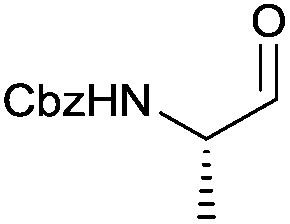
With the optimized protocol in hands, a selection of proteinogenic amino acids with different N-protecting groups was converted to the corresponding aldehydes in excellent yields and high purity based on NMR and gas chromatography analysis (Table 1). The data on enantiomeric purity were determined by gas chromatography using prepared reference racemic samples, while for substrates which could not be separated via chiral GC optical rotations are reported in the ESI.† Boc-valine was converted to the corresponding aldehyde in 84% yield and >99% ee on a 10 gram-scale (entry 1), as well as other Boc-protected amino acids (entries 2–6) were obtained in excellent yields. An informative test substrate for our method was phenylalanine, known as the most racemisation-prone proteinogenic amino acid in peptide synthesis.14 In the event Boc-phenylalaninal (3c) was produced with the CDI/DIBAL-H method in >99% ee as judged by chiral GC (entry 3), and Cbz-phenylalaninal (3i) with >97% ee measured by chiral HPLC after reduction of the isolated aldehyde with NaBH4 (entry 9).15 This result allows us to expect that our method can generally cover the conversion of proteinogenic amino acids to chiral amino aldehydes without racemization (entries 4–6 and 10–11) – even in the cases in which no suitable ee-determination method with chiral GC or HPLC is currently available. In order to compare our improved protocol with the original one by Stammer10a we investigated the reduction of Cbz-leucine to Cbz-leucinal (3h) (entry 8) and could determine >98% ee via chiral GC (compared to the reported 60% ee in their publication, which had been determined via optical rotation). This confirms that by our modifications the one-pot strategy of CDI-activation/DIBAL-H reduction could be developed into a feasible and useful method for the synthesis of N-protected α-amino aldehydes.
| Entry | Product | Yield | ee | |
|---|---|---|---|---|
| a Chiral-GC-FID based measurement. b Chiral HPLC-based measurement. c Diastereomeric excess. d Isolated by flash chromatography on silica due to lower purity of the crude material, to determine the abundance of the desired aldehyde. e 3.35 eq. DIBAL-H used. f 4.00 eq. DIBAL-H used. g 83% ee obtained by adding 0.5 eq. CuCl2 during the activation step, and 72% ee when no additive was used under the same conditions. | ||||
| 1 |
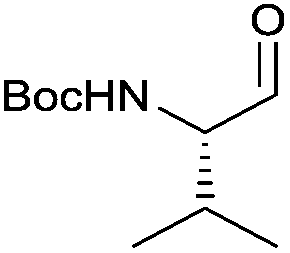
|
3a | 84% | >99%a |
| 2 |
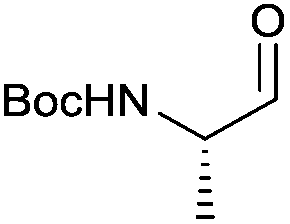
|
3b | 87% | — |
| 3 |
|
3c | 96% | >99%a |
| 4 |
|
3d | 97% | >99%b |
| 5 |
|
3e | 92% | >99%a |
| 6 |
|
3f | 91% | >99%a,c |
| 7 |
|
3g | 62%d | — |
| 8 |
|
3h | 87% | >98%a |
| 9 |
|
3i | 99% | >97%b |
| 10 |
|
3j | 94% | >99%b |
| 11 |
|
3k | 72%d,e | >99%b |
| 12 |
|
3l | 52%d,f | — |
| 13 |
|
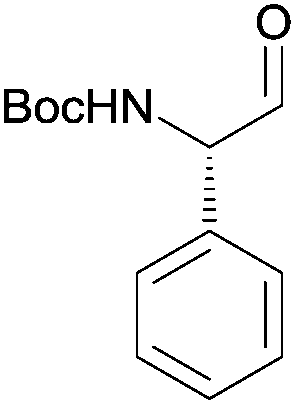
3m | 88% | 83% (72%)a,g |
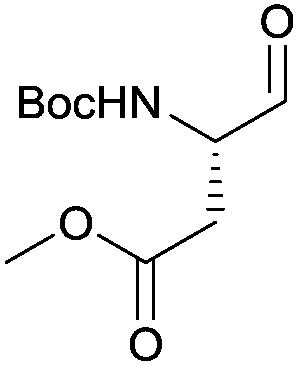
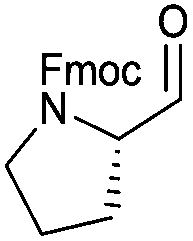
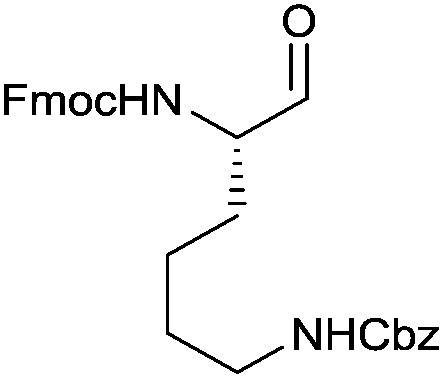
In terms of functional groups in the side chain, we could notice that this method reduces acyl imidazolide significantly faster than methyl ester, as exemplified for protected aspartate aldehyde 3g (entry 7). Notably Fmoc-protection was orthogonal to the reaction conditions, although at least one more equivalent of DIBAL-H had to be added to complete the conversion to the aldehydes 3k and 3l (entries 11–12).
After having established the scope of this robust protocol for the conversion of proteinogenic amino acids, we wanted to check the limitations of our method and apply it to phenylglycine – a very challenging substrate, which racemizes very easily and for which only few stereoselective transformations are known.16 Myers et al. have successfully accessed Fmoc-phenylglycinal via oxidation of Fmoc-phenyglycinol with Dess–Martin-periodinane,17 while Wroblewski and Piotrowska demonstrated the same with Boc- and Bz-phenylglycine.18 When we tested our CDI/DIBAL-H method for Boc-Phg we received the product in good 88% yield, but disappointing enantiopurity of 72% ee. We rationalized that the second equivalent of DIBAL-H is necessary to complete the conversion of the acyl imidazolide intermediate due to the presence of one equivalent of imidazole byproduct from the activation step. Thus imidazole deprotonated by the hydride reagent is thought to be a base contributing to racemization of the product. We hypothesized that by addition of complexing metal salts we could scavenge deprotonated imidazole. Copper(II) salts were found to produce complexes with imidazole in DCM (in 60 min vs. >24 h in case of nickel salts) based on visual observation.19 Consequently, we performed a series of experiments adding copper(II) chloride as a scavenging agent additive after CDI activation and prior to the addition of DIBAL-H. The addition of 0.5 eq. CuCl2 enhanced the ee of produced Boc-phenylglycinal (3m) in each experiment, in contrast to parallel experiments with no additives (Fig. 1). Adding more than 0.5 eq. of CuCl2 did not further increase the enantiomeric purity. Additional factors that improve ee of the resulting Boc-phenylglycinal are precise rate of addition of DIBAL-H and temperatures lower than −50 °C, which are also beneficial for the yield.20
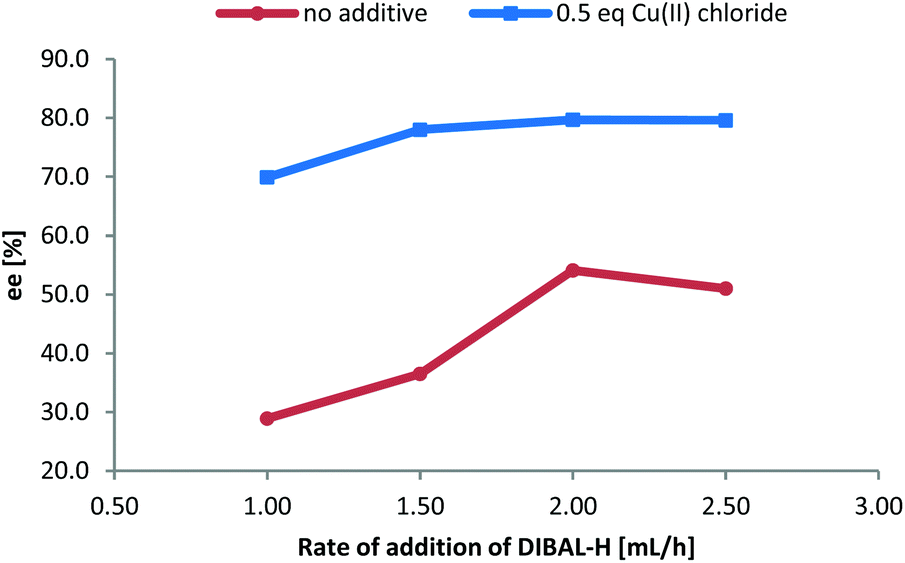 | ||
| Fig. 1 Dependence of ee of Boc-phenylglycinal on the rate of addition of DIBAL-H and enhancement of ee with CuCl2. | ||
Another test case which could define the limits of our method, was the synthesis of peptide aldehydes,21 in particular with the epimerization prone Phe at the C-terminus.14 From the corresponding dipeptide acids, the CDI/DIBAL-H procedure furnished two epimeric aldehydes, 4a and 4b, in good yields and purities (Table 2). In these cases increased amounts of DIBAL-H (3.1–4.5 eq. in total) were necessary to complete the conversion, accompanied by lower reaction concentrations to avoid gelation that occurred otherwise. A similar degree of epimerization was found for both peptide aldehydes (entries 1–2). We reasoned that epimerization occurs already at the activation step with CDI via an oxazolone intermediate, according to a mechanism well known in peptide coupling.22 Consequently, we attempted to suppress epimerization by using additives developed for peptide coupling14,23 and mild acidic buffering with pyridinium p-toluenesulfonate during the activation step (entries 4–8). Among these only the addition of 1.0 eq. of HOBt or HOAt led to an increase in diastereoselecitivity to 90% de, which still reflects the limitation of this method that the rather slow formation of peptide-imidazolides at lower temperatures is accompanied by epimerization presumably via oxazolone formation.
| Entry | Product | Additive | DIBAL-H | Yield | de |
|---|---|---|---|---|---|
| Due to instability of aldehydes in HPLC conditions, de was measured after reduction15 to the corresponding alcohols, which were readily separated by reverse phase HPLC; PPTS: pyridinium p-toluenesulfonate; HOBt: 1-hydroxybenzotriazole; HOAt: 1-hydroxy-7-azabenzotriazole. | |||||
| 1 | Boc-Val-L-Phe-H (4a) | — | 3.1 eq. | 89% | 79% |
| 2 | Boc-Val-D-Phe-H (4b) | — | 3.1 eq. | 95% | 77% |
| 3 | 4b | CuCl2, 1.0 eq. | 4.0 eq. | 52% | 81% |
| 4 | 4b | PPTS, 1.0 eq. | 3.5 eq. | 99% | 69% |
| 5 | 4b | HOBt, 1.0 eq. | 4.5 eq. | 93% | 90% |
| 6 | 4b | HOBt, 1.5 eq. | 4.5 eq. | Quant. | 79% |
| 7 | 4b | HOBt, 1.5 eq., 4 Å mol. sieves | 4.5 eq. | 84% | 85% |
| 8 | 4b | HOAt, 1.0 eq. | 4.5 eq. | Quant. | 90% |
Conclusions
In summary, we have presented an efficient one-pot method for the conversion of N-protected amino acids into chiral N-protected α-amino aldehydes by in situ activation with CDI followed by reduction with DIBAL-H. The advantages of this method compared to established two-step protocols are (1) its operational simplicity, (2) the use of inexpensive reagents, (3) the simple extractive workup and (4) its short overall processing time (typically less than 4 hours) to deliver the product in high purity. While the presented method is excellent for proteinogenic amino acids leading to good yields and preserved stereointegrity, it has its limitations in phenylglycine and dipeptides where epimerization was observed.Acknowledgements
This work was supported by the doctoral program W901-B05 DK Molecular Enzymology, funded by the Austrian Science Fund (FWF) and by NAWI Graz.References
- Reviews: (a) R. Hili, S. Baktharaman and A. K. Yudin, Eur. J. Org. Chem., 2008, 5201–5203 CrossRef CAS PubMed; (b) D. Gryko, J. Chalko and J. Jurczak, Chirality, 2003, 15, 514–541 CrossRef CAS PubMed; (c) M. T. Reetz, Chem. Rev., 1999, 99, 1121–1162 CrossRef CAS PubMed; (d) J. Jurczak and A. Golebiowski, Chem. Rev., 1989, 89, 149–164 CrossRef CAS.
- Selected examples: (a) A. Fässler, G. Bold and H. Steiner, Tetrahedron Lett., 1998, 39, 4925–4928 CrossRef; (b) B. E. Maryanoff, M. N. Greco, H.-C. Zhang, P. Andrade-Gordon, J. A. Kauffman, K. C. Nicolaou, A. Liu and P. H. Brungs, J. Am. Chem. Soc., 1995, 117, 1225–1239 CrossRef CAS.
- As an alternative the asymmetric synthesis of α-amino aldehydes has been described. See for example: (a) G. Bringmann and J.-P. Geisler, Synthesis, 1989, 608–610 CrossRef CAS; (b) F. A. Davis, T. Ramachandar, J. Chai and E. Skucas, Tetrahedron Lett., 2006, 47, 2743–2746 CrossRef CAS PubMed.
- (a) T. Moriwake, S.-i. Hamano, S. Saito and S. Torii, Chem. Lett., 1987, 2085–2088 CrossRef CAS; (b) J. R. Luly, J. F. Dellaria, J. J. Plattner, J. L. Soderquist and N. Yi, J. Org. Chem., 1987, 52, 1487–1492 CrossRef CAS; (c) A. Golebiowski, U. Jacobsson and J. Jurczak, Tetrahedron, 1987, 43, 3063–3066 CrossRef CAS; (d) J. R. Luly, C.-N. Hsiao, N. BaMaung and J. J. Plattner, J. Org. Chem., 1988, 53, 6109–6112 CrossRef CAS; (e) M. Falorni, G. Giacomelli, A. Porcheddu and M. Taddei, J. Org. Chem., 1999, 64, 8962–8964 CrossRef CAS; (f) R. Hili and A. K. Yudin, J. Am. Chem. Soc., 2006, 128, 14772–14773 CrossRef CAS PubMed; (g) F. Coelho, G. Diaz, C. A. M. Abella and W. P. Almeida, Synlett, 2006, 435–439 CrossRef CAS; (h) B. Soto-Cairoli, J. J. de Pomar and J. A. Soderquist, Org. Lett., 2008, 10, 333–336 CrossRef CAS PubMed.
- (a) J.-A. Fehrentz and B. Castro, Synthesis, 1983, 676–678 CrossRef CAS; (b) T. Yasuma, S. Oi, N. Choh, T. Nomura, N. Furuyama, A. Nishimura, Y. Fujisawa and T. Sohda, J. Med. Chem., 1998, 41, 4301–4308 CrossRef CAS PubMed.
- (a) Pyridine*SO3: Y. Hamada and T. Shiori, Chem. Pharm. Bull., 1982, 30, 1921–1924 CrossRef CAS; (b) DMSO, (COCl)2: A. W. Konradi, S. J. Kemp and S. F. Pedersen, J. Am. Chem. Soc., 1994, 116, 1316–1323 CrossRef CAS; (c) TEMPO: J. Jurczak, D. Gryko, E. Kobrycka, H. Gruza and P. Prokopowicz, Tetrahedron, 1998, 54, 6051–6064 CrossRef CAS; (d) MnO2: M. E. Sergeev, V. B. Pronin and T. L. Voyushina, Synlett, 2005, 2802–2804 CrossRef CAS; (e) IBX: M. Ocejo, J. L. Vicario, D. Badia, L. Carrillo and E. Reyes, Synlett, 2005, 2110–2112 CAS; (f) C.-K. Jung and M. J. Krische, J. Am. Chem. Soc., 2006, 128, 17051–17056 CrossRef CAS PubMed; (g) NaIO4 cleavage of diol substrate: R. P. Sharma, M. G. Gore and M. Akhtar, J. Chem. Soc., Chem. Commun., 1979, 875–877 RSC.
- (a) A. Ito, R. Takahashi and Y. Baba, Chem. Pharm. Bull., 1975, 23, 3081–3087 CrossRef CAS; (b) K. E. Rittle, C. F. Homnick, G. S. Ponitcello and B. E. Evans, J. Org. Chem., 1987, 52, 1487–1492 CrossRef.
- T. Morwick, M. Hrapchak, M. De Turi and S. Campbell, Org. Lett., 2002, 4, 2665–2668 CrossRef CAS PubMed.
- H. A. Staab, H. Bauer and K. M. Schneider, Azolides in Organic Synthesis and Biochemistry, Wiley-VCH, Weinheim, 2002 Search PubMed.
- (a) H. Khatri and C. H. Stammer, J. Chem. Soc., Chem. Commun., 1979, 79–80 RSC; (b) For an earlier report of reduction of an acyl imidazolide with LiAlH4 but without reported stereoselectivity, see: B. Shimizu, A. Saito, A. Ito, K. Tokawa, K. Maeda and H. Umezawa, J. Antibiot., 1972, 25, 515–523 CrossRef CAS.
- (a) K. N. Khatri, C. H. Stammer, M. M. Bradford and R. A. McRorie, Biochem. Biophys. Res. Commun., 1980, 96, 163–167 CrossRef; (b) B. Austen, S. Cliffe, D. Grant and J. Hermon-Taylor, WO84/04301, 1984 Search PubMed; (c) B. E. Maryanoff, M. N. Greco, H. Zhang, P. Andrade-Gordon, J. A. Kauffman, K. C. Nicolaou, A. Liu and P.H. Brungs, J. Am. Chem. Soc., 1995, 117, 1225–1239 CrossRef CAS.
- (a) H. A. Staab and K. Wendel, Org. Synth., 1968, 48, 44 CrossRef CAS; (b) K. M. Engstrom, A. Sheikh, R. Ho and R. W. Miller, Org. Process Res. Dev., 2014, 18, 488–494 CrossRef CAS.
- Examples of DIBAL-H quenching procedures involving Rochelle-salt from recent total syntheses: (a) O. Krebs and R. J. K. Taylor, Org. Lett., 2005, 7, 1063–1066 CrossRef CAS PubMed; (b) K. Ishigai, H. Fuwa, K. Hashizume, R. Fukazawa, Y. Cho, M. Yotsu-Yamashita and M. Sasaki, Chem. – Eur. J., 2013, 19, 5276–5288 CrossRef CAS PubMed; (c) A. Arlt, S. Benson, S. Schulthoff, B. Gabor and A. Fürstner, Chem. – Eur. J., 2013, 19, 3596–3608 CrossRef CAS PubMed.
- (a) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed; (b) H. Schröder, G. A. Strohmeier, M. Leypold, T. Nuijens, P. J. M. Quaedflieg and R. Breinbauer, Adv. Synth. Catal., 2013, 355, 1799–1807 CrossRef PubMed; (c) Y. E. Jad, G. A. Costa, S. N. Khattab, B. G. de la Torre, T. Govender, H. G. Kruger, A. El-Faham and F. Albericio, Org. Biomol. Chem., 2015, 13, 2393–2398 RSC; (d) M. Kunishima, A. Kitao, C. Kawachi, Y. Watanabe, S. Iguchi, K. Hioki and S. Tani, Chem. Pharm. Bull., 2002, 50, 549–550 CrossRef CAS; (e) H. Hibino and Y. Nishiuchi, Tetrahedron Lett., 2011, 52, 4947–4949 CrossRef CAS PubMed; (f) M. A. Elsawy, C. Hewage and B. Walker, J. Pept. Sci., 2012, 18, 302–311 CrossRef CAS PubMed.
- A. Dondoni, D. Perrone and T. Semola, Synthesis, 1995, 181–186 CrossRef CAS.
- For a notable exception in peptide synthesis, see: S. Popovic, H. Bieräugel, R. J. Detz, A. M. Kluwer, J. A. A. Koole, D. E. Streefkerk, H. Hiemstra and J. H. van Maarseveen, Chem. – Eur. J., 2013, 19, 16934–16937 CrossRef CAS PubMed.
- (a) A. G. Myers, B. Zhong, M. Movassaghi, D. W. Kung, B. A. Lanman and S. Kwon, Tetrahedron Lett., 2000, 41, 1359–1362 CrossRef CAS; (b) A. G. Myers, B. Zhong, M. Movassaghi, D. W. Kung, B. A. Lanman and S. Kwon, Tetrahedron Lett., 2000, 41, 1359–1362 CrossRef CAS.
- (a) A. E. Wroblewski and D. G. Piotrowska, Tetrahedron: Asymmetry, 2002, 13, 2509–2512 CrossRef CAS; (b) Earlier report about Boc-phenylglycinal: J.-N. Denis, A. Correa and A. E. Greene, J. Org. Chem., 1991, 56, 6939–6942 CrossRef CAS.
- (a) W. Wu, J. Xie and Y. Xuan, J. Chem. Res., 2008, 344–346 CrossRef CAS; (b) W. Wu, Z. Kou, J. Xie and D. Xie, Russ. J. Inorg. Chem., 2010, 55, 384–389 CrossRef.
- For more information see Fig. S2–S5 in the ESI.†.
- Review: A. Moulin, J. Martinez and J. Fehrentz, J. Pept. Sci., 2007, 13, 1–15 CrossRef CAS PubMed.
- M. Bodanszky and A. Bodanszky, Chem. Commun., 1967, 591–593 RSC.
- M. G. Ryadnov, L. V. Klimenko and Y. V. Mitin, J. Pept. Res., 1999, 53, 322–328 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob01838b |
| This journal is © The Royal Society of Chemistry 2015 |